
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHg2+ and Cd2+ binding of a bioinspired hexapeptide with two cysteine units constructed as a minimalistic metal ion sensing fluorescent probe†
Levente I.
Szekeres
a,
Sára
Bálint
a,
Gábor
Galbács
 a,
Ildikó
Kálomista
a,
Tamás
Kiss
a,
Flemming H.
Larsen
b,
Lars
Hemmingsen
c and
Attila
Jancsó
*a
a,
Ildikó
Kálomista
a,
Tamás
Kiss
a,
Flemming H.
Larsen
b,
Lars
Hemmingsen
c and
Attila
Jancsó
*a
aDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, Szeged, H-6720, Hungary. E-mail: jancso@chem.u-szeged.hu
bDepartment of Food Science, University of Copenhagen, Rolighedsvej 30, 1958 Frederiksberg C, Denmark
cDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark
First published on 2nd May 2019
Abstract
Hg2+ and Cd2+ complexation of a short hexapeptide, Ac-DCSSCY-NH2 (DY), was studied by pH-potentiometry, UV and NMR spectroscopy and fluorimetry in aqueous solutions and the Hg2+-binding ability of the ligand was also described in an immobilized form, where the peptides were anchored to a hydrophilic resin. Hg2+ was demonstrated to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with the ligand even at pH = 2.0 while Cd2+ coordination by the peptide takes place only above pH ∼ 3.5. Both metal ions form bis-ligand complexes by the coordination of four Cys-thiolates at ligand excess above pH ∼ 5.5 (Cd2+) and 7.0 (Hg2+). Fluorescence studies demonstrated a Hg2+ induced concentration-dependent quenching of the Tyr fluorescence until a 1:1 Hg2+:DY ratio. The fluorescence emission intensity decreases linearly with the increasing Hg2+ concentration in a range of over two orders of magnitude. The fact that this occurs even in the presence of 1.0 eq. of Cd2+ per ligand reflects a complete displacement of the latter metal ion by Hg2+ from its peptide-bound form. The immobilized peptide was also shown to bind Hg2+ very efficiently even from samples at pH = 2.0. However, the existence of lower affinity binding sites was also demonstrated by binding of more than 1.0 eq. of Hg2+ per immobilized DY molecule under Hg2+-excess conditions. Experiments performed with a mixture of four metal ions, Hg2+, Cd2+, Zn2+ and Ni2+, indicate that this molecular probe may potentially be used in Hg2+-sensing systems under acidic conditions for the measurement of μM range concentrations.
:1 complex with the ligand even at pH = 2.0 while Cd2+ coordination by the peptide takes place only above pH ∼ 3.5. Both metal ions form bis-ligand complexes by the coordination of four Cys-thiolates at ligand excess above pH ∼ 5.5 (Cd2+) and 7.0 (Hg2+). Fluorescence studies demonstrated a Hg2+ induced concentration-dependent quenching of the Tyr fluorescence until a 1:1 Hg2+:DY ratio. The fluorescence emission intensity decreases linearly with the increasing Hg2+ concentration in a range of over two orders of magnitude. The fact that this occurs even in the presence of 1.0 eq. of Cd2+ per ligand reflects a complete displacement of the latter metal ion by Hg2+ from its peptide-bound form. The immobilized peptide was also shown to bind Hg2+ very efficiently even from samples at pH = 2.0. However, the existence of lower affinity binding sites was also demonstrated by binding of more than 1.0 eq. of Hg2+ per immobilized DY molecule under Hg2+-excess conditions. Experiments performed with a mixture of four metal ions, Hg2+, Cd2+, Zn2+ and Ni2+, indicate that this molecular probe may potentially be used in Hg2+-sensing systems under acidic conditions for the measurement of μM range concentrations.
Introduction
Hg2+ and Cd2+ ions can induce serious cytotoxic effects and cancer development in living organisms. Among others, Cd2+ can interfere with DNA repair mechanisms and causes osteoporosis by mimicking Zn2+ and Ca2+ ions.1,2 The high affinity of Hg2+ to the sulfhydryl groups of thiols has long been accepted to play a role in Hg2+ toxicity. Nevertheless, it has also been suggested that the major mechanism of toxicity is related to its interaction with the selenocysteine residues of glutathione peroxidase and thioredoxin reductase, leading to the inhibition of gap junction-mediated intercellular communication and proinflammatory cytokine production.3,4 Cellular defence mechanisms remove these toxic metal ions from the cytosol. In many bacteria the removal of Cd2+ ions, owing to their similarity to Zn2+, can be implemented by multi-cation resistance-mediating CzcABC efflux pumps5 and by the P-type ATPase ZntA or the more specific CadA systems.6 In contrast, the detoxification of Hg2+ takes place in the form of Hg0via the specific mercury resistance mer system.7,8 Cys-abundant sequence motifs can provide efficient Hg2+ and Cd2+ binding for proteins. The CXXC motif often constitutes the metal ion binding site in proteins, such as in rubredoxins,9 P-type ATPases,10 alcohol dehydrogenases,11 zinc fingers,12 metallochaperones,13,14 metallothioneins,15 and in the members of the Hg2+ and Cd2+ selective resistance systems (MerP,16 MerC,17 CadA,6 ZntA6 and NMerA18).Hg2+ can form extremely stable complexes with linear, {HgS2} type coordination mode with thiol ligands, e.g. Cys (logβ2 = 39.4–43.6),19–21 BAL (logβ1 = 44.8),22etc. The formation of this binding mode was observed during the interaction of Hg2+ with many CXXC-type proteins, such as MerP,16 NMerA,23 and Atx1,24 and with cyclic25,26 or linear27,28 metalloprotein model peptides, etc. The presence of amino acids with high rigidity and/or helical penalty (e.g. Pro and Gly) in the positions of X1 and X2 increases the Hg2+ binding efficiency by preorganising the peptide structure. Moreover, the Hg2+ complex of the rigid CDPPC peptide displays an exceptionally high stability constant (logβ = 40.0) even compared to the effective CPPC sequence.29 There are also examples for the formation of bis complexes with a {HgS4} binding mode among proteins bearing a CXXC motif (HAH1,24 rubredoxins,30etc.).
In its complexes, Cd2+, similar to Zn2+, prefers to possess a tetrahedral coordination sphere with two or four thiolate moieties but Cd2+ has a higher thiophilic character. When the coordination sphere of Cd2+ is not saturated by thiolate groups, the complexes are stabilized mostly by complementary binding of other functional groups. Interactions with the carboxylate groups of Glu and Asp units probably play a role in the selectivity of CadA and ZntA.31 Besides monomeric bis complexes with a pure {CdS4} coordination mode, the presence of dimeric species with a mixed Cys-Glu ({CdS3O}) coordination was also observed in a system of Cd2+ and a 71 amino acid long polypeptide, derived from CadA, at pH = 6.0.31
CXXC moieties are potentially applicable as metal ion binding units in Hg2+ and Cd2+ sensing systems, since their interaction is typically characterized by a well-defined stoichiometry, high efficiency and selectivity. Since these metal ions can accumulate in the food chain and therefore ingested by humans, they pose significant health risks, and therefore their fast, selective sensing and remediation are highly prioritised.8 Sensing schemes for these metal ions, based on fluorimetry,32 potentiometry,33–35 impedance measurement,36 protein-functionalized microcantilevers,37 or differential surface plasmon resonance38etc., have already been proposed. Peptide ionophores can be designed with an ability for signalling by introducing fluorescing residues or amino acids bearing fluorescing side chains into their sequence.39–41 Metal ion binding to peptidic probes mostly induces fluorescence quenching, especially with Cu2+ (ref. 42–53) and Hg2+.42,48,54 However, chelation can also enhance the fluorescence (CHEF) via electronic interactions between the fluorophore and the altered coordination sphere. This is very frequent with Zn2+ (ref. 43, 46–49, 51 and 53) and Cd2+ (ref. 43 and 46–49) but examples are also known with Ag+,42,46–48 Pb2+ (ref. 46 and 47) and Hg2+.48 In addition, the metal ion promoted conformational change of the peptide may lead to a “close contact” and thus to a Förster resonance energy transfer (FRET) effect between different fluorophore residues.46,50,52,54–56 Aggregation induced emission (AIE) enhancement can also be utilized for the analysis of metal ions.57,58 The response mechanism and the sensitivity and selectivity of a peptide sensor designed for a specific metal ion is highly influenced by the quality and quantity of the binding groups, as well as the fluorophores themselves, in addition to the amino acid sequence and the higher order peptide structure. Although many fluorescent peptides have already been tested as potential sensor candidates for Hg2+, Cd2+ or other metal ions, examples where the studied ionophore bears a CXXC motif are rather scarce. In the unique case of the short dansyl-CPGCW-NH2 peptide, the intervening amino acids between the two Cys residues provide high backbone rigidity for the ionophore. Upon excitation of either of the two fluorophore units, Cd2+ ions induced a significant enhancement in the light emission of the dansyl group. Rather interestingly, Hg2+ and Zn2+ were shown to have no effect on the fluorescence response of the molecule.56 Another notable, previously studied dicysteinyl peptidic probe possesses a CXXHC binding motif combined with dansyl and Trp fluorophores. This probe displayed a ratiometric turn on response for Hg2+, Cd2+, Pb2+, Zn2+, and Ag+ ions upon the excitation of either of the two fluorophore units.46 In addition, Joshi and co-workers also observed a turn-on response for Zn2+ with two fluorescent peptides containing a Cys and a His residue in a 1,4-arrangement.47,49
Optochemical sensors, constructed by the covalent attachment of fluorescent peptides to the surface of solid carriers (e.g. optical fibers), may allow in situ trace analysis of toxic metal ions with low sample quantity requirements. Such systems may present simple alternatives to currently used reliable, but expensive and robust laboratory-based instrumental methods. Despite the fact that several studies have already reported the successful design of such sensors,32,59–61 there have only been very few attempts to characterize the metal capturing peptide elements both in their dissolved and immobilized forms.47,49
With an aim of exploiting the metal ion binding properties of the CXXC motif, we designed and synthesized the short, DCSSCY hexapeptide in an acetylated and amidated free form (Ac-DCSSCY-NH2 (DY)) and in an immobilized, resin-attached form (DY-NTG). Compared to other aromatic amino acids, Tyr is less hydrophobic and still exhibits a reasonable quantum yield (0.14).62 The fluorescence of Tyr has proved to be applicable in determining the Cu2+ complex stabilities of different peptides,63–65 in spite of the relatively close excitation (λEX = 275 nm) and emission (λEM = 304 nm) maxima of the fluorophore.62 Accordingly, this simple ligand and the obtained data may be relevant in the design of peptide-based metal ion sensing systems.
