
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and magnetic characterization of Ni(II), Co(II), and Fe(II) binuclear complexes on a bis(pyridyl-triazolyl)alkane basis†
Alexey
Gusev
*a,
Ivan
Nemec
 bc,
Radovan
Herchel
b,
Irina
Riush
a,
Ján
Titiš
d,
Roman
Boča
d,
Konstantin
Lyssenko
e,
Mikhail
Kiskin
f,
Igor
Eremenko
ef and
Wolfgang
Linert
*g
bc,
Radovan
Herchel
b,
Irina
Riush
a,
Ján
Titiš
d,
Roman
Boča
d,
Konstantin
Lyssenko
e,
Mikhail
Kiskin
f,
Igor
Eremenko
ef and
Wolfgang
Linert
*g
aCrimean Federal University, Simferopol, 295007, Crimea, Russian Federation
bDepartment of Inorganic Chemistry, Faculty of Science, Palacký University, 77147 Olomouc, Czech Republic
cCentral European Institute of Technology, CEITEC BUT, Technická 3058/10, Brno, Czech Republic
dDepartment of Chemistry, FPV, University of SS Cyril and Methodius, 91701 Trnava, Slovakia
eA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 119991 Moscow, Russian Federation
fN.S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Moscow, 119991, Russian Federation
gInstitute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9/163-AC, A-1060 Vienna, Austria. E-mail: wolfgang.linert@tuwien.ac.at
First published on 24th June 2019
Abstract
Reactions of bis[5-(2-pyridyl)-1,2,4-triazol-3-yl]alkanes (alkane spacers = –CH2– in L2, –C3H6– in L3, –C4H8– in L4) with M(II)A2 salts (M = Ni, Co, Fe) resulted in the preparation of five series of mononuclear ([M(L2)(H2O)2]2+, 1a–c) or binuclear ([M2(L3)2(H2O)4]4+, 2a–c; ([M2(L4)2(H2O)4]4+, 3a–c, [M2(L3)2(μ-ox)]2+, 4a–c; [M2(L4)2(μ-ox)]2+, 5a–c) complexes. The crystal structures of ten complexes were determined by single-crystal X-ray crystallography. Magnetic properties of the compounds were characterized by SQUID magnetometry and were analyzed by fitting on a spin Hamiltonian model. It was revealed that Fe(II) and Co(II) compounds exhibit non-negligible anisotropy and in the case of 2a–c and 3a–c complexes weak ferromagnetic interactions between the metal centers were observed. In the case of complexes containing an {M2(μ-ox)}2+ core strong antiferromagnetic interactions were observed within the dimer. Remarkably, solid state luminescence of Co(II) and Fe(II) complexes (1b, 2b, 3b and 1c, 2c, 3c) was observed.
Introduction
The assembly of polynuclear coordination cages, from a combination of metal ions and relatively simple bridging ligands, has fascinated coordination chemists more over the last two decades.1 These assemblies have elegantly demonstrated how the formation of architecturally complex systems is directed by the interplay between simple parameters such as the stereo-electronic preference of the metal ions and the disposition of the binding sites in the ligand. Establishment of design principles is a reliable starting point for the construction of complex metal–organic assemblies.2 Despite the recent development in strategies of self-assembly of polynuclear cage-structures, the basic question of the predictability of the architecture of self-organization products remains open.3 The main reason is the large number of factors that directly or indirectly affect the mechanism of self-assembly and this results in changes of the complex topology. Partly a question of predictability for complex structures has been settled for clusters on a rigid ligand basis. Polycarboxylate and polypyridyl ligands have appeared as very appealing tectons for the design of a great diversity of transition metal compounds with unusual topology and properties.4 A much more complicated and unpredictable situation develops when ligands with flexible spacers are used in the synthesis. Serendipity continues to play a major role in many examples of self-assembly for such systems. The systematic studies of self-assembled complexes based on flexible polypyridyl ligands, by Lehn and co-workers5 and other groups6,7 allowed determining the strategy of synthesis of such compounds and stimulated much further research. Therefore, Ward and coworkers7 have reported an extensive series of high-nuclearity cages based on the self-assembly of metal cations with edge-bridging (ditopic) or face-capping (tritopic) bridging ligands containing two or three pyrazolylpyridine units. Except for relatively simple mononuclear complexes, an impressive number of polyhedral shapes of polynuclear complexes, e.g. M4L6 tetrahedra, M6L9 trigonal prisms, M8L12 cubes and ‘cuneane’, M12L18 truncated tetrahedra, and M16L24 tetra-capped truncated tetrahedra, have been described.7 In our previous studies, we described a new type of bis-bidentate bridging ligands on a pyridyl-triazole basis,8 which is structurally related to the abovementioned pyrazolyl ligands of Ward's group. We have demonstrated several unusual complexes on a bistriazole basis with a monomethylene linker and in its absence thereof. The coordination chemistry of ligands in which triazolyl-pyridine chelating arms bind by a long chain is less studied. A few examples of 3d-metal complexes with a similar type of ligand indicate that the formation of mononuclear compounds is most probable.8bIn order to continue our research in this paper we report on the crystal structure and magnetic properties of novel complexes Fe(II), Co(II) and Ni(II) containing [5-(2-pyridine-2-yl)-1,2,4-triazole-3-yl]alkane ligands (L2 = 1,2-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)ethane, L3 = 1,3-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)propane, L4 = 1,4-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)butane, Scheme 1). We have prepared and characterized five series of mononuclear and binuclear complexes. Their magnetic properties were studied by SQUID magnetometry and analyzed using the spin Hamiltonian formalism. The analysis was supported by DFT calculations. Furthermore, it was revealed that some of the Co(II) and Fe(II) compounds exhibited solid-state luminescence.
