
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic ligand variation to modulate the electrochemical properties of iron and manganese complexes†
Stefan S.
Rohner
 a,
Niklas W.
Kinzel
ab,
Christophe
Werlé
*b and
Walter
Leitner
*ab
a,
Niklas W.
Kinzel
ab,
Christophe
Werlé
*b and
Walter
Leitner
*ab
aInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: walter.leitner@cec.mpg.de
bMax-Planck-Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: christophe.werle@cec.mpg.de
First published on 8th July 2019
Abstract
A series of iron(+III) and manganese(+II) complexes based on the dpaqR-ligand system (dpaq = 2-[bis(pyridine-2-ylmethyl)]amino-N-quinolin-8-yl-acetamide) were investigated using cyclic voltammetry (CV) to elucidate how the electronic properties of the ligands influence the overpotential and catalytic current in the context of water oxidation catalysis. For the Fe-complexes an electron withdrawing NO2 or CF3 group attached to the 5-position of the quinoline unit increased the catalytic current, but only with a simultaneous increase of the overpotential. However, when a pyrene moiety was attached to the dipicolylamine unit of the ligand, the overpotential decreased with concomitant increase of the catalytic current. Although the manganese complexes showed no reversible formation of a molecular catalytically active species for water oxidation, the variations of the ligand scaffold affected largely the same trends in their electrochemical behavior.
Introduction
Energy generation from renewable energy sources such as wind and solar energy is becoming increasingly important on a global level. Due to their intermittent availability, there is a great need for efficient energy storage, for example in the form of chemical bond energy.1 In this context, the electrochemical splitting of H2O into O2 and H2 plays a crucial role.2 However, the water oxidation half-reaction (eqn (1)) is thermodynamically (E = 1.23 V vs. NHE at pH = 0) as well as kinetically demanding. The development of water oxidation catalysts (WOCs) has become a crucial subject of current research to reduce the overpotential and hence facilitate water splitting.3| 2H2O → O2 + 4H+ + 4e− | (1) |
While ruthenium and iridium catalysts have so far shown the highest activity,4 first-row transition metals are increasingly becoming the focus of current research due to their greater abundances and lower costs.5 Formation of the oxygen–oxygen bond is generally assumed to occur via either water nucleophilic attack (WNA) or radical coupling, but controlling the detailed mechanistic cycles remains a challenge.6 The use of the [Fe(dpaqH)(H2O)](ClO4)2 complex (1a) as a WOC in propylene carbonate (PC) with water as a substrate was reported in 2014 by the group of Meyer.7 The catalysis was reported to take place at 1.58 V vs. NHE, resulting in an overpotential of ca. 760 mV for the water oxidation at pH 7.8 In electrolysis experiments a TON of 29 with a faradaic efficiency of 45% was measured. Furthermore, the stability of the complex under reaction conditions was concluded from UV-Vis spectroscopy and CV measurements before and after the electrolysis. For the oxygen–oxygen bond formation, a WNA mechanism was proposed. In this case, the active high-valent FeV-oxo species acts as an electrophile. This reactivity might be influenced by electronic variation of the ligand framework, provided that the active iron center is within the same coordination sphere at this stage.9
Systematic modification of the ligand backbone to influence the electronic structure and steric environment of metal centers is a typical approach to improve performance of lead structures in organometallic catalysis. Nevertheless, there are only few studies concerning the topic of systematic ligand modifications and their intrinsic effects on activity and overpotential for earth-abundant transition metal complexes.10 In this article, we report the influence that various modifications of the electronic properties of the H-dpaqR ligand system have on the catalytic current of the respective iron(+III) complexes (see Fig. 1a).11 In a second step, we investigate the influences of the ligand modifications on the analogous manganese(+II) complexes (see Fig. 1b).12
