
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphine-substituted 1,2,3-triazoles as P,C- and P,N-ligands for photoluminescent coinage metal complexes†‡
Tim P.
Seifert
a,
Sebastian
Bestgen
 a,
Thomas J.
Feuerstein
a,
Sergei
Lebedkin
b,
Felix
Krämer
a,
Christian
Fengler
a,
Michael T.
Gamer
a,
Manfred M.
Kappes
bc and
Peter W.
Roesky
*a
a,
Thomas J.
Feuerstein
a,
Sergei
Lebedkin
b,
Felix
Krämer
a,
Christian
Fengler
a,
Michael T.
Gamer
a,
Manfred M.
Kappes
bc and
Peter W.
Roesky
*a
aInstitute for Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
bInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Herrmann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cInstitute for Physical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany
First published on 14th May 2019
Abstract
A series of homo- and hetero-polynuclear coinage metal complexes based on a phosphine-substituted 1,2,3-triazole system is presented. Besides the P,N-ligand 1-benzyl-4-(diphenylphosphanyl)-1H-1,2,3-triazole (LPN), the P,C-donor ligand 1-benzyl-4-(diphenylphosphanyl)-3-methyl-1H-1,2,3-triazolium (LPCH+), featuring a potential mesoionic carbene moiety, was obtained by methylation of LPN. Starting from the monogold chloride complexes AuCl(LPN/LPC), the syntheses of a heterotrinuclear Au2Cu complex as well as a digold carbene complex are described. The multinuclear complexes show metallophilic interactions. Their photophysical properties were investigated by temperature-dependent photoluminescence (PL) measurements. In particular, the digold complex shows interesting PL properties including narrow exciton peaks arising in the excitation and emission spectra below 50 K. These might be related to the molecular ‘chains’ in the crystal structure of the digold complex, formed by phenyl ligand stacking.
Introduction
Carbenes and particularly imidazole-based N-heterocyclic carbenes (NHCs) are presently well established ligands which have been widely used in organometallic chemistry for the last three decades.1,2 Their strong σ-donor properties allow the formation and stabilization of organometallic complexes comprising almost every metal or metalloid.3–7 Typically, NHCs serve as spectator ligands, however, reactions transforming the NHC itself (such as a ring expansion) are known as well.8–12 Their great application potential has led to further development of ligands with tailor-made steric and electronic properties derived from classical NHCs. These include remote, abnormal, cyclic alkylamino and mesoionic carbenes (Scheme 1).13–20 The latter are either based on substituted imidazole, pyrazole or 1,2,3-triazole backbones, respectively.21,22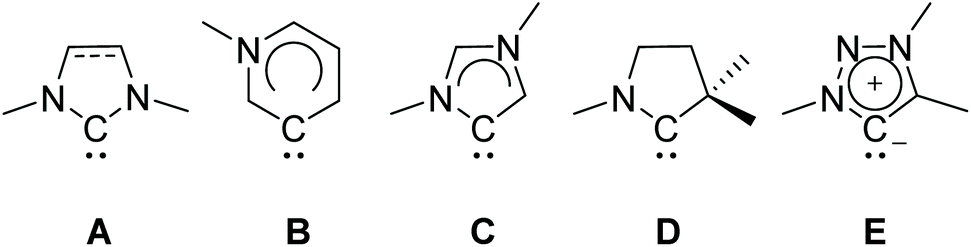 | ||
| Scheme 1 Schematic drawing of NHC types: classical N-heterocyclic (NHCs; A), remote (rNHC; B), abnormal (aNHC; C), cyclic alkylamino (cAAC; D) and mesoionic carbenes (MIC; E). | ||
The 1,2,3-triazol-5-ylidenes (Scheme 1, type E) are typically stronger donors than their 1,2,4-triazolylidene isomers and other five-membered NHCs.23–25 They can be readily prepared on a multi-gram scale by using the well-established CuAAC “click” reaction. The variety of substituents on the carbon and nitrogen atoms, which affect the steric and electronic properties, is very large and practically only limited by the stability of the parent azide and alkyne precursors (Scheme 2).22,26,27
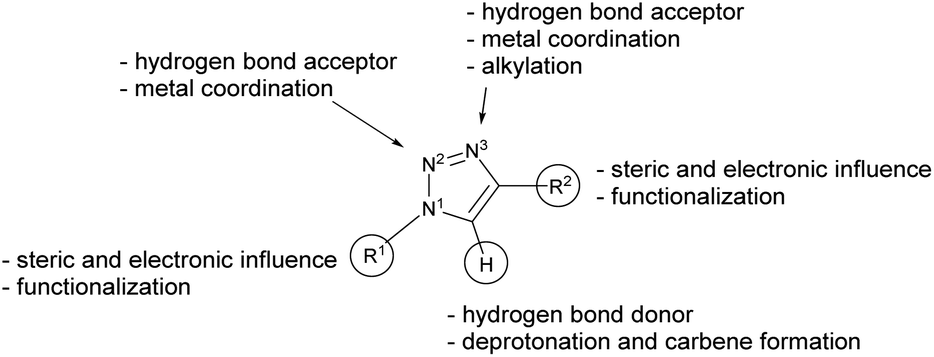 | ||
| Scheme 2 Chemical properties of the triazole scaffold.28 | ||
The free carbenes are generated by deprotonation of (typically N3-alkylated) triazolium salts.22 A donor-substituted triazolium salt thus exhibits orthogonal donor sites for subsequent and controllable coordination of metals. Following this concept, we herein present a methylated phosphine-substituted 1,2,3-triazol-5-ylidene LPC, as well as a phosphine-substituted triazole LPN as ligand systems (Scheme 3). LPN was reported before by C. Alayrac and A.-C. Gaumond, but no further metal coordination has been carried out.29
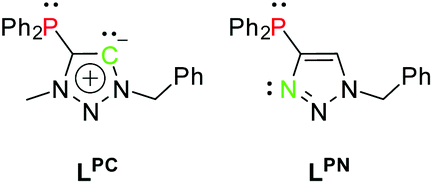 | ||
| Scheme 3 Ligand systems LPC and LPN. | ||
Following Pearson's concept of hard and soft acids and bases,30,31 the nitrogen coordination site in LPN is suitable for the coordination of rather hard metals, whereas the phosphine prefers to coordinate to soft metals. Furthermore, the ligands LPC and LPN exhibit a rigid scaffold and a spatial proximity of their respective donor sites. These are the key factors for preparation of heterometallic complexes with metal–metal interactions, i.e. complexes showing the phenomenon of metallophilicity.32,33 Those interactions are commonly observed in transition metal complexes of heavy metals. The most prominent examples feature d10 electron configurations such as in the monovalent coinage metals.34–37 The strongest interactions arise in multinuclear complexes with Au(I). The term ‘aurophilicity’ has therefore been established as early as 1989 by H. Schmidbaur38,39 and intensive research on aurophilicity from a theoretical and experimental point of view has since been carried out.40–44 However, cooperative effects and metallophilicity in complexes with different metals and the relation between luminescence properties and metal–metal interactions are still actively pursued research fields.45–58 The different coordination sites in LPC and LPN therefore allow the synthesis of multimetallic complexes for the investigation of supported metallophilic interactions, which then can be systematically studied with respect to their photoluminescence properties.