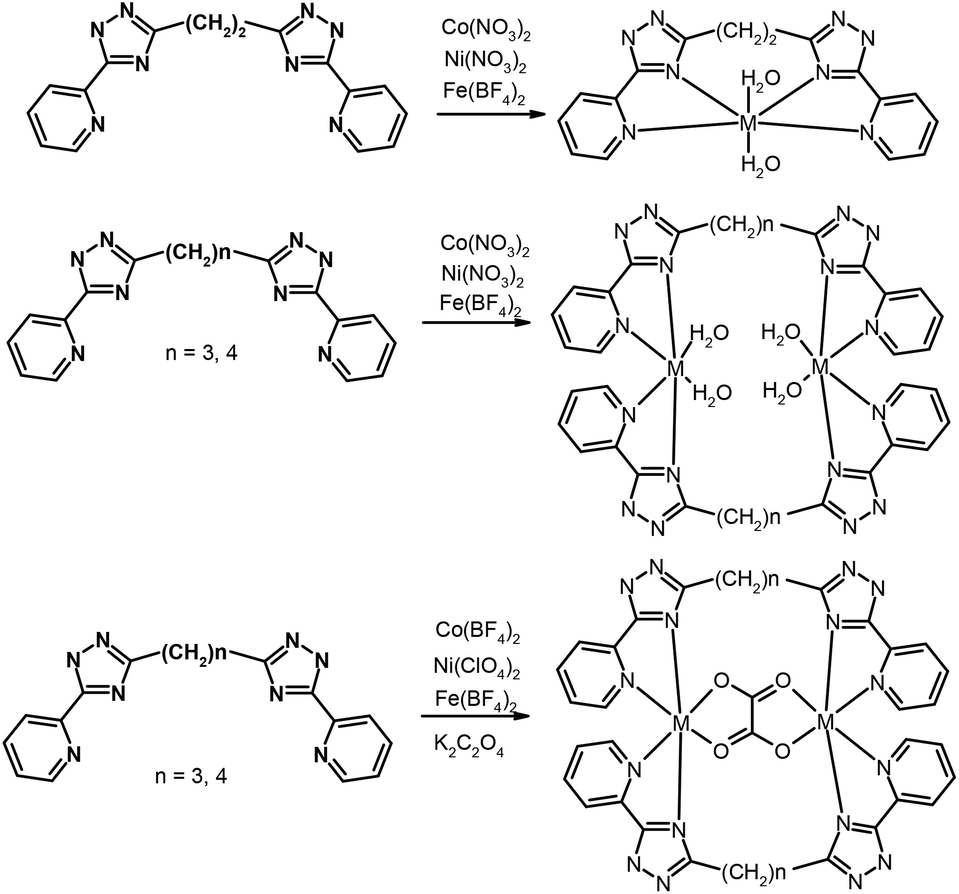 | ||
| Scheme 1 The schematic representation of formation of complexes. | ||
Results and discussion
Synthesis and structure
Previous studies performed on coordination compounds with bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)alkane ligands demonstrated a strong impact of aliphatic chain lengths on the observed coordination modes and nuclearity of the prepared complexes. E.g., it was observed that if two bidentate (5-(pyridine-2-yl)-1,2,4-triazol-3-yl) fragments are linked by monomethylene spacer high-nuclearity complexes with the structures of double helicates and tetra- and octanuclear molecular cages were prepared.8 This study reports on the reactions of Ni(II), Co(II) and Fe(II) salts with bis-pyridyl-triazolyl-alkanes in which the chelating arms are linked by –(CH2)n– chains (n = 2, 3, 4) (Scheme 1). It could be reasonably expected that an increase in the flexibility of a polymethylene spacer due to prolongation of the alkyl chain increases the likelihood of the mononuclear coordination mode. In fact, the reaction between L2 and Ni(II), Co(II) and Fe(II) salts led to the preparation of mononuclear complexes, which were identified by ESI mass spectroscopy (observation of the main peaks corresponding to mononuclear species [M(L2)]+).Furthermore, recrystallisation of the solid products from methanol resulted in the isolation of single crystals, which were measured by single-crystal X-ray diffraction. The determined crystal structures revealed that complexes 1a and 1b are mononuclear. In the molecular structures of both complex cations the ligand L2 is coordinated to the Ni(II) and Co(II) centers in a tetradentate chelate fashion with two other coordination sites (axial positions) occupied by aqua ligands (Fig. 1). The dihedral angle between the least-square planes of coordinated pyridyl-triazolyl moieties adopts significantly different values: 4.88° (1a) and 24.12° (1b). The metal–ligand (M–L) bond lengths are slightly longer in the Co(II) complex 1b, especially in the case of bonds involving pyridyl nitrogen atoms (NPy): Co–NPy = 2.221 Å vs. Ni–NPy = 2.157 and 2.164 Å. The M–L bonds involving triazolyl nitrogen (NTz) and aqua oxygen atoms are shorter (in Å): d(Ni–NTz) = 2.036, 2.043 and d(Ni–O) = 2.064, 2.079 in 1a; d(Co–NTz) = 2.104 and d(Co–O) = 2.026. In summary, the elongation of the spacer from one to two methylene groups between pyridyl-triazolyl fragments increases the flexibility of the ligand and allows it to coordinate with the same metal ion. This contrasts with the related bis(pyridyl-triazolyl)-methane ligands (L1), which typically act as a bridging ligand.8
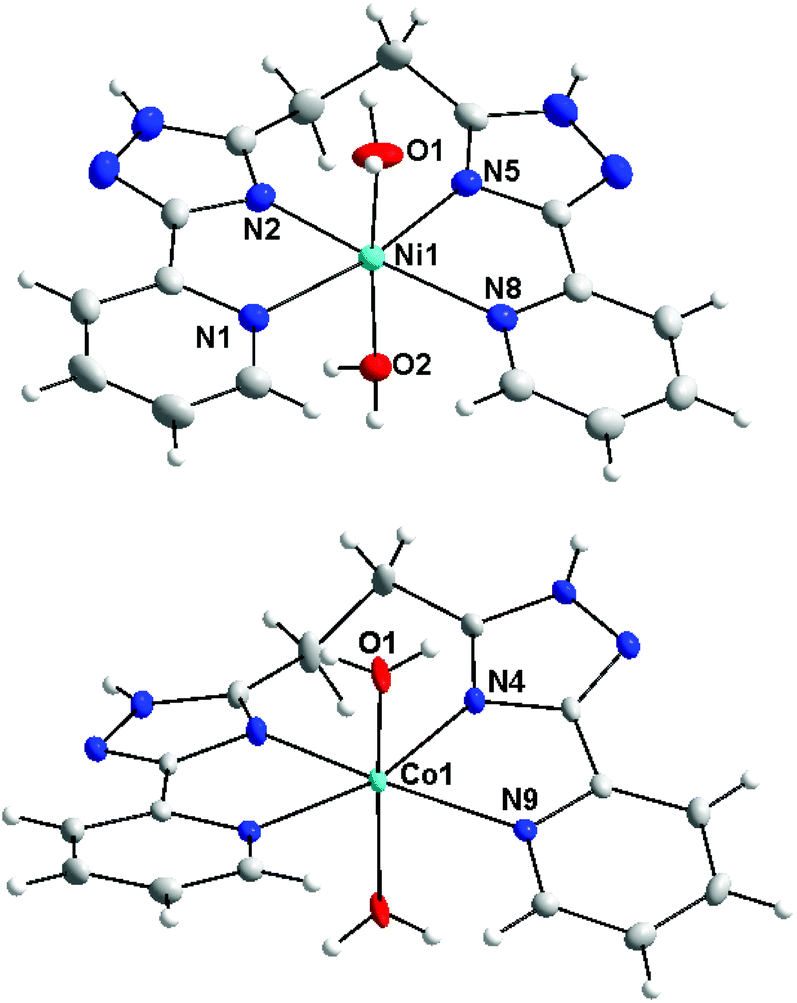 | ||
| Fig. 1 Pictures of molecular structures of complex cations in 1a and 1b. Molecular structures presented as ADP ellipsoids at 50% probability (solvate and counter anions are omitted for clarity). Colour code: C (grey), N (blue), O (red), and Co or Ni (light blue). | ||
We expected that a further increase in the number of methylene groups in the ligand's spacer to three (L3 = 1,3-bis(5-(2-pyridine-2-yl)-1,2,4-triazole-3-yl)propane) and four (L4 = 1,4-bis(5-(2-pyridine-2-yl)-1,2,4-triazole-3-yl)butane) would also make the mononucleating coordination mode more preferable. The reaction of Ni(II), Co(II) and Fe(II) salts with L3 or L4 afforded two series of complexes 2a–c and 3a–c for which a simple formula [M(L3/4)(H2O)2]X4·nH2O (X indicates nitrate (M = Ni, Co) or tetrafluoroborate-anions (M = Fe)) can be derived based on the results of elemental analysis. However, the ESI-mass spectroscopic data clearly suggested that M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L3/4 = 2:2 complexes had formed (see Fig. S1 in the ESI† as an example). Binuclear structures of complexes 2a–c and 3a–c were subsequently confirmed by single-crystal X-ray diffraction determination of their crystal structures. Single-crystals of complexes 2b, 3a and 3b were isolated by recrystallisation of the precipitate from propanol-2 or methanol. The structures of the complexes 2b and 3b are shown in Fig. 2.
:L3/4 = 2:2 complexes had formed (see Fig. S1 in the ESI† as an example). Binuclear structures of complexes 2a–c and 3a–c were subsequently confirmed by single-crystal X-ray diffraction determination of their crystal structures. Single-crystals of complexes 2b, 3a and 3b were isolated by recrystallisation of the precipitate from propanol-2 or methanol. The structures of the complexes 2b and 3b are shown in Fig. 2.
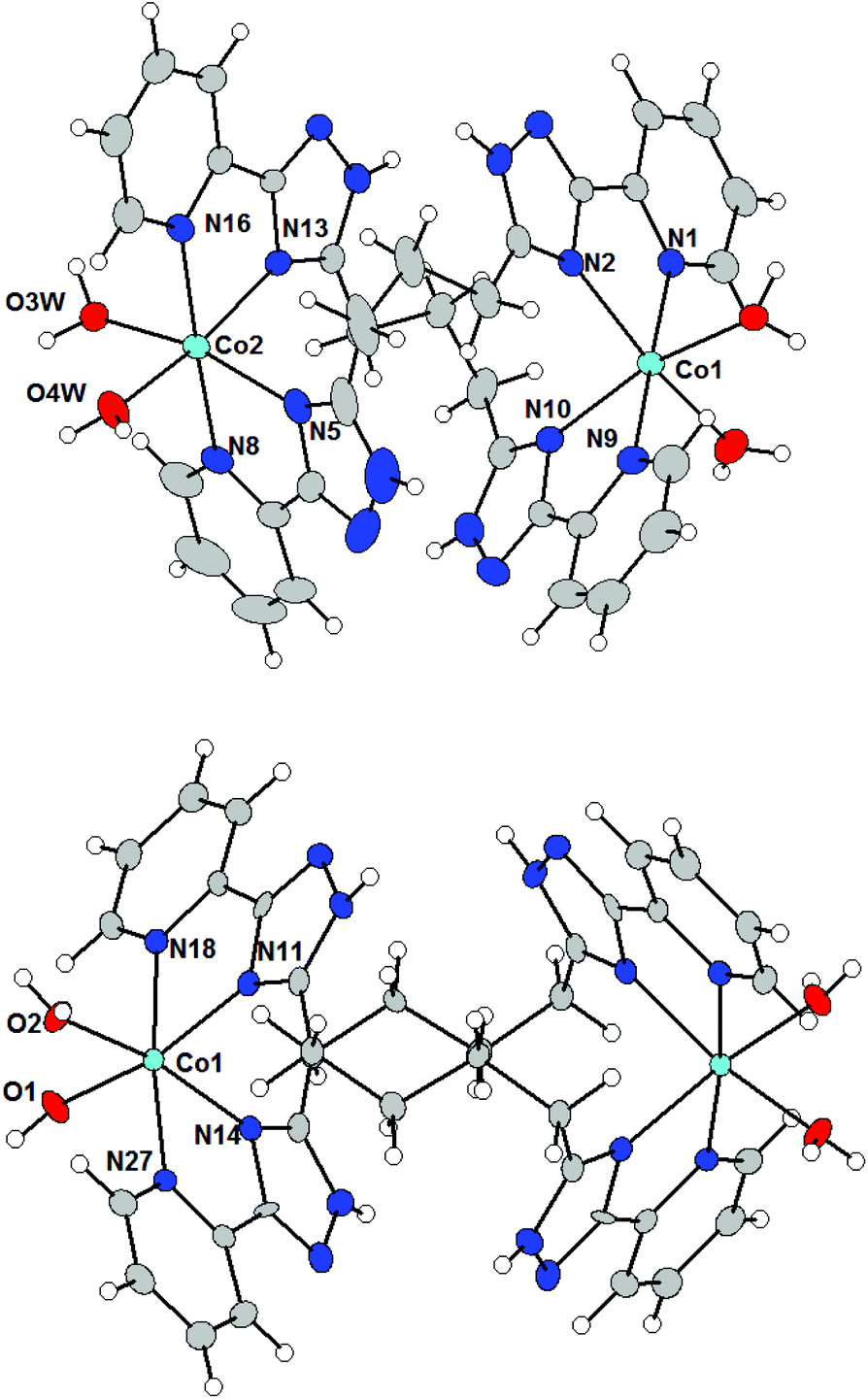 | ||
| Fig. 2 Molecular structures of the complex cations in 2b (above) and 3b (below). Molecular structure presented as ADP ellipsoids at 50% probability (solvate and counter anions are omitted for clarity). Color code: C (grey), N (blue), O (red), and Co (light blue). | ||
Complex cations in 2b, 3a and 3b are dinuclear double-stranded helicates, in which two ligands wrap around two metal atoms, which make the molecules chiral. However, since the space groups are centrosymmetric (2b: P21/n, 3a: Pnma, 3b: P21/c), the two enantiomers co-crystallize within the same crystal, and in 3a, both enantiomers are in the asymmetric unit of the crystal structure.
