
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complex formation between UO22+ and α-isosaccharinic acid: insights on a molecular level†
Hannes
Brinkmann
 *a,
Michael
Patzschke
a,
Peter
Kaden
a,
Manuel
Raiwa
b,
André
Rossberg
ac,
Roger
Kloditz
a,
Karsten
Heim
a,
Henry
Moll
a and
Thorsten
Stumpf
a
*a,
Michael
Patzschke
a,
Peter
Kaden
a,
Manuel
Raiwa
b,
André
Rossberg
ac,
Roger
Kloditz
a,
Karsten
Heim
a,
Henry
Moll
a and
Thorsten
Stumpf
a
aHelmholtz-Zentrum Dresden – Rossendorf, Institute of Resource Ecology, Bautzner Landstraße 400, 01328 Dresden, Germany. E-mail: h.brinkmann@hzdr.de; m.patzschke@hzdr.de; p.kaden@hzdr.de; r.kloditz@hzdr.de; k.heim@hzdr.de; h.moll@hzdr.de; t.stumpf@hzdr.de
bGottfried Wilhelm Leibniz Universität Hannover, Institute of Radioecology and Radiation Protection, Herrenhäuser Str. 2, 30419 Hannover, Germany. E-mail: raiwa@irs.uni-hannover.de
cESRF European Synchrotron, Rossendorf Beamline, CS40220, F-38043 Grenoble, France. E-mail: rossberg@esrf.fr
First published on 23rd August 2019
Abstract
Cellulosic materials present as tissue, paper, wood, or filter materials in low and intermediate level waste will degrade under alkaline conditions if water ingresses in a cementitious backfilled repository. The main degradation product is isosaccharinic acid. Complex formation with isosaccharinic acid may adversely affect the retention of radionuclides by the sorption or formation of solid phases. Hence, this compound is of particular concern in the context of nuclear waste disposal. Structural information of complexes is limited to spherical metal centers and little is known about the interaction of uranyl (UVIO22+) with isosaccharinic acid. Therefore, the interaction of UO22+ with α-isosaccharinate (ISA) was studied under acidic conditions focusing particularly on the structural characterization of the formed complexes. Attenuated total reflection Fourier-transform infrared (ATR-FTIR), nuclear magnetic resonance (NMR), UV-Vis, extended X-ray absorption fine structure (EXAFS) spectroscopy and electrospray-ionization mass spectrometry (ESI-MS) were combined with theoretical calculations to obtain a process understanding on the molecular level. The dominant binding motifs in the formed complexes are 5- and 6-membered rings involving the carboxylic group as well as the α- or β-hydroxy group of ISA. Two concentration dependent complex formation mechanisms were identified involving either mono- ([UO2(ISA)(H2O)3]+) or binuclear ([(UO2)2(ISA)(H2O)6]3+) species. Furthermore, this study unveils the interaction of UO22+ with the protonated α-isosaccharinic acid (HISA) promoting its transformation to the corresponding α-isosaccharinate-1,4-lactone (ISL) and inhibiting the formation of polynuclear UO22+–ISA species. Future studies on related systems will benefit from the comprehensive knowledge concerning the behavior of ISA as a complexing agent gained in the present study.
1 Introduction
Energy production by nuclear power plants and the use of radioactive materials for military, industrial, and medical applications generates nuclear waste. To ensure its safe isolation from the biosphere as well as to minimize the potential risk for humans and the environment of being exposed to harmful compounds, the nuclear waste will be stored in deep geological facilities. Therefore, suitable containers confining the conditioned waste will be collected in vaults and stored in stable host rock formations. Cementitious materials can be used to backfill the remaining cavities providing a physical and chemical barrier to radionuclide release. Additionally, cement-based materials serve as construction materials and for waste conditioning purposes. The ingress of groundwater will promote the degradation of cementitious materials, consequently leading to the development of alkaline conditions, which will in turn impact the chemical composition of the waste and the speciation of radionuclides.1The largest proportion of nuclear waste in terms of volume consists of low and intermediate level waste (LILW). Amongst others, it contains different types of organic polymers like ion exchange resins, halogenated and non-halogenated plastics, rubber, and cellulosic materials. The latter, which are introduced as paper, cotton, tissue, wood, or filter materials, are not stable under alkaline conditions and will degrade to water-soluble, low-molecular-weight organic compounds known as cellulose degradation products (CDPs) with isosaccharinic acid as a main degradation product.2–8 Studies related to the impact of CDPs on the sorption or solubility of RN and other metals relevant in the context of nuclear waste disposal showed that they have an adverse effect while isosaccharinic acid was identified as a responsible complexing agent in many cases.9–13 Hence, this particular organic ligand is of great concern in the context of safety assessment for a nuclear waste repository. Extensive efforts have been made to understand this behavior. This is reflected in the number of studies related to the effect of isosaccharinic acid on the sorption or solubility of RN and other metals. Hummel et al. provided a summary of the related literature up to the year of 2004.15 Gaona et al. and Rai and Kitamura provided comprehensive and critical summaries of studies.16,17 Recently, Tasi et al. reported new insights concerning the interaction of isosaccharinic acid with plutonium while they also provided a detailed overview of experimental studies related to the complexation of that ligand with tri- and tetravalent actinides and lanthanides.18,19
Two diastereomers (α- and β-form) of isosaccharinic acid form in similar quantities.4 However, most studies focused on α-isosaccharinic acid since its synthesis is well described in the literature. Furthermore, Van Loon and Glaus observed a stronger complexation for certain metals compared to the β-form.14 α-Isosaccharinic acid is a polyhydroxy-carboxylic acid with four alcohol functionalities (structures are shown in Fig. 1; the following abbreviations will be used throughout the remaining article: ISL for α-isosaccharinate-1,4-lactone, HISA for α-isosaccharinic acid and ISA for α-isosaccharinate). Under very acidic conditions ISL dominates while the transformation from HISA via a proton catalyzed dehydration is a rather slow reaction.6,20–23 The ring closure occurs between the carboxylic carbon (C1) and the secondary alcohol (C4–OH) resulting in a γ-lactone. Equilibrium constants for deprotonation and lactonization were recently evaluated by Rai and Kitamura.24 The pKa value of the carboxylic group is 3.27 while the deprotonation of alcohol functionalities is expected to occur above pH 12.23,25,26 However, the acidity of alcohol functionalities is expected to increase upon their interaction with Lewis acidic metal centers.23,27 The previously mentioned impact of ISA on the sorption and solubility was generally traced back to the formation of strong complexes in which the hydroxyl groups of the ligand in addition to the carboxylic group play a crucial role. However, while the stoichiometry of the expected complexes can be deduced from the sorption or solubility data, the structural properties and binding motifs were usually assumed. As stated by Randall et al., comprehensive understanding of the structure of the formed complexes is crucial, since the sensitivity of complexation to changing parameters such as pH or ligand concentrations can be confidently understood, if the underlying structure and the structural changes of the dominant complexes are known.10
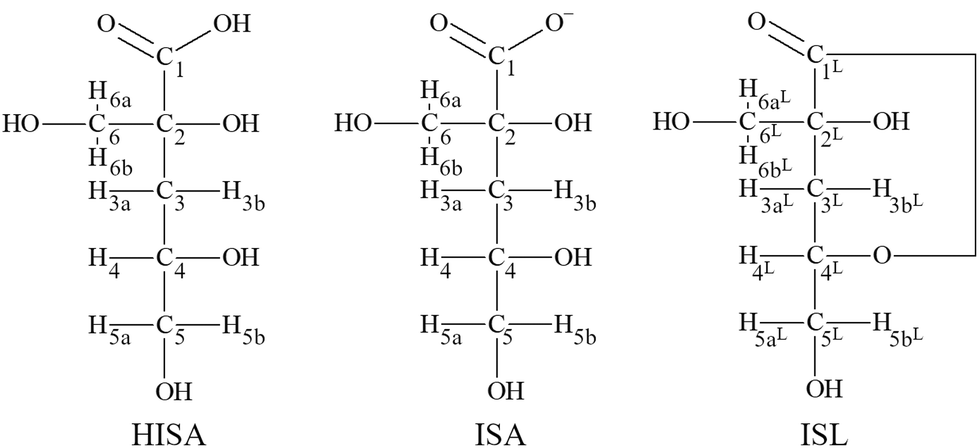 | ||
| Fig. 1 Structural formula of HISA (α-isosaccharinic acid), ISA (α-isosaccharinate) and ISL (α-isosaccharinate-1,4-lactone) with labelling of protons and carbon atoms. | ||
EXAFS measurements in the Th(IV)–ISA system performed by Colàs suggested the coordination of one or more hydroxyl groups while they showed no indication for a bidentate coordination of the carboxylic group.28 NMR-measurements from Dudás et al. revealed that ISA acts as a multidentate ligand for Ca(II) under alkaline conditions.26 They suggested the coordination via the carboxylic group as well as the tertiary alcohol (C2–OH) and the secondary alcohol (C4–OH), of which one is deprotonated. Furthermore, the authors concluded that the primary alcohol in the β-position (C6–OH) to the carboxylic group is not involved in complex formation. Similar conclusions were drawn by Randall et al. based on NMR investigations in the Eu(III)–ISA system.10 A coordination of Eu(III) by the carboxylic group, the secondary alcohol (C4–OH) and the tertiary alcohol (C2–OH) was proposed. Based on the experimental results in combination with DFT-calculations, Tasi et al. identified the same functional groups involved in Pu(IV) complexation under alkaline conditions.18 These findings reveal a coherent picture for the coordination of spherical metal centers nested in a pocket of oxygen atoms from the carboxylic group, and the secondary and tertiary alcohols. However, experimental data for hexavalent actinides possessing a sterically demanding structure with two linearly arranged oxygen atoms are scarce. Rao et al. characterized the uranyl (UVIO22+, hereafter referred to as UO22+)–ISA systems under acidic conditions by potentiometry and calorimetry.29 Their potentiometric measurements suggested the formation of [UO2(ISA)n]2−n complexes with n = 1, 2 and 3. The same system was investigated by Warwick et al. under neutral and alkaline conditions.30 The formation of UO22+–ISA complexes with a 1-to-1 stoichiometry was determined in which four hydroxyl groups are involved in the coordination under alkaline conditions. Both studies discussed the possible involvement of hydroxyl groups of the ligand but no experiments were performed dedicated to that issue. In a DFT-study, Birjkumar et al. investigated the stability of different binding motifs in UO22+–ISA complexes at different pH values.31 Coordination via both oxygens of the carboxylic group (4-membered ring), one oxygen of the carboxylic and one of the α-hydroxy groups (5-membered ring) and one oxygen of the carboxylic and one of the β-hydroxy groups (6-membered ring) was considered. Their calculations revealed that under certain experimental conditions different binding motifs might coexist in solution. However, the involvement of the γ-hydroxy group (C4–OH) was not considered, which was shown to coordinate to spherical metal centers as mentioned earlier.
It is the aim of the present study to investigate the interaction between ISA and UO22+, also as a representative for other actinyl ions, on the molecular level. The focus is on the identification of structural properties of the formed complexes in solution. Since acidic conditions were chosen to avoid the competitive formation of hydrolysis species, the additional question arises whether the presence of the Lewis acidic UO22+ ion has an impact on the transformation of HISA to ISL. Different complementary spectroscopic and spectrometric techniques were combined with theoretical calculations in order to describe this system from the metal and the ligand point of view.
2 Experimental & methods
2.1 Preparation of a NaISA-stock solution
Preparation of the NaISA-stock solution was performed as described previously.32,33 50 g α-lactose monohydrate and 13.6 g Ca(OH)2 were added to 500 mL argon flushed double-distilled (dd) water. The mixture was additionally flushed with argon for 1 h and then carefully sealed. After stirring for 3 days under anaerobic conditions at RT the brown solution was boiled under reflux for 6 h. The hot solution was filtered, and the volume was reduced to 100 mL by boiling. The remaining solution was stored at 4 °C until a white precipitate was formed, which was then filtered and subsequently washed with water and ethanol. After drying at 50 °C, 1.2 g of the crude product was dissolved in 100 mL dd water by boiling and the volume was reduced to 10 mL. The white precipitate was washed twice with water and ethanol and finally dried at 50 °C. In order to exchange Ca2+ with Na+, a cation exchange resin (Chelex® 100, BioRad) was used as described by Van Loon et al.34 An amount of 4 g of the white Ca(ISA)2 was stirred in 100 mL dd water together with 100 g of Chelex-100 resin in the Na+ form for 3 h. The suspension was centrifuged, and the supernatant was filtered to fully remove the resin. The volume was reduced to 45 mL by boiling, resulting in a brownish solution. The concentration of ISA in the stock solution was determined by total organic carbon (TOC) analysis. Total carbon (TC) and total inorganic carbon (TIC) were determined and the TOC was calculated by subtracting the TIC from the TC. Product purity was confirmed by electrospray-ionization mass spectrometry and nuclear magnetic resonance spectroscopy (section 3.1).2.2 General sample preparation
For ultraviolet-visible (UV-Vis), electrospray-ionization mass spectrometry (ESI-MS), extended X-ray absorption fine structure (EXAFS) and nuclear magnetic resonance (NMR) measurements a UO22+–perchlorate and for attenuated total reflection Fourier-transform infrared (ATR-FTIR) measurements a UO22+–chloride stock solution were used. A concentration of 0.1 M for uranium in both stock solutions was determined by ICP-MS. To prepare the UO22+–ISA samples, a 0.6 M NaISA stock solution with a pH of 10.5 was used. To adjust the pH, NaOH, HClO4 or HCl were used. The pH was measured with a SenTix Mic electrode (WTW, Germany).Measurements in different facilities and accompanying transport issues, varying technical efforts as well as different accumulation times to generate evaluable spectra have caused different time spans between preparing the samples and collecting the spectra. The passed times are separately stated for each method in the associated sections below. UV-Vis spectroscopy was used as a reference method to check whether the same dominant species can be expected, if different UO22+ concentrations and background electrolytes were used, or different waiting times were necessary. An overview of the UV-Vis spectra of the measured UO22+-containing samples is shown in ESI section 5.†
2.3 UV-Vis measurements
To determine the number of dominant UO22+–ISA species several test series were prepared differing in their molar ratios of UO22+ and NaISA in solution. The metal-to-ligand (M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L) ratios were 2:1, 1:1, 1:2, 1:6 and 1:16. While varying the NaISA concentration, the UO22+ concentration was kept constant at 15 mM. The ionic strength was adjusted to 1 M with NaClO4. The pH-titration experiments were performed as follows: an initial pH was adjusted and the absorption spectrum was immediately recorded. Then, a small amount of NaOH or HClO4 was added to the solution, the pH was measured, and the spectrum was immediately recorded. This procedure was repeated several times. The UV-Vis measurements were performed with a TIDAS 100 spectrometer (J&M Analytik AG, Germany) in a 1 cm quartz cuvette with dd water as the reference with a resolution of 0.1 nm. All spectra were corrected to zero at the lowest absorption of the corresponding spectrum and each test series was individually evaluated with iterative transformation factor analysis (ITFA, additional information are provided in ESI section 8†) between 390 nm and 480 nm.35
:L) ratios were 2:1, 1:1, 1:2, 1:6 and 1:16. While varying the NaISA concentration, the UO22+ concentration was kept constant at 15 mM. The ionic strength was adjusted to 1 M with NaClO4. The pH-titration experiments were performed as follows: an initial pH was adjusted and the absorption spectrum was immediately recorded. Then, a small amount of NaOH or HClO4 was added to the solution, the pH was measured, and the spectrum was immediately recorded. This procedure was repeated several times. The UV-Vis measurements were performed with a TIDAS 100 spectrometer (J&M Analytik AG, Germany) in a 1 cm quartz cuvette with dd water as the reference with a resolution of 0.1 nm. All spectra were corrected to zero at the lowest absorption of the corresponding spectrum and each test series was individually evaluated with iterative transformation factor analysis (ITFA, additional information are provided in ESI section 8†) between 390 nm and 480 nm.35
The method of continuous variation was used to determine the stoichiometry of the formed UO22+–ISA species. Therefore, the pH of a 15 mM UO22+ solution as well as of a 100 mM NaISA solution was adjusted to 4.0. The latter was added to 2 mL of the UO22+ solution in 40 μL steps. The absorption spectrum was measured immediately after each step as described above.