Results and discussion
The synthesis of the two ligands LPC and LPN was achieved by a stepwise synthetic procedure, starting from ethynyldiphenylphosphine and benzylazide in a CuAAC click reaction.59 As trivalent phosphines are known to react with azides to yield phosphazenes (Staudinger reduction),60,61 the phosphine was first converted to the corresponding ethynyldiphenylphosphine oxide 1, using aqueous H2O2.62 The subsequent click reaction between 1 and benzylazide gave the phosphinoxide-substituted triazole 2 in 78% yield (for crystal structure, see Fig. S2‡). In the next step, 2 was alkylated with methyl trifluoromethanesulfonate (MeOTf) in CH2Cl2 to give the N-methylated triazolium salt 3. Upon methylation, a broad resonance at δ = 15.2 ppm is observed in the 31P{1H} NMR spectrum of compound 3, which is significantly broadened compared to the sharp resonance at δ = 16.6 ppm for 2. The broadening may be attributed to the delocalization of the positive charge. Although minor impurities were detected in the NMR spectra, compound 3 was used without further purification. In the last step, the reduction of 2 and 3 to LPN (4) and LPCH+·OTf− (5) respectively, was conducted with PhSiH3 as reducing agent (Scheme 4).63 In the 31P{1H} NMR spectrum, the two ligands LPN and LPCH+·OTf− exhibit almost identical chemical shifts at δ = −31.9 ppm (LPN) and −31.5 ppm (LPCH+·OTf−).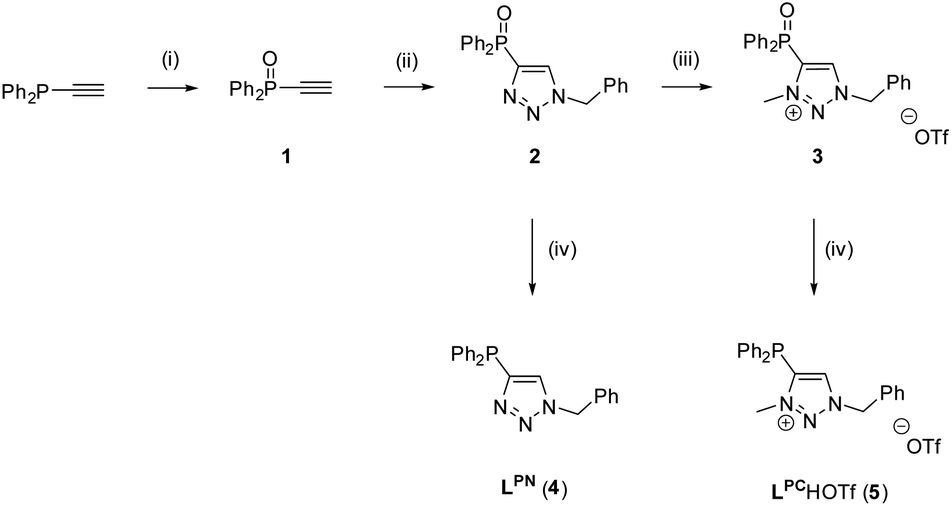 | ||
| Scheme 4 Synthesis of LPN and LPCH+·OTf−. Reaction conditions: (i): H2O2(aq), Et2O; (ii): PhCH2N3, CuSO4, sodium ascorbate, tBuOH/H2O, 3 d; (iii): MeOTf, CH2Cl2; (iv): PhSiH3, toluene, 10 d, 110 °C. | ||
The respective monogold chloride complexes of LPN and LPCH+·OTf− were obtained by reaction with [AuCl(tht)] (tht = tetrahydothiophene) (Scheme 5). After workup, [AuCl(LPN)] (6) and [AuCl(LPC·H)]OTf (7) were obtained as colorless solids in yields of 73% (6) and 68% (7).
 | ||
| Scheme 5 Synthesis of the goldchlorophospine complexes 6 and 7. | ||
The reaction can be easily monitored by 31P{1H} NMR spectroscopy. Chemical shifts of δ = 6.96 (6) and 12.5 ppm (7) indicate the coordination of the corresponding phosphines to the metal atoms. Note that a downfield shift in the range of 30–40 ppm is characteristic upon coordination of arylphosphines to AuCl.64,65
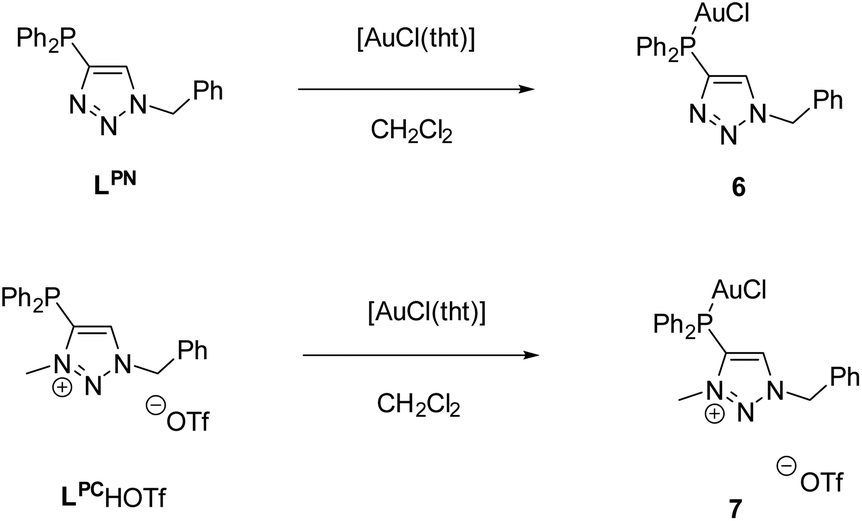
Complex 6 crystallizes in the trigonal space group P3121 with additional solvent molecules (CH2Cl2 and tetrahydrothiophene, for details refer to ESI, Fig. S1‡) in the asymmetric unit (Fig. 1). The Au1–Cl1 (2.270(3) Å) and P1–Au1 (2.213(3) Å) bond lengths are in the typical range for chloro phosphine gold complexes.66 The P^N angle P1–C1–N3 124.1(8)° is nearly identical to the N–C–N angle in, e.g., dinuclear gold amidinates, for which strong aurophilic interactions have been reported.55,67,68
 | ||
| Fig. 1 Molecular structure of 6 in the solid state. Non-coordinating solvents (CH2Cl2, tht) and hydrogen atoms are not displayed. Selected bond lengths [Å] and angles [°]: Au1–Cl1 2.270(3), Au1–P1 2.213(3), C1–C2 1.39(2), P1–Au1–Cl1 177.84(10), P1–C1–N3 124.1(8), P1–C1–C2 127.5(8). | ||
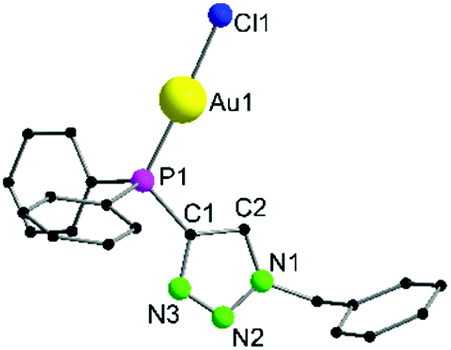