Results and discussion
UV absorption experiments
UV absorption spectra of DY, as a function of pH, were recorded in the absence and presence of metal ions with the purpose of monitoring the effects of Hg2+ and Cd2+ on the deprotonation processes of the ligand. As is expected, the proton dissociation of both the Cys –SH groups and the phenolic –OH group markedly influences the recorded spectra, even in the absence of metal ions (Fig. S1†). Deprotonated thiol groups are characterized by a UV-band centred around λ = 235–236 nm66,67 while the proton release from the Tyr–OH function is known to result in a shift of two characteristic absorption peaks from 223 to 240 nm and from 277 to 295 nm.68 The dissociation of the Asp sidechain carboxyl group does not have a noticeable spectral influence in the λ = 220–400 nm range. Since the Cys and Tyr residues all deprotonate above neutral pH, a continuous evolution of the absorbance is observed between pH ∼ 6.5 and 11.0 around 240 nm while the absorbance at 295 nm starts increasing from a notably higher pH, above pH ∼ 8.5 (Fig. S1†), indicating the exclusive effect of the deprotonation of the Tyr residue. One may estimate the pKa value of the Tyr –OH function from the A295 nmvs. pH plot (Fig. S1†) to be around 10.0 which is in line with the data determined from pH-metric titrations (see later).UV/pH titrations were conducted in the presence of 0.5 and 1.0 equivalents of Hg2+ ions within the range of pH ∼ 1.8–11.0 (Fig. S2†) and representative absorbance traces from selected wavelength values are depicted in Fig. 1A. The significantly larger absorbances observed at pH = 1.8 at λ ∼ 210–230 nm in the presence of Hg2+ relative to the metal ion free sample originate from S−–Hg2+ charge transfer bands and clearly indicate peptide binding to the metal ion, as was also observed previously for other systems of Hg2+ and cysteine containing peptides under acidic conditions.29,69–71 Furthermore, the absorbance increase, induced by Hg2+-binding, seems to be proportional to the applied Hg2+:DY ratio (Fig. 1A and S2B†) between pH ∼ 1.8–7.0. At the same time, absorbances in the higher wavelength regime, i.e. around 295 nm, are almost identical independent of the Hg2+ concentration. These findings suggest an unvarying speciation up to neutral pH at any Hg2+:DY ratio (and even up to pH 11.0 at one equivalent of Hg2+ per peptide) and the high energy charge transfer band, characterized by an absorption maximum below 220 nm (see the calculated difference spectra in Fig. S3†), indicates a {HgS2} type coordination mode in the complexes formed.26,29,69,70,72,73
 | ||
| Fig. 1 Absorbances at selected wavelength values as a function of pH, recorded for Hg2+:DY (A) and Cd2+:DY (B). (cDY = 1 × 10−4 M (A) or 5 × 10−5 M (B), T = 298 K). | ||
A remarkable difference between the speciation in Hg2+:DY 0.5:1 and 1:1 above pH ∼ 7.5 is reflected by the A vs. pH profiles at 295 nm. The absorbances observed for Hg2+:DY 1:1 up to pH = 11.0 are nearly identical to those obtained for the free ligand suggesting that the bound Hg2+ has only a minor influence, if any, on the deprotonation of the Tyr sidechain group. The remarkably different A295 nmvs. pH trace of Hg2+:DY 0.5:1 (Fig. 1A) suggests a fundamental rearrangement in the coordination sphere of Hg2+ above neutral pH, most probably induced by the coordination of a second ligand and the formation of additional S−–Hg2+ bonds. LMCT bands with maxima around 240–250 nm or 280–300 nm in the UV spectra of Hg2+–thiolate complexes were previously assigned to signatures of trigonal {HgS3}74,75 or (pseudo)tetrahedral {HgS4} species, respectively,30,75,76 and this suggests that the two DY ligands bind Hg2+ with both of their thiolates under basic conditions (see Fig. S3†). The slight bathochromic shift of the absorption maximum, observed above pH ∼ 9.5 (Fig. S2A†), is presumably the effect of the deprotonation of the Tyr residues of the bound ligands taking place without metal ion assistance. This interpretation is further supported by the fact that isosbestic points are observed above pH 10 at λ ∼ 264 nm and ∼282 nm (Fig. S2A†), indicating a simple transition between two species.
The pH-dependent series of UV spectra recorded for Cd2+:DY 0.5:1 and 1:1 (Fig. 1B and S4†) indicate metal ion coordination via the formation of thiolate–Cd2+ bonds from pH ∼ 4.5. The A240 nmvs. pH profiles, representing the evolution of S−–Cd2+ charge transfer bands,77–83 suggest that metal ion binding is essentially complete by pH ∼ 6 at 1 eq. of Cd2+ per ligand; however, the curve for Cd2+:DY 0.5:1 is characterized by a narrow plateau near pH 6 and a second step levelling off above pH ∼ 7.5 (Fig. 1B). This indicates a different complexation pathway when the peptide is in excess over Cd2+. One may presume the formation of bis-ligand species, as was also reported for the Cd2+ binding of other short, two Cys-containing peptides.79–81,84 This is also supported by the systematically higher absorbances observed for Cd2+:DY 0.5:1 relative to those observed both for Cd2+:DY 1:1 and for the free ligand (Fig. 1B). Indeed, the molar extinction coefficients for λ = 240 nm that one may estimate for the species present at pH ∼ 8.0 at 0.5 and 1 eq. of Cd2+ per ligand (25100 M−1 cm−1 and 11700 M−1 cm−1) correspond well to four and two thiolate coordinated Cd2+-centres, respectively.77,78 The further increase of absorbance above pH 8 (at any Cd2+:DY ratio) is most likely the consequence of the deprotonation of the Tyr sidechain phenols; nevertheless, other processes that may affect the coordination sphere of the metal ion, e.g. proton release(s) from Cd2+-coordinated water molecules, may also contribute to the observed spectral changes under alkaline conditions.
1H NMR studies
1H NMR spectra of the free peptide were recorded as a function of pH (Fig. S6†) and resonances of DY were assigned as described in the Experimental part and are shown in Fig. S5.† We performed NMR titrations in the Hg2+:DY (Fig. 2) and Cd2+:DY (Fig. S9†) systems by changing the metal ion to ligand ratio at selected pH values or at constant Hg2+:DY ratios (0.5:1 and 1:1) as a function of pH (Fig. S7 and S8†). However, pH-dependent 1H NMR experiments were not executed for Cd2+:DY because of severe line broadening and poorly resolved resonances observed for all of the signals already at 0.25 eq. of Cd2+ per ligand (see Fig. S9†). We had previously observed a similarly strong influence of Cd2+ on the 1H NMR signals of other, relatively short, Cys-containing peptides81,85 and as in those reported cases, the line broadening seen in the spectra of Cd2+:DY indicates ligand exchange and/or conformational dynamics occurring in an intermediate time regime, relative to the NMR time scale.
 | ||
| Fig. 2
1H NMR spectra of DY obtained with varying equivalents of Hg2+ per peptide at pH = 5.7 (A) and 8.5 (B) (H2O:D2O = 90:10% v/v, cDY = 1.0 × 10−3 M, T = 298 K). The symbol ‘*’ denotes 1H resonances of the ligand in its metal bound state. | ||
The deprotonation processes of the carboxyl group of the Asp residue, the two Cys-thiols and the sidechain phenolic function of Tyr in the free DY ligand can be clearly followed by the gradual shifting of the corresponding CβH2 resonances or the CδH and CεH signals of Tyr towards lower chemical shifts in the relevant pH-ranges (Fig. S6†). The presence of Hg2+ has only minor effects on the shape and chemical shift of these resonances (with the exception of the slightly more influenced CβH2 signals of Asp – see later), suggesting that Hg2+ does not bind to these moieties. As opposed to this, below neutral pH Hg2+ binding of the two thiolates is represented by the collapse of the Cys CβH2 signals of the unbound ligand and the emerging new resonances attributed to the bound molecule (see the effect of the increasing Hg2+:DY ratio at pH = 5.7, Fig. 2A). This indicates that the ligand exchange dynamics is moderately slow at this pH, relative to the NMR timescale.
