
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A strategy to convert propane to aromatics (BTX) using TiNp4 grafted at the periphery of ZSM-5 by surface organometallic chemistry†
Walid
Al Maksoud
a,
Lieven E.
Gevers
a,
Jullian
Vittenet
a,
Samy
Ould-Chikh
 a,
Selvedin
Telalovic
a,
Kushal
Bhatte
a,
Edy
Abou-Hamad
b,
Dalaver H.
Anjum
b,
Mohamed N.
Hedhili
b,
Vinu
Vishwanath
c,
Abdulrahman
Alhazmi
c,
Khaled
Almusaiteer
c and
Jean Marie
Basset
*a
a,
Selvedin
Telalovic
a,
Kushal
Bhatte
a,
Edy
Abou-Hamad
b,
Dalaver H.
Anjum
b,
Mohamed N.
Hedhili
b,
Vinu
Vishwanath
c,
Abdulrahman
Alhazmi
c,
Khaled
Almusaiteer
c and
Jean Marie
Basset
*a
aKing Abdullah University of Science and Technology (KAUST), KAUST Catalysis Center (KCC), Thuwal, 23955-6900, Saudi Arabia. E-mail: jeanmarie.basset@kaust.edu.sa
bKing Abdullah University of Science and Technology (KAUST), Core Labs, Thuwal, 23955-6900, Saudi Arabia
cSABIC Corporate Research and Development, Thuwal 23955-6900, Saudi Arabia
First published on 24th April 2019
Abstract
The direct conversion of propane into aromatics (BTX) using modified ZSM-5 was achieved with a strategy of “catalysis by design”. In contrast to the classical mode of action of classical aromatization catalysts which are purely based on acidity, we have designed the catalyst associating two functions: One function (Ti-hydride) was selected to activate the C–H bond of propane by σ-bond metathesis to further obtain olefin by β-H elimination and the other function (Brønsted acid) being responsible for the oligomerization, cyclization, and aromatization. This bifunctional catalyst was obtained by selectively grafting a bulky organometallic complex of tetrakis(neopentyl)titanium (TiNp4) at the external surface (external silanol (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH) group) of [H-ZSM-5300] to obtain [Ti/ZSM-5] catalyst 1. This metal was chosen to activate the C–H bond of paraffin at the periphery of the ZSM-5 while maintaining the Brønsted acid properties of the internal [H-ZSM-5] for oligomerization, cyclization, and aromatization. Catalyst 2 [Ti–H/ZSM-5] was obtained after treatment under H2 at 550 °C of freshly prepared catalyst 1 ([Ti/ZSM-5]) and catalyst 1 was thoroughly characterized by ICP analysis, DRIFT, XRD, N2-physisorption, multinuclear solid-state NMR, XPS and HR-TEM analysis including STEM imaging. The conversion of propane to aromatics was studied in a dynamic flow reactor. With the pristine [H-ZSM-5300] catalyst, the conversion of propane is very low. However, with [Ti–H/ZSM-5] catalyst 2 under the same reaction conditions, the conversion of propane remains significant during 60 h of the reaction (ca. 22%). Furthermore, the [Ti–H/ZSM-5] catalyst shows a good and stable selectivity (55%) for aromatics (BTX) of time on stream. With 2, it was found that the Ti remains at the periphery of the [H-ZSM-5] even after reaction time.