Complex 7 crystallizes in the monoclinic space group P21/n as an Au–Au connected dimer with two additional CH2Cl2 molecules in the asymmetric unit (Fig. 2). The Au1–Au2 contact of 3.1074(2) Å indicates aurophilic interactions of medium strength.69 As already seen for 6, the Au–Cl and P–Au bond lengths (Au1–Cl1 2.2791(11) Å, Au1–P1 2.2262(10) Å, Au2–Cl2 2.2983(10) Å, Au2–P2 2.2254(10) Å) are in the expected range. However, while the P1–Au1–Cl1 bond angle (177.74(4)°) is close to the typical linear coordination, the P2–Au2–Cl2 angle (169.60(4)°) deviates by approximately 10°. This is rather unusual but may be explained by crystal packing effects. Compared to 6, the introduction of positive charge by methylation results in two effects: (i) aurophilic interactions are observed in 7 while 6 remains monomeric in the solid state. This supports the general assumption that the neighbouring atoms, as well as the symmetry and nature of the ligand strongly influence the aurophilicity phenomenon;44,70,71 (ii) the methyl group has a considerable impact on the chemical shift in the 31P{1H} NMR spectrum. A downfield shift of 5.50 ppm relative to the unmethylated derivative 6 is observed for 7.
 | ||
| Fig. 2 Molecular structure of 7 in the solid state. Non-coordinating solvent molecules (2 CH2Cl2), anions (2 OTf−) and hydrogen atoms are not displayed. Selected bond lengths [Å] and angles [°]: Au1–Au2 3.1074(2), Au1–Cl1 2.2791(11), Au1–P1 2.2262(10), Au2–Cl2 2.2983(10), Au2–P2 2.2254(10), P1–Au1–Cl1 177.74(4), P2–Au2–Cl2 169.60(4), P1–C1–C2 132.0(3), P2–C23–C24 130.7(4). | ||
For the synthesis of a defined multimetallic complex, [AuCl(LPN)] (6) was further reacted with [Cu(MeCN)4]PF6 to give the heterotrinuclear complex [Cu(AuCl(LPN))2(thf)2]PF6 (8) (Scheme 6).
 | ||
| Scheme 6 Synthesis of the heterotrinuclear complex 8. | ||
The heterotrinuclear complex 8 crystallizes in the monoclinic space group P21/n with two additional THF molecules in the asymmetric unit (Fig. 3). The central Cu(I) atom is coordinated in a distorted octahedral fashion by the trans-located N3 and N6 atoms of the triazoles (Cu1–N3 1.944(13) Å, Cu1–N6 1.923(13) Å), the two Au(I) centers (Au1–Cu1 3.069(2) Å, Au2–Cu1 3.200(2) Å), as well as two thf molecules in a cis-coordination mode. The Cu1–N3 and Cu1–N6 distances are in the range of comparable Cu(I) triazole complexes.72,73 The copper atom and the two gold atoms form a triangle with interior angles of 54.52(4)° (Cu1–Au1–Au2), 58.10(4)° (Cu1–Au2–Au1) and 67.37(4)° (Au1–Cu1–Au2). The intermetallic distances are close to comparable complexes showing heterometallophilic interactions between Au and Cu.74,75 Due to the large Au1–Au2 contact length (3.4791(9) Å), the aurophilic interactions are assumed to be rather weak. In the 31P{1H} NMR spectrum, one resonance for the phosphines is observed at δ = 12.6 ppm, whereas the PF6− anion shows a characteristic septet at δ = −144.2 ppm. Furthermore, the chemical shift of the triazole protons is observed at δ = 7.95 ppm in the 1H NMR spectrum. Since only one resonance can be observed for these protons (which is the case for the phosphines in the 31P{1H} NMR spectrum as well), the non-crystallographic C2-symmetry of the complex is most likely preserved in solution.
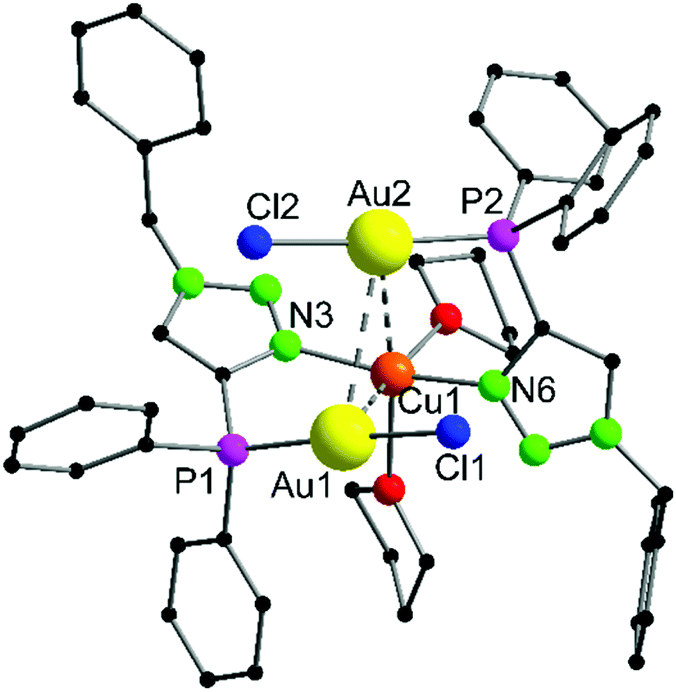 | ||
| Fig. 3 Molecular structure of 8 in the solid state. Non-coordinating solvent molecules (2 THF) and hydrogen atoms are not displayed. Selected bond lengths [Å] and angles [°]: Au1–Au2 3.4791(9), Au1–Cu1 3.069(2), Au2–Cu1 3.200(2), Cu1–N3 1.944(13), Cu1–N6 1.923(13), Cu1–Au2–Au1 54.52(4), Cu1–Au1–Au2 58.10(4), Au2–Cu1–Au1 67.37(4), N3–C1–P1 121.5(12), N6–C22–P2 122.1(12). | ||
While the second donor site of [AuCl(LPN)] (6) is accessible, the formation of a carbene is conducted by deprotonation of the methylated triazolium salt [AuCl(LPCH)]OTf (7). For this purpose, 7 was reacted with AgOAc, whereby AgCl precipitates and the (LPCH)+ moiety is deprotonated by the acetate to form the mesoionic carbene. Intramolecular stabilization of the Au+ is not favoured and thus, dimerization to the digold complex [Au2(LPC)2](OTf)29 occurs (Scheme 7).