The pH-dependence of resonances observed with 0.5 eq. of Hg2+ per peptide agrees excellently with the UV data and with the formation of a bis-ligand complex with a {HgS4} coordination geometry. The fact that the chemical shifts of the Cys protons lie in between those of the free peptide and the resonances of the mono-complex (see the broad peaks around 2.9 and 3.2 ppm in the middle spectrum in Fig. 2B) may simply reflect that Hg2+ quite efficiently attracts electrons in the {HgS2} type species, but that this effect (per cysteine) is less pronounced in the {HgS4} structure. Consequently, the deshielding effect on the Cys protons caused by Hg2+ coordination is weaker for {HgS4} than for {HgS2} structures. It is also interesting to note that the signals of the Cys CβH2 protons are hardly observable around neutral pH at a 0.5:1 Hg2+:DY ratio (Fig. S7A†), possibly as a consequence of severe line broadening. This might imply that the increase of pH and approaching the range where the thiol groups of the unbound ligands deprotonate have an influence on the ligand exchange rate and that ligand exchange dynamics also affects the observed Cys CβH2 resonances.
Complex speciation in the metal ion–peptide systems, unravelled by pH-potentiometric titrations
pH-Potentiometry is, in general, a powerful method for the characterization of complex formation processes that lead to the release of protons from the metal ion binding moieties of the ligand, and the pKa values of the free ligand and its Cd2+ complexes are presented in Table 1. However, Hg2+ ions are bound so efficiently to ligands with thiol groups, including the studied DY peptide, that the free metal ion fraction is negligible even at the very acidic end of the pH-range coverable by a glass electrode.29,69–71 As a consequence, the relevant metal-ion induced deprotonation processes are completed at the initial pH of the titrations and thus the method is not directly applicable to the determination of stability constants in such systems. (An indirect pH-metric approach for the characterization of Hg2+–peptide complex speciation using a competing Hg2+-binding ligand has been previously reported by Iranzo et al.29) In spite of the limitations of this method for the present system, we executed pH-potentiometric titrations aiming to follow the various deprotonation processes occurring at the Hg2+-bound peptide(s) and to determine the proton dissociation constants characterizing the interconversion of the differently protonated complex species. These data, collected in Table S1 in the ESI,† allowed the calculation of species distribution curves for Hg2+:DY 0.5:1 and 1:1 (Fig. 3) based on the plausible assumption that the species present at pH = 2.0 is HgH2L with two Hg2+-coordinated thiolate groups and protonated Asp and Tyr residues of the ligand. Indeed, the calculation of such a speciation scheme required the overall formation constant (logβ) of one of the species to be fixed (conveniently the logβ of HgH2L). Since we did not have an experimentally measured value for logβHgH2L we assumed that the Hg2+ + [H2L]2− ⇌ [HgH2L] process of DY could be characterized by the same stability constant as that of the parent mono-complex (HgL) of the terminally protected CDPPC peptide, determined by Iranzo et al. (logK = 40.0).29 By this assumption we could set a fixed, “arbitrary” logβHgH2L value (see more details in the ESI†) required for data evaluation. Note that it does not affect the conclusions of this work how well this arbitrary stability constant matches the real stability value of the HgH2L species, as long as it is high enough to ensure a complete binding of Hg2+ at the lowest utilized pH. This is also reflected by model calculations where in spite of the different applied logβHgH2L values the resulting pKa data for the deprotonation processes of the complexes turned out to be identical (Table S2†). The calculated distribution of species (Fig. 3) supports well all of our previous conclusions drawn from the spectroscopic data. The pKa values associated with the release of the two protons, leading to [HgHL]− (pKa = 4.1, Table S1†) and then to [HgL]2− (pKa = 9.6, Table S1†), are close to the values determined for the deprotonation processes of the Asp and Tyr units of the free ligand, respectively (pKa,1 = 3.80 and pKa,4 = 10.05; see Table 1). This indicates that the deprotonation of these groups is essentially not affected by the metal ion in the Hg2+-bound peptide. At Hg2+:DY = 0.5:1 the bis-ligand complexes dominate above pH 8. Considering the characteristic pH-range for the presence of the differently protonated bis complexes and the observed position of their S−–Hg2+ charge transfer bands, it is straightforward to suggest three coordinating thiolate groups in [HgH3L2]3− and thus a {HgS4} coordination mode in [HgH2L2]4−. Similar to the deprotonation step of the [HgL]2− parent complex, the two H+-dissociation processes of the [HgH2L2]4− species, occurring in the alkaline pH range (pKa = 10.0 and 11.2 – Table S1†), are assigned to the deprotonation of non-coordinating Tyr sidechains. Proposed schematic structures for the Hg2+:DY complexes are collected in Scheme S1.†
 | ||
| Fig. 3 Distribution of species in the Hg2+:DY 0.5:1 (continuous lines) and 1:1 (dashed lines) systems (cDY = 1.0 × 10−3 M, T = 298 K). | ||
β) of the Cd2+ complexes of DY (with the estimated errors in parentheses, last digit) together with some calculated data (I = 0.1 M NaClO4, T = 298 K)
| Species | logβ |
pKa |
|---|---|---|
|
a pKaCdxHyLz = logβCdxHyLz − logβCdxHy−1Lz.
b Stability constant calculated for Cd2+ + [HL]3− = [CdHL]−, logK1+H = logβ[CdHL]− − logβ[HL]3−.
c Stability constant calculated for [CdHL]− + [HL]3− = [CdH2L2]4−, logK2+2H = logβ[CdH2L2]4− − logβ[CdHL]− − logβ[HL]3−.
d NP = number of points.
e FP = fitting parameter.
|
||
| [HL]3− | 10.05(1) | 10.05 |
| [H2L]2− | 19.29(1) | 9.24 |
| [H3L]− | 27.56(2) | 8.27 |
| [H4L] | 31.36(2) | 3.80 |
| pKaCdxHyLza |
||
| [CdHL]− | 22.61(4) | 9.83 |
| [CdL]2− | 12.78(8) | 11.48 |
| [CdH–1L]3− | 1.3(2) | — |
| [CdH3L2]3− | 47.6(2) | 6.7 |
| [CdH2L2]4− | 40.9(1) | 9.9 |
| [CdHL2]5− | 31.0(2) | 10.6 |
| [CdL2]6− | 20.4(1) | — |
| [Cd2HL]+ | 25.4(2) | — |
| logK1+Hb |
12.56 | |
| logK2+2Hc |
8.21 | |
| log(K1+H/K2+2H) | 4.35 | |
| NP.d | 279 | |
| FPe (cm3) | 0.004 | |
Evaluation of pH-potentiometric data obtained for Cd2+:DY reveals that Cd2+-binding occurs above pH ∼ 3.5 and the first dominant species, formed in parallel with a small amount of a dinuclear species, is a mono-protonated mono-complex independent of the metal ion to ligand ratio (Fig. 4).
 | ||
| Fig. 4 Distribution of species in the Cd2+:DY 0.5:1 (A) and 1:1 (B) systems (cDY = 1.0 × 10−3 M, T = 298 K). | ||
The composition ‘[CdHL]−’ corresponds well to the coordination of both thiolates to Cd2+ and to a protonated Tyr phenol group in this species. Obviously, the Asp carboxyl function is deprotonated in [CdHL]−; however, since the formation of this complex spans the pH-range where the Asp sidechain of the free ligand also deprotonates, pH-metric data are not conclusive with regard to the Cd2+-binding of this group. Previous studies on short oligopeptides containing both Cys and Asp residues reflected, however, that participation of the available carboxylate groups in Cd2+-binding is possible,80,81,83 even if two thiolate donors are also coordinated.80,81 Binding of the Asp sidechain of DY to Cd2+ is supported by the stability constant calculated for the Cd2+ + [HL]3− = [CdHL]− process (logK1+H = 12.56). This value is nearly identical to the stability that may be calculated for the relevant, {CdS2O2} type complex of a short phytochelatin peptide ((γ-Glu-Cys)2-Gly – PC2, logK1+H = 12.57)80 and significantly higher than the stability of the protonated mono-complex of ACSSACS-NH2 with only two coordinating thiolate groups (logK1+H = 10.22).84 The remarkable extra stability observed for the mono-complex of DY is presumably only partly related to the additional binding of the Asp carboxylate group, and may also be a consequence of the more favoured CXXC amino acid pattern vs. the CXXXC sequence in the ACSSACS-NH2 heptapeptide.