Si–OH) group) of [H-ZSM-5300] to obtain [Ti/ZSM-5] catalyst 1. This metal was chosen to activate the C–H bond of paraffin at the periphery of the ZSM-5 while maintaining the Brønsted acid properties of the internal [H-ZSM-5] for oligomerization, cyclization, and aromatization. Catalyst 2 [Ti–H/ZSM-5] was obtained after treatment under H2 at 550 °C of freshly prepared catalyst 1 ([Ti/ZSM-5]) and catalyst 1 was thoroughly characterized by ICP analysis, DRIFT, XRD, N2-physisorption, multinuclear solid-state NMR, XPS and HR-TEM analysis including STEM imaging. The conversion of propane to aromatics was studied in a dynamic flow reactor. With the pristine [H-ZSM-5300] catalyst, the conversion of propane is very low. However, with [Ti–H/ZSM-5] catalyst 2 under the same reaction conditions, the conversion of propane remains significant during 60 h of the reaction (ca. 22%). Furthermore, the [Ti–H/ZSM-5] catalyst shows a good and stable selectivity (55%) for aromatics (BTX) of time on stream. With 2, it was found that the Ti remains at the periphery of the [H-ZSM-5] even after reaction time.
Introduction
Aromatics, especially Benzene, Toluene and Xylenes (BTX), are important chemicals, which are extensively used in the production of styrene, phenol, polymers, plastics, medicines, rubbers and others. Catalytic cracking/reforming of naphtha can lead to aromatics processes using ZSM-5 catalysts1–5 The aromatization reaction of light alkanes, such as ethane, propane or n-butane, are important catalytic reactions for the academic community as it represents an example of a complex and multistep reaction requiring a multi-functional catalyst. This process is also used industrially to upgrade light alkanes such as ethane, propane and butane in the petroleum refinery.6–9ZSM-5 is known to be one of the best catalysts for the aromatization reaction mostly due to its controllable acidity, its high-temperature stability and its shape selectivity due to its pore structure. Most of the reported studies indicate that the yield of aromatics can be increased mainly by using a doping metal.10–12
Several metals have been added to zeolites such as Ga, Mo, and Zn. The results indicate that Ga modified zeolites are the most efficient catalysts for light alkane aromatization.10,13–16 An industrial process has been successfully developed by BP and UOP and is known as the Cyclar™ process.2,17 There is a consensus that Ga+ is involved in the dehydrogenation step leading to olefins by an oxidative addition step and that the acid sites of zeolites facilitate catalysis, oligomerization, cyclization and aromatization steps.18 Various studies showed that the catalytic performance of the aromatization of propane mainly depends on the repartition between the Brønsted and Lewis acid sites present on the heterogeneous catalysts.14,19 Wetness impregnation has been the principal method to modify ZSM-5 by Ga, Mo, Pt, or Zn. Also, the mechanical mixing of oxides and chemical vapor deposition of precursors like GaMe3 are also reported.20 Nevertheless, a pre-treatment step before the reaction is required to obtain an active form of the catalyst prepared by this method.
Another tool for the preparation of the bifunctional catalyst is to prepare them by Surface Organometallic Chemistry (SOMC). This method usually leads to a well-defined single-site catalyst with a uniform distribution of active sites. But the choice of organometallic compounds must come from an in-deep analysis of the mechanism:21–23 Based on the assumption that alkane aromatization occurs by dehydrogenation of the paraffin into an olefin followed by the aromatization (oligomerization, cyclisation, dehydrogenation) of the obtained olefin by an acid-catalyzed mechanism, the choice and the localization of the metal for the first step are crucial. The selection of Ti was based on the observation that among oxide-supported group IV metal hydrides, titanium hydrides are the best catalysts for low-temperature hydrogenolysis of paraffin.24 This hydrogenolysis is assumed to come from a Sigma bond metathesis step between the C–H bond of the paraffin and the d0 metal hydride.25–28 In order to fully control these steps, we decided to immobilize Ti outside the pore system of the [ZSM-5] which could lead to olefin by Sigma bond metathesis. Then, it was expected that the formed olefin could migrate into the pore system and undergo oligomerization, cyclisation and aromatization inside the cavities. In order to selectively graft titanium outside cavities, we chose a bulky organometallic complex of Ti (TiNp4) (kinetic diameter of 9–10 Å) with the expectation, based on modeling, that it could not enter the pores of [H-ZSM-5] (ca. = 5–6 Å) based on the van der Waals radius. We were also expecting that the grafting of Ti was neutralizing the external acidity (mostly OH) so as to fully control the bifunctional nature of the catalyst, respectively, at the periphery for dehydrogenation and inside the pores for the aromatization steps (oligomerization, cyclisation, and aromatization).