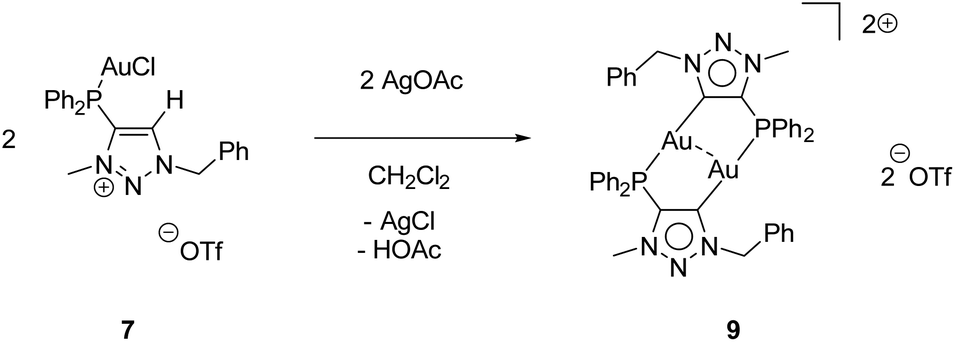 | ||
| Scheme 7 Synthesis of the digold complex 9. | ||
Complex 9 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with half of a molecule in the asymmetric unit (Fig. 4). As expected, the formation of the dinuclear arrangement with a linear coordination of phosphines and carbenes to the gold atoms (C2–Au1–P1 176.87(10)°) is observed. The carbene–gold bond length (Au1–C2 2.037(3) Å) is in the common range. The intermetallic contact Au1–Au1′ 2.9513(3) Å is short, indicating strong aurophilic interactions.76–78 In the 31P{1H} NMR spectrum of 9, a chemical shift of δ = 21.1 ppm is detected, which is shifted downfield by 8.6 ppm compared to the precursor 7, due to the change in the chemical environment, primarily in the trans-located ligand (Cl−vs. carbene). In gold carbene complexes, the carbene carbon atoms typically exhibit a chemical shift of around 180–210 ppm in the 13C{1H} NMR spectrum. For 9, unfortunately no resonance could be detected above 135 ppm.56,79
with half of a molecule in the asymmetric unit (Fig. 4). As expected, the formation of the dinuclear arrangement with a linear coordination of phosphines and carbenes to the gold atoms (C2–Au1–P1 176.87(10)°) is observed. The carbene–gold bond length (Au1–C2 2.037(3) Å) is in the common range. The intermetallic contact Au1–Au1′ 2.9513(3) Å is short, indicating strong aurophilic interactions.76–78 In the 31P{1H} NMR spectrum of 9, a chemical shift of δ = 21.1 ppm is detected, which is shifted downfield by 8.6 ppm compared to the precursor 7, due to the change in the chemical environment, primarily in the trans-located ligand (Cl−vs. carbene). In gold carbene complexes, the carbene carbon atoms typically exhibit a chemical shift of around 180–210 ppm in the 13C{1H} NMR spectrum. For 9, unfortunately no resonance could be detected above 135 ppm.56,79
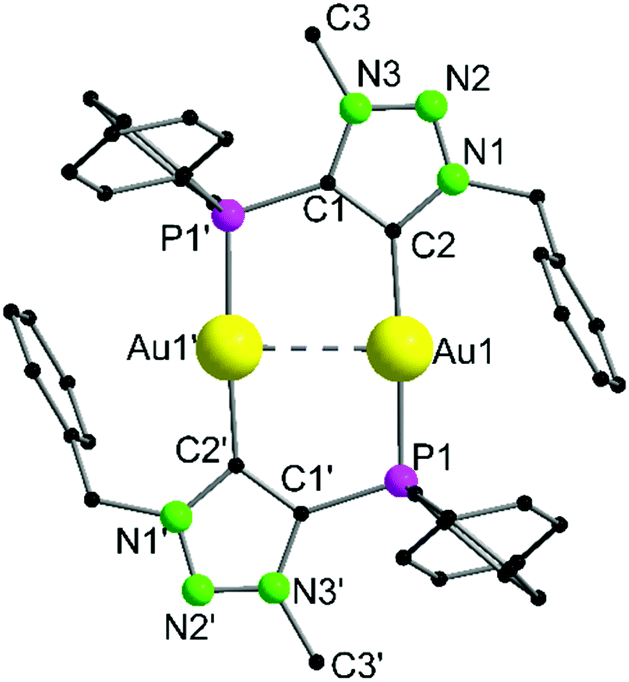 | ||
| Fig. 4 Molecular structure of 9 in the solid state. Anions (2× OTf−) and hydrogen atoms are not displayed for clarity. Selected bond lengths [Å] and angles [°]: Au1–Au1′ 2.9513(3), Au1–P1 2.2836(8), Au1–C2 2.037(3), C2–Au1–P1 176.87(10), P1–Au1–Au1′ 90.82(2), C2–Au1–Au1′ 86.15(10), C2–C1–P1′ 125.6(2). | ||
In a parallel experiment, the starting compound ethynyldiphenylphosphine (Ph2P–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H) was reacted to the known gold chloride complex [AuCl(Ph2P–CC–H)]80 instead of oxidizing the phosphine for the subsequent CuAAC reaction. Besides the ‘click product’ [AuCl(LPN)] (6), a dinuclear species, consisting of a dimeric gold triazolide complex (10), was identified as well (Scheme 8).