Furthermore, the additional coordination of one of the internal peptide carbonyl groups, as suggested for PC2, is also possible in the [CdHL]− complex of DY resulting in a {S−,S−,COO−,CO} donor group environment around the metal ion. The pKa value determined for the [CdHL]− = [CdL]2− + H+ process (pKa = 9.83) is very close to the pKa of the Tyr residue in the free ligand indicating that the sidechain phenolate is not bound to Cd2+, similar to what was concluded for the Hg2+ complexes of DY (suggested schematic structures for the Cd2+:DY complexes are shown in Scheme S2†). Calculation of the apparent dissociation constant for the Cd2+:DY mono-complexes at pH = 7.0, according to the equation Kd = ([Cd2+]free × [Ligand]free)/[Cd2+]complexed, may allow the comparison of the Cd2+-binding affinity of DY to other relevant cysteine containing peptides or proteins. The calculated Kd = 9.1 × 10−10 M value indicates a slightly larger Cd2+-binding affinity of DY compared to that of the PC2 phytochelatin peptide (Kd = 1.7 × 10−9 M)80 but a substantially stronger Cd2+-binding relative to the reduced glutathione (Kd = 1.4 × 10−5 M).86 However, the average dissociation constants determined for the α and β domains of MT-2 metallothioneins (Kd = 1.0 × 10−15 M (α) and 0.1–1.0 × 10−12 M (β))87 clearly reflect a more efficient Cd2+-coordination by these proteins, provided by the thiolate-clusters.
At twofold DY excess over Cd2+ the bis-ligand complexes start to form from ca. pH 5 and become fully dominant by pH ∼ 8 (Fig. 4A), as also suggested by the pH dependent evolution of the S−–Cd2+ charge transfer band and the calculated molar extinction coefficient for λ = 240 nm at Cd2+:DY 0.5:1, vide supra. The two proton release processes, leading from the four-thiolate coordinated [CdH2L2]4− complex to [CdHL2]5− and then to [CdL2]6−, are assigned to dissociation processes of the non-coordinating Tyr phenol sidechains (see Table 1). The stability constant calculated for the protonated bis complex ([CdHL]− + [HL]3− = [CdH2L2]4−, logK2CdH2L2 = 8.21) and the relative stability of the protonated mono- and bis-ligand species (log(K1+H/K2+2H) = 4.35) indicate that binding of the second ligand is substantially less favoured than that of the first one. Very similar data have been reported for the bis complex formation of the phytochelatin pentapeptide PC2 (log(K1+H/K2+2H) = 4.39)80 leading to a four thiolate coordinated species. The relative positions of the two Cys residues in DY and PC2 are different (CXXC and CXC) just as the overall charges of their relevant mono- and bis complexes (DY: −1/−4, PC2: −2/−6); nevertheless, the two peptides are of similar sizes. This suggests that the relative binding strength of the second DY or PC2 ligand is determined by similar factors, i.e. the notably high stability of their mono-complex and steric hindrance (bulkiness of the peptides) but the log(K1+H/K2+2H) values are probably not large enough to assume a major change in the coordination geometry of Cd2+.
Influence of metal ions on the fluorescence of the ligand
The effect of the metal ions on the fluorescence of the Tyr residue, as a function of the metal ion to DY ratio, was monitored at selected pH values, applying excitation at λ = 278 nm. Gradual addition of Cd2+ ions to a solution of the peptide at pH = 7.1 (Fig. S10†) showed a partial quenching (∼35% drop) of the original emission intensities, levelling off at a ca. 0.6–0.7:1 Cd2+:DY ratio. This I308 nmvs. Cd2+:DY ratio profile perfectly correlates with the disappearance of the free ligand, occurring slightly above a 0.5:1 Cd2+:peptide ratio at pH = 7.1 (see Fig. 4A), especially under the small concentrations used for the fluorimetric studies. Since further increase of the Cd2+:DY ratio was found to have no influence on the observed emission intensity, Cd2+ seems to exert a very similar quenching effect on the fluorescence of the Tyr residue, independent of the number of coordinated DY molecules. In a double (Trp and dansyl) labelled peptide containing two Cys residues, Cd2+ was found to induce a turn-on response; nevertheless, the chelation enhanced fluorescence also reached a maximum around a 0.5:1 Cd2+:peptide ratio, and accordingly, it was attributed to the presence of a bis complex.56
For the experiments in the Hg2+:DY system pH = 6.0 was chosen to apply a close-to-neutral condition while avoiding the appearance of bis complexes and thus maintaining a relatively simple speciation that should be desirable for a molecular probe. In view of the outstanding affinity of DY towards Hg2+ that, in contrast to Cd2+, results in a complete binding of this metal ion even at rather low pH values, we recorded Hg2+:DY ratio dependent emission spectra at pH = 2.0, as well.
As Fig. 5 clearly shows, the increase of the concentration of Hg2+ leads to a significant drop of emission intensities, following a linear trend until reaching a 1:1 Hg2+:DY ratio both at pH = 6.0 and 2.0. This correlates well with the calculated complex speciation (Fig. 3) and indicates a strong and complete binding of 1 eq. Hg2+ per DY. However, it also suggests that the ligand cannot capture a second Hg2+ ion in solution, which is also a desirable property for a Hg2+ sensor. Although different species dominate at the two selected pH values (HgH2L at pH = 2.0 and [HgHL]− at pH = 6.0), the observed trends in the decreasing fluorescence emission intensities are nearly identical in the two cases. This suggests that the protonation state of the uncoordinated Asp carboxyl group has no impact on the Hg2+-induced quenching of the Tyr fluorescence.
 | ||
| Fig. 5 Fluorescence titration of DY with Hg2+ at pH = 2.0 (circles) and pH = 6.0 (triangles) in the absence (filled symbols) and presence (open symbols) of Cd2+ ions. Fitted lines represent the trends in the decrease of intensity and the breakpoint at 1.0 eq. of Hg2+ (cDY = 3.0 × 10−5 M, λEM = 308 nm, λEX = 278 nm). The inset shows the recorded spectra at pH = 6.0 in the absence of Cd2+ ions. | ||
Different possibilities may be considered for the mechanism of the metal ion induced quenching of the fluorescence of DY, as suggested by Chen's in-depth overview of the interaction of Hg2+ with proteins containing aromatic amino acids.88 Indeed, Hg2+ was shown to quench the fluorescence of the vast majority of tested proteins, especially of those that contained Cys residues near the aromatic amino acids.88 Although direct interactions of both of the studied metal ions with the Tyr residue of DY have been excluded in our systems, an overlap between the thiolate-related absorption bands and the Tyr emission might cause quenching via energy transfer. Besides, metal ion promoted conformational changes in the peptide structure may also play a role in the altered fluorescence.
We also tested the Hg2+-induced quenching of the fluorescence of DY in the presence of 1.0 eq. of Cd2+. Since the latter metal ion is not bound to the peptide at pH = 2.0 (Fig. 4) it has no influence on the emission intensities at any Hg2+:DY ratio. Nevertheless, 1.0 eq. Cd2+ per DY at pH = 6.0 induces a notable fluorescence quenching, as compared to the Cd2+-free sample, reflecting that Cd2+–DY complexes are present at the start of the titration. When this sample is titrated with Hg2+ the observed fluorescence is gradually quenched down to the same level as seen in the absence of Cd2+, suggesting that the DY-bound Cd2+ is completely displaced by Hg2+.
In view of the potential analytical applicability of such a molecular probe the most promising findings of the above experiments are the significant fluorescence response of the ligand, proportional to the Hg2+-concentration up to a 1:1 Hg2+:DY ratio, as well as the remarkable metal ion binding selectivity that is also manifested in the Hg2+-mediated changes in emission intensities. In this context, the data series recorded at pH 2 (Fig. 5) demonstrates selective binding of Hg2+ at low pH, which may be used to eliminate interferences from other metal ions, such as Cd2+, in a fluorophore based Hg2+ assay. A quick assessment of the potential analytical figures of merit for Hg2+ sensing under our measurement conditions yields an estimated limit of detection of ca. 2 × 10−7 M and a linear dynamic range exceeding 2 orders of magnitude.
Hg2+ binding of the DY peptide immobilized to a resin
Next we aimed to explore Hg2+ binding to the DY ligand in an immobilized form. To achieve this, we synthesized a construct carrying the peptide anchored to a hydrophilic resin (DY-NTG). Hg2+ binding to DY-NTG was studied as a function of the contact time, pH and metal ion concentration at pH = 2.0. As a reference system, the peptide-free resin with acetylated terminal amino groups (Ac-NTG) was also tested at pH = 2.0 by adding 1.5 eq. of Hg2+ per available blocked peptide anchoring site. The determined 0.0354 mmol Hg2+ per g Ac-NTG metal ion binding capacity represents ca. 13% of the theoretical loading of the resin (quantity of available functionalization sites on the resin beads, L = 0.267 mmol g−1). It seems that a small but non-negligible amount of Hg2+ may be bound by Ac-NTG, probably at the polyethylene-glycol chains. However, such binding sites in the peptide functionalized resin, DY-NTG, may play a role in metal ion binding only under metal ion excess conditions.Since metal ion complexation processes could be slower at the resin bead-supported binding sites as compared to the solution phase, first we determined the contact time necessary for a complete equilibration in the reaction of Hg2+ and DY-NTG. Samples were prepared at pH = 2.0 by adding Hg2+ ions in concentrations corresponding to 1.5, 2.0 and 3.0 eq. of Hg2+ relative to the theoretical loading of the resin, i.e. to the available immobilized peptide chains (0.227 mmol g−1; see the sample preparation protocol in the Methods section). The excess of metal ions ensured that the concentration of Hg2+ could be conveniently and accurately measured by ICP-MS after the termination of the reaction. The studied samples were shaken for 30, 45, 60, 120, 180 and 300 minutes. Three additional samples, prepared with 0.33, 0.66 and 1.0 eq. of Hg2+ per peptide, were also tested by using 60 and 120 min contact times. Analysis of the remaining Hg2+ concentrations in all these samples showed that up to 1.0 eq. of Hg2+ per ligand the concentration of the metal ion was stable after 60 min (or even less, as suggested by other preliminary tests); however, a 120 min equilibration time seemed to be necessary for samples containing metal ion excess. Accordingly, a contact time of 120 min was used for all further experiments, including those performed with the reference compound, Ac-NTG.