The molecular structures of complex cations in 2b, 3a and 3b consist of two M(II) atoms, which are hexacoordinated by four donor nitrogen atoms originating from two pyridyl-triazolyl chelating subunits from each of the two different ligands and the remaining coordination sites are occupied by two aqua ligands in the cis-position to give a pseudo-octahedral shape to the coordination polyhedra. The M–NPy bond lengths are again longer than those of M–NTz and also Co complexes 2b and 3b adopt slightly longer M–L bonds than the Ni complex 3a (in Å): d(M–NPy) = 2.14–2.19 in 2b, 2.11–2.16 in 3a, and 2.15–2.20 in 3b; d(M–NTz) = 2.13–2.17 in 2b, 2.11–2.13 in 3a, and 2.11–2.16 in 3b. The Co–O distances are very similar in all three compounds (in Å): d(M–O) = 2.07–2.14 in 2b, 2.07–2.12 in 3a and 2.06–2.10 in 3b. The M⋯M distances within a binuclear helicate are significantly shorter for 2b than those for 3a and 3b which agrees with the length of the alkyl spacer in 2b (L3) vs.3a and 3b (L4), (in Å): d(M⋯M) = 7.2989(6) in 2b, 7.891 in 3a, and 8.720 and 8.532 in 3b.
Remarkably, the main difference between the molecular structures of complex cations with ligands L2 and L3 or L4, e.g. between 1a and 3a, 2b or 3b and 1b, is due to the different degrees of rotation of the pyridyl-triazolyl arms. Two chelating arms from each ligand in 2b, 3a and 3b are rotated relative to each other by the angles of 49.4–74.7° due to the spacer's flexibility. Most likely, the larger conformational flexibility of the ligands L3 and L4 leads to the thermodynamically more stable binucleating coordination mode of these ligands, which is in contrast to compounds involving ligand L2.
A notable feature of the molecular structures of the complexes 2a–3c is the formation of a metallocycle M–L2–M allowing us to possibly encapsulate small guest molecules in its cavity. A rational design of host molecules that can selectively recognize guests is one of the main achievements of supramolecular chemistry; therefore the studies of such objects are of current interest in some research groups.9 Despite the appropriate M⋯M distances in the studied complexes, and a relatively large cavity, our attempts to bind different ligands such as pyrazine, azide and oxalate-anions inside of 2a–3c resulted in the isolation of the “empty” host complexes or led to the precipitation of unidentified insoluble products. As an alternative approach, we explored whether a guest molecule can be trapped inside the host during self-assembly. We treated Ni(II), Co(II) and Fe(II) salts with L3 or L4 and potassium oxalate in a 2:2:1 ratio in methanol or acetone solutions. These preparation routes produced complexes 4a–4c and 5a–5c in which the oxalate-anion is bound as an internal bridging ligand. The ESMS spectra of the 4a–4c and 5a–5c complexes formed with the ditopic ligands L3 and L4 contain peaks corresponding to [M2(L)2(ox)A]+ and [M2(L)2(ox)]2+, respectively (A – BF4−, ClO4−), indicating the formation of binuclear metallocyclic structures in solution (Fig. S2 in the ESI†).
The Co(II), Ni(II) and Fe(II) complexes 4a–4c and 5a–5c were structurally characterized by single-crystal X-ray diffraction and all have the same basic structure, though there are several structural variations observed. The obtained data reveal that isostructural complexes 4a–4c and 5a–5c are formed with L3 and L4 ligands, respectively.
The molecular structures of 4a and 4c (Fig. 3) shows that L3 acts as a bridging ligand between the two metal centers in a ‘face-to-face’ (non-helical) arrangement and they coordinate metal centers by two bidentate pyridyl-triazolyl chelating moieties (one from each ligand L3). The formation of a binuclear complex is further supported by the bridging oxalate ligand (ox2−) producing an {M2(μ-ox)}2+ core. Each metal center is therefore hexacoordinate with a distorted octahedral geometry of the coordination polyhedron.
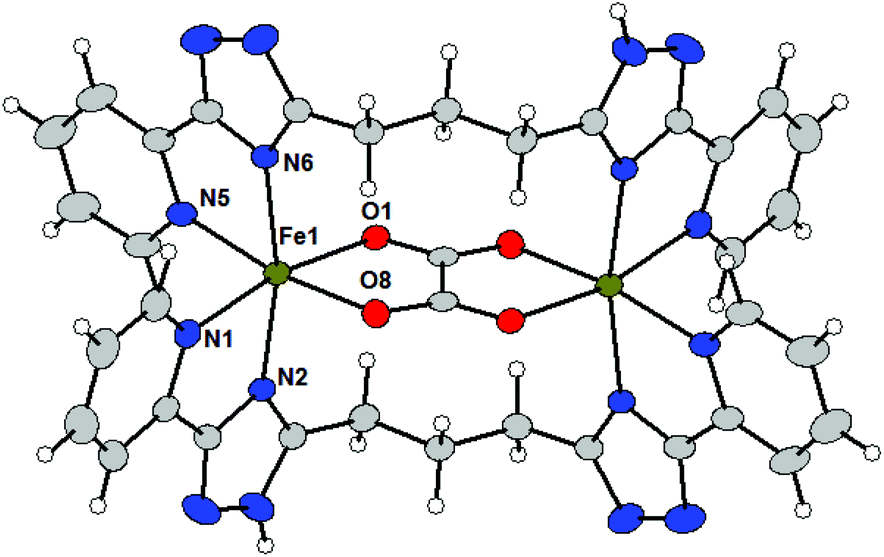 | ||
| Fig. 3 Molecular structures of the complex cations in 4c. Molecular structure presented as ADP ellipsoids at 50% probability (solvate and counter anions are omitted for clarity). Color code: C (grey), N (blue), O (red), Ni (green), and Fe (green). | ||
In 4a all M–L bond lengths lie in the range of 2.07–2.13 Å, whereas Ni–NPy has the longest (2.121 and 2.123 Å) and Ni–O the shortest (2.070 Å) bond lengths. Formation of the {Ni2(μ-ox)}2+ core leads to a rather short Ni⋯Ni separation of 5.392 Å.
The Fe–N bond lengths in 4c (2.15–2.21 Å) are indicative of a high-spin state of the Fe(II) atom of 4c, which is not surprising given the nature of the ligand donor set.10 The two Fe–O bonds are slightly shorter (2.11–2.12 Å). Again, the formation of the {Fe2(μ-ox)}2+ core leads to a rather short Fe⋯Fe distance of 5.515 Å. The mutual rotation of two bidentate triazolyl-pyridine arms is different when compared to the previously discussed structures of complex cations (vide supra). The angle between the least-square planes of the triazolyl-pyridine moieties located on the same ligand adopt rather large values: 90.0° in 4a and 88.6° in 4c.
The crystal structure was determined also for compounds 5a–c and it was revealed that these compounds belong to an isostructural series (space group P21/c). Increasing the length of the spacer from three methylene groups in L3 to four in L4 does not affect the type of the coordination mode. All complex cations within this series consist of two L4 ligands, one oxalate ligand bridging two metal atoms and thus forming binuclear [M2(L4)2(μ-ox)]2+ molecules, which are very similar to [M2(L3)2(μ-ox)]2+ cations from 4a–c. In the Fig. 4 the molecular structure of the [Ni2(L4)2(μ-ox)]2+ cation (5a) is shown as a typical example within the 5a–c series.
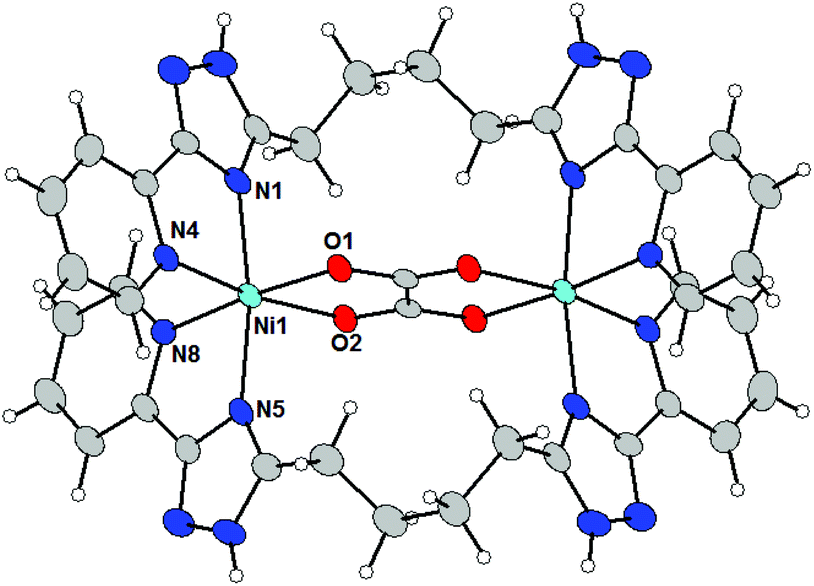 | ||
| Fig. 4 Molecular structures of the complex cations in 5a. Molecular structure presented as ADP ellipsoids at 50% probability (solvate and counter anions are omitted for clarity). Color code: C (grey), N (blue), O (red), and Ni (green). | ||
The metal atoms adopt the expected distorted octahedral coordination geometry in which they are coordinated by four nitrogen atoms (from two triazolyl-pyridine moieties of ligand L4) and two oxygen atoms from the oxalate-anion. The bond lengths around Co(II) and Fe(II) are consistent with those expected for the high-spin state and the average M–L bond lengths decrease in the order Fe (2.162 Å) > Co (2.131 Å) > Ni (2.089 Å). The intradimer M⋯M distances are rather short (in Å): 5.436 Å in 5a, 5.522 Å in 5b and 5.569 Å in 5c. Coordinated ligands L4 are arranged in a ‘butterfly-like’ shape arising from the face-to-face arrangement. The longer length of the spacer (when compared with L3) and the rigidity of the {M2(μ-ox)}2+ core led to the formation of this atypical conformation of the aliphatic chain (Fig. 5). The angles between the least-square planes of the triazolyl-pyridine moieties located on the same ligand L4 are very similar to those observed in the 4a–c series: 89.9° in 5a, 89.0° in 5b and 88.9° in 5c.