C–H) was reacted to the known gold chloride complex [AuCl(Ph2P–CC–H)]80 instead of oxidizing the phosphine for the subsequent CuAAC reaction. Besides the ‘click product’ [AuCl(LPN)] (6), a dinuclear species, consisting of a dimeric gold triazolide complex (10), was identified as well (Scheme 8).
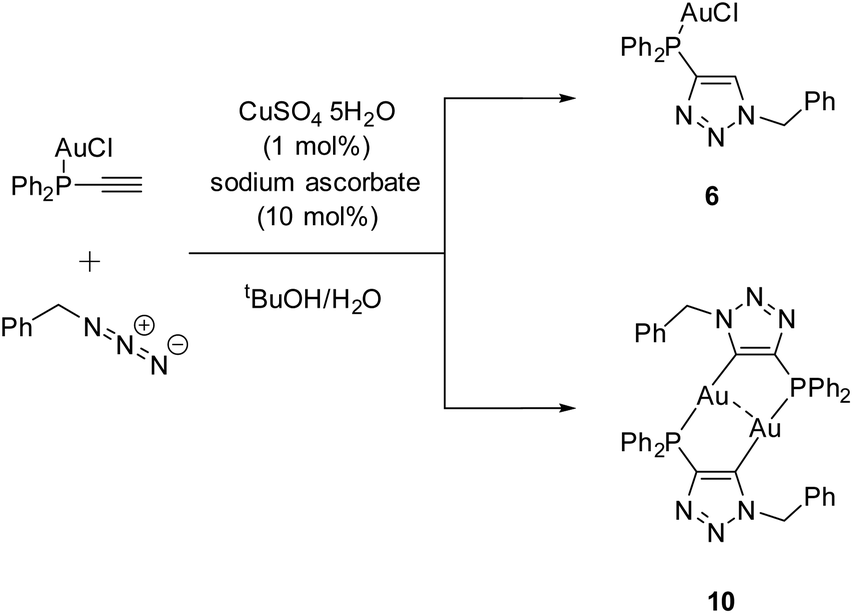 | ||
| Scheme 8 CuAAC reaction between [AuCl(Ph2P–CC–H)] and benzylazide. | ||
During the reaction, the precipitation of a large amount of a colorless solid is observed, which is not soluble in common organic solvents and could not be identified unambiguously. Slow evaporation of the supernatant results in the crystallization of products 6 and 10. A rational synthesis of complex 10, however, including several attempts from different starting materials (including 6 and 4 (LPN)) and varying conditions, failed so far. Thus 10 was characterized by single crystal X-ray diffraction only.
Complex 10 crystallizes in the monoclinic space group P21/c with half of a molecule in the asymmetric unit (Fig. 5). The bimetallic complex displays the neutral and non-methylated analogue to 9. Since the triazole scaffold is not alkylated, 10 is not a gold carbene complex, but a gold triazolide. The gold carbon bond lengths of 9 (Au1–C2 2.037(3) Å) and 10 (Au1–C2 2.036(4) Å), however, do not differ significantly. The intermetallic distance Au1–Au1′ 2.9615(7) Å is close to that in 9, indicating strong aurophilic interactions as well.
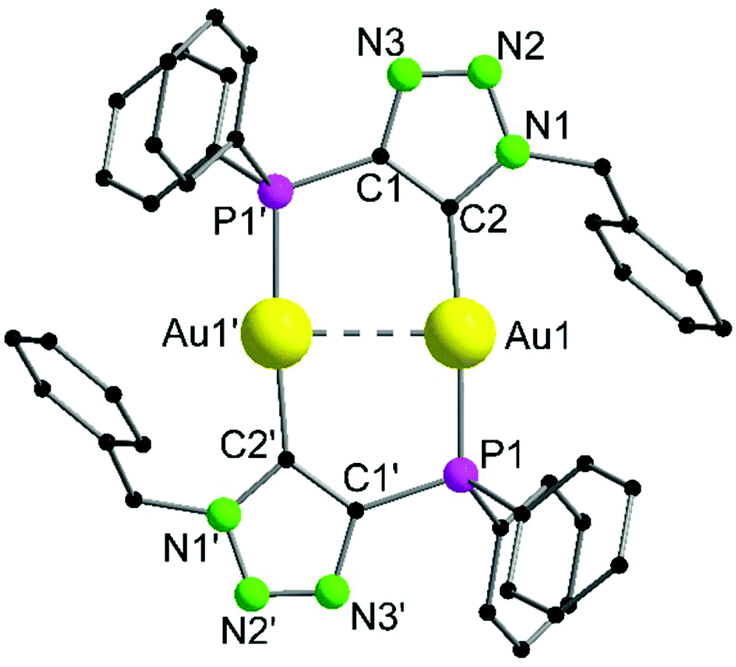 | ||
| Fig. 5 Molecular structure of 10 in the solid state. Hydrogen atoms are omitted. Selected bond lengths [Å] and angles [°]: Au1–Au1′ 2.9629(3), Au1–P1 2.2830(9), Au1–C2 2.036(4), P1–Au1–Au1′ 90.06(2), C2–Au1–Au1′ 85.70(10), C2–Au1–P1 175.76(11). | ||
Photoluminescence properties
The heterotrimetallic complex 8 and the digold complex 9 show, respectively, relatively bright yellow and blue-white photoluminescence (PL) in the solid state at ambient temperature, while the other presented compounds were found to be less efficient or virtually non-luminescent under similar UV excitation.The PL properties of 8 and 9 were therefore studied in more detail. PL spectra of 8 show a broad, featureless emission centred at 580 nm, which is typical of charge-transfer transitions. It moderately enhances in intensity and practically does not change spectrally by decreasing the temperature from 295 to 20 K (Fig. 6). Under nanosecond-pulsed laser excitation at 337 nm, the emission decays monoexponentially with τ = 14 and 8 μs at T = 20 and 295 K, respectively (Fig. S9‡), indicating phosphorescence. A PL quantum yield, ϕ(295 K), of 9% was determined at ambient temperature using an integrating sphere and excitation at 350 nm. It increases to about 35% at low temperatures, according to the temperature-dependent PL spectra (Fig. 6). Compound 9 emits a broad phosphorescence centred at around 440 (480) nm at 18 K (295 K), with a weak vibronic structure at low temperatures, as presented in Fig. 7. The PL decay approximately follows monoexponential curves with τ ≈ 15 and 0.7 μs at 18 and 295 K, respectively (Fig. S10‡). The PL intensity monotonically decreases by a factor of ∼25 by increasing the temperature from 18 to 295 K, thus correlating with the faster decay. Correspondingly, a smaller quantum yield ϕ(295 K) = 4% was determined for 9 in comparison to 8. However, it approaches ∼100% below 50 K as estimated from the temperature-dependent spectra (Fig. 7). For both 8 and 9, the PL may be assigned to triplet metal-to-ligand charge transfer (MLCT) states. In a first approximation, these excited states are localized within the corresponding molecular entities.