 | ||
| Fig. 1 Relevant iron (left) and manganese (right) dpaq complexes. | ||
Results and discussion
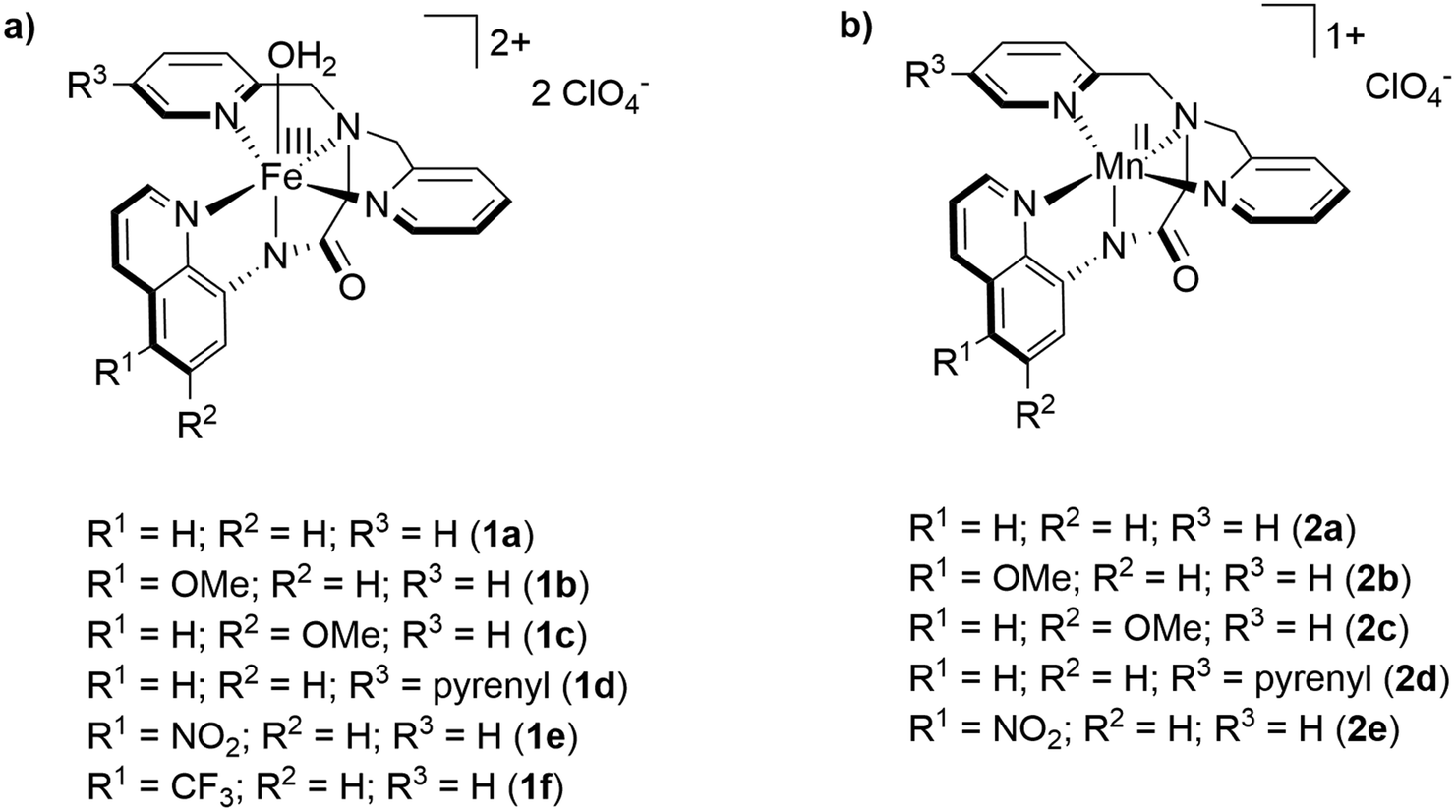
As starting point for the ligand variation, we chose complex 1a as it has been proven to be an active water oxidation catalyst with a TON of 29.7 We reproduced and confirmed the presence of a catalytic current for 1a at 1.58 V vs. NHE in a mixture of propylene carbonate and water (3.2 vol%, Fig. 3). Using this benchmark, we studied complex (1e) in which the proton at the 5-position of the quinoline ring was replaced by an electron withdrawing nitro group,11a to evaluate the effect of increased electrophilicity at the iron center. As shown by cyclic voltammetry (CV) in Fig. 3, complex 1e leads to a catalytic current icat almost twice as high as that of 1a. Further CV investigations support the assumption that the ligand modification did not affect the principle reactivity of the iron core and that the observed catalytic current reflects water oxidation. The square of the catalytic current icat2 of 1e increased linearly with the water concentration (Fig. 2) and a linear dependence of icat on the concentration of 1e was confirmed (Fig. S18†). These data are fully consistent with a WNA mechanism as proposed by the group of Meyer for the unsubstituted catalyst 1a.7 In addition, the presence of the reduction wave FeIII(OH2)2+/FeII(OH2)+ of 1e in the backward scan underlines that the formation of the catalytically active species is reversible (Fig. S18†).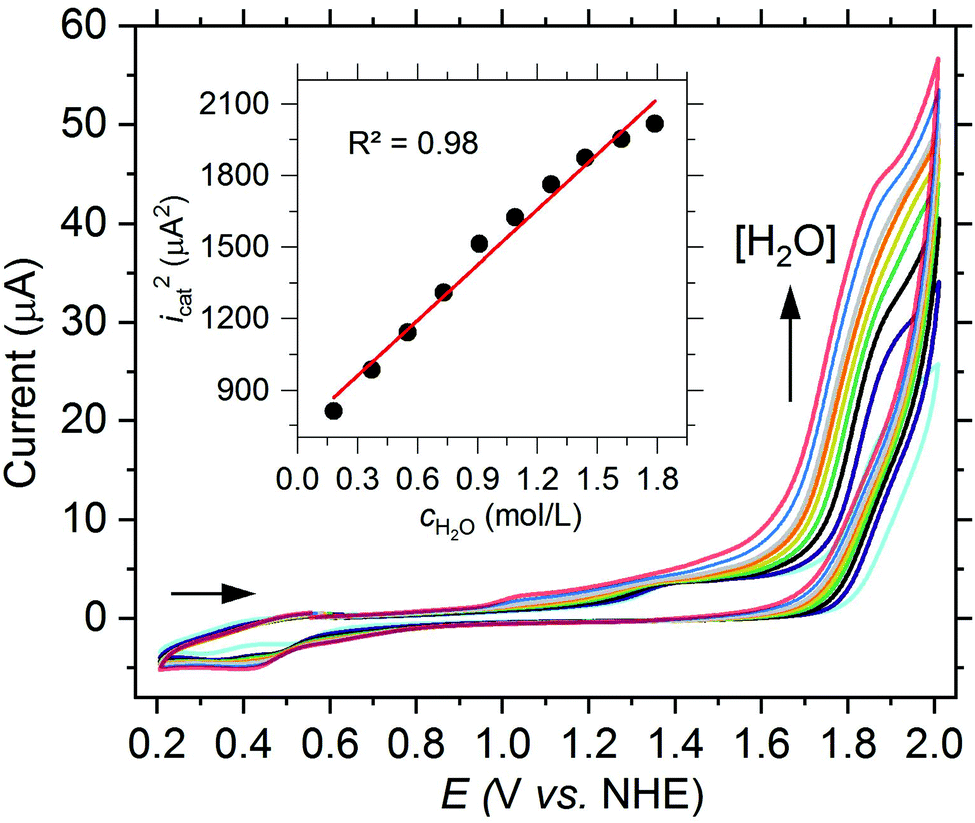 | ||
| Fig. 2 CV of [Fe(dpaq5-NO2)(H2O)](ClO4)2 complex (1e, 1.0 mM) in PC (3.2 vol%, 200 mM LiClO4) with an increasing water concentration (0.2 M–1.8 M) at a scan rate of 75 mV s−1. Inset: Plot of icat2vs. the concentration of water. | ||
 | ||
| Fig. 3 CV of [Fe(dpaq5-R)(H2O)](ClO4)2 complexes (1.0 mM) in PC (3.2 vol% H2O (1.85 M), 200 mM LiClO4) at a scan rate of 75 mV s−1 with R = H (1a, red), OMe (1b, orange), NO2 (1e, blue) or CF3 (1f, green) and in the absence of a complex (grey). Inset: Plot of Eicatvs. the Hammett substitution constants σp of R.13 | ||
Although the catalytic current could be increased by the ligand modification with an electron withdrawing NO2 group, the oxidation of the FeIII(OH2)2+ to the FeV(O)2+ species becomes less favorable due to the lower electron density located at the iron center. This results in a potential of Eicat = 1.89 V vs. NHE, corresponding to a high overpotential of 1.07 V and an increase of more than 300 mV compared to 1a. The influence of the NO2 group can also be observed for the reduction wave FeIII(OH2)2+/FeII(OH2)+ in the backward scan. Compared to 1a the respective potential for 1e is shifted by 130 mV to 0.43 V vs. NHE. To further probe whether the observed effects are based on the electron withdrawing property of the NO2 group, we synthesized a new H-dpaqCF3 ligand (5f, see ESI† for a detailed synthetic procedure) and obtained the corresponding [Fe(dpaqCF3)(H2O)](ClO4)2 complex (1f). A catalytic current at Eicat = 1.82 V vs. NHE further confirms the electronic effect of the para-substituent (see Fig. 3). Conversely, the overpotential should decrease with an increasing electron density at the metal. Hence, in the next step, we used the H-dpaqOMe ligand (5b) with an electron donating methoxy group at the 5-position of the quinoline moiety and investigated the electrochemical properties of the corresponding [Fe(dpaqOMe)(H2O)](ClO4)2 complex (1b). As expected, the oxidation of FeIII(OH2)2+ to FeV(O)2+ is shifted to a more negative potential compared to 1a, but in contrast almost no catalytic current can be observed (see Fig. 3).