The effect of pH on the Hg2+ binding ability of DY-NTG was monitored between pH = 1.0 and 6.0 by preparing samples of ∼1.0 eq. Hg2+ per DY ligand. In good correlation with the complex speciation determined for the Hg2+:DY system in solution (Fig. 3), no variation in the bound metal ion quantity was observed. Consequently, the immobilized peptide resembles well the Hg2+ binding ability of DY in solution up to a 1:1 Hg2+:DY ratio, as indicated by the binding of ca. 95% of the added Hg2+ ions to DY-NTG independent of the applied pH (Fig. S11†).
Hg2+ binding to DY-NTG was investigated at pH = 2.0 as a function of the Hg2+ concentration in a range of 0.774–6.49 μmol/10.0 mL sample representing ca. 0.34–2.86 eq. of metal ions per available immobilized peptide (Fig. 6). The bound quantity of Hg2+, calculated from the remaining metal ion concentrations of the samples in contact with DY-NTG, increases linearly up to ca. 1.0 eq. Hg2+ per peptide. At this specific point the bound quantity of Hg2+, as measured by ICP-MS, indicates that nearly 95% (2.07 μmol) of the added Hg2+ ions (2.20 μmole) are bound. As seen in Fig. 6, the bound Hg2+ quantity further increases in parallel with the increasing Hg2+ concentration of the samples and the obtained curve follows a saturation-like trend converging towards what appears to be ∼2.0 eq. bound Hg2+ per peptide. This suggests that other, weaker binding sites also participate in metal ion binding and the quantity of Hg2+ ions, captured at these sites, cannot be explained by the metal ion binding ability of the non-peptidic parts of the resin, considering the value measured for the reference Ac-NTG (0.0354 mmol g−1). In some of the previously published studies on the metal ion binding abilities of cysteine containing peptides,89–91 immobilized to various matrices, the authors also reported about the presence of binding sites with different affinities in their constructs.91 Since solution phase experiments with the DY peptide showed that it was able to bind only one Hg2+ ion per molecule, the coordination of the second metal ion must be attributed to binding positions originating from the immobilization of the ligands. These positions are probably less accessible than the primary binding positions and might require a slow structural change of the solid supported ligands, as suggested by the longer contact times needed for equilibration when metal ion excess is used.
 | ||
| Fig. 6 Hg2+ binding (μmol) to the immobilized ligand at pH = 2.0 plotted as a function of the original Hg2+ content (μmol) of samples (Vsample = 10.0 mL; mDY-NTG = 10.0 mg). The dashed line is a linear fit of data up to 1.0 eq. of added Hg2+ relative to the theoretical Hg2+ binding capacity of DY-NTG (L = 2.27 μmol/10 mg). The intercept of the narrow dotted lines represents an expected data point if 1.0 eq. of added Hg2+ is fully captured. | ||
These results demonstrate that DY-NTG efficiently binds Hg2+ even at low pH (pH = 2.0) and the bound quantity linearly increases with the increasing metal ion concentration in samples where the Hg2+ content does not exceed the theoretical Hg2+-binding capacity of the resin.
In order to test whether DY-NTG displays a selectivity in the binding of Hg2+, two types of samples containing the resin and four selected metal ions, Hg2+, Cd2+, Zn2+ and Ni2+, were prepared. Since data for Cd2+:DY showed that this metal ion cannot be bound by the ligand at pH = 2.0, which most probably also holds for Zn2+ or Ni2+, the metal ion selectivity experiments were performed at pH 6.0 by applying 1.0 or 2.0 equivalents of all the four metal ions, relative to the available immobilized peptide, in both types of samples. The results obtained reveal a dominance of Hg2+ in occupying the available binding sites of DY-NTG, especially at twofold excess of the metal ions relative to the peptide where only Hg2+ is bound by DY-NTG. However, a significant amount of Cd2+ (0.59 μmol), besides 1.96 μmol Hg2+, was also captured from the sample containing 1.0 eq. of all the four metal ions (Fig. 7). These data represent ca. 90% and 24% binding of the added Hg2+ and Cd2+ ions, respectively. Consequently, at pH = 6.0, Cd2+ can successfully occupy presumably mainly some of the lower affinity sites of the resin, even in the presence of 1.0 eq. of Hg2+ per peptide. Since Cd2+ induces a notable quenching of the fluorescence of DY in solution it may potentially act as an interference in the Hg2+-binding of this molecular probe at pH = 6.0 and in samples with sub-stoichiometric M2+:peptide ratios. However, Zn2+ and Ni2+ cannot compete with Hg2+ or Cd2+ in binding at the lower affinity sites.
 | ||
| Fig. 7 Binding of different M2+ ions to the immobilized ligand (μmol) at pH = 6.0 (cMES = 0.02 M) from two sample solutions containing 1.0 eq. (2.27 μmol) (A) or 2.0 eq. (4.54 μmol) (B) of all the four metal ions relative to the theoretical M2+ binding capacity of DY-NTG. (Vsample = 10.0 mL; mDY-NTG = 10.0 mg). | ||
Conclusions
In an attempt to investigate the possibility of using simple, very short, bioinspired peptides as fluorescent molecular probes for heavy metal ion sensing, we studied the interaction of a hexapeptide, Ac-DCSSCY-NH2 (DY), with Cd2+ and Hg2+ ions in the solution phase and characterized the Hg2+-binding ability of the ligand in the immobilized form, too. The outstanding affinity of Hg2+ to the Cys thiolates results in a complete binding of 1.0 eq. of Hg2+ to the peptide even at pH = 2.0, whereas Cd2+ complexation starts only at ca. pH 3.5. Both mono- and bis-ligand complexes, with the participation of two and four thiolates, respectively, as the key donors in metal ion binding, are present with Cd2+ and Hg2+ above pH ∼ 5.5 (Cd2+) and 7 (Hg2+), depending on the applied metal ion to ligand ratio. Both metal ions were found to quench the fluorescence of the Tyr residue of DY at pH = 6.0; nevertheless, this effect is significantly stronger with Hg2+, resulting in a 60–65% drop of the original emission intensity until 1.0 eq. of Hg2+ is added to the ligand. The decrease of intensity follows a linear trend until reaching a 1:1 Hg2+:DY ratio even in the presence of 1.0 eq. Cd2+ which reflects a complete displacement of the peptide-bound Cd2+ by Hg2+. Based on experimental data in solution and related to the fluorescence emission detected at 308 nm, an estimated 2 × 10−7 M value for the limit of detection for Hg2+ and a linear dynamic range exceeding 2 orders of magnitude were obtained.
The strong Hg2+-binding affinity of DY in acidic samples (pH = 2.0) is preserved even in the resin-supported form. The immobilized DY displays a nearly complete Hg2+-binding ability up to 1.0 eq. of Hg2+ per immobilized peptide is added. However, a further, saturation-type increase in the amount of bound Hg2+ is observed from samples containing Hg2+ ions in excess (from 1.0 to 3.0 eq. per ligand), suggesting a role of lower affinity binding positions in metal ion binding. This is in slight contrast to the Hg2+-coordinating features of the peptide in the solution phase where no signs of dinuclear/oligonuclear Hg2+-complex formation are seen. Indeed, binding of Hg2+ beyond 1.0 eq. per immobilized DY possibly occurs via structures that may exist only under the ligand-rich conditions present in the swollen resin beads and/or by the participation of the PEG chains in metal ion coordination. The presented experimental data, including the remarkable, concentration-dependent quenching effect of Hg2+ on the fluorescence of the ligand and the efficient Hg2+-binding of DY either in a solution or immobilized form show the potential of such simple bio-inspired systems in metal ion sensing. In addition to this, our metal ion selectivity experiments suggest that interference from other metal ions may be avoided by using acidic pH.
In our view, this work presents a fine example for the advantages of a combined study of metal binding molecular probes in solution and immobilized forms as by this method the altering effects of immobilization on the metal ion binding characteristics can also be unravelled, thereby promoting the development of practical metal ion sensing constructs.