Herein, we demonstrate that this strategy works with Ti by exhibiting a good and relatively stable activity on the conversion of propane to aromatics (BTX). As we will show, the reason for that is the stability of Ti on the periphery of [H-ZSM-5] during the reaction. Additional experiments have also been performed on the spent catalysts to gain an insight into the reasons for the catalytic deactivation of these catalysts.
Results and discussion
The [H-ZSM-5300] (Si/Al = 11) was obtained after the calcination of commercial [NH4-ZSM-5] at 550 °C under air for six hours followed by dehydration of the resulting [H-ZSM-5] at 300 °C under vacuum (10−5 bar/12 h). Free surface silanols (Si–OH) were obtained from adsorbed water or adjacent silanols subsequently used as a grafting site.29 The concentration of silanol [H-ZSM-5300] was estimated by titration with MeLi (quantitative evolution of methane) and found to be 0.4 ± 0.1 mmol of Si–OH per gram of [H-ZSM-5300]. An excess of TiNp4 (Np: –CH2C(CH3)3) was then reacted at RT with [H-ZSM-5300] in a Schlenck tube for 8 h in n-pentane. The catalyst 1 was thus obtained after repeated washing with n-pentane and dried at 80 °C under vacuum (10−5 bar). The treatment of catalyst 1 under H2 at 550 °C for two hours yields catalyst 2 as expected and was confirmed by different characterization methods described further (Scheme 1).
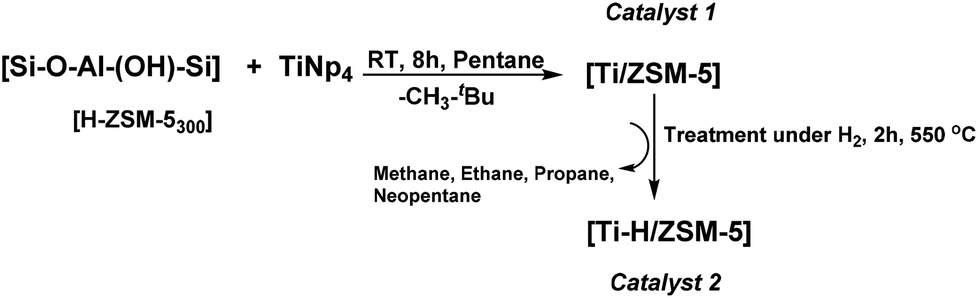 | ||
| Scheme 1 Grafting of TiNp4 on [H-ZSM-5300] to give catalyst 1 ([Ti/ZSM-5]), following by the treatment of catalyst 1 at 550 °C under H2 for 2 h to give catalyst 2 ([Ti–H/ZSM-5]). | ||
The elemental analysis of [Ti/ZSM-5] (1) indicated the presence of 0.8 wt% of Ti (∼0.17 mmol of Ti grafted on 1 g of [H-ZSM-5300]) (Table 1). The atomic ratio Ti/OH was found to be 0.42. This means that only a part of the silanols (less than 50%) has been consumed by the grafting reaction. This is expected because (i) the size of the organometallic precursor is in the 9–10 Å range being larger than the pore diameter of zeolite (5.0–6.0 Å), and (ii) the silanols lying on the external particle surface are the only accessible for grafting.25 This will be subsequently confirmed below by EDX elemental mapping.