2.4 Attenuated total reflection Fourier-transform infrared (ATR-FTIR)-spectroscopy
ATR-FTIR spectroscopy can be used to identify structural changes in the ligand due to complex formation, but also in the uranyl moiety by analysis of the bands of the IR active asymmetric stretching vibration of UO22+. Measurements were performed as described by Müller et al.36 The ATR-FTIR spectra of aqueous solutions were recorded in the range between 1800 and 700 cm−1 on a Bruker Vertex 80/v vacuum spectrometer equipped with a mercury cadmium telluride (MCT) detector. The spectral resolution was 4 cm−1 and spectra were averaged from 256 scans. A horizontal diamond crystal with nine internal reflections (DURA SamplIR II, Smiths Inc.) was used as the ATR accessory. The ATR unit was continuously purged with a current of dry air. To ensure adequate background subtraction, an ATR flow cell was used.Since relatively large volumes were needed for these measurements, slightly lower concentrations of UO22+ were used compared to UV-Vis, NMR or EXAFS measurements. A pH series from pH = 1.1 to 4.0 with a fixed molar ratio of UO22+ (11.25 mM) and NaISA (90 mM) in solution was prepared. Furthermore, a concentration series was prepared at a fixed pH of 4.0. Here, the UO22+ concentration was constant at 11.25 mM and the M:L ratios were 2:1, 3:2, 1:1, 2:3, 1:2 and 1:4. To identify the spectral changes related to complex formation with UO22+, all samples were prepared in the absence and presence of UO22+. The concentration of the background electrolyte NaCl was 1 M in all samples and the spectra were recorded one day after preparation.
To assign the spectral changes to certain functional groups, single component spectra of ISL, HISA and ISA were determined, and vibrational modes were assigned to functional groups based on these spectra. To this end, the UO22+-free samples of the pH series and an additional sample at pH 9.2 were evaluated with ITFA.
2.5 NMR-spectroscopy
NMR spectra were recorded either on a Varian Inova 400 spectrometer equipped with an AutoX ID probe head with z-gradients operating at 399.89 MHz for 1H or on an Agilent DD2-600 spectrometer equipped with an Agilent One probe operating at 599.80 MHz for 1H frequency, respectively. For 1D and 2D spectra standard pulse programs were used. D2O was purchased from Deutero GmbH (Kastellaun, Germany) and used as is. All spectra were referenced to TMSP.To check the purity of the synthesized NaISA stock solution and to generate the reference data, 1H, 13C and 1H,13C-HSQC (heteronuclear single quantum correlation) spectra of 50 mM solutions at pH 2.2, 4.2 and 10.0 were measured immediately after preparation. The sample having a pH of 2.2 was additionally measured after eight days to obtain reliable data for ISL. Samples were prepared by diluting an aliquot of the alkaline NaISA stock solution with D2O. The pH was adjusted by adding appropriate amounts of HClO4.
To investigate the influence of UO22+ on the formation of ISL under acidic conditions, two samples were prepared with a NaISA concentration of 60 mM. Solutions were prepared from the alkaline NaISA stock, which contains a fully deprotonated acid (ISA). One of the two samples contained 15 mM UO22+. After adjusting the pH to 2.2 (t = 0 min), a series of 1H-NMR spectra was recorded for both samples until equilibrium was reached (all points in time are listed in Tables SI 6 and 7;† spectra are shown in Fig. SI 19 and 20†). The integrals of the 3a and 3b 1H-signals of ISL (IISL) and HISA (IHISA) were used to calculate their relative concentrations by using the following equations.
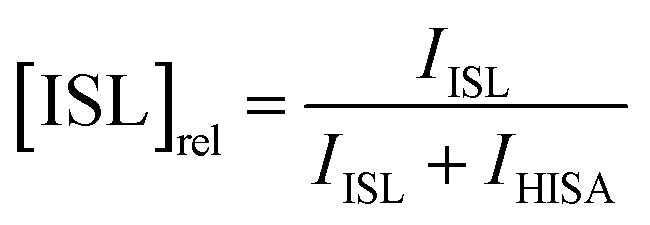
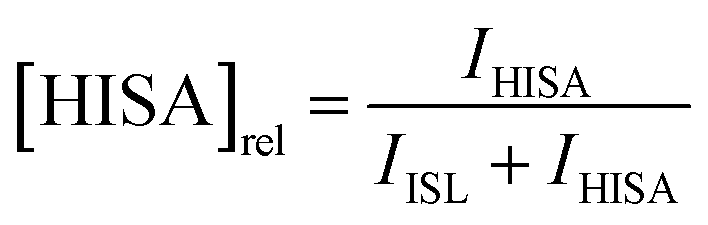
To investigate whether the lactone formation has an influence on the speciation of UO22+, UV-Vis spectra were obtained from an aliquot of the UO22+-containing sample after different points in time.
To determine the dominant binding sites of ISA in the formed UO22+–ISA complexes, a sample was prepared containing 15 mM UO22+ and 30 mM NaISA in D2O. HClO4 was added until the pH was 4.2. 1H, 13C and 1H,13C-HSQC spectra were recorded 1 hour, 3 days and 11 hours after preparation, respectively.
2.6 Electrospray-ionization mass spectrometry (ESI-MS)
ESI-MS was used to determine the composition of UO22+–ISA complexes formed in solution as well as to check the purity of the synthesized NaISA stock solution. Measurements were performed with a Velos Pro Orbitrap Elite (Thermo Fisher Scientific Inc., Waltham, MA, USA) using a Nanospray Flex Source. 10 μL of solution were loaded into GlassTip™ emitters from New Objective Inc. (Woburn, MA, USA) and analyzed with an Orbitrap mass analyzer. Full-MS scans from m/z 200 to 2000 were recorded and averaged for 5 minutes. Mass accuracy around 1 ppm was ensured with known lock masses from ambient air. The temperature of the transfer capillary was fixed at 240 °C. All measurements were performed in the positive ionization mode with an applied voltage to the nanospray emitter of 1.8 kV. Four samples were evaluated (six days after preparation), including a NaISA stock solution (15 mM, pH 4) and three UO22+–NaISA samples with 1.5 mM UO22+ and M:L ratios of 2:1, 1:1 and 1:4. Lower UO22+-concentrations were used to avoid artefacts in the form of two initially separated molecules measured at their combined mass. The probability of this effect increases with increasing metal and ligand concentrations. Treatment of the data was done with the Xcalibur and Freestyle software from Thermo Fisher.
2.7 DFT calculations
Structure optimizations were performed using the PBE exchange–correlation functional in combination with the TZVPP basis sets for C, N, H and O as implemented in the quantum-chemical program-suite Turbomole, v.7.1.37–39 For U only the valence electrons were explicitly treated with the TZVPP basis set since a scalar-relativistically corrected 60-electron effective core potential (RECP) was used.40 The fast and reliable dispersion corrections by Grimme et al. as well as the conductor-like screening model (COSMO) with an infinite dielectric constant were used to include dispersion and solvent effects, respectively.41,42 All structures were identified as minima on the potential energy surface by performing harmonic frequency analyses.The density difference plot is a helpful tool to identify regions in which electron density is accumulated and depleted during the process of complex formation. The higher the displacement of electrons into bonding regions the stronger the interactions between the respective bonding partners. Calculating a density difference requires a useful decomposition of the complex into single components between which the interactions are to be investigated. The complexes, for which the density difference was calculated, were decomposed while each UO22+, each HISA or ISA molecule and the water molecules coordinated to a certain UO22+ were considered as single components. The electron densities ρ of the whole complex as well as of the single components (with exactly the same coordinates as in the complex) are extracted by single-point calculations with the aforementioned settings (without COSMO) and the electron density difference ρdiff is calculated as:
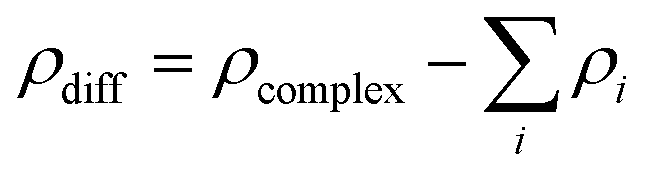
If ρdiff is positive in a specific region, then the electron density is higher in the complex than in the single components. This is interpreted as an accumulation of electrons during the artificial complex formation. Accordingly, a negative ρdiff shows a depletion of electrons during complex formation.
All representations of optimized structures were created with the Visual Molecular Dynamics (VMD) program version 1.9.3 (http://www.ks.uiuc.edu/Research/vmd/).43
A great number of different structures were optimized. The structures were selected according to the different binding motifs that are expected. These include 1:1, 2:1, 2:3 and 1:2 UO22+–ISA complexes with five- and six-membered rings. The structures shown here are the ones agreeing best with the experimental data. For many of these complexes alternative structures were also optimized. These differed in the direction in which the parts of the ISA molecule that were not directly involved in UO22+-coordination were pointing. Such changes did not affect the UO22+–ISA binding motif. Whenever possible, the isomer with the lowest energy was chosen.