Experimental
Materials
The hexapeptide N-acetyl-Asp-Cys-Ser-Ser-Cys-Tyr-NH2 (abbreviated as DY) was purchased from CASLO ApS with a purity of 98.03%. All other chemicals and solvents, such as N-α-Fmoc-O-tert-butyl-L-tyrosine, N-α-Fmoc-S-trityl-L-cysteine, N-α-Fmoc-O-tert-butyl-L-serine, N-α-Fmoc-L-aspartic acid β-tert-butyl ester, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), N-hydroxybenzotriazole (HOBt), Novasyn TG amino resin (all Novabiochem products), pyridine (Merck), N,N-diisopropylethylamine (DIPEA), diethylether, triisopropylsilane (TIS), 1,2-ethanedithiol (EDT), piperidine, acetic anhydride, trifluoroacetic acid (TFA), phenol, NaClO4, HgCl2, ZnCl2, CdCl·xH2O, NiCl2·6H2O, Hg(ClO4)2·nH2O, potassium hydrogen phthalate, NaOH solution (all from Sigma-Aldrich), 1-methyl-2-pyrrolidone (NMP), dichloromethane, methanol (Molar Chemicals products), acetonitrile (BDH Prolabo Chemicals) and Cd(ClO4)2·6H2O (Alfa-Aesar) were also of commercial origin and used without further purification. Hg(ClO4)2·nH2O stock solutions were prepared by dissolving the salt in 0.0600 M perchloric acid. Metal ion stock solutions (Hg2+, Cd2+, Zn2+, and Ni2+) were standardized complexometrically except that of HgCl2. Stock solutions of HgCl2 of the desired concentrations were prepared from precise weights of the high purity mercury salt. pH-Potentiometric titrations were performed using a NaOH titrant, standardized with potassium hydrogen phthalate.Synthesis of the immobilized ligand
The hexapeptide in an immobilized form was synthesized on a Novasyn TG amino resin (NTG, 90 μm beads, loading: 0.26 mmol g−1) by solid phase peptide synthesis using the Fmoc methodology (Fmoc = 9-fluorenylmethoxycarbonyl). The polyethylene glycol (PEG) polystyrene composite resin was chosen owing to its excellent swelling characteristics in aqueous media. The synthesized peptide molecules were tightly anchored to the resin via amide bonds between the C-terminal Tyr carboxylic groups and the amino functions of the resin which prevented the cleavage of the peptides off the resin under the conditions of the performed experiments. The notation “DY-NTG”, referring to the immobilized ligand, is used throughout the text. As a reference compound, “Ac-NTG” (a peptide-free resin with acetylated amino functions) was also prepared and tested in the metal-ion binding studies. The synthesis of the immobilized peptide, DY-NTG, and the reference material, Ac-NTG, was executed according to the steps described in detail earlier.92Methods
UV absorption and fluorescence measurements
UV–Visible (UV–Vis) spectra were measured on a Thermo Scientific Evolution 220 spectrophotometer in the wavelength range of 210–400 nm, using a quartz cell with a 1.0 cm optical path length, equipped with a Teflon cap. The concentration of the ligand was 1.0 × 10−4 M and the metal ion concentration varied between 5.0 × 10−5 and 2.0 × 10−4 M. In order to avoid the eventual oxidation of the ligand, weighed amounts of the peptide were initially dissolved in a 1.0 × 10−2 M HClO4 solution. The concentrations of the stock solutions of the ligand were determined by pH-potentiometric titrations. The pH of the samples was adjusted with NaOH solutions and measured using an Orion 710A precision digital pH-meter equipped with a Metrohm Micro pH glass electrode. The water baseline was subtracted from the raw spectra and the spectra were normalized to a 1.0 × 10−4 M peptide concentration to correct for the possible dilution occurring in the titrations.Fluorescence emission spectra were recorded on a Hitachi-F4500 spectrofluorimeter in the wavelength range of 285–400 nm applying an excitation wavelength of 278 nm and slit widths of 5 nm (excitation beam) and 10 nm (emission beam), using a 1.0 cm × 1.0 cm quartz cell equipped with a PTFE cap. For samples of Hg2+–DY, the experiments were performed at two pH values (pH = 2.0 and 6.0; T = 298 K) by varying the Hg2+-concentration (c = 0.0–75.0 μM) and, accordingly, the Hg2+ to peptide ratio. The initial concentration of the ligand (cDY = 28.0 μM) varied only as a result of the slight dilution during the titrations. The pH of the samples at pH = 6.0 was monitored during the titrations and corrected, when necessary, by the addition of small volumes of a 0.01 M NaOH solution. There was no need for pH-correction in samples adjusted to pH = 2.0. Experiments were also performed in the presence of Cd2+ ions where a constant 1:1 concentration ratio of Cd2+ and DY was maintained. Variation of the fluorescence intensity as a function of pH was monitored in samples containing Cd2+ and DY in different concentration ratios (0:1, 0.5:1 and 1:1).
The background-subtracted spectra were corrected for the inner filter effects according to the equation:62
| Fcorr = Fobs × 10(Aex+Aem)/2 |
NMR experiments
1H NMR measurements were performed on a Bruker Avance DRX 500 spectrometer operating at 500.132 MHz. Samples were prepared in H2O containing D2O in 10% (v/v) and in a few cases in pure D2O and the spectra were recorded at T = 298 K. The zgpr or zgcppr pulse sequences were applied for the presaturation of the H2O/HDO resonances. The typical concentration of the peptide was 1.3 × 10−3 M. The chemical shifts were referenced to TSP-d4 at 0.0 ppm. 1D NMR spectra were recorded by using a recycle delay of 5 s, an acquisition time of 1.64 s, a spectral width of 5 or 10 kHz and 64–128 scans. For samples prepared in D2O the pH-meter readings (uncorrected by the deuterium effect), pH*, were adjusted to the desired values with NaOD. The recorded spectra were processed using the ACD/Spectrus Processor software.93 Assignment of the 1H resonances of the peptide was accomplished by executing 2D COSY, TOCSY and ROESY experiments. All 2D spectra were acquired with 2048(F2) × 256(F1) complex points, covering a spectral range of 6 kHz both in F2 and F1. The TOCSY experiments were executed by using the MLEVPHPR sequence with a mixing time of 70 ms while the ROESY spectrum was recorded with a mixing time of 200 ms. The obtained data were processed using the Bruker software Topspin 3.1.pH-Potentiometric measurements
(De)protonation and complex formation processes were studied in aqueous solutions degassed with high purity Ar gas and by applying an Ar atmosphere throughout the experiments to avoid the oxidation of the ligand. The pH-potentiometric titrations were carried out with a PC-controlled automatic titration set including a Metrohm Dosimat 765 autoburette and an Orion 710A pH/ISE meter equipped with a Thermo Scientific Orion 8103BNUWP Ross Ultra pH electrode (165 × 6 mm). Relative mV values, as pH-meter readings, were converted to hydrogen ion concentrations as described earlier.85 The protonation and complex formation equilibria were characterized by the following general equilibrium process and the relevant overall formation constant:| pM + qH + rL ⇌ MpHqLr |
where M stands for the metal ion, L for the deprotonated ligand molecule, and H for the protons. Charges are omitted in the above equations for simplicity but can be calculated from the composition of the fully protonated, neutral ligand form, H4L. Overall formation constants (βMpHqLr) defined by the above equation were calculated using the PSEQUAD software.94 Four parallel titrations (60–70 data points per titration) were performed in the absence of metal ions (cpeptide ∼ 1.0 × 10−3 M) for determining the (de)protonation constants of the ligand. The purpose of the titration of the ligand stock solutions was also to obtain the accurate concentration of the peptide. Complex formation constants were evaluated from 3–4 independent titrations in the different metal ion ligand systems (60–100 data points per titration). The applied ratios of the metal ion (Cd2+ or Hg2+) and the ligand were 0.5
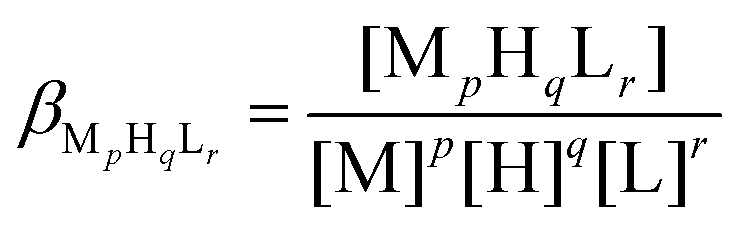 :1, 1:1 and 2:1 with the ligand concentrations varying between 7.3 × 10−4–1.0 × 10−3 M. In the Hg2+:DY 2:1 system precipitation occurred even under acidic conditions that prevented performing titrations under such conditions with this metal ion.
:1, 1:1 and 2:1 with the ligand concentrations varying between 7.3 × 10−4–1.0 × 10−3 M. In the Hg2+:DY 2:1 system precipitation occurred even under acidic conditions that prevented performing titrations under such conditions with this metal ion.