| Catalysts | Tia (wt%) | C (wt%) | S BETb (m2 g−1) | Micropore areac (m2 g−1) | External surface areac (m2 g−1) | Micropore volumec (cm3 g−1) | Pore volumed (cm3 g−1) |
|---|---|---|---|---|---|---|---|
| a ICP-AES analysis of Ti. b BET, according to Rouquerol criteria. c t-Plot. d Single point at P/P0 = 0.1. | |||||||
| [H-ZSM-5300] | — | — | 453 | 254 | 89 | 0.14 | 0.17 |
| [Ti/ZSM-5] (1) | 0.8 | ∼3.9 | — | — | — | — | — |
| [Ti–H/ZSM-5] (2) | 0.8 | ∼0.2 | 390 | 222 | 79 | 0.12 | 0.14 |
The percentage of C was found to be 3.9 wt% (3.25 mmol of C) for catalyst 1 (Table 1). The ratio C/Ti was ca. 16.2 (expected 15 C for ((Si–O–)TiNp3)). The excess of carbon may be tentatively attributed to the presence of pentane which was still adsorbed on the support ([H-ZSM-5300]) during the grafting stage and not completely removed after drying of the catalyst under vacuum at 80 °C. In the 2 ([Ti–H/ZSM-5]), that is after the treatment of 1 under H2 at the 550 °C, the percentage of Ti was found not to be varied compared to 1 (∼0.8 wt%), which is logical because Ti is very oxophilic and once it is linked to the Si–O–moiety, it cannot move anymore (the bonding energy of Ti–O bond is 665 kj mol−1) (as demonstrated further by electron microscopy). However, the % of C decreases to 0.2%, and again this is an expected result. This is due to the formation of Ti–H by removing the organic fragment after treatment under H2 at 550 °C and realized of methane, ethane propane and neopentane in the gas phase as detected by GC.30 The formation of Ti–H was confirmed by the DRIFT spectra of catalysts 1 and 2 described further (Fig. 1.)
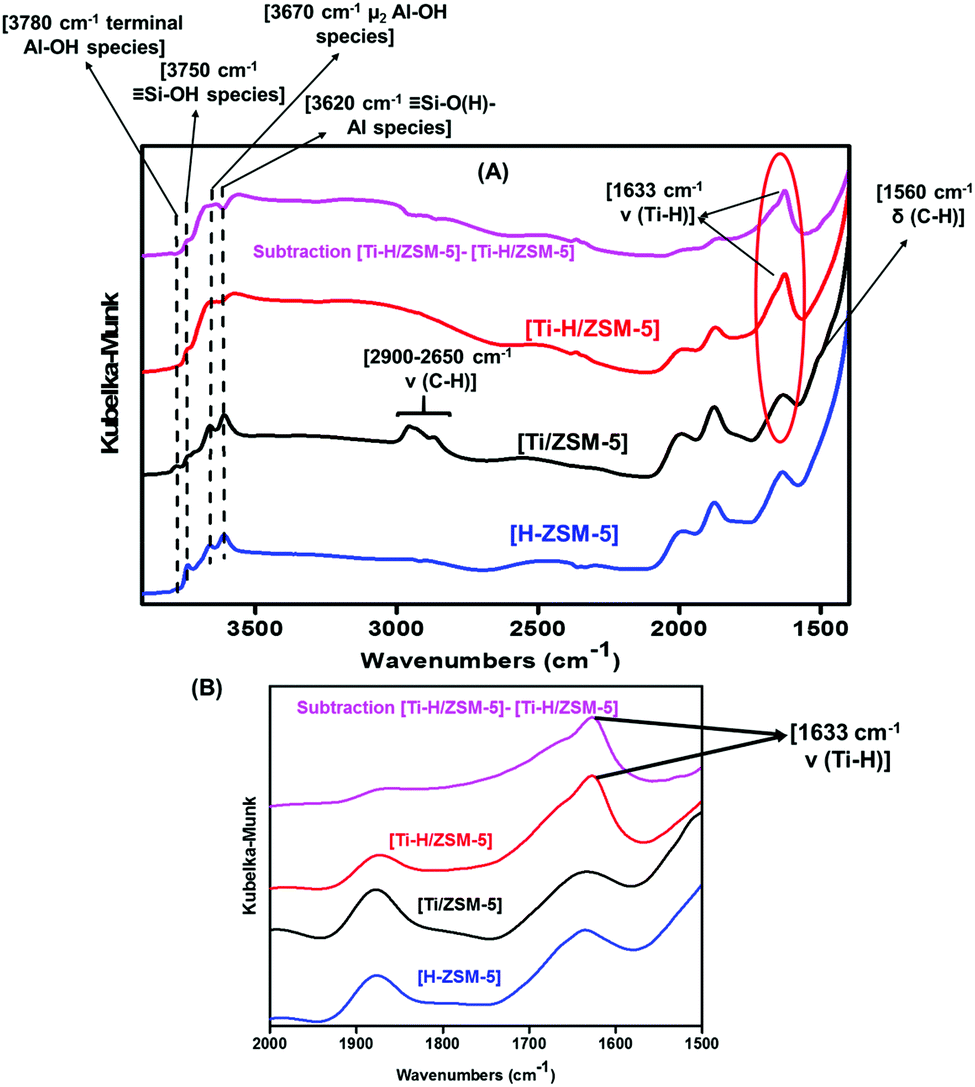 | ||