2.8 Extended X-ray absorption fine structure (EXAFS)
The U LIII-edge (17166 eV) EXAFS spectra of a pH series (pH 1.0 to 4.1, 90 mM of NaISA, 15 mM UO22+) and a concentration series (NaISA concentration of 3.5 to 50 mM, pH 3, 15 mM UO22+) (Table SI 9†) were recorded (10 to 13 days after preparation) at the Rossendorf Beamline (ROBL) at the European Synchrotron Radiation Facility (ESRF).44 Higher harmonics were rejected by using two Rh-coated mirrors, while a liquid nitrogen cooled Si(111) double crystal monochromator was used for performing the energy scans in a channel cut mode. For each sample multiple absorption scans were measured with gas filled ionization chambers at room temperature and the Y K-edge (17038 eV) of a Y metal foil was recorded simultaneously for the calibration of the incident photon energy to the absolute energy scale. In order to increase the signal-to-noise ratio for each sample, four to six scans were measured and averaged. The U LIII-edge ionization potential was arbitrarily defined as E0 = 17185 eV, whereas the shift in energy threshold (ΔE0) was defined as a free parameter with ΔE0 = E0 − Et (Et – theoretical ionization potential) for the shell fit procedure. The softwares WinXAS (Version 3.11), Sixpack/SamView (Version 0.59) and EXAFSPAK were used for averaging of the multiple sample scans, energy calibration, background subtraction, isolation of the EXAFS signal, and the shell fit.45–47 For the shell fit, the theoretical scattering phase and amplitude functions were calculated with the ab initio scattering code FEFF 8.20 by using a DFT calculated structure of the 2:1 UO22+:ISA complex, which contains the structural motif of a five- and a six-membered ring (Fig. SI 39†).48 From each EXAFS sample a UV-Vis spectrum was recorded (immediately and 15 days after preparation), which are depicted in Fig. SI 37.† ITFA was used for the calculation of the qualitative fractionation of the structurally different complexes in the EXAFS and UV-Vis (spectra immediately measured after preparation were used) spectral mixtures (a short description of ITFA is provided in ESI section 8†).35
3 Results
3.1 Preparation of NaISA
Since this study focusses on the interaction of UO22+ with ISA, the discussion of ESI-MS and NMR data of UO22+-free NaISA samples, which will be later used as references, was placed in the ESI of this article (ESI section 1†). In conclusion, no impurities were detected in the NaISA stock solution by ESI-MS and NMR spectroscopy.3.2 NMR spectroscopy
The 1H-signals of the methylene group (C3) of HISA (3a/3b at 1.93/1.73 ppm) and ISL (3aL/3bL at 2.34/2.32 ppm) are well separated and can be used to monitor the lactonization over time (integrated intensities are provided in Tables SI 6 and 7;†1H-NMR spectra are depicted in Fig. SI 19 and 20†). The transformation of HISA to ISL in the absence and presence of UO22+ is shown in Fig. 2. The experimental data could be reproduced by using a monoexponential function (R2 ≥ 99.7%), indicating the first-order kinetic behavior of this reaction (all fit results are listed in Table SI 8†). Whether UO22+ was present or not had no significant effect on the equilibrium concentrations of HISA and ISL (Table SI 8†). In contrast, a crucial impact on the rate constant (k) and consequently on the half-life (t1/2) of the lactone formation reaction was observed. In the presence of UO22+t1/2 decreased from 3294 to 2058 min while k increased from 2.10 × 10−4 to 3.37 × 10−4 min−1. Previous studies from Ekberg et al. and Brown et al. reported much faster kinetics for the reaction at lower pH, in good agreement with the postulated acid catalyzed reaction mechanism.20,21
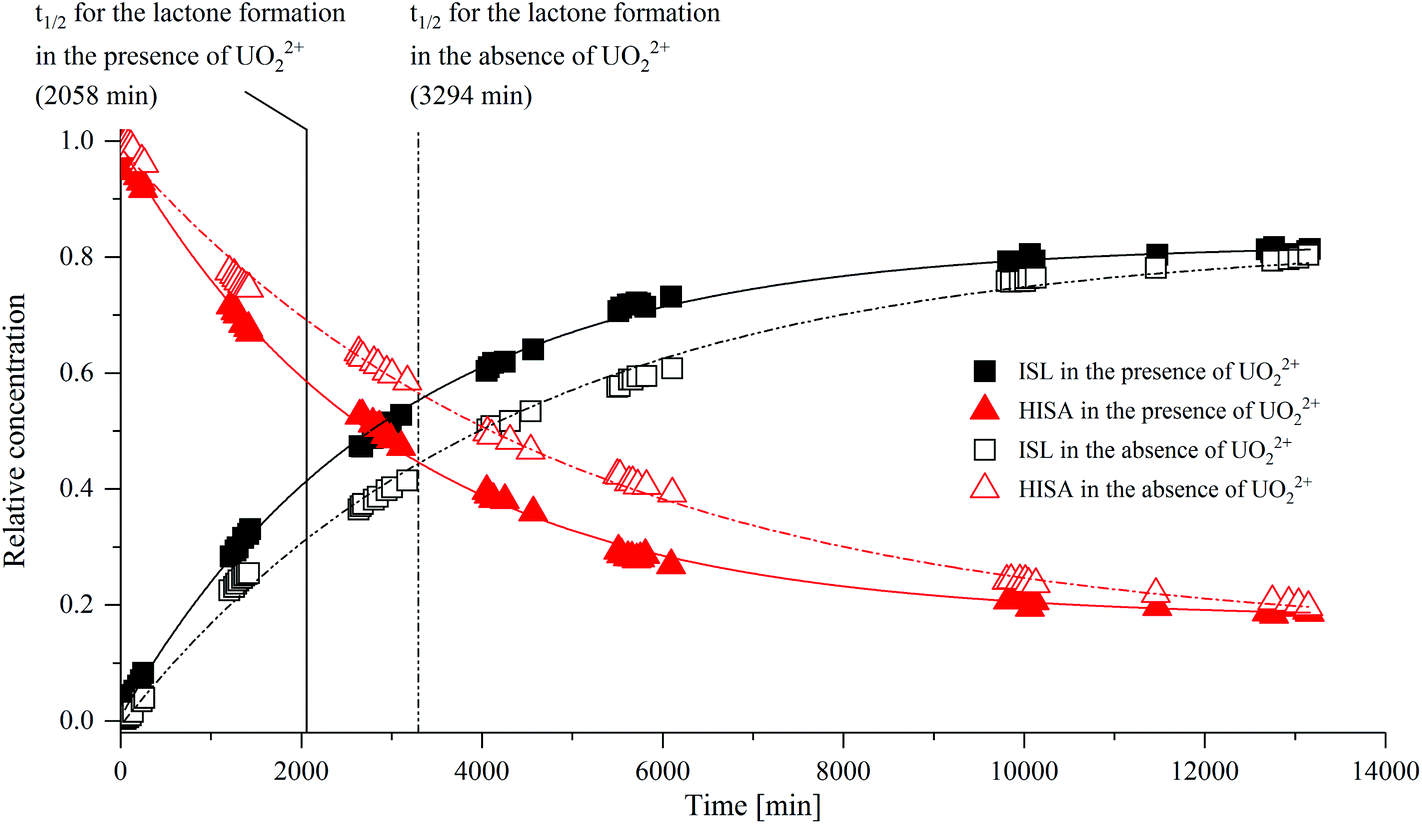 | ||
| Fig. 2 Influence of UO22+ on the transformation of HISA to ISL at pH 2.2. Relative concentrations of HISA and ISL were determined based on the ratio of their integrated 1H-NMR signals of 3a/3b and 3aL/3bL (see section 2.5). Experimental data were fitted with a monoexponential function. | ||
To identify the origin of the faster reaction kinetics, the positions of 1H-signals of HISA and ISL in the absence and presence of UO22+ were compared and are summarized in Table 1 (the corresponding spectra are shown in Fig. SI 21–24†). The presence of UO22+ has no discernable influence on the signals associated with ISL, however shifts could be observed for the HISA signals, suggesting an interaction between the protonated acid and UO22+. The values for HISA in the presence of UO22+ were extracted from the spectrum, which was obtained 102 min after preparation, since the partly overlapping ISL signals were not yet dominant after that relatively short time. However, the spectrum obtained after 10116 min (Fig. SI 23 and 24†) reveals that the signals corresponding to protons 3a, 3b and 4 of HISA were further shifted downfield to 2.08, 1.90 and 4.00 ppm. This indicates that the exchange of HISA molecules, which are coordinated to UO22+, is fast and hence the positions extracted after 102 min are averaged signals of the free and coordinated HISA molecules.
| Proton | δ(1H) of HISA [ppm] | δ(1H) of ISL [ppm] | ||||
|---|---|---|---|---|---|---|
| Without UO22+a |
With UO22+b |
Δδ(1H) | Without UO22+a |
With UO22+c |
Δδ(1H) | |
|
a Reference data for HISA and ISL were obtained from Table SI 3.†
b 102 min after preparation.
c 10116 min after preparation (proton 4 could not be assigned due to the overlap with the strong water signal); Δδ was calculated by subtracting δ without UO22+ from δ with UO22+.
|
||||||
| 3a(L)/3b(L) | 1.93/1.73 | 2.00/1.81 | 0.07/0.08 | 2.34/2.32 | 2.34/2.31 | 0.00/−0.01 |
| 4(L) | 3.96 | 3.98 | 0.02 | 4.87 | — | — |
| 5a(L)/5b(L) | 3.55/3.47 | 3.56/3.48 | 0.01/0.01 | 3.92/3.68 | 3.92/3.68 | 0.00/0.00 |
| 6a(L)/6b(L) | 3.85/3.59 | 3.91/3.67 | 0.06/0.08 | 3.78/3.72 | 3.79/3.72 | 0.01/0.00 |
Within the first 10000 min, in which the HISA concentration considerably changed with increasing time, the absorption spectra of the UO22+-containing sample revealed only small changes (Fig. SI 33†). A slightly increased absorption as well as a shift of the initial maximum at 422.3 nm to higher wavelengths occurred. When the equilibrium was almost reached (after 10000 min) and consequently the HISA concentration remained nearly constant, the absorption spectra did not change. This in combination with the shifted 1H-signals of HISA and the impact on the lactone formation are striking indications that the protonated acid has to be considered, besides ISA, as a ligand for UO22+.
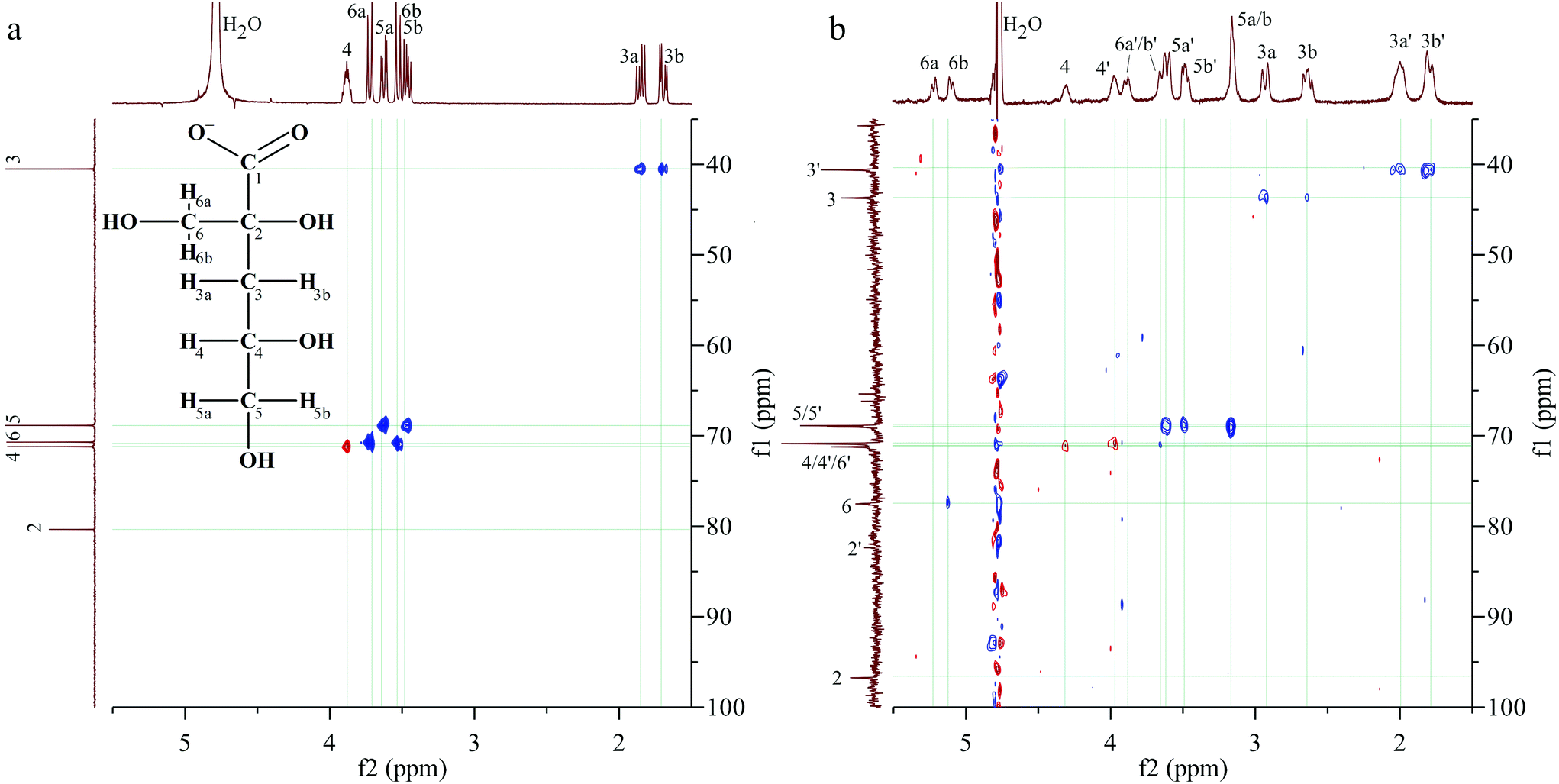 | ||
| Fig. 3 1H, 13C-HSQC spectra of (a) the NaISA stock solution (50 mM, pH 10) and (b) a UO22+–ISA sample (30 mM NaISA, 15 mM UO22+, pH 4.2). | ||
Table 2 summarizes the 1H- and 13C-signals in the absence and presence of UO22+ as well as the corresponding shifts, which are discussed in this section. Two signals are present in the range, where the signal of the carboxylic carbon was expected. The more intense signal at 191.3 ppm is clearly shifted by 8.5 ppm, compared to the signal in the absence of UO22+. Further assignments are based on the 1H,13C-HSQC spectrum in Fig. 3b. Since the carbon signal at 96.8 ppm shows no correlation with proton signals, it corresponds to carbon 2. A rather weak signal without correlations is present at 82.4 ppm, which is close to the position of carbon 2 in the absence of UO22+. The smallest carbon shifts are present at 40.6 and 43.7 ppm, which were consequently assigned to carbon 3. The corresponding proton signals were identified based on the following correlations. Two opposite phased (red) signals must be caused by two spatially differently arranged protons attached to carbon 4. The corresponding carbons show no considerably changed chemical shifts. A pair of doublets, caused by the protons 6a and 6b, which are attached to the carbon of the β-hydroxy group (C6), is located at 5.23 and 5.13 ppm. The HSQC spectrum shows a correlation of the second signal with the 13C signal at 77.5 ppm, which is consequently carbon 6. Two intense signals result from the coupling of carbon signals at 69.1 and 68.9 ppm with proton signals at 3.62, 3.50 and 3.17 ppm. The carbon shifts match perfectly with the positions of carbon 5 in the absence of UO22+. The two remaining proton signals at 3.90 and 3.65 ppm correlate with a carbon signal at 70.8 ppm. These are similar positions for the carbon and protons of position 6 in the absence of UO22+.
| Carbon; proton | δ(13C) [ppm] | δ(1H) [ppm] | ||||
|---|---|---|---|---|---|---|
| Reference data ISAa and HISAb | with UO22+ (set 1c and 2d) | Δδ | Reference data ISAa and HISAb | with UO22+ (set 1c and 2d) | Δδ | |
| Reference data for HISA and ISA were obtained from Tables SI 2 and 3;† Δδ was calculated by subtracting a from c and b from d. | ||||||
| 1; — | 182.8a | 191.3c | 8.5 | — | — | — |
| 180.5b | 181.4d | 0.9 | ||||
| 2; — | 80.4a | 96.8c | 16.4 | — | — | — |
| 79.4b | 82.4d | 3.0 | ||||
| 3; 3a/3b | 40.5a | 43.7c | 3.2 | 1.85/1.69a | 2.94/2.66c | 1.09/0.97 |
| 40.4b | 40.6d | 0.2 | 1.93/1.73b | 2.01/1.81d | 0.08/0.08 | |
| 4; 4 | 71.2a | 71.2c | 0.0 | 3.88a | 4.32c | 0.44 |
| 70.0b | 70.8d | 0.8 | 3.96b | 3.99d | 0.03 | |
| 5; 5a/5b | 68.9a | 69.1c | 0.1 | 3.63/3.46a | 3.17c | −0.46/−0.29 |
| 68.7b | 68.9d | 0.2 | 3.55/3.47b | 3.62/3.50d | 0.07/0.03 | |
| 6; 6a/6b | 70.9a | 77.5c | 6.6 | 3.72/3.53a | 5.23/5.13c | 1.51/1.60 |
| 70.7b | 70.8d | 0.1 | 3.85/3.59b | 3.90/3.65d | 0.05/0.06 | |
This analysis revealed that two sets of signals are present for each carbon and proton signal under these experimental conditions, when UO22+ was in solution. Comparing the 1H-signals of the set, which is marked with an apostrophe in Fig. 3b (set 2 in Table 2), with reference data for HISA, it becomes apparent that the differences are comparable to those observed earlier in the previous paragraph (Table 1). Therefore, this set can be assigned to HISA interacting with UO22+. The shifts of the signals observed for the other set are considerably larger, relative to uncoordinated ISA, indicating a stronger interaction between UO22+ and ISA compared to HISA. Most significant changes occurred at carbons 1, 2 and 6.