When comparing the different values for Eicat in more detail, the potential and the associated Hammett substitution constants (σpara) exhibit a strong correlation, suggesting that the change in onset potential is due to the electronic modification by the different ligand substituents (see inset of Fig. 3). This inductive electronic substitution effect can only be observed at the 5-position of the quinoline moiety which is para to the anionically charged nitrogen of the amide functionality in the dpaq-ligand system. If the methoxy group is incorporated at the neighboring 6-position of the H-dpaq6-OMe-ligand (5c) in [Fe(dpaq6–OMe)(H2O)](ClO4)2 (1c), almost no difference in the onset potential and only a small decrease of the catalytic current is observed compared to the unsubstituted complex 1a (see Fig. S19†).
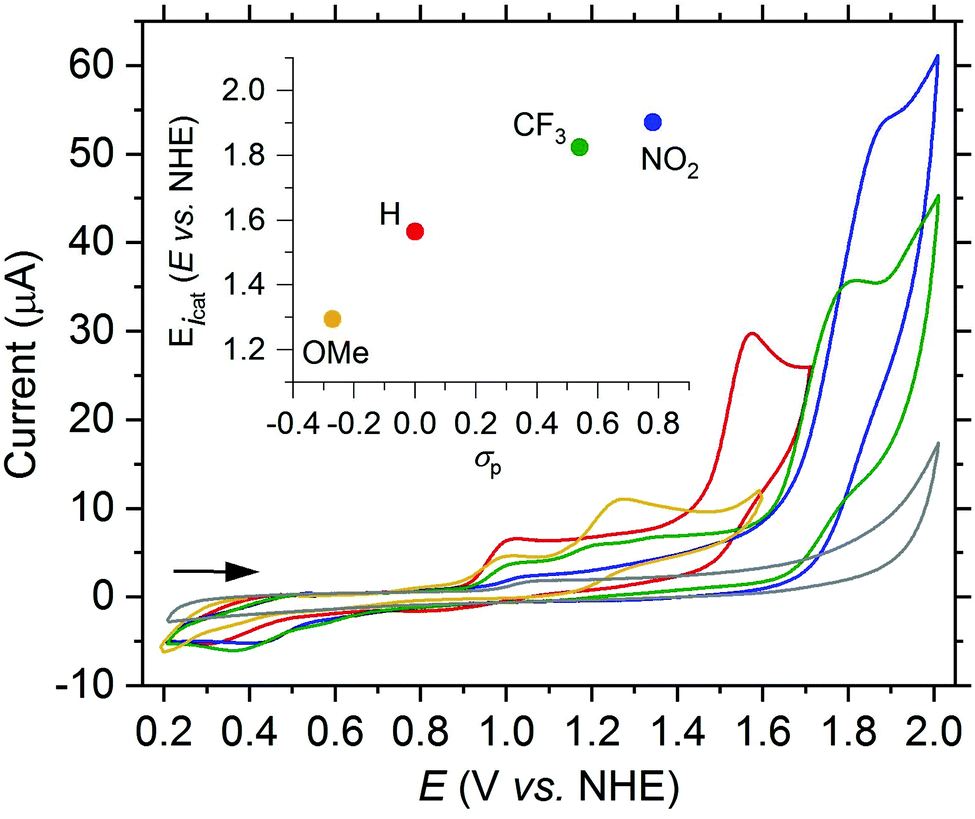
Since higher catalytic currents achieved by electron withdrawing substituents at the quinoline moiety also led to higher onset potentials, hence, the overall electrochemical properties could not be improved substantially relative to the parent ligand toward the aim of water oxidation catalysis. Therefore, as a next step, we modified the dipicolylamine unit of the ligand. We successfully synthesized a new H-dpaq-based ligand with an additional pyrene moiety covalently bound to one of the pyridine rings (5d, see ESI† for a detailed synthetic procedure) and obtained the corresponding [Fe(dpaqPyr)(H2O)](ClO4)2 complex (1d). The pyrene moiety increases the electronic π-conjugation of the ligand and hence may have a positive effect on the overpotential.14 Indeed, a cathodic shift of the onset potential of 70 mV is observed for the pyrene-modified complex 1d compared to 1a as well as a higher catalytic peak current (see Fig. 4). The catalytic current is linearly dependent on the concentration of 1d (see Fig. S20†). Even after running 50 cycles the linear relation remains valid, supporting the conclusion that a species in solution rather than adsorbed at the electrode is responsible for the activity (see Fig. S21,† left). Furthermore, the catalytic current of 1d is slowly decreasing with an increasing number of scans (see Fig. S21,† right), confirming that the higher current is not caused by a partial attachment of the pyrene moiety onto the electrode over time that would result in a concomitant better conductivity.