Experimental conditions and instrumentation utilized in studying Hg2+-binding to the immobilized peptide
Metal ion binding properties of DY-NTG were investigated in 10.00 mL volume samples containing 10.0 mg (±1%) of the resin-supported peptide and the metal ion (typically from a HgCl2 stock solution) in a concentration range of 7.57 × 10−5–6.81 × 10−4 M depending on the type of experiment. The pH of the samples varied between pH = 1.0 and 6.0 and adjusted with perchloric acid solutions (c = 1.0 M or 0.1 M) or with AcOH/NaOAc (pH = 4.0) or MES/NaOH (pH = 6.0) buffer systems. All solutions used for sample preparation were carefully degassed with Ar to prevent the potential oxidation of the peptide. The 10.0 mL samples, placed in glass vessels, were vertically rotated for 120 min with a vertical rotating shaker to allow complete equilibration in the metal ion binding process under any conditions. The resin beads were settled by centrifugation for 15 min at 5000 rpm and 2.0 mL aliquots of the solutions were precisely diluted and analyzed for determining the residual metal ion concentrations in the liquid phase of the resin contacted samples.The applied concentrations of the metal ions were calculated on the basis of the theoretical peptide loading of the resins and the increase of resin mass during the synthesis, according to
All of the presented data are the results of duplicate experiments except that of the Hg2+ binding capacity of Ac-NTG obtained from five repetitions.
Determination of metal ion concentrations in the solutions was accomplished on an Agilent 7700x inductively coupled plasma mass spectrometer (ICP-MS). The instrument was used in the helium mode of the OSR3 collisional cell. Calibrating solutions were prepared from a certified monoelemental Hg stock standard solution (CaPurAn M324.5NP.L1) and trace quality deionized lab-water (Millipore Elix Advantage 5 + Synergy) to perform a multi-point calibration. The mass peaks of 201Hg and 111Cd isotopes were utilized for quantification with 209Bi and 103Rh signals, respectively, as internal standards (using the Agilent no. 5188-6525 internal standard mix). Prior to all measurements, the autotuning of the ICP-MS instrument was performed according to the manufacturer's specifications, using standard solutions supplied by Agilent. All labware was cleaned before use with ultratrace grade nitric acid and hydrochloric acid (BDH Aristar Ultra), thoroughly rinsed with the above trace quality deionized lab-water and finally dried under a laminar flow clean bench.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Hungarian National Research, Development and Innovation Office-NKFIH through project GINOP-2.3.2-15-2016-00038 and grant no. K_16/120130. LSz wishes to thank the Balassi Institute for a fellowship within the frames of the Campus Hungary Programme, contract no. 47607.Notes and references
- D. F. Flick, H. F. Kraybill and J. M. Dimitroff, Environ. Res., 1971, 4, 71–85 CrossRef CAS PubMed.
- G. Bertin and D. Averbeck, Biochimie, 2006, 88, 1549–1559 CrossRef CAS PubMed.
- C. M. L. Carvalho, E.-H. Chew, S. I. Hashemy, J. Lu and A. Holmgren, J. Biol. Chem., 2008, 283, 11913–11923 CrossRef CAS PubMed.
- R. Zefferino, C. Piccoli, N. Ricciardi, R. Scrima and N. Capitanio, Oxid. Med. Cell. Longevity, 2017, 7028583 Search PubMed.
- D. H. Nies, J. Bacteriol., 1995, 177, 2707–2712 CrossRef CAS PubMed.
- C. Rensing, B. Mitra and B. P. Rosen, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14326–14331 CrossRef CAS PubMed.
- E. Aguilar-Barajas, M. I. Ramírez-Díaz, H. Riveros-Rosas and C. Cervantes, in Pseudomonas, ed. J. L. Ramos and A. Filloux, Springer, Dordrecht, 2010, ch. 9, pp. 255–282 Search PubMed.
- T. Barkay, S. M. Miller and A. O. Summers, FEMS Microbiol. Rev., 2003, 27, 355–384 CrossRef CAS PubMed.
- F. Bonomi, S. Iametti, D. M. Kurtz, E. M. Ragg and K. A. Richie, J. Biol. Inorg. Chem., 1998, 3, 595–605 CrossRef CAS.
- I. Voskoboinik, D. Strausak, M. Greenough, H. Brooks, M. Petris, S. Smith, J. F. Mercer and J. Camakaris, J. Biol. Chem., 1999, 274, 22008–22012 CrossRef CAS PubMed.
- A. J. Sytkowski and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 344–348 CrossRef CAS PubMed.
- C. Frauer, A. Rottach, D. Meilinger, S. Bultmann, K. Fellinger, S. Hasenöder, M. Wang, W. Qin, J. Söding, F. Spada and H. Leonhardt, PLoS One, 2011, 6, e16627 CrossRef CAS PubMed.
- T. V. O'Halloran and V. C. Culotta, J. Biol. Chem., 2000, 275, 25057–25060 CrossRef PubMed.
- A. C. Rosenzweig, Chem. Biol., 2002, 9, 673–677 CrossRef CAS PubMed.
- C. A. Blindauer, J. Biol. Inorg. Chem., 2011, 16, 1011–1024 CrossRef CAS PubMed.
- R. A. Steele and S. J. Opella, Biochemistry, 1997, 36, 6885–6895 CrossRef CAS PubMed.
- T. K. Misra, N. L. Brown, L. Haberstroh, A. Schmidt, D. Goddette and S. Silver, Gene, 1985, 34, 253–262 CrossRef CAS PubMed.
- R. Ledwidge, B. Patel, A. Dong, D. Fiedler, M. Falkowski, J. Zelikova, A. O. Summers, E. F. Pai and S. M. Miller, Biochemistry, 2005, 44, 11402–11416 CrossRef CAS PubMed.
- W. E. Van Der Linden and C. Beers, Anal. Chim. Acta, 1974, 68, 143–154 CrossRef CAS PubMed.
- J. Starý and K. Kratzer, J. Radioanal. Nucl. Chem. Lett., 1988, 126, 69–75 CrossRef.
- W. Stricks and I. M. Kolthoff, J. Am. Chem. Soc., 1953, 75, 5673–5681 CrossRef CAS.
- J. S. Casas and M. M. Jones, J. Inorg. Nucl. Chem., 1980, 42, 99–102 CrossRef CAS.
- R. Ledwidge, B. Hong, V. Dötsch and S. M. Miller, Biochemistry, 2010, 49, 8988–8998 CrossRef CAS PubMed.
- A. C. Rosenzweig, D. L. Huffman, M. Y. Hou, A. K. Wernimont, R. A. Pufahl and T. V. O'Halloran, Structure, 1999, 7, 605–617 CrossRef CAS PubMed.
- O. Sénèque, S. Crouzy, D. Boturyn, P. Dumy, M. Ferrand and P. Delangle, Chem. Commun., 2004, 770–771 RSC.
- P. Rousselot-Pailley, O. Sénèque, C. Lebrun, S. Crouzy, D. Boturyn, P. Dumy, M. Ferrand and P. Delangle, Inorg. Chem., 2006, 45, 5510–5520 CrossRef CAS PubMed.
- G. Veglia, F. Porcelli, T. DeSilva, A. Prantner and S. J. Opella, J. Am. Chem. Soc., 2000, 122, 2389–2390 CrossRef CAS.
- T. M. DeSilva, G. Veglia, F. Porcelli, A. M. Prantner and S. J. Opella, Biopolymers, 2002, 64, 189–197 CrossRef CAS PubMed.
- S. Pires, J. Habjanič, M. Sezer, C. M. Soares, L. Hemmingsen and O. Iranzo, Inorg. Chem., 2012, 51, 11339–11348 CrossRef CAS PubMed.
- P. Faller, B. Ctortecka, W. Tröger, T. Butz, ISOLDE Collaboration and M. Vašák, J. Biol. Inorg. Chem., 2000, 5, 393–401 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, X. C. Su, R. Miras, N. Bal, E. Mintz, P. Catty, J. E. Shokes and R. A. Scott, J. Mol. Biol., 2006, 356, 638–650 CrossRef CAS PubMed.
- L. Prodi, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS.
- M. Capdevila, A. González-Bellavista, M. Muñoz, S. Atrian and E. Fàbregas, Chem. Commun., 2010, 46, 2040–2042 RSC.
- A. González-Bellavista, S. Atrian, M. Muñoz, M. Capdevila and E. Fàbregas, Talanta, 2009, 77, 1528–1533 CrossRef PubMed.
- H. Ju and D. Leech, J. Electroanal. Chem., 2000, 484, 150–156 CrossRef CAS.
- K. K. Tadi, I. Alshanski, E. Mervinetsky, G. Marx, P. Petrou, K. M. Dimitrios, C. Gilon, M. Hurevich and S. Yitzchaik, ACS Omega, 2017, 2, 8770–8778 CrossRef CAS PubMed.
- S. Cherian, R. K. Gupta, B. C. Mullin and T. Thundat, Biosens. Bioelectron., 2003, 19, 411–416 CrossRef CAS PubMed.
- E. S. Forzani, H. Zhang, W. Chen and N. Tao, Environ. Sci. Technol., 2005, 39, 1257–1262 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- M. Dutta and D. Das, TrAC, Trends Anal. Chem., 2012, 32, 113–132 CrossRef CAS.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- J.-M. Kim, C. R. Lohani, L. N. Neupane, Y. Choi and K.-H. Lee, Chem. Commun., 2012, 48, 3012–3014 RSC.
- J.-M. Kim, B. P. Joshi and K.-H. Lee, Bull. Korean Chem. Soc., 2010, 31, 2537–2541 CrossRef CAS.