3.3 ATR-FTIR spectroscopy
ATR-FTIR spectroscopy was used as a complementary method for NMR spectroscopy to investigate the structure of ISA in the absence and presence of UO22+. Furthermore, the shift of the asymmetric stretching mode of the UO22+ unit (ν3) provides information related to the speciation of UO22+. To interpret the spectra in the presence of UO22+ it was necessary to identify the relevant vibrational modes of isosaccharinic acid. Therefore, the UO22+-free samples of the pH series including an additional sample at pH 9.2 were measured (spectra are shown in Fig. SI 29† and as dashed lines in Fig. 4b) and evaluated with iterative transformation factor analysis (ITFA). This program is based on principal component analysis and can be used to estimate the number of independent components within a spectroscopic data set as well as to calculate the single component spectra. Furthermore, the distribution of the components within a certain test series can be calculated. A detailed description was given by Rossberg et al.35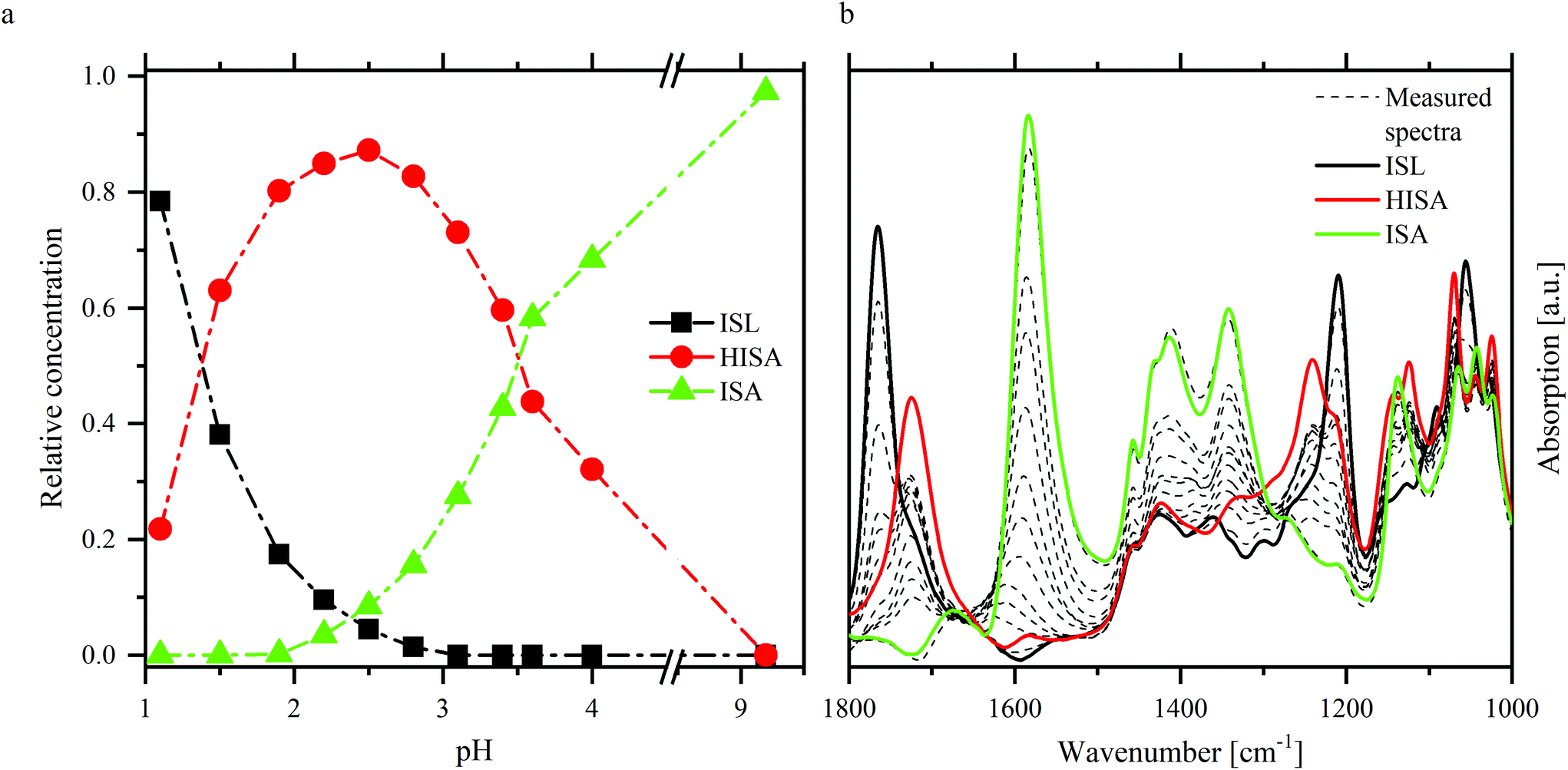 | ||
| Fig. 4 ITFA evaluation of NaISA ATR-FTIR spectra (90 mM NaISA, 1 M NaCl): distribution of the components (a) and single component spectra (b) of ISL, HISA and ISA. | ||
Fig. 4 shows the results of ITFA-evaluation. Three components were necessary to reproduce the measured spectra, the relative distribution of which is shown in Fig. 4a allowing a reasonable assignment to the three possible forms of isosaccharinic acid. ISL dominates under very acidic conditions and gradually transforms to HISA with increasing pH > 1. At higher pH values, HISA is then deprotonated to ISA. Based on the calculated relative concentrations for HISA and ISA between pH 2.8 and 4, a pKa of 3.6 ± 0.1 was determined, which is comparable to the values reported by Rai and Kitamura.17 The single component spectra of ISL, HISA and ISA are shown in Fig. 4b and Table 3 summarizes their important vibrational modes (further information concerning the assignments is provided in ESI section 4.2†).
| Compound | Wavenumber [cm−1] | Assignment |
|---|---|---|
| ac: acid; alc: alcohol; δ: deformation; ν: stretching; as: asymmetric; s: symmetric. | ||
| ISL | 1765 |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
| 1209 | ν as(C–O–C) | |
| 1055 | ν sym(C–O–C) | |
| HISA | 1724 |
ν(CO) |
| 1240 | δ(C–OH) + ν(C–OH) (ac) | |
| 1000–1150 | ν(C–OH) (alc) | |
| ISA | 1583 | ν as(COO−) |
| 1413 | ν s(COO−) | |
| 1000–1150 | ν(C–OH) (alc) | |
Changes occurring due to the presence of UO22+ were analyzed based on difference spectra. Therefore, a spectrum of a sample without UO22+ was subtracted from the corresponding spectrum with UO22+ while both had the same pH and NaISA concentration. This procedure was, amongst others, described by Heller et al.49 The difference spectra of the pH series are shown in Fig. 5a–c and of the concentration series in Fig. 5d–f.
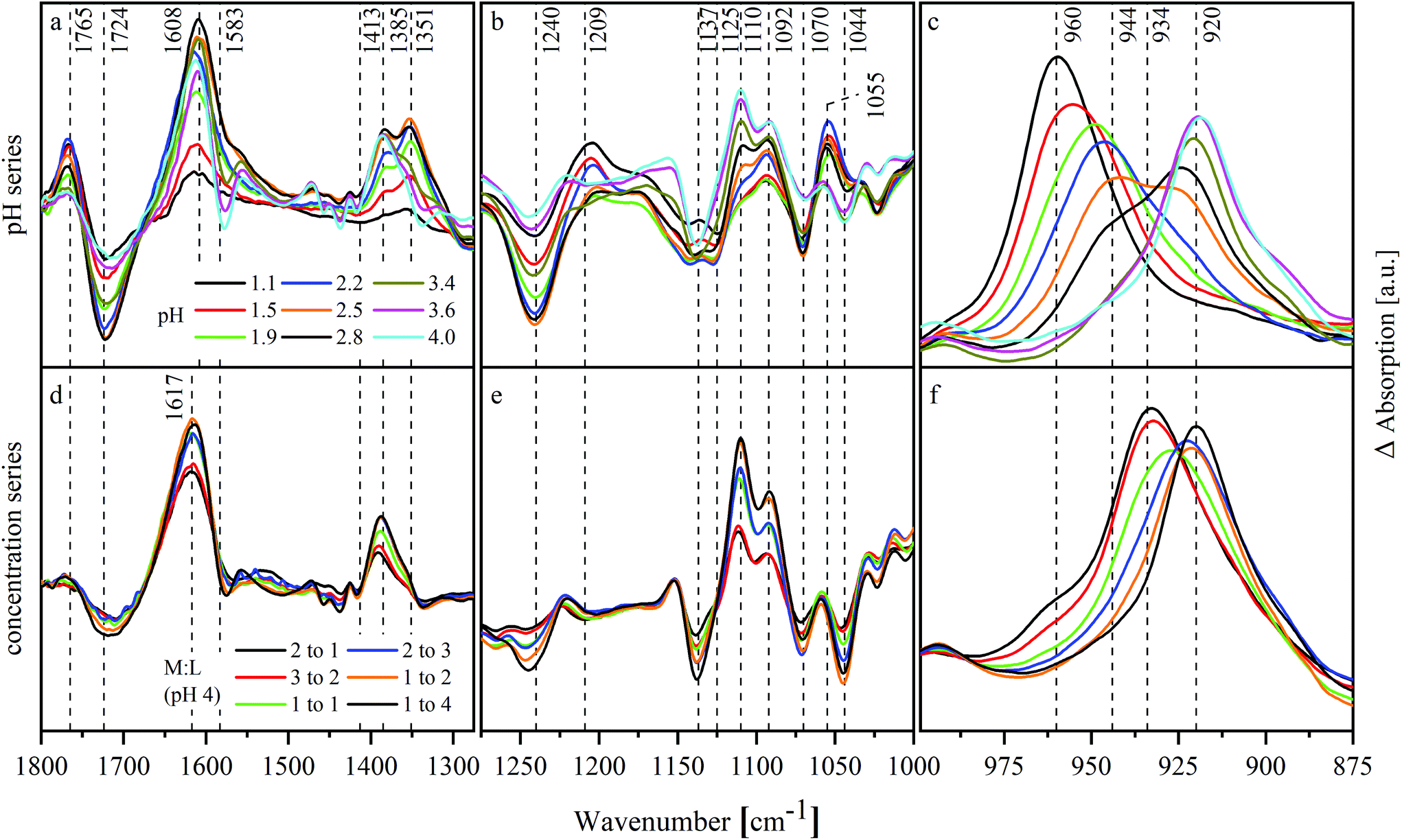 | ||
| Fig. 5 ATR-FTIR difference spectra of UO22+–ISA test series between 1800 cm−1 and 875 cm−1: (a, b, c) pH series (11.25 mM UO22+, 90 mM NaISA, 1 M NaCl); (d, e, f) concentration series (11.25 mM UO22+, pH 4, 1 M NaCl). | ||
Changes in the spectra, which were caused by the interaction of the ligand with UO22+ or by the shift of chemical equilibria, occur mainly in the range between 1800 and 1000 cm−1 (Fig. 5a, b, d and e). The spectra of the pH series show negative modes at 1724, 1240, 1125 and 1070 cm−1 especially below pH 3.4. These modes belong to HISA reflecting a decreased amount of this compound in the presence of UO22+. Additionally, positive modes occur in the same pH range at 1765, 1209 and 1055 cm−1 corresponding to ISL. This indicates a positive effect of UO22+ on the lactone formation. The mode at 1351 cm−1 correlates with the positive modes of ISL. In the light of the previous results, this mode might be caused by HISA interacting with UO22+. Further positive modes occur at 1608, 1385 and 1110 cm−1. Since they are neither present in the spectra of ISL and HISA nor in ISA, they probably originate from UO22+–ISA complexes. Their appearance coincides with a shift of the asymmetric stretching of the UO22+ unit (ν3) indicating its complexation. The modes at 1608 and 1385 cm−1 result from νas and νs of the deprotonated carboxylic group, which were shifted due to the interaction with UO22+. Their difference (Δν) increased in the pH series from 170 cm−1 in the absence of UO22+ to 223 cm−1 in the presence of UO22+.
Δν in the concentration series is 232 cm−1 (νas at 1617 and νs at 1385 cm−1) slightly larger. The positive modes of ISL as well as the mode at 1351 cm−1 are not present within this series. Both series show significant changes in the region between 1150 and 1000 cm−1 (Fig. 5b and e). Since this is the region where the C–O stretching modes of the alcohol functionalities occur, this indicates their participation in complex formation.
Fig. 5c and f show the shift of ν3 of the UO22+ entity. In both series, a shift to lower wavenumbers with increasing pH or mole ratio in solution can be observed reflecting the complex formation. The mode shifts successively from 960 to 944 and finally to 920 cm−1 in the pH series. At a mole ratio of 2:1 in the concentration series, the mode at 960 cm−1 is only present as a shoulder while a mode at 934 cm−1 dominates. This mode shifts then to 920 cm−1.