 | ||
| Fig. 4 CV of 1a (red) and 1d (black) (both 1.0 mM) in PC (3.2 vol% H2O (1.85 M), 200 mM LiClO4) at a scan rate of 75 mV s−1. | ||
To expand the investigations of the influence of the ligand modifications on the catalytic current to other metals with potential for WOCs, we turned our attention from iron to the respective manganese analogues. In a first step, we focused on the unsubstituted [Mn(dpaqH)](ClO4) complex (2a).12 When dissolving the pale yellowish complex in acetonitrile and stirring the solution under air, the color changes to dark brown within a few minutes suggesting an oxidation to a MnIII species similar to what was recently reported for the [Mn(dpaqH)](OTf) complex by the group of Jackson.15 By detailed NMR investigations they showed that an equilibrium between a μ-O bridged dimer and two mononuclear [MnIII(OH)(dpaq)]+ species is formed. In the presence of water, the equilibrium is shifted to the mononuclear species.15
The cyclic voltammogram of 2a dissolved in MeCN shows three quasi-reversible oxidation waves at E1/2 = 0.62 V, 1.33 V and 1.58 V vs. NHE at a scan rate of 300 mV s−1 (see Fig. S22†). At slower scan rates, the third oxidation wave becomes irreversible, showing that the oxidized species is stable only for a short time. Upon addition of water to the solution, a large increase in current can be observed following the third oxidation wave, suggesting water oxidation taking place (see Fig. S23,† left). The square of the peak current at E = 1.58 V vs. NHE increases linearly with [H2O] (see Fig. S23,† right). Furthermore, the current increases linearly with [Mn] (see Fig. S24†) which is in accordance with a reaction with first-order kinetics.16
At low water concentrations the oxidation wave at E1/2 = 1.33 V vs. NHE remains reversible. However, at higher water concentrations the reversibility decreases, as shown by an increasing ΔEp. The oxidation wave at 0.62 V vs. NHE, which is visible in pure acetonitrile (see Fig. S22†), disappears almost completely upon addition of water. In acetonitrile this oxidation wave presumably corresponds to the MnII/III couple. Like mentioned above, the dissolved MnII species can be oxidized under air to a μ-O bridged MnIII-dimer, which then forms a [MnIII(OH)(dpaq)]+ species in the presence of water. If this species was not reducible to a MnII species, it would explain why the MnII/III oxidation wave disappears after addition of water.
A large current increase can be observed not only in acetonitrile but also in aqueous buffer systems at a potential of 1.56 V vs. NHE when using the [Mn(dpaqH)](ClO4) complex (2a) pre-dissolved in MeCN. Scan rate dependent CVs show that the normalized current (i/ν0.5) at 1.56 V vs. NHE decreases with increasing scan rate, consistent with a rate-determining step (see Fig. S25†). Just as in acetonitrile, the current at 1.56 V vs. NHE in a borate buffer linearly increases with the complex concentration, indicating a reaction with first-order kinetics with respect to the complex (see Fig. 5).
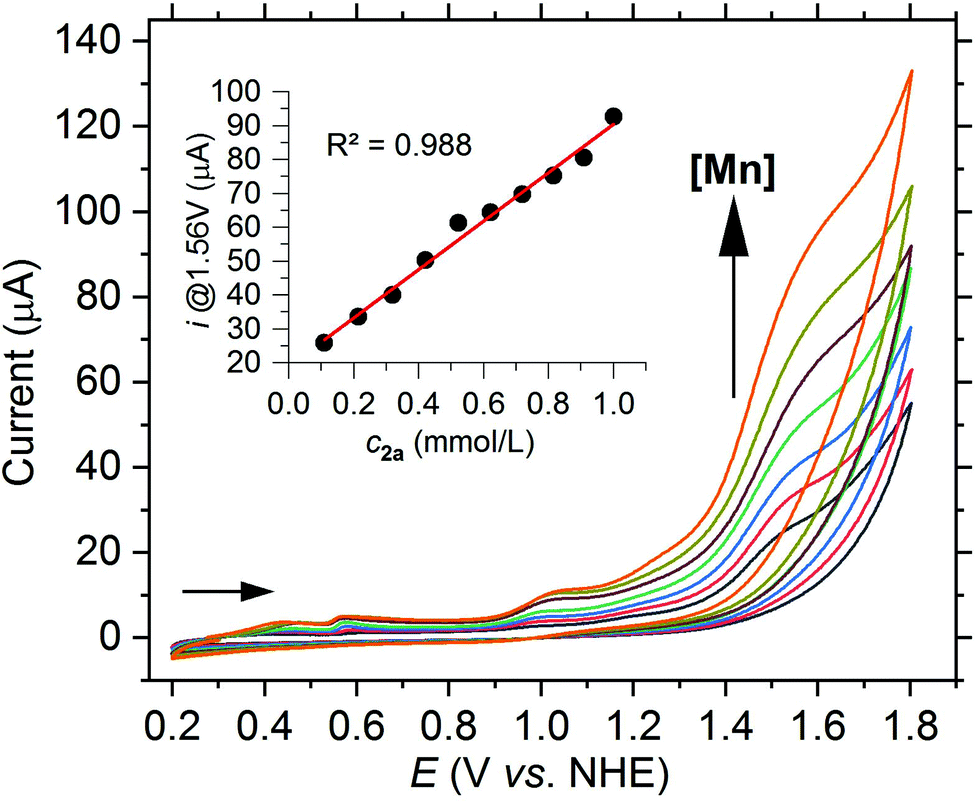 | ||
| Fig. 5 CV of 2a with increasing concentration from 0.1 mM up to 1.0 mM in borate buffer (0.2 M, pH 8) at a scan rate of 50 mV s−1. Inset: Plot of the current at 1.56 V vs. the concentration. | ||
However, in contrast to the [Fe(dpaqR)(H2O)](ClO4)2 complexes (1a–f) in propylene carbonate and the [Mn(dpaqH)](ClO4) complex (2a) in acetonitrile, 2a in borate buffer does not show a reversible reduction wave in the reverse scan. Therefore, it must be assumed that the structure of the complex changes irreversibly under the given reaction conditions.
In a next step, the pH-dependence of the oxidation waves of complex 2a was investigated by CV measurements in a borate buffer solution.17 The oxidation wave at 1.04 V vs. NHE shifts to a more negative potential with increasing pH (see Fig. 6). If the potential is plotted vs. the pH value in a Pourbaix diagram, a slope of −67 mV pH−1 is achieved, which agrees with a proton-coupled electron transfer (PCET) reaction (see Fig. S26†). We thus attribute the wave to the oxidation of [MnIII–OH]+ to a [MnIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]+ species. In contrast, the potential of the current at 1.56 V vs. NHE is independent of the pH value. However, the current significantly increases with the pH value (see Fig. 6).
O]+ species. In contrast, the potential of the current at 1.56 V vs. NHE is independent of the pH value. However, the current significantly increases with the pH value (see Fig. 6).