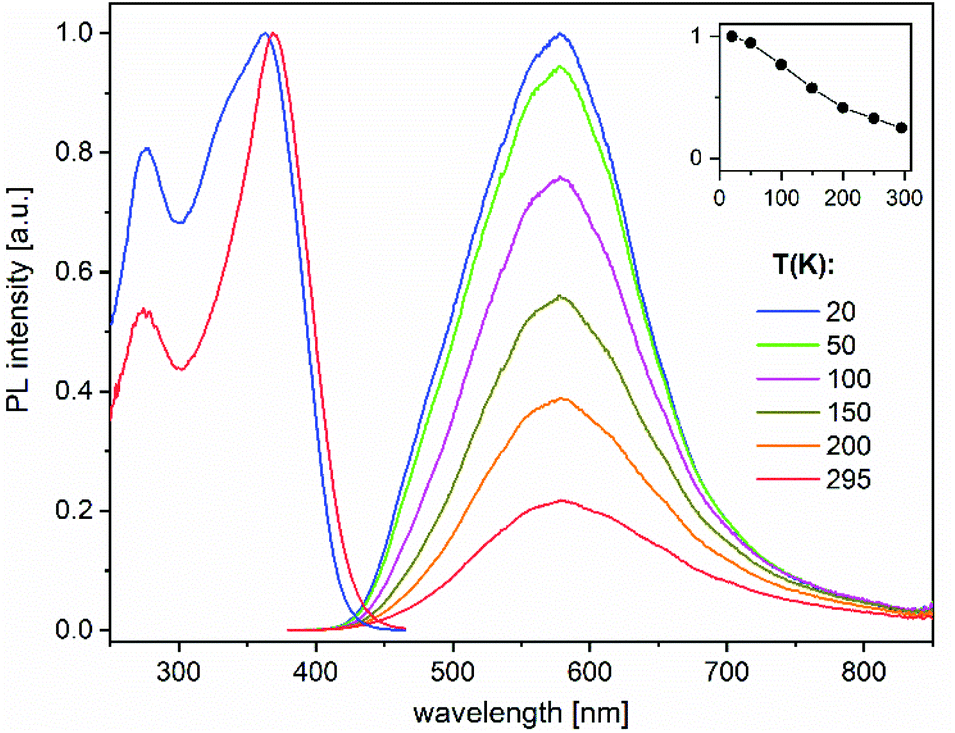 | ||
| Fig. 6 Photoluminescence excitation (PLE, left) and emission (PL, right) spectra of compound 8 in a temperature range between 20 and 295 K. The PLE and PL spectra were recorded and excited at 580 and 350 nm, respectively. The insert shows a temperature dependence of the integrated PL intensity. | ||
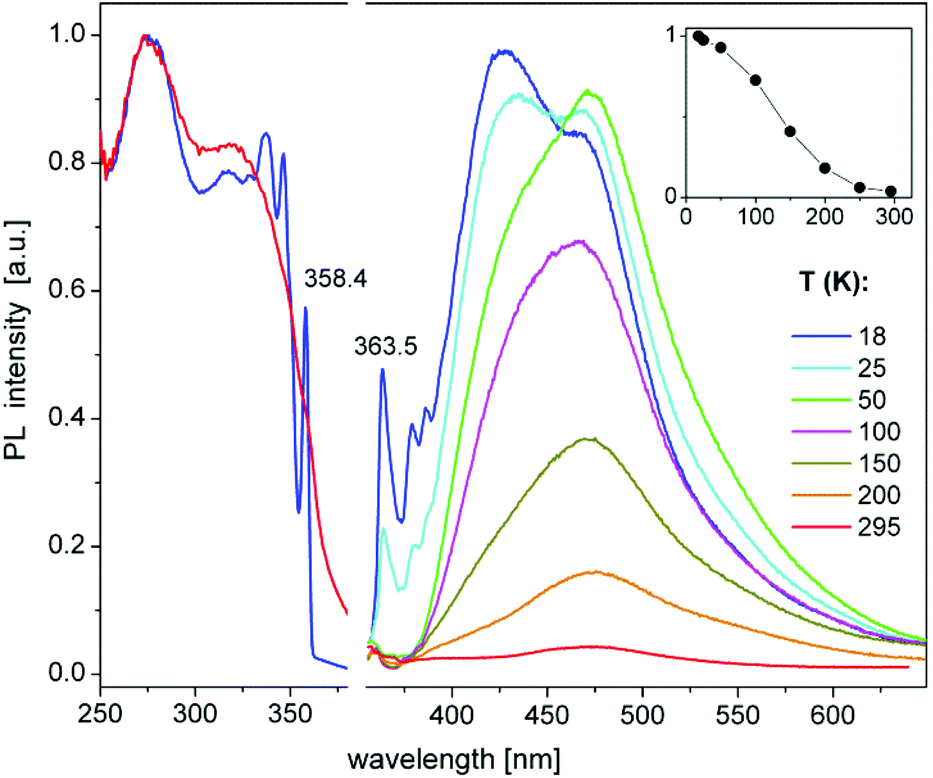 | ||
| Fig. 7 Photoluminescence excitation (PLE, left) and emission (PL, right) spectra of compound 9 in a temperature range between 18 and 295 K. The spectra were recorded and excited at 430 and 345 nm, respectively. The insert shows a temperature dependence of the integrated PL intensity. | ||
Perhaps the most interesting PL feature of 9 are narrow bands arising at the onsets of excitation and emission spectra at temperatures below 50 K (Fig. 7). These bands peak at 358.4 and 363.5 nm, respectively (T = 18 K), corresponding to a small Stokes shift of ca. 50 meV. The width (FWHM) of these PLE and PL bands is about 2–3 and 5–6 nm (20–30 and 50–60 meV), respectively. The decay of the 363 nm emission under pulsed laser excitation at 337 nm also proceeds on a microsecond time scale, but, in difference to the major PL band at ∼440 nm, is clearly nonexponential. It can be well fit by the stretched exponential function of the form y = y0 × t−n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ),81 with τ = 70 μs and n = 0.51 at 18 K. The decay at 363 nm is notably slowed down by further cooling of 9 down to 3.7 K (Fig. S10‡), whereas the PL spectra and decay of the broad band remain nearly unchanged (Fig. S11‡). The low-temperature PLE and PL spectra demonstrate sidebands to the peaks at 358.4 and 363.5 nm, which can be assigned to the phonon replica. Their frequencies are indicated in Fig. S11.‡ The sharp PLE and PL bands of 9 resemble those observed for free excitons in some low-dimensional materials, for instance, in 1D and 2D lead halide perovskites.82,83 Accordingly, we tentatively assign these features to free (or relatively delocalized) excitons in 9. A delocalization might be related to the π-stacking between the phenyl groups of the neighbouring molecules in solid 9 (Fig. S7‡). Especially at low temperatures, this may provide for a rigid, chain-like intermolecular structure. The microsecond-long emission decay indicates triplet excitons. The excitation–emission Stokes shift (see above) thus corresponds to the singlet–triplet exciton energy separation of ca. 50 meV.