3.4 UV-Vis spectroscopy
UV-Vis spectroscopy was used to estimate the number of dominant UO22+–ISA complexes, formed in solution. Therefore, the changes in the absorption spectra of UO22+–ISA samples with increasing pH were measured at different mole ratios of UO22+ to NaISA in solution (M:L = 2:1, 1:1, 1:2, 1:6 and 1:16). The measured spectra (dashed lines in Fig. 6) were individually evaluated with ITFA. Fig. 6 shows the resulting single component spectra (a–e) and the corresponding distribution of the components (f–j) within the different test series.
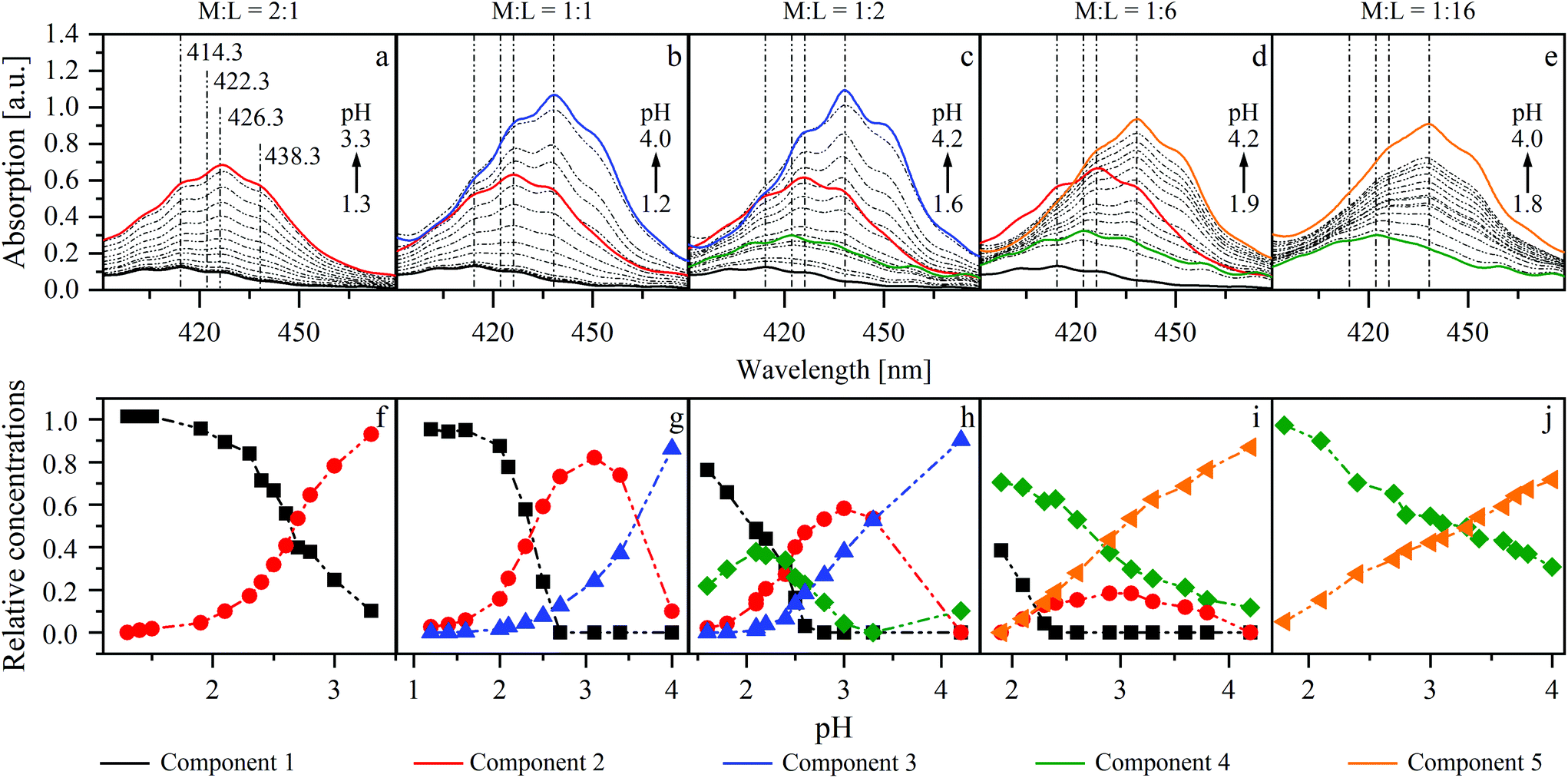 | ||
| Fig. 6 ITFA evaluation of UV-Vis series at different mole ratios of UO22+ (15 mM) and ISA in solution (M:L) (a and f = 2:1; b and g = 1:1; c and h = 1:2; d and i = 1:6; e and j = 1:16). (a–e) Measured spectra (dashed lines) and calculated single component spectra (solid lines); (f–j): calculated relative concentrations of components within a series. | ||
Two components (1 and 2) were necessary to reproduce the measured spectra in the first series (M:L = 2:1). Increasing the pH is accompanied by an increased absorption and a shift of the maximum from 414.3 to 426.3 nm (Fig. 6a). The isolated single component spectra of this series were used as references for components 1 and 2 for the evaluation of the test series with M:L ratios of 1:1, 1:2 and 1:6. If ISA and UO22+ are present in equimolar concentrations, an additional shift from 426.3 to 438.3 nm was observed at higher pH values (Fig. 6b). Therefore, an additional component (3) was necessary to reproduce the measured spectra. Component 4 was needed to describe the spectra of the remaining series, in which the concentration of UO22+ was lower than that of ISA. The proportion of this component, which has a maximum at 422.3 nm, increases with increasing excess of ISA and simultaneously suppresses the formation of component 2 (Fig. 6c–e). Therefore, at the largest investigated excess (M:L was 1:16), again only two components (4 and 5) were necessary to reproduce the measured spectra. The limiting absorption maximum in all series, except the first, is at 438.3 nm. This might emphasize the conclusion that the corresponding components belong to the same chemical species. But the single component spectra of the second and the third series (M:L = 1:1 and 1:2; Fig. 6b and c) show a larger extinction, compared to the fourth and the fifth series (M:L = 1:6 and 1:16; Fig. 6d and e). Furthermore, the limiting species were either formed from component 2 or 4. This leads to the conclusion that different limiting species were formed (components 3 and 5), depending on the initial mole ratio.
The averaged single component spectra of all components are shown in Fig. SI 32 (left).† The spectrum of component 1, showing a maximum at 414.3 nm and a corresponding extinction coefficient of 8.5 l mol−1 cm−1, is in good agreement with the spectrum of a pure UO22+ solution without NaISA at pH 2 (Fig. SI 32 right†) and with values reported by Bell and Biggers as well as Meinrath et al. for the [UO2(H2O)5]2+ complex.50,51 Therefore, these results suggest the formation of four dominant UO22+–ISA complexes. The spectral similarities of components 3 and 5 may be an indication for structural similarities of these species.
UV-Vis spectroscopy was used as an accompanying method for other techniques to check whether the same dominant UO22+ species can be expected even though different concentrations or mole ratios were used in certain cases. All additional UV-Vis spectra are summarized in ESI section 5.† No additional absorption maxima were observed and spectral developments in the ATR-FTIR as well as EXAFS pH series are comparable to that of the UV-Vis series with a UO22+ to NaISA mole ratio of 1 to 6 (Fig. 6d). Therefore, the results of the different applied techniques can be complementarily discussed to describe the structural properties as well as formation mechanisms of the formed complexes, since no different dominant species can be expected from the UV-Vis data.
The method of continuous variation, which is commonly known as a Job plot, was used to estimate the stoichiometry in one of the limiting complexes (component 3). This method, which was comprehensively described by Renny et al., was used for that purpose in several other studies.52–55 In the present case, the absorption maximum of component 3 at 438.3 nm was used. A titration was performed, where a NaISA solution was stepwise added to a UO22+ solution (for details see section 2.3). The absorption spectra were measured after each step (Fig. 7a) and the absorption at 438.3 nm was plotted against the ratio of the deprotonated ligand in solution (Ldeprot) to UO22+ (M) (Fig. 7b). Ldeprot was calculated based on the pKa value which was determined from ATR-FTIR measurements (section 3.3). The inflection at 1.43 indicates a stoichiometry of ISA and UO22+ of 1.5:1. Consequently, the stoichiometry in the formed complex is expected to be 2:3 (UO22+:ISA).
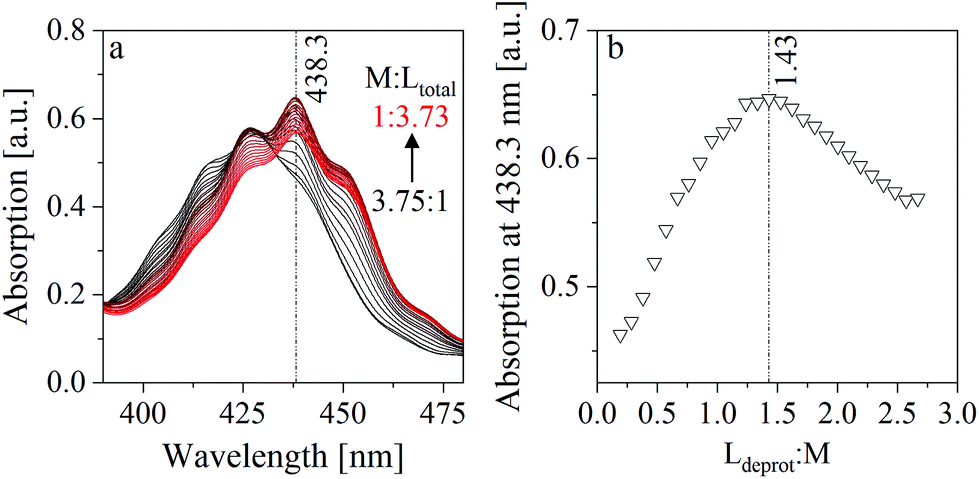 | ||
| Fig. 7 Method of continuous variation: (a) measured UV-Vis spectra (titration of a 15 mM UO22+ solution with a 100 mM NaISA solution at pH 4), (b) Job plot (absorption at 438.3 nm plotted against the ratio of deprotonated ligand (Ldeprot) to UO22+ (M)). | ||
3.5 ESI-MS
Different UO22+, ISA and UO22+–ISA components were detected by ESI-MS (Fig. SI 38†). For samples 1 and 2 all signals up to 5% of the base peak, whereas for sample 3 all signals up to 2% were considered. To obtain more information concerning the dominant stoichiometries in the formed complexes, the relative proportions of UO22+ to ISA ratios in the detected compounds were calculated. Therefore, the measured relative intensity of a detected compound was multiplied with the number of containing UO22+ units. This was then related to the total intensity of all UO22+-containing compounds. The results are summarized in Table 4. In the first two samples, where [M] ≥ [L], UO22+–ISA compounds with a stoichiometry of 2:1 are dominant. The amount of that compound decreased in the third sample, where stoichiometries of 2:1, 2:3, 1:1, and 1:2 were present in similar proportions between 17.6 and 26.8%. Considering this and the 2:3 complex stoichiometry, which was determined by the method of continuous variation at similar molar ratios of UO22+ and NaISA in solution (section 3.4), a reasonable explanation may be the fragmentation of the 2:3 complex. Even though the ionization process is relatively soft, fragmentation could be caused by the instability of larger complexes in the gas phase and might be further induced by collision with N2. Consequently, the 1:1 and 1:2 units can be interpreted as fragments of a 2:3 complex, which was initially formed in solution.
| M:L for sample preparation |
Relative abundancy of UO22+–ISA compounds | |||||
|---|---|---|---|---|---|---|
| 1:0 [%] |
2:1 [%] |
1:1 [%] |
2:3 [%] |
1:2 [%] |
1:3 [%] |
|
| 2:1 |
62.9 | 34.8 | 2.3 | — | — | — |
| 1:1 |
31.0 | 56.3 | 12.7 | — | — | — |
| 1:4 |
0 | 24.0 | 26.8 | 17.6 | 23.9 | 7.7 |
3.6 EXAFS
ITFA was applied on the EXAFS spectral mixtures (Fig. SI 42 and Table SI 9†), while the indicator function (IND) was used for the estimation of the number of components, representing structurally different environments of UO22+. The IND has a minimum at n = 3 (Table SI 10†), indicating the presence of three dominant components. The VARIMAX procedure results in a qualitative distribution of the components depending on the pH or the NaISA concentration, which is shown in Fig. 8a. However, five components were identified by ITFA based on UV-Vis measurements (section 3.4, Fig. SI 40†) and the resulting relative concentrations within the EXAFS samples are shown in Fig. SI 41.† The different number of detected components can be explained by the different sensitivities of the spectroscopic methods towards structural changes. Since the EXAFS signals depend solely on the atomic near range structure, the lower number of complexes can be caused by structural similarities of certain complexes, which were identified by UV-Vis. Thus, the structurally similar complexes can be identified by comparing the VARIMAX determined distribution of the EXAFS components with the relative concentrations of the components determined by UV-Vis. Only if the UV-Vis determined relative concentrations of components 2 and 4 and of components 3 and 5 are summed, the resulting distribution shows the same trend as observed by the VARIMAX procedure (Fig. 8). Consequently, components 2 and 4 and components 3 and 5 represent complexes with a similar first coordination shell of UO22+.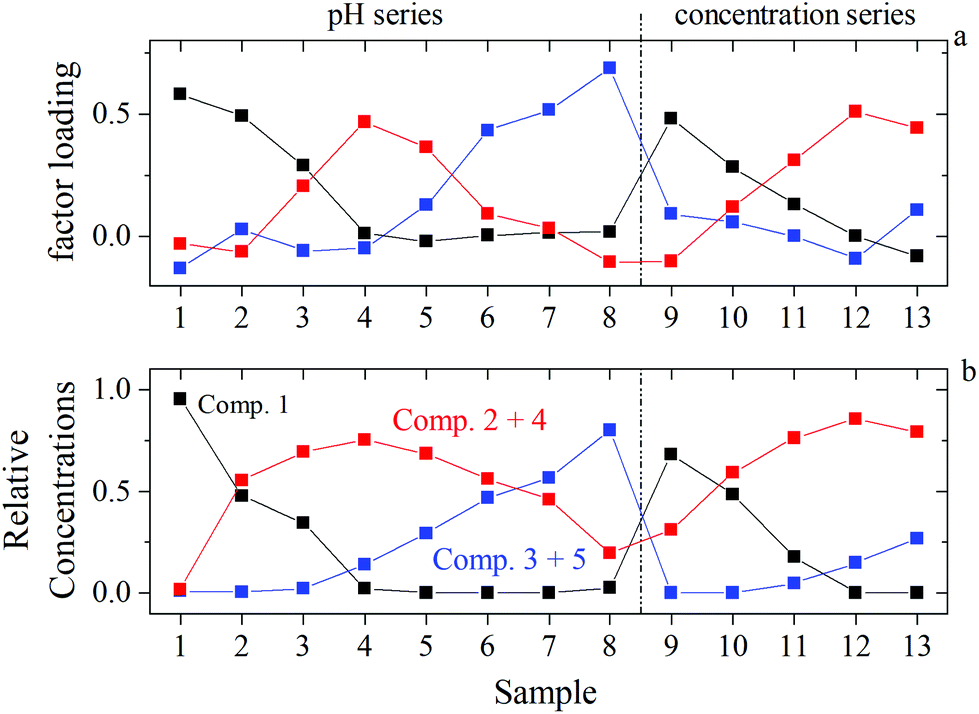 | ||
| Fig. 8 Identification of structurally similar components by comparing EXAFS and UV-Vis data of identical samples: (a) EXAFS: VARIMAX factor loadings of components 1 (black), 2 (blue), and 3 (red); (b) UV-Vis: ITFA derived relative concentrations of component 1 (black), sum of components 2 and 4 (red), sum of components 3 and 5 (blue) (according to Fig. SI 41†). | ||
In order to isolate the EXAFS spectra of the three components, an iterative target test (ITT, further information is provided in ESI section 8†) was performed by using the UV-Vis determined relative concentrations of the three components, which were kept constant during the iteration. The linear combinations of the isolated spectra weighted by their relative concentrations are in favorable agreement with the measured spectra (Fig. SI 42†), underpinning the validity in the treatment of the UV-Vis data. The shell fits of the isolated spectra and the resulting EXAFS structural parameter are shown in Fig. SI 43† and given in Table 5. The multiple scattering path U–Oax1–U–Oax2 (MS of Oax) along the axial O atoms (Oax) was included in the fit, while the radial distance (R) and the Debye–Waller factor (σ2) was set twice the R and σ2 of Oax.