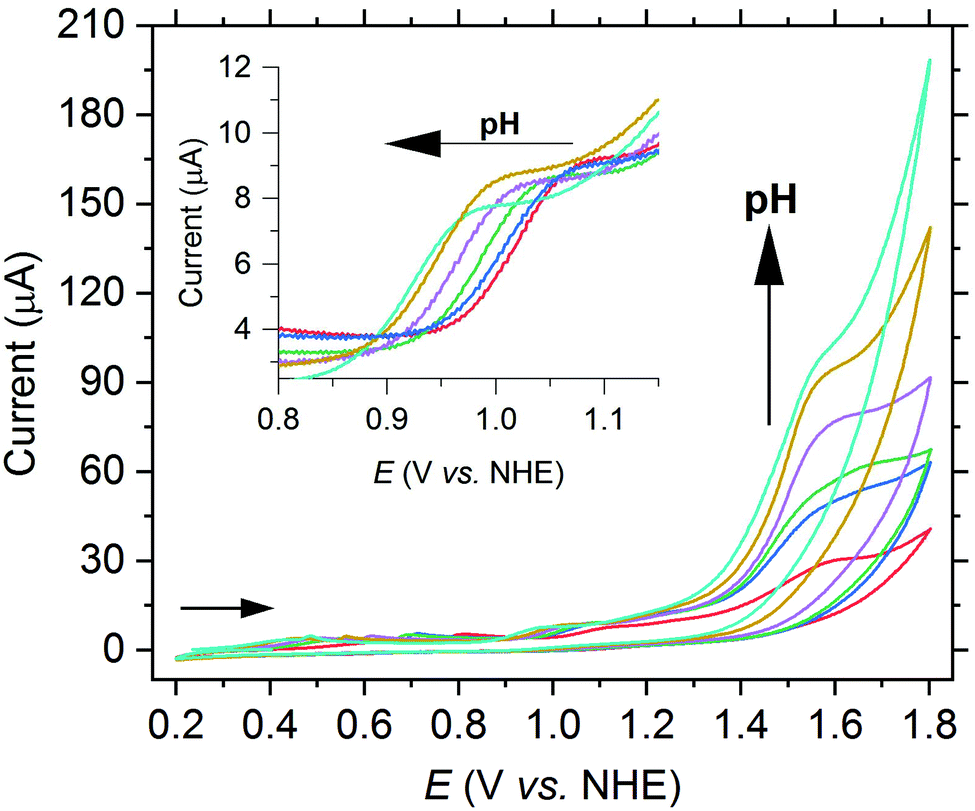 | ||
| Fig. 6 CV of 2a (1.0 mM) with increasing pH from 7.0 up to 9.4 in borate buffer (0.2 M) at a scan rate of 50 mV s−1. | ||
Thereafter, we probed the effects of modifying the electronic properties of the dpaq-ligand system on the behavior of the [Mn(dpaqR)](ClO4) complexes in analogy to the iron complexes (see Fig. 7).
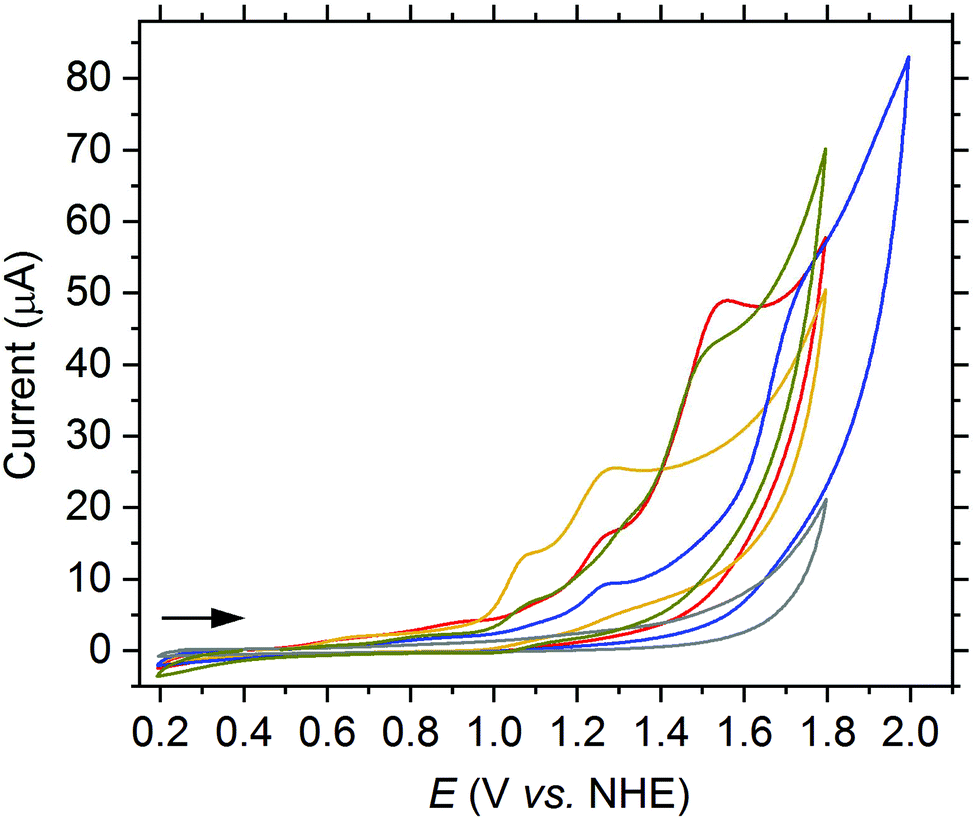 | ||
| Fig. 7 CV of [Mn(dpaqR)](ClO4) complexes (1.0 mM) in phosphate buffer (0.1 M, pH 7) at a scan rate of 50 mV s−1 with R = H (2a, red), 5-OMe (2b, orange), 6-OMe (2c, green) or NO2 (2e, blue) and in the absence of a complex (grey). | ||
Interestingly, the modifications at the quinoline moiety of the dpaq-ligand showed the same trend in the electrochemical response of the Mn-complexes as in the Fe-complexes. The electron withdrawing NO2 group of 2e elevates the potential of the current increase by about 210 mV compared to 2a. In contrast, the electron donating OMe group at the 5-position of 2b reduces the oxidation potential of the second oxidation wave by approximatively 270 mV. However, all complexes show an irreversible degradation under the test conditions in aqueous solution, irrespective of the ligand modification.