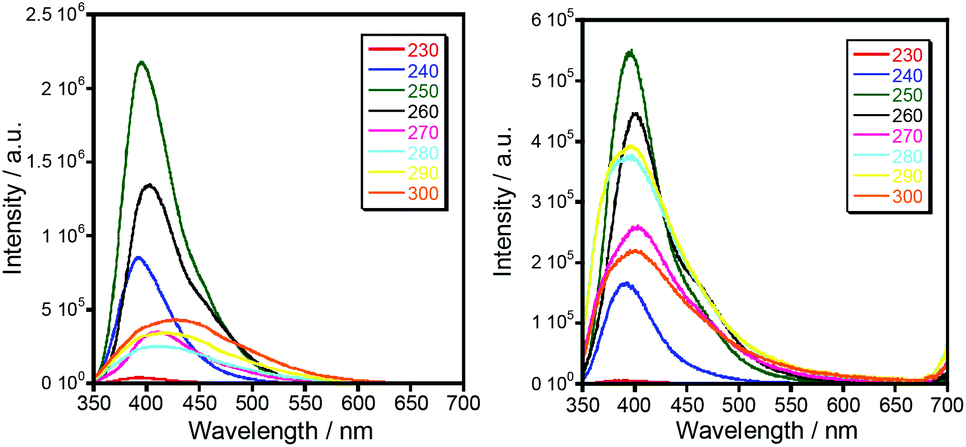 | ||
| Fig. 5 Luminescence spectra of 1b (left) and 1c (right) at room temperature with excitation at different wavelengths. | ||
Luminescence properties
Luminescent complexes are attracting more and more attention due to their potential applications for producing new optic materials. Compared to the extensive research on magnetic properties, the luminescence properties of triazole compounds are relatively limitedly explored. It should also be noted that the coordination of paramagnetic ions usually leads to the quenching of the photoluminescence of organic fluorophores. To our surprise, the coordination compounds of iron(II) (1c, 2c and 3c) and cobalt(II) (1b, 2b, and 3b) exhibit fairly intense photoluminescence clearly observed with the naked eye. The solid state photoluminescence properties of the complexes 1b and 1c are shown in Fig. 5 as a typical example for this series. Intense fluorescence emissions are observed at 399–406 nm (λex = 250 nm), which can be probably assigned to the intraligand charge transition of Ln due to their similar emission peaks. In addition, the fluorescence intensity of Co(II) complexes are weaker than that of the corresponding Fe(II) complexes, which indicates that the quenching effect of iron ions is less pronounced. The quantum yield of luminescence, however, turned out to be insignificant, QY < 0.3%.Magnetic properties
Static magnetic properties of compounds 1a–5c were measured by SQUID magnetometry as the temperature dependence of the magnetic moment in a weak external magnetic field (B = 0.5 T), which was transformed to an effective magnetic moment (μeff/μB).Compounds 1a and 1b are mononuclear compounds, which are not connected directly by strong non-covalent interactions between the coordinating atoms or aromatic rings of the ligands. The shortest M⋯M distances are rather long: d(Ni⋯Ni) = 7.6483(7) Å in 1a and d(Co⋯Co) = 7.338(2) Å in 1b. Therefore, no significant interactions between the spin carriers could be expected. The measured data (Fig. 6) show in the case of 1a behaviour close to that of an ideal paramagnet with a g value larger than that for an isolated electron (2.0023).11 At T = 293 K the μeff/μB adopts a value of 3.1 which corresponds in the case of non-interacting spin S = 1 to g = 2.2. The μeff/μB product remains constant with a decrease in temperature and only at T close to 2 K a tiny decrease is observed. The magnetic data were fitted with the spin Hamiltonian involving an axial magnetic anisotropy parameter D, molecular field correction zJ and a Zeeman term:
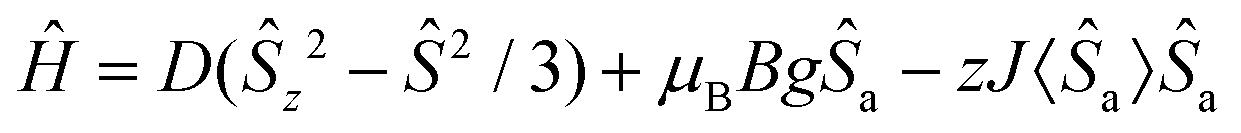 | (1) |
 | (2) |
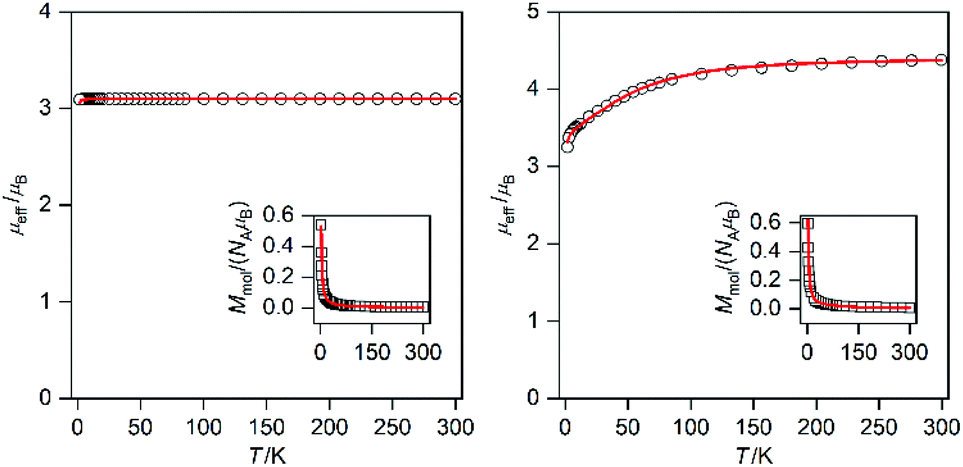 | ||
| Fig. 6 Magnetic data for 1a (left) and 1b (right) depicted as temperature dependence of the magnetic moment calculated from the molar magnetization measured at B = 0.5 T shown in the inset. Experimental data are shown as empty circles, and fitted data are shown as red lines. | ||
Obviously, the magnetic data for 1a can be fitted without using zJ and the tiny drop of μeff/μB at very low temperatures resulted in a negligibly small |D| value (0.002 cm−1).
Magnetic data for 1b (Fig. 6) show completely different behavior from 1a; when at room temperature the value of μeff/μB is 4.3 and this is larger than the spin only value (S = 3/2, μeff/μB = 3.87). The μeff/μB product is almost constant down to 100 K and then a large decrease of its value down to ca. 3.2 is observed. This behavior is typical of Co(II) complexes with very large magnetic anisotropy.12 Therefore, the spin Hamiltonian (eqn (1)) was used to model these data and fit resulted in a very large D (+53.6 cm−1) and g = 2.20. Thus, 1b exhibits easy-plane magnetic anisotropy.
Compounds 2a–2c and 3a–3c are binuclear complexes (Fig. 7) in which the magnetic centers are not interconnected by a short covalent pathway, but the central atoms are held together by bridging L3 or L4 ligands. This means that a possible super-exchange pathway involves also the alkyl chains and therefore, as in the case of 1a and 1b, no strong magnetic exchange interactions could be expected. In addition, there are intramolecular π–π interactions between triazole moieties observed in 2b and 3a, which can also contribute through the spin polarization mechanism to the superexchange interactions.12a In contrast, the magnetic intermolecular interactions via hydrogen bonding can be excluded, because hydrogen bonds in 2a–b and 3a–b are formed only between the aqua ligands and nitrate anions and direct hydrogen bonding between the aqua ligands coordinating metal centers (which could mediate magnetic exchange interactions of significant strength)12b–d is missing.