- A. Torrado, G. K. Walkup and B. Imperiali, J. Am. Chem. Soc., 1998, 120, 609–610 CrossRef CAS.
- B. R. White and J. A. Holcombe, Talanta, 2007, 71, 2015–2020 CrossRef CAS PubMed.
- B. P. Joshi, J. Park, W. I. Lee and K.-H. Lee, Talanta, 2009, 78, 903–909 CrossRef CAS PubMed.
- B. P. Joshi, J.-Y. Park and K.-H. Lee, Sens. Actuators, B, 2014, 191, 122–129 CrossRef CAS.
- B. P. Joshi, C. R. Lohani and K.-H. Lee, Org. Biomol. Chem., 2010, 8, 3220–3226 RSC.
- B. P. Joshi, W. M. Cho, J. Kim, J. Yoon and K.-H. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 6425–6429 CrossRef CAS PubMed.
- X. Pang, L. Gao, H. Feng, X. Li, J. Kong and L. Li, New J. Chem., 2018, 42, 15770–15777 RSC.
- B. P. Joshi and K.-H. Lee, Bioorg. Med. Chem. Lett., 2008, 16, 8501–8509 CrossRef CAS PubMed.
- P. Wang, J. Wu, P. Zhou, W. Liu and Y. Tang, J. Mater. Chem. B, 2015, 3, 3617–3624 RSC.
- P. Wang, L. Liu, P. Zhou, W. Wu, J. Wu, W. Liu and Y. Tang, Biosens. Bioelectron., 2015, 72, 80–86 CrossRef CAS PubMed.
- B. R. White, H. M. Liljestrand and J. A. Holcombe, Analyst, 2008, 133, 65–70 RSC.
- J. Yang, H. Rong, P. Shao, Y. Tao, J. Dang, P. Wang, Y. Ge, J. Wu and D. Liu, J. Mater. Chem. B, 2016, 4, 6065–6073 RSC.
- Y. Li, L. Li, X. Pu, G. Ma, E. Wang, J. Kong, Z. Liu and Y. Liu, Bioorg. Med. Chem. Lett., 2012, 22, 4014–4017 CrossRef CAS PubMed.
- X. He, X. Wang, L. Zhang, G. Fang, J. Liu and S. Wang, Sens. Actuators, B, 2018, 271, 289–299 CrossRef CAS.
- K. Tomar, G. Kaur, S. Verma and G. Ramanathan, Tetrahedron Lett., 2018, 59, 3653–3656 CrossRef CAS.
- B. Imperiali, D. A. Pearce, J.-E. Sohna Sohna, G. Walkup and A. Torrado, Proc. SPIE, 1999, 3858, 135–143 CrossRef CAS.
- Q. Liu, J. Wang and B. J. Boyd, Talanta, 2015, 136, 114–127 CrossRef CAS PubMed.
- L. Choulier and K. Enander, Sensors, 2010, 10, 3126–3144 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, New York, 3rd edn, 2006 Search PubMed.
- B. Alies, E. Renaglia, M. Rózga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85, 1501–1508 CrossRef CAS PubMed.
- T. T. Tominaga, H. Imasato, O. R. Nascimento and M. Tabak, Anal. Chim. Acta, 1995, 315, 217–224 CrossRef CAS.
- J. Makowska, K. Żamojć, D. Wyrzykowski, D. Uber, M. Wierzbicka, W. Wiczk and L. Chmurzyński, Spectrochim. Acta, Part A, 2016, 153, 451–456 CrossRef CAS PubMed.
- F. Jalilehvand, B. O. Leung, M. Izadifard and E. Damian, Inorg. Chem., 2006, 45, 66–73 CrossRef CAS PubMed.
- A. Kolozsi, A. Lakatos, G. Galbács, A. Ø. Madsen, E. Larsen and B. Gyurcsik, Inorg. Chem., 2008, 47, 3832–3840 CrossRef CAS PubMed.
- J. M. Antosiewicz and D. Shugar, Biophys. Rev., 2016, 8, 151–161 CrossRef CAS PubMed.
- E. Mesterházy, C. Lebrun, S. Crouzy, A. Jancsó and P. Delangle, Metallomics, 2018, 10, 1232–1244 RSC.
- D. Szunyogh, B. Gyurcsik, F. H. Larsen, M. Stachura, P. W. Thulstrup, L. Hemmingsen and A. Jancsó, Dalton Trans., 2015, 44, 12576–12588 RSC.
- D. Szunyogh, H. Szokolai, P. W. Thulstrup, F. H. Larsen, B. Gyurcsik, N. J. Christensen, M. Stachura, L. Hemmingsen and A. Jancsó, Angew. Chem., Int. Ed., 2015, 54, 15756–15761 CrossRef CAS PubMed.
- M. Łuczkowski, B. A. Zeider, A. V. H. Hinz, M. Stachura, S. Chakraborty, L. Hemmingsen, D. L. Huffman and V. L. Pecoraro, Chem. – Eur. J., 2013, 19, 9042–9049 CrossRef PubMed.
- O. Sénèque, P. Rousselot-Pailley, A. Pujol, D. Boturyn, S. Crouzy, O. Proux, A. Manceau, C. Lebrun and P. Delangle, Inorg. Chem., 2018, 57, 2705–2713 CrossRef PubMed.
- A. M. Pujol, C. Lebrun, C. Gateau, A. Manceau and P. Delangle, Eur. J. Inorg. Chem., 2012, 3835–3843 CrossRef CAS.
- M. Łuczkowski, M. Stachura, V. Schirf, B. Demeler, L. Hemmingsen and V. L. Pecoraro, Inorg. Chem., 2008, 47, 10875–10888 CrossRef PubMed.
- J. G. Wright, M. J. Natan, F. M. MacDonnel, D. M. Ralston and T. V. O'Halloran, in Progress in Inorganic Chemistry, ed. B. S. J. Lippard, Wiley and Sons, New York, 1990, vol. 38, pp. 323–412 Search PubMed.
- C. J. Henehan, D. L. Pountney, M. Vašák and O. Zerbe, Protein Sci., 1993, 2, 1756–1764 CrossRef CAS PubMed.
- D. L. Pountney, S. M. Fundel, P. Faller, N. E. Birchler, P. Hunziker and M. Va
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) ák, FEBS Lett., 1994, 345, 193–197 CrossRef CAS PubMed.
ák, FEBS Lett., 1994, 345, 193–197 CrossRef CAS PubMed. - K. Kulon, D. Woźniak, K. Wegner, Z. Grzonka and H. Kozłowski, J. Inorg. Biochem., 2007, 101, 1699–1706 CrossRef CAS PubMed.
- V. Dorčák and A. Krężel, Dalton Trans., 2003, 2253–2259 RSC.
- A. Jancsó, D. Szunyogh, F. H. Larsen, P. W. Thulstrup, N. J. Christensen, B. Gyurcsik and L. Hemmingsen, Metallomics, 2011, 3, 1331–1339 RSC.
- A. G. Tebo, L. Hemmingsen and V. L. Pecoraro, Metallomics, 2015, 7, 1555–1561 RSC.
- N. Lihi, M. Lukács, D. Szűcs, K. Várnagy and I. Sóvágó, Polyhedron, 2017, 133, 364–373 CrossRef CAS.
- N. Lihi, Á. Grenács, S. Timári, I. Turi, I. Bányai, I. Sóvágó and K. Várnagy, New J. Chem., 2015, 39, 8364–8372 RSC.
- A. Jancsó, B. Gyurcsik, E. Mesterházy and R. Berkecz, J. Inorg. Biochem., 2013, 126, 96–103 CrossRef PubMed.
- M. J. Walsh and B. A. Ahner, J. Inorg. Biochem., 2013, 128, 112–123 CrossRef CAS PubMed.
- A. Drozd, D. Wojewska, M. D. Peris-Díaz, P. Jakimowicz and A. Krężel, Metallomics, 2018, 10, 595–613 RSC.
- R. F. Chen, Arch. Biochem. Biophys., 1971, 142, 552–564 CrossRef CAS PubMed.
- A. Denizli, H. Yavuz, C. Arpa, S. Bektas and Ö. Genç, Sep. Sci. Technol., 2003, 38, 1869–1881 CrossRef CAS.
- L. Malachowski, J. L. Stair and J. A. Holcombe, Pure Appl. Chem., 2004, 76, 777–787 CAS.
- J. L. Stair and J. A. Holcombe, Microchem. J., 2005, 81, 69–80 CrossRef CAS.
- G. Galbács, H. Szokolai, A. Kormányos, A. Metzinger, L. Szekeres, C. Marcu, F. Peter, C. Muntean, A. Negrea, M. Ciopec and A. Jancsó, Bull. Chem. Soc. Jpn., 2016, 89, 243–253 CrossRef.
- ACD/Spectrus Processor, version 2012 (Build 61851, 24 Jan 2013), Advanced Chemistry Development, Inc., Toronto, 2012 Search PubMed.
- I. Zékány, I. Nagypál and G. Peintler, PSEQUAD for Chemical Equilibria, Technical Software Distributors, Baltimore, 1991 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt01141b |
| This journal is © The Royal Society of Chemistry 2019 |