We then used the H-dpaqPyr ligand (5d) to synthesize the [Mn(dpaqPyr)]ClO4 complex (2d). With 2d the potential of the peak current is reduced by 25 mV in a PC solution with water (3.2 vol%) compared to 2a, while the current itself is slightly increased (see Fig. S27†). Hence, a higher electronic π-delocalization of the pyrene moiety shows the same effect on the electrochemical behavior as for the analogous iron complex 1d, albeit the influence is significantly lower.
In summary, we studied the electrocatalytic properties of a series of iron and manganese complexes based on the H-dpaqR ligand system in the context of electrochemical water oxidation. Modification of electronic properties by variation of substituents in the ligand framework of the iron complexes was found to exhibit a big influence on the overpotential as well as the catalytic current. Electron withdrawing groups at the 5-position of the quinoline moiety, such as NO2 and CF3 increase the catalytic current due to the higher electrophilicity at the active species, albeit on the expense to increase the overpotential simultaneously. In contrast, electron donating groups, such as OMe, increase the electron density at the metal center to a point where nucleophilic attack by water can no longer take place. However, by incorporating a pyrene moiety into one of the pyridine groups, the overpotential for the [Fe(dpaqPyr)(H2O)](ClO4)2 complex (1d) was reduced and the catalytic current was increased at the same time. This may be at least partly due to the greater electronic π-conjugation of pyrene. Although, the investigated manganese complexes showed no reversible catalytic water oxidation behavior, the ligand modifications affected the electrochemical properties in the same way as it was the case with the iron complexes. While the stability of the ligand framework under oxidative conditions poses severe limitations for the systems discussed herein, these results substantiate the possibility to control and improve the catalytic performance of first-row metal-based WOCs by systematic variation of their coordinative environment. This may eventually provide design criteria that can be translated into catalytic materials.18
Experimental section
General considerations
All syntheses involving air sensitive compounds were carried out under argon using flame-dried glassware, ensuring rigorously inert conditions. The chemicals were purchased from Sigma-Aldrich, Alfa-Aesar, abcr, Acros Organics, TCI chemicals and used without further purification. The H-dpaqR ligands (5a–f) were synthesized based on modified literature procedures (for further details see ESI†). The different solvents were purified using solvent purification-systems and were stored and handled appropriately under argon. NMR-spectra were recorded on Bruker AV400, AS400 or AS600 at the indicated temperature. Chemical shifts (δ) are given in ppm and were referenced against the solvent signal (acetonitrile-d3: δH = 1.94 ppm and δC = 118.3 ppm; CDCl3: δH = 7.26 ppm and δC = 77.1 ppm; CD2Cl2: δH = 5.32 ppm and δC = 53.8; CD3OD: δH = 3.31 ppm and δC = 49.0).19 The coupling constants (J) are given in Hz. Infrared (IR) spectra were recorded using a Bruker Alpha–P FT-IR spectrometer. ESI-HRMS spectra were recorded on a ThermoFisher Scientific LTQ-Orbitrap XL spectrometer. Elemental analyses (C, H, N) were recorded at the Mikroanalytisches Laboratorium Kolbe in Oberhausen, Germany. Cyclic voltammetry (CV) was performed with a Metrohm Autolab PGSTAT101 potentiostat and processed using Metrohm Autolab Nova software 2.1. A three-electrode setup constituted of a glassy-carbon working electrode (3 mm diameter), a platinum-wire counter electrode and an Ag/AgCl reference electrode (3 M NaCl) was used. All voltammograms were referenced against the normal hydrogen electrode (NHE) by adding 0.208 V to the measured potential against the Ag/AgCl reference electrode. If the catalytic current was not pronounced as a clear peak but only as a shoulder, the current was measured at the inflection point of the curve. The glassy-carbon working electrode was conditioned prior to each experiment using a 0.05 μm alumina paste on a polishing pad and washed with ultra-pure water.General procedure for the synthesis of [Fe(dpaqR)(H2O)](ClO4)2 complexes (1a–f)
Caution! Perchlorate salts of metal complexes are potentially explosive. The complexes should be prepared only in small quantities, and should be handled with great care. The syntheses were performed according to a modified reported procedure.11b A solution of Fe(ClO4)3·6H2O in methanol was added to a solution of the H-dpaqR ligand and triethylamine in methanol or dichloromethane. The mixture was allowed to stand overnight at room temperature before the precipitate was recovered by filtration over fritted glass. The residue was washed with diethyl ether (10 mL) and dried under high vacuum.
1a. Fe(ClO4)3·6H2O (110 mg, 0.24 mmol) in methanol (1 mL), H-dpaqH ligand (5a, 99.7 mg, 0.26 mmol) and triethylamine (0.036 mL, 0.13 mmol) in methanol (1 mL). Yield = 58.1 mg, 0.09 mmol, 34%. Dark green solid. Anal. calcd for [Fe(dpaqH)(H2O)](ClO4)2(H2O)(MeOH)0.5: C: 40.95; H, 3.80; N, 10.16; found: C: 40.84; H, 3.48; N, 9.82. HRMS-ESI (m/z): calcd for C23H20FeN5O+: 438.10173; found: 438.09970. FT-IR: 1616 cm−1 (amide CO).
1b. Fe(ClO4)3·6H2O (110 mg, 0.24 mmol) in methanol (1 mL), H-dpaq5-OMe ligand (5b, 107.5 mg, 0.26 mmol) and triethylamine (0.036 mL, 0.13 mmol) in methanol (1 mL). Yield = 58.2 mg, 0.09 mmol, 32%. Dark green solid. Anal. calcd for [Fe(dpaq5–OMe)(H2O)](ClO4)2(H2O)0.4(MeOH)0.3: C: 41.57; H, 3.73; N, 9.98; found: C: 41.61; H, 3.76; N, 10.04. HRMS-ESI (m/z): calcd for C24H22FeN5O2+: 468.11174; found: 468.11139. FT-IR: 1609 cm−1 (amide CO).
1c. Fe(ClO4)3·6H2O (110 mg, 0.24 mmol) in methanol (1 mL), H-dpaq6-OMe ligand (5c, 107.5 mg, 0.26 mmol) and triethylamine (0.036 mL, 0.13 mmol) in methanol (1 mL). Yield = 46.9 mg, 0.06 mmol, 25%. Dark green solid. Anal. calcd for [Fe(dpaq6–OMe)(H2O)](ClO4)2(MeOH)0.7: C: 41.92; H, 3.82; N, 9.90; found: C: 41.91; H, 3.87; N, 9.95. HRMS-ESI (m/z): calcd for C24H22FeN5O2+: 468.11174; found: 468.11209. FT-IR: 1611 cm−1 (amide CO).