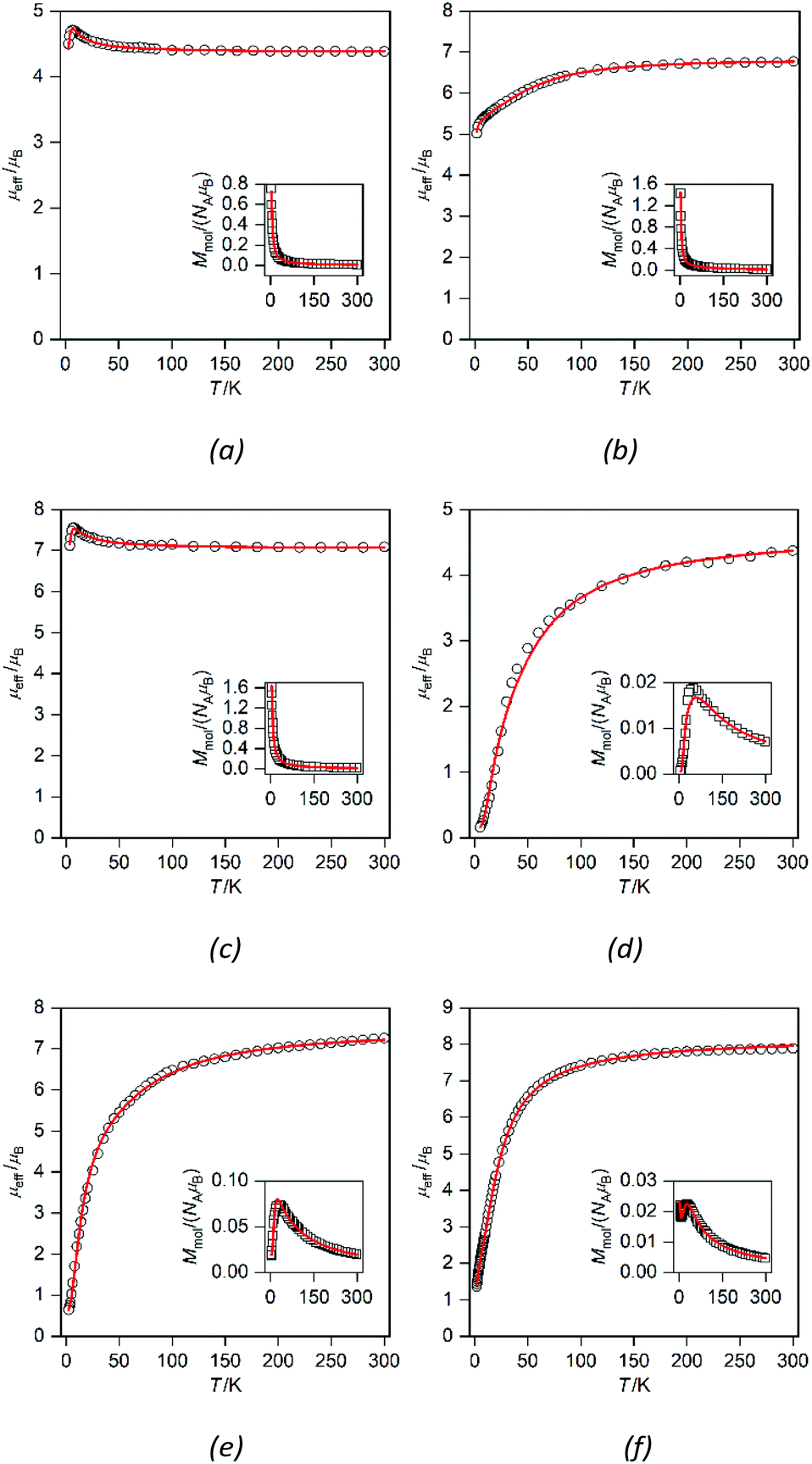 | ||
| Fig. 7 Magnetic data for 2a (a), 2b (b), 2c (c), 4a (d), 4b (e) and 4c (f) depicted as temperature dependence of the magnetic moment calculated from the molar magnetization measured at B = 0.5 T shown in the inset. Experimental data are shown as empty circles, and fitted data are shown as red lines. | ||
The magnetic data for 2a–2c are shown in Fig. 7a–c; the magnetic data for 3a–3c are analogous and therefore they are shown only in the ESI (Fig. S3†). At room temperature the value of μeff/μB is slightly larger than the spin-only value for two uncoupled spins (for g = 2.0 and S = 1, μeff/μB = 4.0; for g = 2.0 and S = 3/2, μeff/μB = 5.5; and for g = 2.0 and S = 2, μeff/μB = 6.9): μeff/μB = 4.4 for 2a and 3a, μeff/μB = 6.8 for 2b and 3b, and μeff/μB = 7.1–7.2 for 2c and 3c. The temperature dependence of μeff/μB is very similar for the Ni(II) and Fe(II) compounds. Upon lowering the temperature, a very small increase of the μeff/μB product is observed below ca. 50 K, which can be explained only as a very weak ferromagnetic interaction between the magnetic centers. The magnetic data were fitted using the following spin Hamiltonian involving the isotropic exchange interaction J, axial magnetic anisotropy and Zeeman terms:
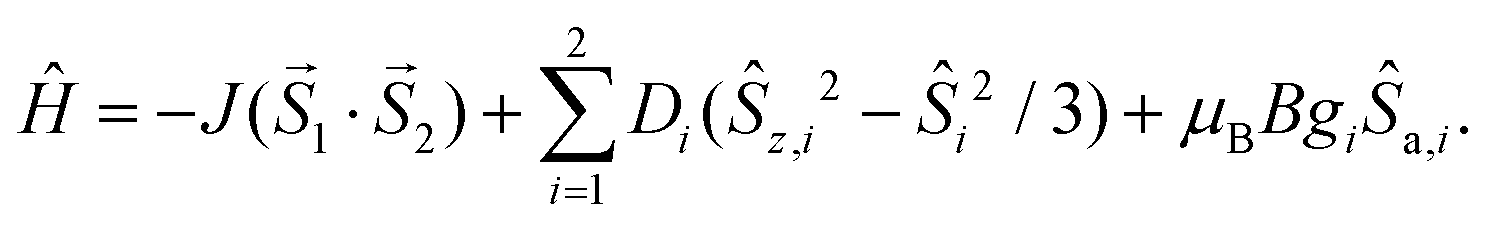 | (3) |
Satisfactory fits were obtained for Ni(II) compounds by assuming small positive D parameters (5.25 cm−1 in 2a and 5.22 cm−1 in 3a) and rather weak ferromagnetic exchange interactions (J = +2.27 cm−1 in 2a and +1.65 cm−1 in 3a, Table 1). In the case of Fe(II) compounds the fitting results also confirmed weak ferromagnetic exchange and small axial ZFS parameters (in cm−1): D = 5.68, J = +0.84 in 2c and D = 5.93, J = +0.89 in 3c. In the case of Co(II) compounds 2b and 3b, there is an evident drop of the effective magnetic moment due to the large magnetic anisotropy. The fitting procedure revealed for 2b and 3b very large easy-plane anisotropy (in cm−1, D = +66.1 for 2b and +61.5 for 3b) and only very weak antiferromagnetic exchange was needed to model the magnetic data satisfactorily (Table 1). However, due to the small value of |J| and very large ZFS dominating the low temperature data, the weak ferromagnetic interaction in 2b and 3b cannot be excluded.
| Compound | D (cm−1) | g | zJ or J (cm−1) |
|---|---|---|---|
| [Ni(L2)(H2O)2](NO3)2·2H2O, (1a) | 0.0 | 2.19 | — |
| [Co(L2)(H2O)2](NO3)2, (1b) | 67.6 | 2.27 | zJ = −0.058 |
| [Ni2(L3)2(H2O)4](NO3)4·4H2O, (2a) | 5.3 | 2.18 | J = +2.27 |
| [Co2(L3)2(H2O)4](NO3)4·5H2O, (2b) | 66.1 | 2.49 | J = −0.082 |
| [Fe2(L3)2(H2O)4](BF4)4·4H2O, (2c) | 5.7 | 2.03 | J = +0.84 |
| [Ni2(L4)2(H2O)4](NO3)4·4H2O, (3a) | 5.2 | 2.20 | J = +1.65 |
| [Co2(L4)2(H2O)4](NO3)4·4H2O, (3b) | 61.5 | 2.48 | J = −0.035 |
| [Fe2(L4)2(H2O)4](BF4)4·4H2O, (3c) | 5.9 | 2.06 | J = +0.89 |
| [Ni2(L3)2(ox)](ClO4)2·4MeOH, (4a) | 14.0 | 2.35 | J = −40.0 |
| [Co2(L3)2(ox)](BF4)2·4MeOH, (4b) | 75.8 | 2.74 | J = −9.4 |
| [Fe2(L3)2(ox)](BF4)2·4MeOH, (4c) | −5.1 | 2.37 | J = −6.60 |
| [Ni2(L4)2(ox)](ClO4)2·4H2O·2C3H6O, (5a) | 15.6 | 2.28 | J = −36.0 |
| [Co2(L4)2(ox)](BF4)2·4H2O·2C3H6O, (5b) | 46.3 | 2.57 | J = −10.8 |
| [Fe2(L4)2(ox)](BF4)2·4H2O·2C3H6O, (5c) | −70.2 | 2.09 | J = −2.8 |
Compounds 4a–c and 5a–c are binuclear compounds involving in their structure {M2(μ-ox)}2+ cores, which are well known for the mediation of strong magnetic exchange interactions.8b The temperature dependence of molar magnetization for 4a–c (Fig. 7d–f; magnetic data for 5a–c are shown in the ESI, Fig. S4†) clearly shows maxima of molar magnetization at low temperatures indicating strong antiferromagnetic interactions between the metal atoms.
The values of μeff/μB for these compounds are again larger than the spin only values at room temperature thus indicating a significant orbital contribution: μeff/μB = 4.6 for 4a and 5a, 7.1 for 4b, 7.0 for 5b, 7.9 for 4c, and 7.0 for 5c. In compounds 4a–c and 5a–c, a steep decrease of μeff/μB starts at ca. 100 K and it can be extrapolated at μeff/μB = 0 at 0 K indicating thus the ground spin state S = 0 and rather strong antiferromagnetic coupling (Fig. 7). Therefore, the magnetic data of 4a–c and 5a–c were fitted using the spin Hamiltonian for a dimer (eqn (3)). The fitting procedure revealed negative isotropic exchange coupling constants and large magnetic anisotropy (in cm−1): J = −40.0 for 4a, −36.0 for 5a, −9.4 for 4b, −10.8 for 5b, −6.60 for 4c, and −2.8 for 5c; D = +14.0 for 4a, +15.6 for 5a, +75.8 for 4b, +46.3 for 5b, −5.1 for 4c, and −70.2 for 5c (Table 1).
The rather large value of D for complex 5c can be rationalized by the fact that it possesses a more symmetric bond length within the {FeN4O2} chromophore than complex 4c; thus the splitting of the T2g ground state of an idealized Oh symmetry of the 3d6 electronic configuration is less pronounced leading to a larger contribution of the orbital angular momentum of the excited states to the zero-field splitting tensor.
In addition to DC magnetic measurement, the AC susceptibility measurements were performed for one of the weakly coupled Co(II) dimers 2b in order to explore the possibility of the slow relaxation of the magnetization (Fig. 8). First, the AC susceptibility was measured at T = 2 K and the static magnetic field (BDC) was varied between 0 and 0.25 T, which showed that a small non-zero BDC is needed to suppress the quantum tunneling of the magnetization. Therefore, the temperature-dependent AC susceptibility data were acquired at BDC = 0.1 T, and the analysis based on one-component Debye's model resulted in relaxation times (τ). First, the Arrhenius law was used to fit the temperature-dependence of τ, which resulted in τ0 = 1.0 × 10−5 s, Ueff = 6 K (Fig. S5†). Moreover, the relaxation times were fitted to the combination of Orbach and direct terms providing A = 3624 K−1 s−1, τ0 = 6.4 × 10−6 s, Ueff = 11.4 K (Fig. S5†). However, these Ueff values are rather small in comparison with the derived D-value, thus suggesting other relaxation processes taking the leading role.