exp(−t/τ),81 with τ = 70 μs and n = 0.51 at 18 K. The decay at 363 nm is notably slowed down by further cooling of 9 down to 3.7 K (Fig. S10‡), whereas the PL spectra and decay of the broad band remain nearly unchanged (Fig. S11‡). The low-temperature PLE and PL spectra demonstrate sidebands to the peaks at 358.4 and 363.5 nm, which can be assigned to the phonon replica. Their frequencies are indicated in Fig. S11.‡ The sharp PLE and PL bands of 9 resemble those observed for free excitons in some low-dimensional materials, for instance, in 1D and 2D lead halide perovskites.82,83 Accordingly, we tentatively assign these features to free (or relatively delocalized) excitons in 9. A delocalization might be related to the π-stacking between the phenyl groups of the neighbouring molecules in solid 9 (Fig. S7‡). Especially at low temperatures, this may provide for a rigid, chain-like intermolecular structure. The microsecond-long emission decay indicates triplet excitons. The excitation–emission Stokes shift (see above) thus corresponds to the singlet–triplet exciton energy separation of ca. 50 meV.
Conclusions
By a facile synthetic procedure, two imidazole-based ligands LPN and LPCH+·OTf− were obtained. They display different donor site combinations for selective metal coordination while having a similar structural geometry. Both ligands were converted to their respective mono gold chloride complexes [AuCl(LPN)] (6) and [AuCl(LPC·H)]OTf (7). Compound 6 was used for further coordination to Cu(I) to yield a heterotrimetallic Au2Cu complex 8, exhibiting moderate metallophilic interactions. However, strong aurophilic interactions were observed in the dinuclear gold carbene complex 9, obtained by deprotonation and chloride abstraction of the triazolium salt 7. Furthermore, the photoluminescence (PL) properties of the multimetallic complexes 8 and 9 were investigated in the solid state. These compounds show relatively bright, spectrally broad, yellow and blue-white phosphorescence, respectively. In addition, the low-temperature PL spectra of 9 demonstrate narrow excitation and emission bands with a linewidth of about 20–30 and 50–60 meV, respectively, and a Stokes shift of about 50 meV. These were tentatively assigned to free (or relatively delocalized) excitons in 9. To the best of our knowledge, this is the first observation of such features for gold and other coinage metal complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the DFG-funded transregional collaborative research center SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)” projects C3 and C7 is gratefully acknowledged. S. B. gratefully acknowledges the Funds of the Chemical Industry (FCI) as well as the Deutsche Forschungsgemeinschaft (DFG, BE 6401/1-1) for generous fellowships.Notes and references
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- T. Weskamp, V. P. W. Böhm and W. A. Herrmann, J. Organomet. Chem., 2000, 600, 12–22 CrossRef CAS.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed.
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef.
- S. K. Bose, K. Fucke, L. Liu, P. G. Steel and T. B. Marder, Angew. Chem., Int. Ed., 2014, 53, 1799–1803 CrossRef CAS PubMed.
- M. Eck, S. Würtemberger-Pietsch, A. Eichhorn, J. H. J. Berthel, R. Bertermann, U. S. D. Paul, H. Schneider, A. Friedrich, C. Kleeberg, U. Radius and T. B. Marder, Dalton Trans., 2017, 46, 3661–3680 RSC.
- P. Hemberger, A. Bodi, J. H. J. Berthel and U. Radius, Chem. – Eur. J., 2015, 21, 1434–1438 CrossRef CAS PubMed.
- V. Nair, S. Bindu and V. Sreekumar, Angew. Chem., Int. Ed., 2004, 43, 5130–5135 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2012, 51, 2098–2100 CrossRef CAS PubMed.
- B. Borthakur, T. Rahman and A. K. Phukan, J. Org. Chem., 2014, 79, 10801–10810 CrossRef CAS PubMed.
- P. L. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596–609 CrossRef CAS.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS.
- D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304–5313 CrossRef CAS PubMed.
- F. E. Hahn, Chem. Rev., 2018, 118, 9455–9456 CrossRef CAS PubMed.
- A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209–3210 CrossRef CAS PubMed.
- G. Ung and G. Bertrand, Chem. – Eur. J., 2011, 17, 8269–8272 CrossRef CAS PubMed.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759–4762 CrossRef CAS PubMed.
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- D. Schweinfurth, N. Deibel, F. Weisser and B. Sarkar, Nachr. Chem., 2011, 59, 937–941 CrossRef CAS.
- R. Veillard, E. Bernoud, I. Abdellah, J.-F. Lohier, C. Alayrac and A.-C. Gaumont, Org. Biomol. Chem., 2014, 12, 3635–3640 RSC.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643 CrossRef CAS.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93–98 CrossRef CAS.
- L. Cao, S. Huang, W. Liu and X. Yan, Organometallics, 2018, 37, 2010–2013 CrossRef CAS.
- E. Kühnel, I. V. Shishkov, F. Rominger, T. Oeser and P. Hofmann, Organometallics, 2012, 31, 8000–8011 CrossRef.
- P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- H. L. Hermann, G. Boche and P. Schwerdtfeger, Chem. – Eur. J., 2001, 7, 5333–5342 CrossRef CAS.
- W.-F. Fu, X. Gan, C.-M. Che, Q.-Y. Cao, Z.-Y. Zhou and N. N.-Y. Zhu, Chem. – Eur. J., 2004, 10, 2228–2236 CrossRef CAS PubMed.
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- P. Pyykkö, N. Runeberg and F. Mendizabal, Chem. – Eur. J., 1997, 3, 1451–1457 CrossRef.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- P. Pyykkö, Annu. Rev. Phys. Chem., 2012, 63, 45–64 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- M. Kim, T. J. Taylor and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 6332–6333 CrossRef CAS PubMed.