1d. Fe(ClO4)3·6H2O (55 mg, 0.12 mmol) in methanol (0.3 mL), H-dpaqPyr (5d, 76.0 mg, 0.13 mmol) and triethylamine (0.018 mL, 0.13 mmol) in dichloromethane (0.5 mL). The mixture was allowed to stand at −20 °C overnight. Yield = 47.2 mg, 0.06 mmol, (46%). Dark green solid. Anal. calcd for [Fe(dpaqPyr)(H2O)](ClO4)2(H2O)2: C: 52.55; H, 3.84; N, 7.86; found: C: 52.54; H, 3.68; N, 7.70. HRMS-ESI (m/z): [M + OMe]+ calcd for C39H28FeN5O2+: 669.18327; found: 669.18250. FT-IR: 1606 cm−1 (amide CO).
1e. Fe(ClO4)3·6H2O (110 mg, 0.24 mmol) in methanol (1 mL), H-dpaqNO2 ligand (5e, 111.4 mg, 0.26 mmol) and triethylamine (0.036 mL, 0.13 mmol) in methanol (1 mL). Yield = 130.1 mg, 0.19 mmol, 72%. Dark green-brownish solid. Anal. calcd for [Fe(dpaqNO2)(H2O)](ClO4)2: C: 39.45; H, 3.02; N, 12.00; found: C: 40.26; H, 3.99; N, 12.87. HRMS-ESI (m/z): calcd for C23H19FeN6O3+: 483.08681; found: 483.08484. FT-IR: 1611 cm−1 (amide CO).
1f. Fe(ClO4)3·6H2O (110 mg, 0.24 mmol) in methanol (1 mL), H-dpaqCF3 ligand (5f, 117.4 mg, 0.26 mmol) and triethylamine (0.036 mL, 0.13 mmol) in methanol (1 mL). Yield = 46.9 mg, 0.065 mmol, 39%. Dark green-purple solid. Anal. calcd for [Fe(dpaqCF3)(H2O)](ClO4)2: C: 39.86; H, 2.93; N, 9.68; found: C: 40.04; H, 3.11; N, 9.75. HRMS-ESI (m/z): calcd for C24H19F3FeN5O+: 506.08911; found: 506.08868. FT-IR: 1609 cm−1 (amide CO).
General procedure for the synthesis of [Mn(dpaqR)](ClO4) complexes (2a–e)
The syntheses were performed according to a modified reported procedure.12 A solution of Mn(ClO4)2·6H2O in acetonitrile was added to a solution of the H-dpaqR ligand and triethylamine in dichloromethane and/or acetonitrile. The reaction mixture was stirred at room temperature for 2 h before the precipitate was recovered by filtration over fritted glass. The residue was washed with acetonitrile (5 mL) and diethyl ether (5 mL) and dried under high vacuum.
2a. Mn(ClO4)2·6H2O (110 mg, 0.31 mmol) in acetonitrile (0.5 mL), H-dpaqH ligand (5a, 99.7 mg, 0.26 mmol) and triethylamine (0.050 mL, 0.36 mmol) in acetonitrile (2 mL). Yield = 89.3 mg, 0.17 mmol, 64%. Pale beige solid. Anal. calcd for [Mn(dpaqH)](ClO4)(H2O)1.3: C: 49.31; H, 4.07; N, 12.50; found: C: 49.46; H, 4.27; N, 12.50. HRMS-ESI (m/z): calcd for C23H20MnN5O+: 437.10483; found: 437.10275. FT-IR: 1542 cm−1 (amide CO).
2b. Mn(ClO4)2·6H2O (110 mg, 0.31 mmol) in acetonitrile (0.5 mL), H–dpaq5–OMe ligand (5b, 107.5 mg, 0.26 mmol) and triethylamine (0.050 mL, 0.36 mmol) in acetonitrile (2 mL). The reaction mixture had to be stored at −20 °C for several days before the precipitate could be recovered. Yield = 85.5 mg, 0.15 mmol, 58%. Pale beige-yellowish solid. HRMS-ESI (m/z): calcd for C24H22MnN5O2+: 467.11485; found: 467.11456. FT-IR: 1549 cm−1 (amide CO).
2c. Mn(ClO4)2·6H2O (110 mg, 0.31 mmol) in acetonitrile (0.5 mL), H–dpaq6–OMe ligand (5c, 107.5 mg, 0.26 mmol) and triethylamine (0.050 mL, 0.36 mmol) in acetonitrile (2 mL). Yield = 82.5 mg, 0.15 mmol, 56%. Pale beige-yellowish solid. Anal. calcd for [Mn(dpaq6–OMe)](ClO4)(H2O)3.4: C: 45.89; H, 4.62; N,11.15; found: C: 45.52; H, 4.12; N,11.05. HRMS-ESI (m/z): calcd for C24H22MnN5O2+: 467.11485; found: 467.11429. FT-IR: 1542 cm−1 (amide CO).
2d. Mn(ClO4)2·6H2O (110 mg, 0.31 mmol) in acetonitrile (0.5 mL), H–dpaqPyr (5d, 76.0 mg, 0.13 mmol) and triethylamine (0.025 mL, 0.18 mmol) in a mixture of dichloromethane (0.5 mL) and acetonitrile (2 mL). Yield = 62.7 mg, 0.09 mmol, 65%. Dark brownish solid. Anal. calcd for [Mn(dpaqPyr)](ClO4)(H2O)2.8: C: 59.48; H, 4.30; N, 8.89; found: C: 59.43; H, 4.17; N, 8.87. HRMS-ESI (m/z): calcd for C39H28MnN5O+: 637.16744; found: 637.16681. FT-IR: 1539 cm−1 (amide CO).
2e. Mn(ClO4)2·6H2O (110 mg, 0.31 mmol) in acetonitrile (0.5 mL), H-dpaqNO2 ligand (5e, 111.4 mg, 0.26 mmol) and triethylamine (0.050 mL, 0.36 mmol) in acetonitrile (2 mL). Yield = 105.9 mg, 0.18 mmol, 70%. Orange solid. Anal. calcd for [Mn(dpaqNO2)](ClO4)(H2O)3.6: C: 42.72; H, 4.08; N, 13.00; found: C: 42.15; H, 3.41; N, 12.87. HRMS-ESI (m/z): calcd for C23H20MnN6O3+: 482.08991; found: 482.08755. FT-IR: 1531 cm−1 (amide CO).