| Component | Shell | CN | R [Å] | σ 2 [Å2] | ΔE0 [eV] |
|---|---|---|---|---|---|
| 1 | Oax | 2* | 1.762(1) | 0.0010(1) | 3.1(3) |
| 1.764a | 0.0013a | ||||
| MS of Oax | /2 | /3.524 | /0.002 | /3.1 | |
| Oeq | 5.2(3) | 2.414(3) | 0.0073(5) | /3.1 | |
| 5.1a | 2.403a | 0.0067a | |||
| 2 | Oax | 2* | 1.782(2) | 0.0015(2) | 1.8(6) |
| MS of Oax | /2 | /3.564 | /0.0030 | /1.8 | |
| Oeq | 4.7(5) | 2.364(7) | 0.0011(1) | /1.8 | |
| 3 | Oax | 2* | 1.772(2) | 0.0020(2) | 3.2(4) |
| MS of Oax | /2 | /3.544 | /0.0040 | /3.2 | |
| Oeq | 4.9(3) | 2.380(4) | 0.0079(7) | /3.2 |
In the case of component 1, the structural parameters agree within error with those previously observed for [UO2(H2O)5]2+.56 Hence, component 1 represents the UO22+ aqua ion. The coordination number (CN) in the equatorial plane (Oeq) is five for all components. However, the bond distance ROeq observed for components 2 and 3 (2.36–2.38 Å) is significantly shorter than ROeq of 2.41 Å, measured for [UO2(H2O)5]2+. Thus, for components 2 and 3 an interaction with ISA can be assumed. Furthermore, the observed shortening in ROeq is in line with the presence of chelates as dominant binding motifs, in which the ligands coordinate UO22+via the formation of 5- and 6-membered rings (detailed discussion in section 4.2).35,57 For components 2 and 3 a feature between 3.35–4.46 Å is present in the Fourier-transform (Fig. SI 43†), which could be explained by various single scattering (SS) and MS events (e.g. O3, C3,4,5 and U–O1–C3, U–C1–C3, U–O2–O3, U–C2–O3 according to Fig. SI 39†). However, it was not shell fitted due to the high number of overlapping scattering effects.
4 Discussion
In a system containing UO22+ and isosaccharinic acid, the metal center acts as strong Lewis acid while the oxygen atoms of ISA act as Lewis bases. Consequently, the interaction is accompanied by changes in electron density. This was calculated for complexes expected to be formed in solution (further information in section 2.7; figures are shown in ESI section 9† and Fig. 9). Generally, increased electron density is present between U and the coordinating oxygen atoms, which is caused by the electron withdrawing effect of the UO22+ unit. Additionally, adjacent carbon or proton atoms of the ligand still feel this effect. The coordination of organic ligands in the equatorial plane of the UO22+ unit decreases the electron density along the U–Oyl bond reflecting a weakening of these bonds (causing the shift of ν3(UO22+) towards lower energies observed in the IR spectra, Fig. 5c and f). Furthermore, the two oxygens of the UO22+-entity show also an increased electron density, leading to an increased Lewis basicity. This behavior was previously observed for other UO22+-ligand systems.58,59 These changes in the electron distribution within the involved compounds consequently lead to a changed reactivity, which will be further discussed in the following sections.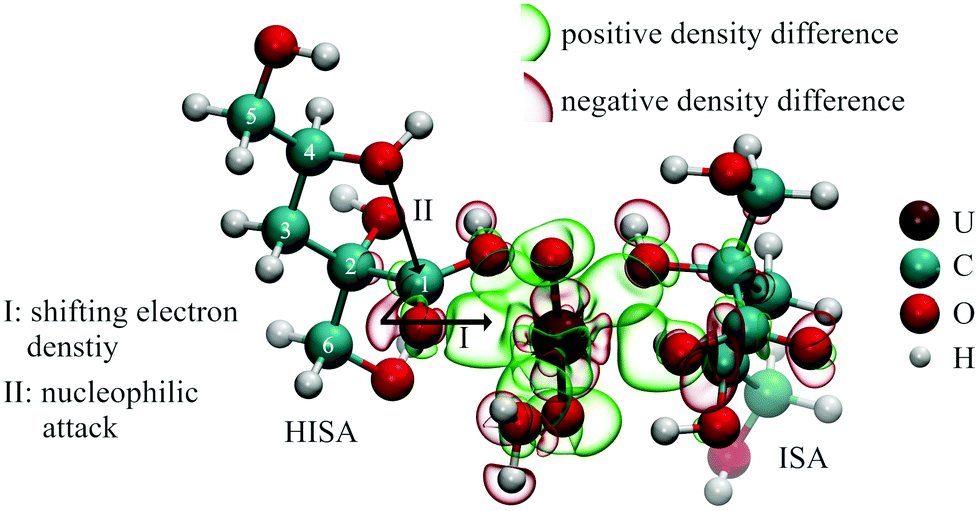 | ||
| Fig. 9 Calculated electron density changes in a [UO2(HISA)(ISA)(H2O)]+ complex and the impact on the lactone formation reaction (isovalues for the representation of electron densities were −0.005 (red) and +0.005 (green). | ||
4.1 Influence of UO22+ on the lactone formation
1H-NMR spectroscopic measurements revealed the interaction of UO22+ with HISA at low pH values. Major changes were observed for signals of protons, which are in the surrounding of the carboxylic group (3a/3b and 6a/6b; see Table 1) suggesting a coordination of HISA to UO22+ in this region. Fig. 9 shows the theoretically calculated electron density changes of a complex involving HISA revealing the previously described general pattern of changing electron density. A clear decrease can be observed between the carboxylic carbon (C1) of HISA and the corresponding carbonyl oxygen. In other words, this chemical bond is weakened and consequently the corresponding vibrational mode should be shifted to lower wavenumbers. Accordingly, the mode observed at 1351 cm−1 in the pH series (Fig. 5a) can be assigned to the stretching vibration of the carbonyl group of HISA coordinated to UO22+.NMR measurements revealed that lactone formation is significantly accelerated in the presence of UO22+ (Fig. 2) while an effect on the equilibrium concentrations of HISA and ISL was not observed. The nucleophilic attack of the secondary alcohol (C4–OH) is likely facilitated by electron withdrawal from the carboxylic carbon (C1) upon complexation by UO22+ as shown in Fig. 9, similar to the nucleophilic substitution mechanism postulated for the metal free reaction (Fig. SI 18†). A similar effect was demonstrated by Takao and Akashi showing the promotion of a nucleophilic acyl substitution by a UO22+ induced carbonyl carbon activation.55 From this the overall conclusion can be drawn that UO22+ interacts with HISA and the electrophilic character of U catalyzes the lactone formation reaction under acidic conditions.
4.2 Structural properties of UO22+–ISA complexes
The interaction of ISA with UO22+ causes significant shifts of some 13C signals, which is a consequence of a changed electron density around the corresponding carbon nuclei. The largest shifts were observed for C1, C2, and C6 (Table 2), corresponding to the carboxylic carbon (C1) as well as the carbons of the α- (C2–OH) and β-hydroxy group (C6–OH). All these signals were shifted downfield, which indicates a decreased electron density. This can be traced back to the previously discussed electron withdrawing effect of the UO22+ entity and therefore reflects the participation of these functional groups in complex formation. Since no considerable shifts were measured for C4 and C5, the γ- (C4–OH) and δ-hydroxy groups (C5–OH) are not expected to be involved.ATR-FTIR results add more general information concerning the binding motifs present in UO22+–ISA complexes. Changes in the difference between the νas and νs of the carboxylic group, Δν, were often used as a first approximation to describe the coordination behavior in metal–carboxylate complexes.60–63 A decrease of Δν compared to the non-coordinated carboxylate anion is associated with a bidentate coordination (both oxygen atoms are coordinated to the metal center) while an increase is associated with a monodentate coordination (only one oxygen is coordinated to the metal center). According to the theoretical investigation of Sutton et al., this behavior can be traced back to different impacts on the geometry of the carboxylic group.64 Monodentate coordination strongly influences the C–O bond lengths while bidentate coordination particularly affects the O–C–O angle, causing these different changes in Δν. For UO22+–ISA the increased Δν in both, the pH series (+53 cm−1) as well as in the concentration series (+62 cm−1) strongly suggests the monodentate coordination of the carboxylic group. In the case of a bridging motif, where each of the two carboxylate oxygens coordinates to different metal units, it was claimed that Δν remains constant or behaves like in the case of bidentate coordination. However, Deacon and Phillips also showed examples of asymmetric bridging motifs, where Δν was increased.61 Therefore, this motif cannot be excluded at this point. The changes in the region between 1150 cm−1 and 1000 cm−1 indicate the participation of hydroxy groups in complex formation, which agrees with NMR results. The shortening of the U–Oeq bond lengths, which were observed by EXAFS measurements, underpins the conclusion that chelates are formed.
All these findings lead to the conclusion that three binding motifs can be expected in UO22+–ISA complexes, which are visualized in Fig. 10: (a) monodentate coordination of the carboxylic and the α-hydroxy group (5-membered ring), (b) monodentate coordination of the carboxylic and the β-hydroxy group (6-membered ring), (c) the carboxylic group acts as a bridge between two UO22+ units with one UO22+ additionally binding to the α- and the other UO22+ binding to the β-hydroxy group (5- and 6-membered rings are simultaneously present).
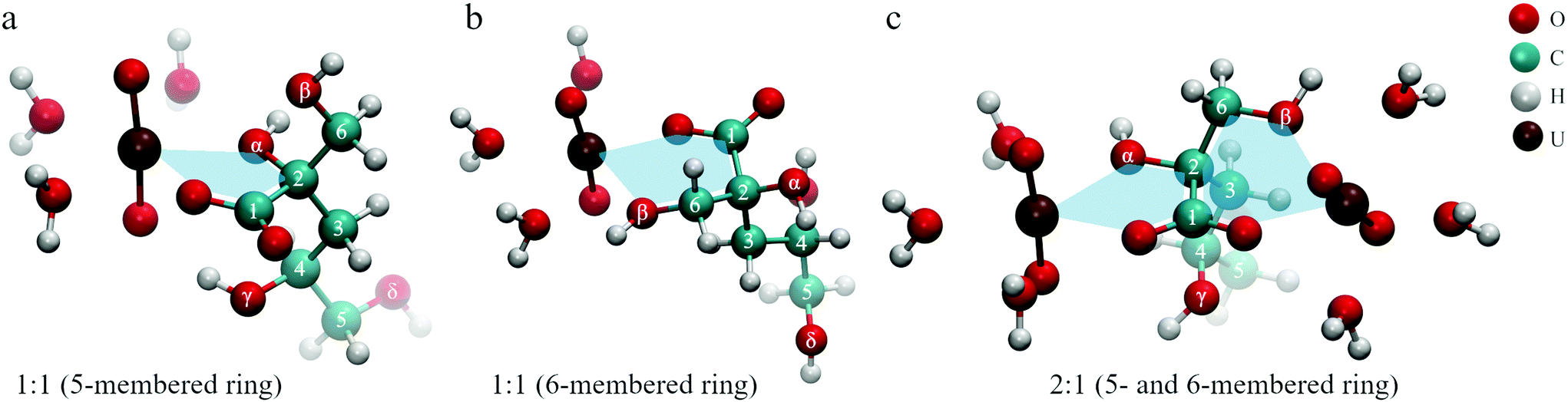 | ||
| Fig. 10 Dominant binding motifs in UO22+–ISA complexes (pictures represent optimized DFT structures): (a) 5-membered ring, (b) 6-membered ring, (c) bridging (5- and 6-membered ring). | ||
The binding motifs shown in Fig. 10 agree with the consensus in the literature, that the carboxylic and hydroxy groups coordinate the metal center in concert. Birjkumar et al. included the 5- and 6-membered rings as binding motifs in their theoretical study.31 They concluded that the energy difference between these coordination modes is not sufficient to rule out that both coexist. This agrees with the findings of the present study. Experimental studies related to the structural properties of ISA complexes in general are limited and none was found for UO22+ or other actinyl ions. Randall et al. reported NMR results from studies concerning the identification of functional groups involved in metal coordination.10 They concluded that both Ca2+ and Eu3+ form complexes with ISA via the carboxylic as well as the α- and γ-hydroxy groups. Similar results were obtained for Ca2+ by Dudás et al.26 Apart from the identification of the carboxylic, and α- and γ-hydroxy groups as coordinating functionalities, their NMR results showed that the β-hydroxy group is not involved. Tasi et al. concluded from DFT optimizations of Pu(IV)–ISA complexes that again the carboxylic, and α- and γ-hydroxy groups are the coordinating functionalities of the ISA molecule.18 In all described cases, the preference for the α- over the β-hydroxy group is related to the additional binding to the γ-hydroxy group, which is only possible on the side of the α-hydroxy group.