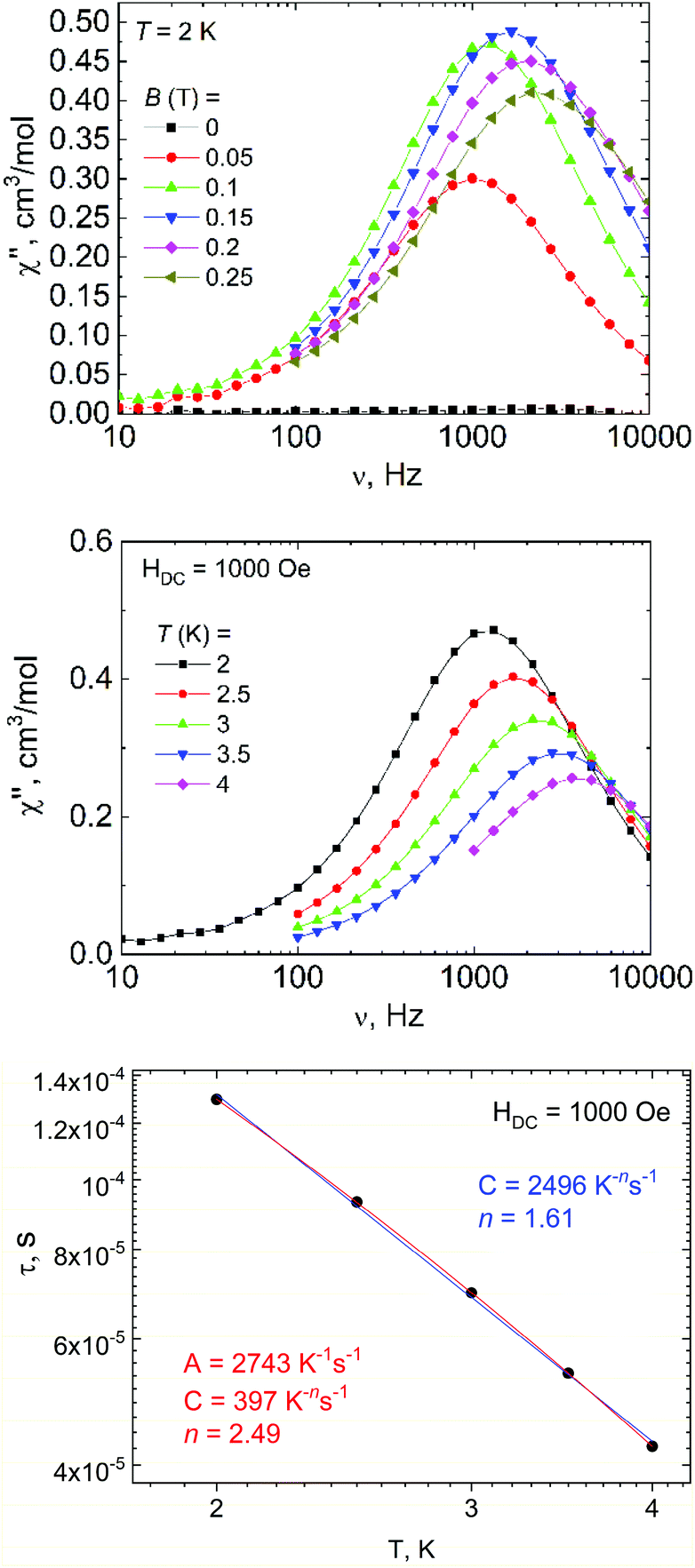 | ||
| Fig. 8 AC susceptibility data for 2b. The out-of-phase susceptibility χ′′ at various static magnetic fields (above) and as a function of temperature measured at BDC = 0.1 T (middle). The analysis of the relaxation times derived from the AC susceptibility data for 2b with the Raman relaxation process (blue line) or with a combination of direct and Raman processes (red line) (below). | ||
Therefore, the data were analyzed with the Raman process using the following equation
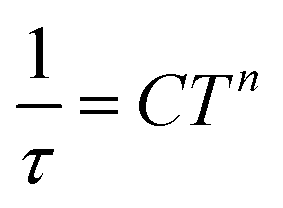 | (4) |
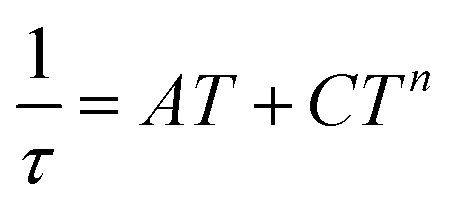 | (5) |
Theoretical calculations
To support the theoretical analysis of the magnetic properties of herein reported compounds we performed Broken-Symmetry Density Functional Calculations (BS-DFT) to estimate the magnetic coupling between the metal centers for systems with magnetic coupling (2b–c, 3a–c, 4a–c and 5a–c). The DFT calculations were performed on molecular structures of particular complexes as determined by X-ray diffraction (the positions of hydrogen atoms were optimized – for details see the Experimental section). The ORCA computation package 4.1 was employed for all the calculations together with the B3LYP functional and def2-TZVP basis set. The energy of the high spin (HS) state was calculated and compared with the broken-symmetry (BS) spin state energy and by using Ruiz's approach,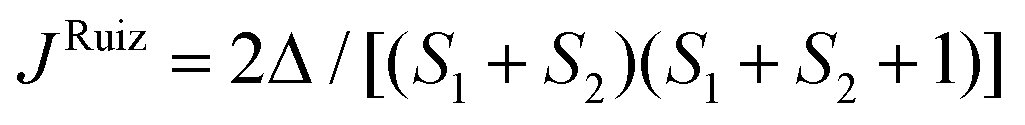 | (6) |
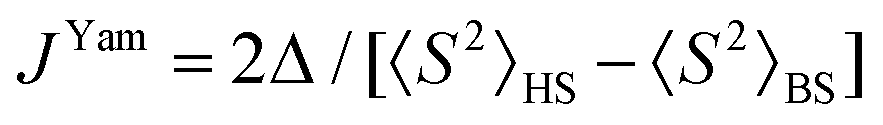 | (7) |
In the case of weakly coupled systems 2a–c and 3a–c the calculations were performed for complexes 2b, 3a and 3b and in all three cases very weak ferromagnetic coupling was predicted (Table 2) using both Ruiz's and Yamaguchi's approaches and this conforms to the experimentally observed weak magnetic exchange.
| Complex cation | J Ruiz (cm−1) | J Yam (cm−1) | J exp (cm−1) |
|---|---|---|---|
| [Co2(L3)2(H2O)4]4+ of 2b | +0.10 | +0.14 | −0.082 |
| [Ni2(L4)2(H2O)4]4+ of 3a | +0.006 | +0.009 | +1.65 |
| [Co2(L4)2(H2O)4]4+ of 3b | +0.12 | +0.16 | −0.035 |
| [Ni2(L3)2(ox)]2+ of 4a | −42.1 | −63.1 | −40.0 |
| [Fe2(L3)2(ox)]2+ of 4c | −8.0 | −10.0 | −6.6 |
| [Ni2(L4)2(ox)]2+ of 5a | −41.1 | −61.6 | −36.0 |
| [Co2(L4)2(ox)]2+ of 5b | −13.8 | −18.4 | −10.8 |
| [Fe2(L4)2(ox)]2+ of 5c | −7.6 | −9.5 | −2.8 |
In the case of oxalate bridged compounds (4a–c and 5a–c) the calculations were performed for complexes 4a, 4c and 5a–c and by using both Ruiz's and Yamaguchi's approaches strong antiferromagnetic coupling was predicted and from the obtained coupling constants (Table 2) it is clear that Ruiz's approach agrees with the experiment fairly well. The spin density plots showing the BS spin states for [Co2(L3)2(H2O)4]4+ of 2b and for [Ni2(L4)2(ox)]2+ of 5a are presented in Fig. 9.
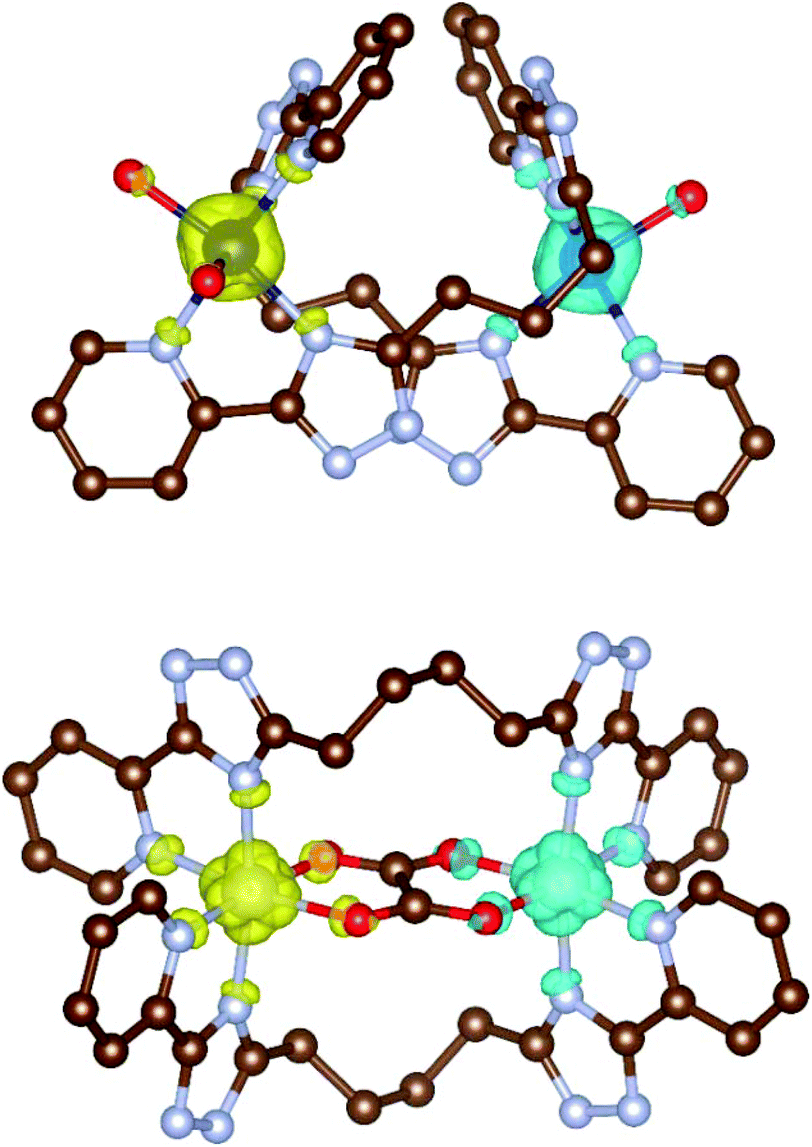 | ||
| Fig. 9 The calculated spin density distribution of the broken-symmetry spin states for [Co2(L3)2(H2O)4]4+ of 2b (left) and for [Ni2(L4)2(ox)]2+ of 5a (right) using B3LYP/de2-TZVP. Positive and negative spin densities are represented by yellow and cyan surfaces, respectively. The isodensity surfaces are plotted with a cut-off value of 0.01 ea0−3. Hydrogen atoms are not shown for clarity. | ||
Conclusions
In this article we reported on the synthesis, crystal structures, luminescence and magnetic properties of five series of various compounds prepared by reactions between bis(pyridyl-triazolyl)-alkane ligands L2 (1,2-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)ethane, mononuclear complexes 1a–c), L3 (1,2-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)propane, binuclear complexes 2a–c), L4 (1,2-bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)butane, binuclear complexes 3a–c) and Ni(II), Co(II), and Fe(II) salts. In the case of series 4a–c and 5a–c the reactions between the L3 (4a–c) or L4 (5a–c) ligands and aforementioned metal salts, and also the oxalate bridging anions were used for the synthesis of binuclear compounds containing an {M2(μ-ox)}2+ core.Interestingly, compounds 1b, 2b, and 3b and 1c, 2c and 3c exhibit luminescence, which was the most intense when they were excited by irradiation with λex = 250 nm.