| Fig. 1 DRIFT spectra of [H-ZSM-5300] (blue), fresh catalyst 1 (black) [Ti/ZSM-5] and after treatment under H2 at 550 °C for 2 h [Ti-H/ZSM-5]; (A) in the 4000–1450 cm−1 region and (B) in the 2000–1500 cm−1 region. | ||
The XRD analysis of [H-ZSM-5300] (Si/Al = 11) and [Ti–H/ZSM-5] 2 was performed after pre-treatment at 550 °C under H2 for 2 h. The XRD patterns showed that the MFI framework was preserved as there was no substantial loss of crystallinity (Fig. 2-ESI†).
The N2-physisorption isotherm for the [H-ZSM-5300] and the catalyst 2 indicated a decrease of the surface area from 453 m2 g−1 for [H-ZSM-5300] to 390 m2 g−1 for the catalyst 2 ((Table 1) and (Fig. 1-ESI†)). The total pore volume, reported in Table 1, has slightly decreased from 0.17 cm3 g−1 for H-ZSM-5300 to 0.14 cm3 g−1 for the catalyst 2. Textural properties were similar for catalyst 2 which suggests the uniform distribution of metals utilizing the SOMC approach regardless of the metal precursor supported on [H-ZSM-5] during this study.
The DRIFT spectra of the grafted TiNp4 [Ti/ZSM-5] 1 compared to the [H-ZSM-5300] indicated a noticeable consumption of the isolated silanols (Si–OH). This was demonstrated by the strong attenuation of the intensity of the isolated silanol (Si–OH) band obtained at 3740 cm−1 as reported in Fig. 1A. Moreover, different bands at 2900–2650 cm−1 and 1560–1530 cm−1 appeared (TiNp4 grafted on [H-ZSM-5300]) which are attributed, respectively, to ν(C–H) and δ(C–H) vibrations of the alkyl fragments (Fig. 1A).
After treatment of the catalyst 1 under H2 at 550 °C for 2 h, a new peak at 1633 cm−1 corresponding to the formation of Ti–H appeared and the bands at 2900–2650 cm−1 and 1560–1530 cm−1 totally disappeared (Fig. 1B).26,30 The formation of Ti–H confirms our strategy to immobilize the Ti–H strongly on the surface of [H-ZSM-5300].