Such a coordination is not feasible for an actinyl moiety, due to its linear O–U–O arrangement. The γ-hydroxy group cannot bind to the uranium at the same time as the carboxylic group and instead can form a hydrogen bond with one of the UO22+ oxygen atoms, which can be seen in Fig. 10a. Consequently, the energetic difference between the 5-membered ring involving the α-hydroxy group and the 6-membered ring involving the β-hydroxy group is negligible for UO22+, which can explain the coexistence of the two binding sites. The bridging motif in Fig. 10c cannot be directly proven based on NMR or ATR-FTIR data, as it is essentially both motifs shown in Fig. 10a and b occurring in the same molecule. The experimentally determined complex stoichiometries of 2:1 and 2:3, however, suggest the presence of such a bridging motif in these binuclear complexes. Furthermore, the presence of two binding sites for each ISA molecule is the key information to understand the mechanisms on a molecular level.
4.3 Complex-formation mechanism
Both UV-Vis and ATR-FTIR measurements suggested the formation of different species depending on the mole ratio of UO22+ and ISA in solution. If a twofold excess of UO22+ was present, a species showing an absorption maximum at 426.3 nm and a asymmetric stretching mode of the UO22+ (ν3) unit at 934 cm−1 was formed in solution (Fig. 5f and 6a/f). Furthermore, Fig. 6f shows that the initially present UO22+ was almost completely transformed into that compound. Considering that each ISA molecule can bind two UO22+ cations, the metal-to-binding site ratio is 1:1, if the molar ratio of UO22+ and NaISA is 2:1. This suggests that complexes with the stoichiometry 2:1 (UO22+:ISA) were formed. This hypothesis is confirmed by the ESI-MS measurements, which show that a 2:1 stoichiometry dominates the complex species for samples showing the absorption maximum at 426.3 nm in UV-Vis experiments (Table 4 and Fig. SI 36†). According to the general conclusion from EXAFS measurements that the coordination number of the UO22+ unit was slightly larger than five for all compounds, the initially formed species under these conditions can be written as [(UO2)2(ISA)(H2O)6]3+, whose structure is shown in Fig. 10c (and Fig. SI 50/51†).
These initially formed [(UO2)2(ISA)(H2O)6]3+ complexes can be bridged by additional ISA molecules, if enough ligand molecules are present in solution, leading to the formation of polynuclear chains. Such a chain-like pattern was observed in the structure of [UO2(C2O4)2H2O]·2H2O.65 This process is shown in Scheme 1. The additional coordination of a second ISA molecule in the equatorial plane of UO22 can be recognized by a further shift of ν3(UO22+) to 920 cm−1 as well as an additional component in the UV-Vis spectra with an absorption maximum at 438.3 nm (Fig. 5f and 6b/g). Consequently, the species formed under these conditions can be assigned to complexes with the general formula [(UO2)m(ISA)n(H2O)x]2m−n (with m > 2 and n = m + 1, m or m − 1).
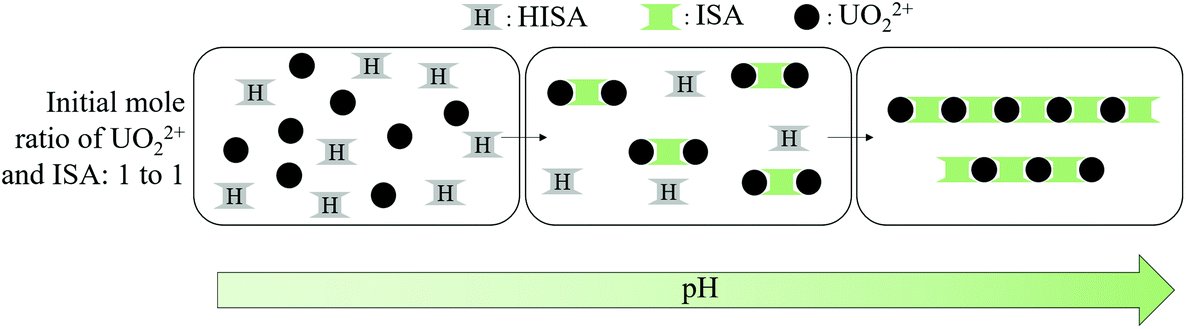 | ||
| Scheme 1 Simplified complex formation mechanism at equimolar concentrations of UO22+ and ISA (ISL and coordinated water molecules were neglected for clarity): formation of [(UO2)2(ISA)(H2O)6]3+ complexes in the first and bridging of those in the second step. | ||
If the initial amount of ISA exceeds that of UO22+, the formation of a different species suppresses the formation of the [(UO2)2(ISA)(H2O)6]3+ complex at low pH values. This can be clearly seen in Fig. 6h–j and c–e, in which component 4 becomes more dominant with increasing excess of NaISA. As described in section 4.1, HISA is also able to coordinate to UO22+. Therefore, the suppressed formation of polynuclear species at lower pH values can be traced back to the interaction of HISA with UO22+ as the competing process. In other words, the higher the amount of HISA present in solution the more is the formation of polynuclear chains suppressed and smaller molecules are formed. This is in agreement with the complex-stoichiometry of 2 to 3 suggested from the method of continuous variation and ESI-MS measurements at higher initial concentrations of ISA compared to UO22+ in solution (Fig. 7 and Table 4). An optimized structure of such a [(UO2)2(ISA)3(H2O)2]+ complex (2:3) is shown in Fig. 11a. The absorption spectra of the EXAFS- and ATR-FTIR pH series show a maximum at 414.3 nm at the lowest pH (1.1) (Fig. SI 35 left and 37 left†) corresponding to the [UO2(H2O)5]2+ complex. Since HISA and ISL dominate at pH 1.1, the species showing the absorption at 422.3 nm and ν3(UO22+) at 944 cm−1, most likely includes a stronger complexing ISA molecule. Consequently, the initially formed species under conditions where the initial concentration of ISA in solution strongly exceeds that of UO22+ can be assigned to a [UO2(ISA)(H2O)3]+ complex (1:1), while some of the water molecules can be exchanged by one or more HISA molecules. An optimized structure with one attached HISA molecule is shown in Fig. 9 and Fig. SI 52.† Once this species is formed and the pH is increased, the attached HISA molecule gets deprotonated leading to the formation of [UO2(ISA)2(H2O)] complexes (1:2). This mechanism is shown in Scheme 2. The optimized structure of a [UO2(ISA)2(H2O)] complex showing a 5- and 6-membered ring as a binding motif is shown in Fig. 11b.
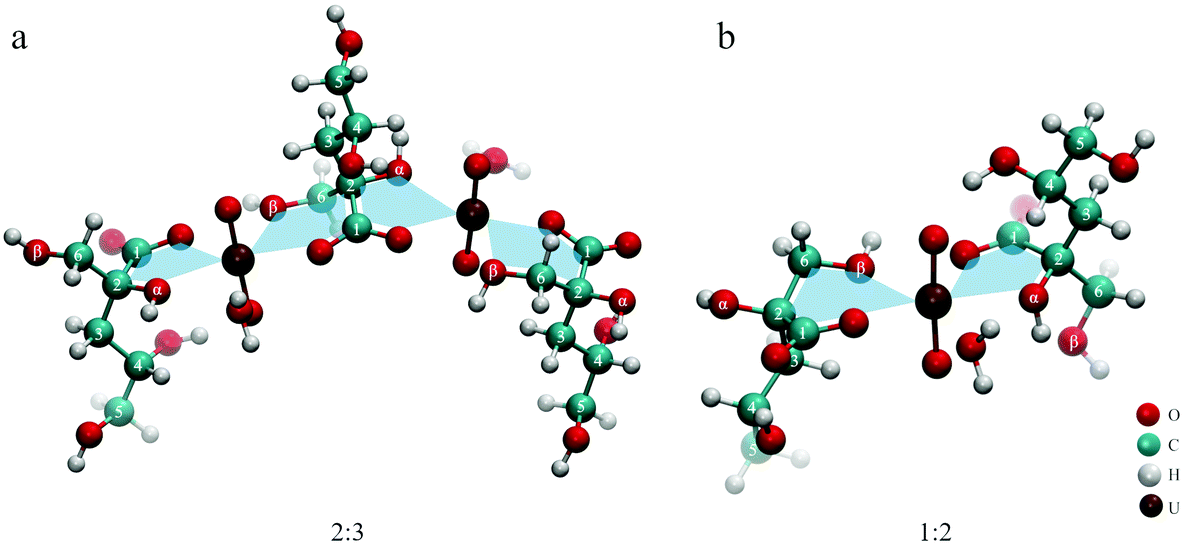 | ||
| Fig. 11 Optimized structures of 2:3 (a) and 1:2 (b) UO22+–ISA complexes. | ||
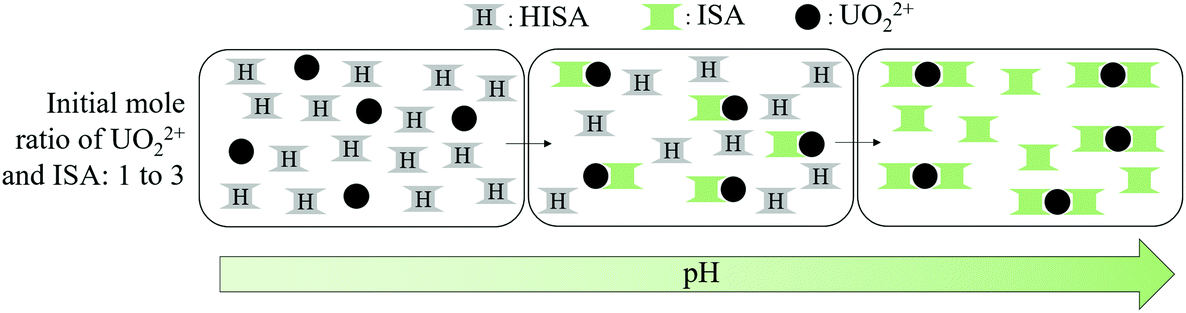 | ||
| Scheme 2 Simplified complex formation mechanism at an excess of ISA (ISL and coordinated water molecules were neglected for clarity): formation of [UO2(ISA)(H2O)3]+ complexes (the formation of polynuclear complexes is inhibited by interaction with HISA molecules) in the first and [UO2(ISA)2(H2O)] complexes in the second step. | ||
The UO22+ units in the polynuclear complexes [(UO2)m(ISA)n(H2O)x]2m−n and also in the [UO2(ISA)2(H2O)] complex have two binding partners in their equatorial plane. These structural similarities provide a reasonable explanation for their spectroscopic similarities in terms of absorption maxima and position of ν3(UO22+). This conclusion was supported by the combination of EXAFS and UV-Vis spectroscopy (see section 3.6). Components 3 and 5, which correspond to the polynuclear [(UO2)m(ISA)n(H2O)x]2m−n and the [UO2(ISA)2(H2O)] complexes, represent two species in the UV-Vis spectra but cannot be distinguished by EXAFS. The five dominant components identified in the present study, their assignments as well as their spectral properties are summarized in Table 6.
| Component | Assigned complex | Absorption maximum [nm] (ε [l·mol−1·cm−1]) | ν 3(UO22+) [cm−1] |
|---|---|---|---|
| a m > 2, n = m + 1, m or m − 1. b Associated with one or more weakly bound HISA molecules. | |||
| 1 | [UO2(H2O)5]2+ | 414.3 (8.5) | 960 |
| 2 | [(UO2)2ISA(H2O)6]3+ | 426.3 (42.8) | 934 |
| 3 | [(UO2)m(ISA)n(H2O)x]2m−na |
438.3 (72.0) | 920 |
| [(UO2)2(ISA)3(H2O)2]+ | |||
| 4 | [UO2(ISA)(H2O)3]+b |
422.3 (20.5) | 944 |
| 5 | [UO2(ISA)2(H2O)] | 438.3 (61.4) | 920 |
The lactone form of the ligand has no direct impact on the complex formation, other than reducing the amount of HISA available for the interaction with UO22+ at low pH. This subtle effect of lactone formation can be seen in Fig. SI 37,† showing the UV-Vis spectra of the EXAFS samples measured immediately and 15 days after preparation. The extinction is slightly increased while the absorption maxima remain constant. A similar effect was observed for the UV-Vis spectra of the long-term lactone formation NMR-experiment (Fig. SI 33†). Furthermore, the absorption maximum slightly shifted from 422.3 nm, corresponding to [UO2(ISA)(H2O)3]+ associated with HISA, to higher wavelengths. According to the previously discussed complex formation mechanisms, the formation of polynuclear species (having their maxima at 426.3 and 438.3 nm) was less suppressed in the NMR-experiment with increasing time causing the observed spectral changes. Since the same absorption maxima were observed and the previously mentioned changes in the UV-vis spectra are rather small, no impact of the lactone formation on the dominant species formed in solution is observed. However, the kinetic aspect of the lactone formation has to be appropriately considered, if the experiments are intended to determine complex formation constants.