The magnetic measurement and subsequent analysis of reported compounds revealed large magnetic anisotropy in both Co(II) monomeric and dimeric compounds. The L3 and L4 ligands seem to mediate weak ferromagnetic exchange between Ni(II) and Fe(II) ions despite a quite long super-exchange pathway. In contrast, the incorporation of second bridging ligand, oxalate, resulted in strong antiferromagnetic exchange between all metal ions. The theoretical calculations based on DFT theory and B3LYP functional corroborated these findings and Ruiz's approach to the evaluation of the energy difference between BS and HS spin states seems to provide better numerical estimates of the isotropic exchange parameters. The dynamic magnetic measurements were performed for weakly coupled Co(II) dimer 2b and confirmed the field-induced slow relaxation of the magnetization typical of the single-molecule magnets, and the analysis of the relaxation times is in favour of direct and Raman relaxation processes.
Experimental
General
The reagents and solvents employed were commercially available and used as received without further purification. The C, H, and N microanalyses were carried out with a EuroVector EUROEA elemental analyzer. The FT-IR spectra were recorded with KBr pellets in the range 4000–400 cm−1 on a Bruker spectrometer. Electrospray mass spectra of complexes were measured on a Finnigan TSQ 700 mass spectrometer in positive ion mode. Samples were prepared at a concentration of ∼2 mg ml−1 MeOH. Spectra were acquired over an m/z range of 50–2000; several scans were averaged to provide the final spectrum. Magnetic measurements of complexes 1a–1c and 2a–c were carried out on a Quantum Design PPMS–9SQUID magnetometer in the temperature range of 2–300 K. The magnetic data for 3a–c, 4a–c and 5a–c were measured with SQUID apparatus (MPMS-XL7, Quantum Design) using the RSO mode of detection with ca. 20 mg of the sample encapsulated in a gelatin-made sample holder. The data were corrected for the underlying diamagnetism and converted to the effective magnetic moment. The diamagnetic contributions of the samples were estimated from Pascal's constants. The commercially available metal salts Ni(NO3)2·6H2O, Ni(ClO4)2·6H2O, Co(NO3)2·6H2O, Co(BF4)2·6H2O, and Fe(BF4)2·6H2O were used as reactants. The ligands L1–L3 were prepared from pyridine-2-carbonitrile and hydrazide of succinic (for L2), glutaric (L3) and adipoic (L4) acids according to the previously described methods.5Synthetic procedures
:1 molar ratio were carried out in methanol–water solutions. The following preparation is provided as a detailed description of the general method.
1,2-Bis(5-(pyridine-2-yl)-1,2,4-triazol-3-yl)ethane (L2, 318 mg, 1 mmol) was placed in an Erlenmeyer flask and suspended in 15 ml methanol and 10 ml of water. Ni(NO3)2·6H2O (290 mg, 1 mmol) was added to the ligand suspension. The mixture was stirred with heating (50 °C) for one hour to obtain a clear blue solution, which was stirred at room temperature for 6 hours. Then, the solution was filtered through paper filter and left to evaporate slowly for several days. This resulted in the precipitation of 1a as a blue solid, which was filtered off, washed with MeOH and dried in air. The same synthetic procedure was used for the preparation of complexes 1b–c, 2a–c and 3a–c. Single crystals suitable for X-ray studies were obtained by recrystallization from MeOH–water or i-PrOH–water solution (2:1, v:v).
For [Ni(L2)(H2O)2](NO3)2·2H2O (1a): yield 41%. Anal. Found: C, 33.34; H, 4.47; N, 24.03. Required for C16H24NiN10O10: C, 33.41; H, 4.21; N, 24.35%. ESI-MS (m/z): calculated for [NiL2NO3]+ 439.03. IR νmax (cm−1): 3240, 1615, 1555, 1504, 1478, 1371 (NO3), 1321 (NO3), 1072, 1016, 755.
For [Co(L2)(H2O)2](NO3)2 (1b): yield 55%. Anal. Found: C, 35.77; H, 3.60; N, 25.93. Required for C16H20CoN10O8: C, 35.63; H, 3.74; N, 25,97%. ESI-MS (m/z): calculated for [CoL2NO3]+ 439.27. IR νmax (cm−1): 3215, 1617, 1561, 1505, 1478, 1373, 1325, 1071, 1016, 754.
For [Fe(L2)(H2O)2](BF4)2·2H2O (1c): yield 37%. Anal. Found: C, 31.01; H, 4.04; N, 17.94. Required for C16H24B2F8FeN8O4: C, 30.90; H, 3.89; N, 18.02%. ESI-MS (m/z): calculated for [FeL2BF4]+ 460.98. IR νmax (cm−1): 3240, 1612, 1549, 1501, 1473, 1054, 1016, 754.
For [Ni2(L3)2(H2O)4](NO3)4·4H2O (2a): yield 55%. Anal. Found: C, 34.52; H, 3.99; N, 23.92. Required for C34H48Ni2N20O20: C, 34.77; H, 4.12; N. 23.85%. ESI-MS (m/z): calculated for [Ni2(L3)2(NO3)2]2+ 453.05. IR νmax (cm−1): 1609, 1562, 1473, 1391, 1283, 1067, 1016, 752.
For [Co2(L3)2(H2O)4](NO3)4·5H2O (2b): yield 53%. Anal. Found: C, 34.11; H, 4.41; N, 23.43. Required for C34H50Co2N20O21: C, 34.25; H, 4.23; N, 23.49%. ESI-MS (m/z): calculated for [Co2(L3)2(NO3)2]2+ 453.07. IR νmax (cm−1): 1609, 1564, 1473, 1394, 1282, 1067, 1016, 752.
For [Fe2(L3)2(H2O)4](BF4)2·4H2O (2c): yield 42%. Anal. Found: C, 32.27; H, 3.51; N, 17.80. Required for C34H48B4F16Fe2N16O8: C, 32.21; H, 3.82; N, 17.68%. ESI-MS (m/z): calculated for [Fe2(L3)2(BF4)2]2+ 475.00. IR νmax (cm−1): 1611, 1561, 1478, 1055, 1016, 754.
For [Ni2(L4)2(H2O)4](NO3)4·4H2O (3a): yield 65%. Anal. Found: C, 36.04; H, 4.57; N, 23.19. Required for C36H52Ni2N20O20: C, 35.96; H, 4.36; N, 23.30%. ESI-MS (m/z): calculated for [Ni2(L4)2(NO3)2]2+ 467.08. IR νmax (cm−1): 1609, 1564, 1473, 1394, 1282, 1067, 1016, 752.
For [Co2(L4)2(H2O)4](NO3)4·4H2O (3b): yield 65%. Anal. Found: C, 35.83; H, 4.16; N, 23.12. Required for C36H52Co2N20O20: C, 35.95; H, 4.36; N, 23.29%. ESI-MS (m/z): calculated for [Co2(L4)2(NO3)2]2+ 467.33. IR νmax (cm−1): 1609, 1564, 1473, 1394, 1282, 1067, 1016, 752.
For [Fe2(L4)2(H2O)4](BF4)4·4H2O (3c): yield 31%. Anal. Found: C, 32.46; H, 4.13; N, 17.40. Required for C36H52B4F16Fe2N16O8: C, 33.37; H, 4.04; N. 17.29%. ESI-MS (m/z): calculated for [Fe2(L4)2(BF4)2]2+ 489.04. IR νmax (cm−1): 1611, 1558, 1474, 1054, 1015, 754.
The same synthetic procedure was used for the preparation of complexes 4a–c and 5a–c. For the complexes 5a–c an acetone–water mixture (2:1, v:v) was used as the solvent. In the case of the Co(II) and Fe(II) complexes the Co(BF4)2·6H2O and Fe(BF4)2·6H2O salts were used instead of perchlorates. The Fe(II) complexes were obtained under an inert atmosphere of argon by using Schlenk techniques.
For [Ni2(L3)2(ox)](ClO4)2·4MeOH (4a): yield 69%. Anal. Found: C, 40.03; H, 3.60; N, 18.94. Required for C40H44Cl2N16O16Ni2: C, 40.25; H, 3.72; N, 18.77%. ESI-MS (m/z): calculated for [Ni2(L3)2OxClO4]+ 969.10, calculated for [Ni2(L3)2Ox]2+ 434.07. IR νmax (cm−1): 1640, 1612, 1562, 1477, 1083, 622.
For [Co2(L3)2(ox)](BF4)2·4MeOH (4b): yield 72%. Anal. Found: C, 41.47; H, 3.95; N, 19.50. Required for C40H48B2F8N16O8Co2: C, 41.20; H, 4.14; N, 19.22%. ESI-MS (m/z): calculated for [Co2(L3)2OxClO4]+ 969.12, calculated for [Co2(L3)2Ox]2+ 434.11. IR νmax (cm−1): 1642, 1611, 1562, 1478, 1083, 626.
For [Fe2(L3)2(ox)](BF4)2·4MeOH (4c): yield 62%. Anal. Found: C, 40.08; H, 3.88; N, 19.64. Required for C40H48B2F8N16O8Fe2: C, 40.99; H, 4.13; N, 19.12%. ESI-MS (m/z): calculated for [Fe2(L3)2OxBF4]+ 950.22, calculated for [Fe2(L3)2Ox]2+ 432.22. IR νmax (cm−1): 1642, 1612, 1562, 1475, 1055, 622.