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge generous financial supports by the MPG and the RWTH Aachen University. This work was supported by the Bundesministerium für Bildung und Forschung as part of the project “MANGAN” (03SF0508). The authors acknowledge Yannick Meiners and Maurice W. Vennewald for their experimental support and Dr Markus Hölscher for fruitful discussion. Open Access funding provided by the Max Planck Society.References
- (a) R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 4436–4439 CrossRef PubMed; (b) R. Schlögl, Top. Catal., 2016, 59, 772–786 CrossRef.
- (a) H. Dau, E. Fujita and L. Sun, ChemSusChem, 2017, 10, 4228–4235 CrossRef CAS PubMed; (b) D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS PubMed.
- (a) X. Du, Q. Shao and X. Zhang, Dalton Trans., 2019, 48, 1322–1331 RSC; (b) X. Q. Du, X. S. Zhang, Y. Li and M. Zhao, Int. J. Hydrogen Energy, 2018, 43, 19955–19964 CrossRef CAS; (c) X. Q. Du, Z. Yang, Y. Li, Y. Q. Gong and M. Zhao, J. Mater. Chem. A, 2018, 6, 6938–6946 RSC; (d) J. Li, R. Güttinger, R. Moré, F. Song, W. Wan and G. R. Patzke, Chem. Soc. Rev., 2017, 46, 6124–6147 RSC; (e) T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148–6169 RSC; (f) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed; (g) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed; (h) D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218–227 RSC.
- (a) J. Creus, R. Matheu, I. Penafiel, D. Moonshiram, P. Blondeau, J. Benet-Buchholz, J. Garcia-Anton, X. Sala, C. Godard and A. Llobet, Angew. Chem., Int. Ed., 2016, 55, 15382–15386 CrossRef CAS; (b) J. M. Thomsen, D. L. Huang, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2015, 44, 12452–12472 RSC; (c) S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef CAS PubMed.
- (a) N. Wang, H. Q. Zheng, W. Zhang and R. Cao, Chin. J. Catal., 2018, 39, 228–244 CrossRef CAS; (b) M. D. Karkas and B. Akermark, Dalton Trans., 2016, 45, 14421–14461 RSC; (c) M. Kondo and S. Masaoka, Chem. Lett., 2016, 45, 1220–1231 CrossRef CAS; (d) M. A. Asraf, H. A. Younus, M. Yusubov and F. Verpoort, Catal. Sci. Technol., 2015, 5, 4901–4925 RSC; (e) A. R. Parent and K. Sakai, ChemSusChem, 2014, 7, 2070–2080 CrossRef CAS PubMed; (f) W. Schöfberger, F. Faschinger, S. Chattopadhyay, S. Bhakta, B. Mondal, J. A. Elemans, S. Müllegger, S. Tebi, R. Koch, F. Klappenberger, M. Paszkiewicz, J. V. Barth, E. Rauls, H. Aldahhak, W. G. Schmidt and A. Dey, Angew. Chem., Int. Ed., 2016, 55, 2350–2305 CrossRef PubMed; (g) W. T. Lee, S. B. Muñoz 3rd, D. A. Dickie and J. M. Smith, Angew. Chem., Int. Ed., 2014, 53, 9856–9859 CrossRef CAS PubMed.
- I. Funes-Ardoiz, P. Garrido-Barros, A. Llobet and F. Maseras, ACS Catal., 2017, 7, 1712–1719 CrossRef CAS.
- M. K. Coggins, M. T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS PubMed.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- We note that secondary ligand effects resulting from the formation of active catalytic clusters might equally apply.
- (a) P. Xu, S. Hu, H. D. Zhang and X. Zheng, Phys. Chem. Chem. Phys., 2018, 20, 14919–14926 RSC; (b) J. Shen, M. Wang, J. Gao, H. Han, H. Liu and L. Sun, ChemSusChem, 2017, 10, 4581–4588 CrossRef CAS PubMed; (c) T. Chantarojsiri, Y. Sun, J. R. Long and C. J. Chang, Inorg. Chem., 2015, 54, 5879–5887 CrossRef CAS PubMed; (d) Z. Codola, I. Garcia-Bosch, F. Acuna-Pares, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 8042–8047 CrossRef CAS PubMed; (e) W. A. Hoffert, M. T. Mock, A. M. Appel and J. Y. Yang, Eur. J. Inorg. Chem., 2013, 2013, 3846–3857 CrossRef CAS.
- (a) Y. Hitomi, K. Arakawa and M. Kodera, Chem. – Eur. J., 2013, 19, 14697–14701 CrossRef; (b) Y. Hitomi, K. Arakawa, T. Funabiki and M. Kodera, Angew. Chem., Int. Ed., 2012, 51, 3448–3452 CrossRef CAS PubMed.
- Y. Hitomi, Y. Iwamoto and M. Kodera, Dalton Trans., 2014, 43, 2161–2167 RSC.
- D. H. Mcdaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS.
- P. Garrido-Barros, C. Gimbert-Surinach, D. Moonshiram, A. Picon, P. Monge, V. S. Batista and A. Llobet, J. Am. Chem. Soc., 2017, 139, 12907–12910 CrossRef CAS PubMed.
- (a) D. B. Rice, S. D. Jones, J. T. Douglas and T. A. Jackson, Inorg. Chem., 2018, 57, 7825–7837 CrossRef CAS PubMed; (b) G. B. Wijeratne, B. Corzine, V. W. Day and T. A. Jackson, Inorg. Chem., 2014, 53, 7622–7634 CrossRef CAS PubMed.
- (a) E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS; (b) C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat, F. M'Halla and J. M. Savéant, J. Electroanal. Chem., 1980, 113, 19–40 CrossRef CAS.
- Starting at pH 7, the pH value was adjusted by adding small amounts of NaOH (0.2 M) to keep the overall ion concentration in solution at a constant level.
- J. Q. Guan, Z. Y. Duan, F. X. Zhang, S. D. Kelly, R. Si, M. Dupuis, Q. G. Huang, J. Q. Chen, C. H. Tang and C. Li, Nat. Catal., 2018, 1, 870–877 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of all used ligands and complexes as well as supporting figures (PDF). See DOI: 10.1039/c9dt01343a |
| This journal is © The Royal Society of Chemistry 2019 |