4.4 Comparison with literature data for the UO22+–ISA system
Warwick et al. characterized complexes between selected divalent metals (Cd2+, Co2+ and UO22+) and ISA under near-neutral and alkaline conditions.30 Furthermore, the stoichiometries of metals, hydroxide ions and ISA molecules as well as stability constants for the identified species were determined. The ratio of metal ions and ISA molecules in the formed complexes was determined by conductometric titrations. The procedure of this particular experiment was the same for all investigated metals. For that purpose, 1 mL aliquots of a 0.1 M metal chloride solution, which corresponds to 0.1 mmol of metal per aliquot, were stepwise added to 20 mL of a 0.1 M NaISA solution, which corresponds to 2 mmol ISA. The conductivity was measured after each titration step. Changes in the gradient of the measured conductance and the corresponding volume of the added metal solution were then used to derive the stoichiometry. For Cd and Co, inflexions were observed at 20 mL added metal solution corresponding to 2 mmol of metal. Consequently, equimolar concentrations of ISA and metal were present at this point leading to the conclusion that the stoichiometry of the formed complex between Cd and Co with ISA is 1 to 1. The same ratio was proposed for UO22+ even though the inflexion in the conductance was already observed at 10 mL of added metal solution. This corresponds to 1 mmol of UO22+ giving a metal to ligand ratio of 1:2 (1 mmol UO22+ to 2 mmol ISA) at this point. This result is in line with one of the proposed species identified in the present study, which was formed when ISA is present in excess.
Another study was performed by Rao et al., where the UO22+–ISA system was characterized under acidic conditions by potentiometry and calorimetry.29 They determined the formation of three complexes with UO22+ to ISA ratios of 1 to 1, 1 to 2 and 1 to 3. Even though the experiments were performed in a similar concentration range, no indication was found for the latter complex in the present study. A possible explanation for that apparent discrepancy may be found in the data evaluation approach. The average number of ISA molecules bound to UO22+ (![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) ) from potentiometric data was calculated by the following equation:
) from potentiometric data was calculated by the following equation:
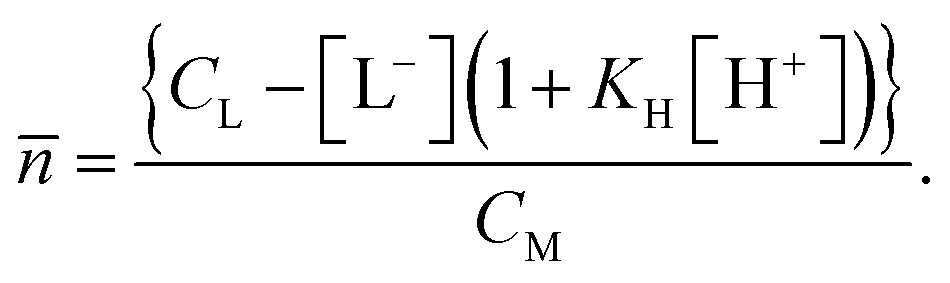
In this approach, the acid constant (KH) for HISA was assumed to be a constant. Our results, however, clearly show the interaction between UO22+ and HISA. The electron withdrawal from the protonated carboxylic group may increase the acidity of the proton. Larger values for KH would in turn lead to smaller values for . The potentiometric analysis would then require a correction for this effect. This emphasizes the requirement, if possible, for the combination of potentiometry with spectroscopic techniques as complementary methods.
5 Conclusion
The present work highlights the complex interaction of mutual influence on the chemical speciation of metals and ligands. Only by the collective interpretation of data of a number of complementary analytical methods, it was possible to understand the interaction of UO22+ and isosaccharinic acid on the molecular level. This approach allows the characterization of the system from the metal and the ligand point of view. This study shows that (1) ISA is able to bridge UO22+ units under certain experimental conditions and (2) UO22+ interacts with the protonated form of the ligand, which changes its chemical reactivity, and in turn affects the complex formation mechanism. These findings for the first time give a complete picture of the interaction of UO22+ with ISA over a wide range of solution compositions and extend the understanding of UO22+ interaction with organic compounds.On the basis of the reaction mechanisms and complex stoichiometries determined in this study, thermodynamic data for the complex formation process can be determined, either by new experiments or by reevaluation of the existing data. In this process, the kinetic effect of the ISL–HISA–ISA equilibrium altered by the presence of UO22+ can also be considered. This will have a significant impact on the reliability of the assessment of the impact of ISA on the mobility of UO22+ and other actinyl ions in the context of nuclear waste disposal research.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the Euratom research and training programme 2014–2018 under Grant Agreement no. 61880. Manuel Raiwa acknowledges the financial support by the German Federal Ministry of Education and Research (BMBF) in the joint project SIRIUS (02NUK044A). The authors would like to acknowledge Carola Eckardt for TOC measurements. We also thank Moritz Schmidt for proofreading the article.References
- U. R. Berner, Waste Manag., 1992, 12, 201–219 CrossRef CAS.
- M. H. Bradbury and L. R. Van Loon, Cementitious Near-Field Sorption Data Bases for Performance Assessment of a L/ILW Repository in a Paifris Marl Host Rock, 1997, vol. PSI-Berich Search PubMed.
- L. Abrahamsen, T. Arnold, H. Brinkmann, N. Leys, M. Merrous, K. Mijnendonckx, H. Moll, P. Polvika, J. Sevcu, J. Small, M. Vikman and K. Wouters, A review of anthropogenic organic wastes and their degradation behaviour, 2015 Search PubMed.
- M. A. Glaus and L. R. Van Loon, Environ. Sci. Technol., 2008, 42, 2906–2911 CrossRef CAS PubMed.
- I. Pavasars, J. Hagberg, H. Borén and B. Allard, J. Polym. Environ., 2003, 11, 39–47 CrossRef CAS.
- M. Glaus, L. R. Van Loon, S. Achatz, A. Chodura and K. Fischer, Anal. Chim. Acta, 1999, 398, 111–122 CrossRef CAS.
- L. R. Van Loon and M. A. Glaus, J. Environ. Polym. Degrad., 1997, 5, 97–109 CrossRef CAS.
- P. N. Humphreys, A. Laws and J. Dawson, A Review of Cellulose Degradation and the Fate of Degradation Products Under Repository Conditions, 2010, vol. 001 Search PubMed.
- L. R. Van Loon, M. A. Glaus, A. Laube and S. Stallone, Radiochim. Acta, 1999, 86, 183–189 CAS.
- M. Randall, B. Rigby, O. Thomson and D. Trivedi, Assessment of the effects of cellulose degradation products on the behaviour of europium and thorium, 2013 Search PubMed.
- G. M. N. Baston, J. A. Berry, K. A. Bond, K. A. Boult, M. Brownsword and C. M. Linklater, J. Alloys Compd., 1994, 213–214, 475–480 CrossRef.
- G. M. N. Baston, J. A. Berry, K. A. Bond, K. A. Boult and C. M. Linklater, Radiochim. Acta, 1994, 66–67, 437–442 Search PubMed.
- G. M. N. Baston, M. M. Cowper, T. G. Heath, T. A. Marshall and S. W. Swanton, Mineral. Mag., 2012, 76, 3381–3390 CrossRef.
- L. R. Van Loon and M. A. Glaus, Experimental and Theoretical Studies on Alkaline Degradation of Cellulose and its Impact on the Sorption of Radionuclides, 1998 Search PubMed.
- W. Hummel, G. Anderegg, L. Rao, I. Puigdomènech and O. Tochiyama, Chemical Thermodynamics Of Compounds And Complexes Of U, Np, Pu, Am, Tc, Se, Ni and Zr With Selected Organic Ligands, 2005 Search PubMed.
- X. Gaona, V. Montoya, E. Colàs, M. Grivé and L. Duro, J. Contam. Hydrol., 2008, 102, 217–227 CrossRef CAS PubMed.
- D. Rai and A. Kitamura, J. Chem. Thermodyn., 2017, 114, 135–143 CrossRef CAS.
- A. Tasi, X. Gaona, D. Fellhauer, M. Böttle, J. Rothe, K. Dardenne, R. Polly, M. Grivé, E. Colàs, J. Bruno, K. Källström, M. Altmaier and H. Geckeis, Appl. Geochem., 2018, 98, 247–264 CrossRef CAS.
- A. Tasi, X. Gaona, D. Fellhauer, M. Böttle, J. Rothe, K. Dardenne, R. Polly, M. Grivé, E. Colàs, J. Bruno, K. Källstrom, M. Altmaier and H. Geckeis, Appl. Geochem., 2018, 98, 351–366 CrossRef CAS.
- P. L. Brown, S. Allard and C. Ekberg, J. Chem. Eng. Data, 2010, 55, 5207–5213 CrossRef CAS.
- S. Ekberg, C. Ekberg and Y. Albinsson, J. Solution Chem., 2004, 33, 465–477 CrossRef CAS.
- H. Cho, D. Rai, N. J. Hess, Y. Xia and L. Rao, J. Solution Chem., 2003, 32, 691–702 CrossRef CAS.
- D. Rai, N. J. Hess, Y. Xia, L. Rao, H. M. Cho, R. C. Moore and L. R. Van Loon, J. Solution Chem., 2003, 32, 665–689 CrossRef CAS.
- D. Rai and A. Kitamura, J. Nucl. Sci. Technol., 2016, 53, 459–467 CrossRef CAS.
- K. Vercammen, M. A. Glaus and L. R. Van Loon, Radiochim. Acta, 2001, 89, 393–401 CAS.
- C. Dudás, B. Kutus, É. Böszörményi, G. Peintler, Z. Kele, I. Pálinkó and P. Sipos, Dalton Trans., 2017, 46, 13888–13896 RSC.
- M. R. González-Siso, X. Gaona, L. Duro, M. Altmaier and J. Bruno, Radiochim. Acta, 2018, 106, 31–45 Search PubMed.
- E. Colàs Anguita, PhD thesis, Universitat Politècnica de Catalunya, 2014, http://hdl.handle.net/2117/95404.
- L. Rao, A. Y. Garnov, D. Rai, Y. Xia and R. C. Moore, Radiochim. Acta, 2004, 92, 575–581 CAS.
- P. Warwick, N. Evans and S. Vines, Radiochim. Acta, 2006, 94, 363–368 CAS.
- K. H. Birjkumar, N. D. Bryan and N. Kaltsoyannis, Dalton Trans., 2012, 41, 5542 RSC.
- L. R. Whistler and J. N. BeMiller, in Methods in carbohydrate chemistry, 1963, vol. 2, pp. 477–479 Search PubMed.
- N. M. Bassil, N. Bryan and J. R. Lloyd, ISME J., 2015, 9, 310–320 CrossRef CAS PubMed.
- L. R. Van Loon, M. A. Glaus, S. Stallone and A. Laube, Environ. Sci. Technol., 1997, 31, 1243–1245 CrossRef CAS.
- A. Rossberg, T. Reich and G. Bernhard, Anal. Bioanal. Chem., 2003, 376, 631–638 CrossRef CAS PubMed.
- K. Müller, V. Brendler and H. Foerstendorf, Inorg. Chem., 2008, 47, 10127–10134 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- TURBOMOLE GmbH, Turbomole, 2016 Search PubMed.
- X. Cao and M. Dolg, J. Mol. Struct. (THEOCHEM), 2004, 673, 203–209 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Reich, G. Bernhard, G. Geipel, H. Funke, C. Hennig, A. Rossberg, W. Matz, N. Schell and H. Nitsche, Radiochim. Acta, 2000, 88(9–11), 633–637 CAS.
- T. Ressler, J. Synchrotron Radiat., 1998, 5, 118–122 CrossRef CAS PubMed.
- S. M. Webb, Phys. Scr., 2005, 1011 CrossRef CAS.
- G. N. George and I. J. Pickering, EXAFSPAK: A Suite of Computer Programs for Analysis of X-ray Absorption Spectra, Stanford Synchrotron Radiation Laboratory, Stanford, CA, USA, 1995 Search PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- A. Heller, A. Barkleit, H. Foerstendorf, S. Tsushima, K. Heim and G. Bernhard, Dalton Trans., 2012, 41, 13969–13983 RSC.
- J. Bell and R. Biggers, J. Mol. Spectrosc., 1965, 18, 247–275 CrossRef CAS.
- G. Meinrath, D. Kwiatek, Z. Hnatejko and S. Lis, Monatsh. Chem. - Chem. Mon., 2014, 145, 1689–1696 CrossRef CAS PubMed.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998–12013 CrossRef CAS PubMed.
- D. Kim, K. Park, M. Yang, T. Kim, R. Mahajan and J. Kim, Talanta, 2007, 74, 223–228 CrossRef CAS PubMed.
- S. Bashir, S. R. Maqsood, G. M. Peerzada, B. Khan and M. A. Rizvi, J. Inorg. Chem., 2014, 2014, 1–11 CrossRef.
- K. Takao and S. Akashi, RSC Adv., 2017, 7, 12201–12207 RSC.
- C. Lucks, A. Rossberg, S. Tsushima, H. Foerstendorf, A. C. Scheinost and G. Bernhard, Inorg. Chem., 2012, 51, 12288–12300 CrossRef CAS PubMed.
- L. Rao, J. Jiang, P. Zanonato, P. Di Bernardo, A. Bismondo and A. Y. Garnov, Radiochim. Acta, 2002, 90, 581–588 CAS.
- M. J. Sarsfield and M. Helliwell, J. Am. Chem. Soc., 2004, 126, 1036–1037 CrossRef CAS PubMed.
- N. Miyamoto, T. Tsukahara and Y. Ikeda, Chem. Lett., 2012, 41, 513–515 CrossRef CAS.
- T. J. Strathmann and S. C. B. Myneni, Geochim. Cosmochim. Acta, 2004, 68, 3441–3458 CrossRef CAS.
- G. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- M. B. Hay and S. C. B. Myneni, Geochim. Cosmochim. Acta, 2007, 71, 3518–3532 CrossRef CAS.
- M. Kakihana, T. Nagumo, M. Okamoto and H. Kakihana, J. Phys. Chem., 1987, 91, 6128–6136 CrossRef CAS.
- C. C. R. Sutton, G. da Silva and G. V. Franks, Chem. – Eur. J., 2015, 21, 6801–6805 CrossRef CAS PubMed.
- J. Leciejewicz, N. W. Alcock and T. J. Kemp, in Coordination Chemistry, Springer-Verlag, Berlin/Heidelberg, 1995, vol. 82, pp. 43–84 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt01080g |
| This journal is © The Royal Society of Chemistry 2019 |