- I. O. Koshevoy, L. Koskinen, M. Haukka, S. P. Tunik, P. Y. Serdobintsev, A. S. Melnikov and T. A. Pakkanen, Angew. Chem., Int. Ed., 2008, 47, 3942–3945 CrossRef CAS PubMed.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- R. Echeverría, J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, Chem. Sci., 2015, 6, 2022–2026 RSC.
- X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS.
- I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, M. Haukka, T. Pakkanen and P.-T. Chou, J. Am. Chem. Soc., 2012, 134, 6564–6567 CrossRef CAS PubMed.
- A. P. Marchenko, H. N. Koidan, A. N. Hurieva, O. V. Gutov, A. N. Kostyuk, C. Tubaro, S. Lollo, A. Lanza, F. Nestola and A. Biffis, Organometallics, 2013, 32, 718–721 CrossRef CAS.
- P. Ai, A. A. Danopoulos, P. Braunstein and K. Y. Monakhov, Chem. Commun., 2014, 50, 103–105 RSC.
- P. Ai, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2015, 54, 3722–3724 CrossRef CAS PubMed.
- P. Ai, M. Mauro, C. Gourlaouen, S. Carrara, L. De Cola, Y. Tobon, U. Giovanella, C. Botta, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2016, 55, 8527–8542 CrossRef CAS PubMed.
- T. J. Feuerstein, M. Poß, T. P. Seifert, S. Bestgen, C. Feldmann and P. W. Roesky, Chem. Commun., 2017, 53, 9012–9015 RSC.
- C. Kaub, S. Lebedkin, A. Li, S. V. Kruppa, P. H. Strebert, M. M. Kappes, C. Riehn and P. W. Roesky, Chem. – Eur. J., 2018, 24, 6094–6104 CrossRef CAS PubMed.
- F. Zhang, X.-M. Cao, J. Wang, J. Jiao, Y. Huang, M. Shi, P. Braunstein and J. Zhang, Chem. Commun., 2018, 54, 5736–5739 RSC.
- A. Marchenko, H. Koidan, A. Hurieva, O. Kurpiieva, Y. Vlasenko, A. Kostyuk, C. Tubaro, A. Lenarda, A. Biffis and C. Graiff, J. Organomet. Chem., 2014, 771, 14–23 CrossRef CAS.
- V. Huc, A. Balueva, R.-M. Sebastian, A.-M. Caminade and J.-P. Majoral, Synthesis, 2000, 726–730 CrossRef CAS.
- H. A. van Kalkeren, J. J. Bruins, F. P. J. T. Rutjes and F. L. van Delft, Adv. Synth. Catal., 2012, 354, 1417–1421 CrossRef CAS.
- A. Watzke, M. Köhn, M. Gutierrez-Rodriguez, R. Wacker, H. Schröder, R. Breinbauer, J. Kuhlmann, K. Alexandrov, C. M. Niemeyer, R. S. Goody and H. Waldmann, Angew. Chem., Int. Ed., 2006, 45, 1408–1412 CrossRef CAS PubMed.
- R. J. P. Corriu, C. Guérin, B. J. L. Henner and A. Jolivet, J. Organomet. Chem., 1997, 530, 39–48 CrossRef CAS.
- S. G. A. van Assema, C. G. J. Tazelaar, G. B. de Jong, J. H. van Maarseveen, M. Schakel, M. Lutz, A. L. Spek, J. C. Slootweg and K. Lammertsma, Organometallics, 2008, 27, 3210–3215 CrossRef CAS.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2015, 21, 601–614 CrossRef CAS PubMed.
- S. Bestgen, C. Schoo, C. Zovko, R. Köppe, R. P. Kelly, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2016, 22, 7115–7126 CrossRef CAS.
- T. Tanase, R. Otaki, T. Nishida, H. Takenaka, Y. Takemura, B. Kure, T. Nakajima, Y. Kitagawa and T. Tsubomura, Chem. – Eur. J., 2014, 20, 1577–1596 CrossRef CAS PubMed.
- H. E. Abdou, A. A. Mohamed, J. M. López-de-Luzuriaga, M. Monge and J. P. Fackler, Inorg. Chem., 2012, 51, 2010–2015 CrossRef CAS PubMed.
- H. E. Abdou, A. A. Mohamed, J. M. López-de-Luzuriaga and J. P. Fackler, J. Cluster Sci., 2004, 15, 397–411 CrossRef CAS.
- P. Schwerdtfeger, A. E. Bruce and M. R. M. Bruce, J. Am. Chem. Soc., 1998, 120, 6587–6597 CrossRef CAS.
- C. Bartolomé, M. Carrasco-Rando, S. Coco, C. Cordovilla, J. M. Martín-Alvarez and P. Espinet, Inorg. Chem., 2008, 47, 1616–1624 CrossRef PubMed.
- C. Croix, A. Balland-Longeau, H. Allouchi, M. Giorgi, A. Duchêne and J. Thibonnet, J. Organomet. Chem., 2005, 690, 4835–4843 CrossRef CAS.
- Z. Benkhellat, M. Allali, M. Beley, E. Wenger, M. Bernard, N. Parizel, K. Selmeczi and J.-P. Joly, New J. Chem., 2014, 38, 419–429 RSC.
- E. P. McCarney, C. S. Hawes, S. Blasco and T. Gunnlaugsson, Dalton Trans., 2016, 45, 10209–10221 RSC.
- V. J. Catalano, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Dalton Trans., 2014, 43, 16486–16497 RSC.
- J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. A. Rodríguez-Castillo, Organometallics, 2012, 31, 3720–3729 CrossRef.
- A. L. Johnson, A. M. Willcocks and S. P. Richards, Inorg. Chem., 2009, 48, 8613–8622 CrossRef CAS PubMed.
- Y. Inoguchi, B. Milewski-Mahrla and H. Schmidbaur, Chem. Ber., 1982, 115, 3085–3095 CrossRef CAS.
- J. Zank, A. Schier and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 1999, 415–420 RSC.
- C. Kaub, S. Lebedkin, S. Bestgen, R. Köppe, M. M. Kappes and P. W. Roesky, Chem. Commun., 2017, 53, 9578–9581 RSC.
- M. Bardají, P. G. Jones and A. Laguna, J. Chem. Soc., Dalton Trans., 2002, 3624–3629 RSC.
- R. Leonelli and J. L. Brebner, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8649–8656 CrossRef CAS.
- G. C. Papavassiliou, G. A. Mousdis and I. B. Koutselas, Adv. Mater. Opt. Electron., 1999, 9, 265–271 CrossRef CAS.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Dr Annie K. Powell on the occasion of her 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, additional photoluminescence data, crystallographic appendix, NMR, IR and MS spectra. CCDC 1901789–1901794. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01239g |
| This journal is © The Royal Society of Chemistry 2019 |