For [Ni2(L4)2(ox)](ClO4)2·4H2O·2C3H6O (5a): yield 69%. Anal. Found: C, 40.92; H, 4.31 3; N, 17.75. Required for C44H56Cl2N16O18Ni2: C, 41.10; H, 4.39; N, 17.43%. ESI-MS (m/z): calculated for [Ni2(L4)2OxClO4]+ 997.13, calculated for [Ni2(L4)2Ox]2+ 448.09. IR νmax (cm−1): 1707, 1634, 1615, 1560, 1474, 1091, 620.
For [Co2(L4)2(ox)](BF4)2·4H2O·2C3H6O (5b): yield 66%. Anal. Found: C, 41.52; H, 4.23; N, 18.04. Required for C44H56B2F8N16O10Co2: C, 41.93; H, 4.47; N, 17.78%. ESI-MS (m/z): calculated for [Co2(L4)2OxClO4]+ 997.17, calculated for [Co2(L4)2Ox]2+ 448.12. IR νmax (cm−1): 1703, 1638, 1614, 1562, 1476, 1089, 621.
For [Fe2(L4)2(ox)](BF4)2·4H2O·2C3H6O (5c): yield 68%. Anal. Found: C, 42.40; H, 4.64; N, 17.94. Required for C44H56B2F8N16O10Fe2: C, 42.13; H, 4.50; N, 17.87%. ESI-MS (m/z): calculated for [Fe2(L4)2OxBF4]+ 978.31, calculated for [Fe2(L4)2Ox]2+ 445.26. IR νmax (cm−1): 1711, 1640, 1612, 1559, 1472, 1092, 620.
Crystallography
Data for title complexes were collected using an Oxford diffraction Xcalibur diffractometer with a Sapphire CCD detector installed at a fine-focus sealed tube (MoKα radiation, λ = 0.71073 Å) and equipped with Oxford Cryosystems nitrogen gas-flow apparatus, and a Bruker APEX II diffractometer equipped with a CCD detector and a graphite-monochromated MoKα radiation source (MoKα radiation, λ = 0.71073 Å). All structures were solved and refined (full-matrix least-squares on Fo2 − Fc2) by using SHELXS-2014.13,14 For some structures, their space group was checked by the ADSYMM procedure of the PLATON15 software. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed into the calculated positions and they were included in the riding-model approximation with Uiso = 1.2 or 1.5Ueq (atom of attachment). The crystallographic parameters and X-ray diffraction experimental-parameters are given in Table S1.† Selected bond lengths for the reported compounds are given in Table S2.† More details of the refinement of crystal structures are provided in the ESI and in CCDC 1907068–1907077.†Theoretical calculations
All theoretical computation was conducted with the ORCA 4.1 computational package.16 The B3LYP DFT functional17 was used for calculations of the isotropic exchange parameter J according to Ruiz18 and Yamaguchi19 approaches, by comparing the energies of high-spin (HS) and broken-symmetry spin (BS) states. The polarized triple-ζ quality basis set def2-TZVP proposed by Ahlrichs and co-workers was used for all atoms.20 The calculations utilized the RI approximation with the decontracted auxiliary def2/J Coulomb fitting basis set21 and the chain-of-spheres (RIJCOSX) approximation to exact exchange22 as implemented in ORCA. Increased integration grids (Grid6 and Gridx6 in ORCA convention) and tight SCF convergence criteria were used in all calculations. The molecular fragment used in the calculations was extracted from the experimental X-ray structures and the atomic positions of the hydrogen atoms were optimized with the BP86 functional.23 The calculated spin density was visualized with the VESTA 3 program.24Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from the Russian foundation for basic research (project 16-03-00386), Russian science foundation (project 18-13-00024) and the Ministry of Education, Youth and Sports of the Czech Republic (LO1305). R. H. and I. N. acknowledge the financial support from institutional sources of the Department of Inorganic Chemistry, Palacky University Olomouc, Czech Republic. I. N. would like to acknowledge the project CEITEC 2020 (LQ1601) with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustainability Programme II. M. K. and I. E. are grateful for financial support from the State Assignment on Fundamental Research of the Kurnakov Institute of General and Inorganic Chemistry of RAS (IGIC RAS) and thank the User Facilities Centers of IGIC RAS for the facility to perform single crystal X-ray measurements.Notes and references
- (a) J.-M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed; (b) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763 CrossRef CAS.
- (a) S. O. Scott, E. L. Gavey, S. J. Lind, K. C. Gordon and J. D. Crowley, Dalton Trans., 2011, 40, 12117 RSC; (b) U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Dalton Trans., 2013, 42, 14064 RSC; (c) R. A. S. Vasdev, D. Preston and J. D. Crowley, Dalton Trans., 2017, 46, 2402 RSC; (d) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed; (e) N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354–1379 RSC; (f) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (g) M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860–6909 RSC.
- (a) L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Chem. Soc. Rev., 2009, 38, 2334 RSC; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed; (c) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS PubMed; (d) T. D. Hamilton and L. R. MacGillivray, Cryst. Growth Des., 2004, 4, 419 CrossRef CAS; (e) A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846 RSC; (f) S. Alvarez, Dalton Trans., 2006, 2209 Search PubMed.
- (a) J. G. Hardy, Chem. Soc. Rev., 2013, 42, 7881 RSC; (b) Q. Yue and E.-Q. Gao, Coord. Chem. Rev., 2019, 382, 1 CrossRef CAS; (c) N. Ahmada, A. H. Chughtaia, H. A. Younusa and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1 CrossRef.
- (a) N. Fatin-Rouge, S. Blanc, A. Pfeil, A. Rigault, A. M. Albrecht-Gary and J.-M. Lehn, Helv. Chim. Acta, 2001, 84, 1694 CrossRef CAS; (b) B. Hasenknopf, J.-M. Lehn, N. Boumediene, E. Leize and A. van Dorsselaer, Angew. Chem., Int. Ed., 1998, 37, 3265 CrossRef CAS.
- (a) M. Liu, W. Liao, C. Hu, S. Du and H. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585 CrossRef CAS PubMed; (b) Q. F. Sun, S. Sato and M. Fujita, Nat. Chem., 2012, 4, 330 CrossRef CAS PubMed; (c) A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- (a) M. D. Ward, Chem. Commun., 2009, 4487 RSC; (b) B. R. Hall, L. E. Manck, I. S. Tidmarsh, A. Stephenson, B. F. Taylor, E. J. Blaikie, D. A. Vander Griend and M. D. Ward, Dalton Trans., 2011, 40, 12132 RSC; (c) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2011, 133, 858 CrossRef CAS PubMed; (d) H. Fenton, I. S. Tidmarsh and M. D. Ward, Dalton Trans., 2009, 4199 RSC; (e) S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding, W. Clegg, R. W. Harrington and M. D. Ward, Dalton Trans., 2006, 2996 Search PubMed; (f) S. P. Argent, H. Adams, L. P. Harding, T. Riis-Johannessen, J. C. Jeffery and M. D. Ward, New J. Chem., 2005, 29, 904 RSC; (g) A. Stephenson, D. Sykes and M. D. Ward, Dalton Trans., 2013, 42, 6756 RSC.
- (a) A. N. Gusev, V. F. Shulgin, E. Beyjyyev, G. G. Alexandrov, I. L. Eremenko and W. Linert, Polyhedron, 2015, 85, 525 CrossRef CAS; (b) A. N. Gusev, I. Nemec, R. Herchel, E. Bayjyyev, G. A. Nyshchimenko, G. G. Alexandrov, I. L. Eremenko, Z. Travnicek, M. Hasegawa and W. Linert, Dalton Trans., 2014, 43, 7153 RSC; (c) A. N. Gusev, M. Hasegawa, G. A. Nishchymenko, V. F. Shul'gin, S. B. Meshkova, P. Doga and W. Linert, Dalton Trans., 2013, 42, 6936 RSC; (d) A. N. Gusev, V. F. Shulgin, I. O. Ryush, M. Hasegawa, M. A. Kiskin, N. N. Efimov, K. A. Lyssenko, I. L. Eremenko and W. Linert, Eur. J. Inorg. Chem., 2017, 704 CrossRef CAS; (e) A. Gusev, I. Nemec, R. Herchel, V. Shulgin, I. Ryush, M. Kiskin, N. Efimov, E. Ugolkova, V. Minin, K. Lyssenko, I. Eremenko and W. Linert, Dalton Trans., 2019, 48, 3052 RSC.
- (a) U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Nat. Commun., 2014, 5, 5883 CrossRef CAS PubMed; (b) U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Dalton Trans., 2013, 42, 14064 RSC.
- U. García-Couceiro, O. Castillo, J. Cepeda, A. Luque, S. Pérez-Yáñez and P. Román, Inorg. Chim. Acta, 2009, 362, 4212 CrossRef.
- R. Boča, A Handbook of Magnetochemical Formulae, Elsevier, Amsterdam, 2012 Search PubMed.
- (a) J.-W. Zhang, H.-S. Wang and Y. Song, Inorg. Chem. Commun., 2011, 14, 56–60 CrossRef CAS; (b) I. Nemec, R. Herchel and Z. Trávníček, Dalton Trans., 2018, 47, 1614 RSC; (c) R. Herchel, L. Váhovská, I. Potočňák and Z. Trávníček, Inorg. Chem., 2014, 53, 5896 CrossRef CAS PubMed; (d) I. Nemec, H. Liu, R. Herchel, X. Zhang and Z. Trávníček, Synth. Met., 2016, 215, 158 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- (a) A. L. Speck, PLATON, a multipurpose crystallographic tool, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed; (b) Y. Le Page, J. Appl. Crystallogr., 1988, 21, 983 CrossRef.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391 CrossRef CAS.
- (a) K. Yamaguchi, Y. Takahara and T. Fueno, in Appl. Quantum Chem, ed. V. H. Smith, Reidel, Dordrecht, 1986, p. 155 Search PubMed; (b) T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- (a) F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS; (b) R. Izsak and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed; (c) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1907068–1907077. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01391a |
| This journal is © The Royal Society of Chemistry 2019 |