The bands at 3620 cm−1 and 3670 cm−1, corresponding to (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) Si–O(H)–Al) species (Brønsted acid site) and to extra-framework aluminum OH-μ2-Al species for which AI is connected with the zeolite framework, respectively, are also preserved. The band at 3780 cm−1 corresponding to terminal OH-μ1-Al species is also kept intact (Fig. 1A).1,31–34
Si–O(H)–Al) species (Brønsted acid site) and to extra-framework aluminum OH-μ2-Al species for which AI is connected with the zeolite framework, respectively, are also preserved. The band at 3780 cm−1 corresponding to terminal OH-μ1-Al species is also kept intact (Fig. 1A).1,31–34
The fresh catalyst [Ti/ZSM-5] (1) was further analyzed by multinuclear solid-state NMR to provide detailed information on the molecular structure of the grafted Ti complex.35 The 1H NMR spectrum exhibited a significant broad signal at 1.2 ppm arising from protons of the neopentyl ligand: the methyl (–CH2C(C![[H with combining low line]](https://www.rsc.org/images/entities/b_i_char_0048_0332.gif) 3)3) fragments of the neopentyl ligand and the methylene protons (–C2C(CH3)3) (Fig. 3-ESI B†).30 The peaks appearing at 2.7 and 4.3 ppm were assigned, respectively, to the bridging hydroxyl species (Si–O(H)–Al), corresponding to the Brønsted acidic sites and to the extra framework aluminum hydroxyls (OH-μ2-Al).36 Therefore, the obtained result shows clearly that grafting the TiNp4 complex on the surface of [H-ZSM-5300] does not affect the Brønsted acid site by reacting only with the isolated silanols on the surface of [H-ZSM-5300]. This NMR result is consistent with the DRIFT spectrum result shown in Fig. 1, where the intensity of the stretching band at 3600 cm−1 of Brønsted acidity is not perturbed by the grafting.
3)3) fragments of the neopentyl ligand and the methylene protons (–C2C(CH3)3) (Fig. 3-ESI B†).30 The peaks appearing at 2.7 and 4.3 ppm were assigned, respectively, to the bridging hydroxyl species (Si–O(H)–Al), corresponding to the Brønsted acidic sites and to the extra framework aluminum hydroxyls (OH-μ2-Al).36 Therefore, the obtained result shows clearly that grafting the TiNp4 complex on the surface of [H-ZSM-5300] does not affect the Brønsted acid site by reacting only with the isolated silanols on the surface of [H-ZSM-5300]. This NMR result is consistent with the DRIFT spectrum result shown in Fig. 1, where the intensity of the stretching band at 3600 cm−1 of Brønsted acidity is not perturbed by the grafting.
The 13C CP/MAS SS NMR spectrum shows an intense peak at 32 ppm assigned to the methyl groups –CH2C(CH3)3 and two weak peaks at 24 and 92.7 ppm, corresponding to the quaternary carbon –CH2CMe3 and the methylenic carbons of a neopentyl ligand –CH2CMe3, respectively (Fig. 3-ESI (C)†).
FT-IR of pyridine adsorption for the pristine [H-ZSM-5300] and [Ti/ZSM-5] (1) catalyst was carried out at 200 °C to identify and quantify the Brønsted and Lewis acidic sites (BAS, LAS) (Fig. 6 ESI†). It is noticed that there are three adsorption bands of pyridine at 1450 cm−1 assigned to the Lewis acid site (LAS), the band at 1500 cm−1 corresponds to co-contribution of LAS and BAS, and the latest band at 1550 cm−1 was assigned to BAS as shown in Fig. 6-ESI.†37–39
Table 2 summarizes the calculated concentration of BAS and LAS obtained after the FT-IR pyridine adsorption for the [Ti/ZSM-5] and [H-ZSM-5300] catalysts.37,40–42Table 2 shows that the number of Brønsted acid sites (BAS) increases from 69 μmol g−1 for [H-ZSM-5300] to 146 μmol g−1 for the grafted catalysts Ti. The increase of the number of Brønsted acid site maybe due to the generation of new acid species after the grafting of TiNp4. There is a possibility that Si–O–Ti is in the vicinity of Si–O–Ti–OH with a strong effect Ti on the Si–OH Brønsted acidity.
| Catalysts | BAS (μmol g−1) | LAS (μmol g−1) | Total acid sites (μmol g−1) |
|---|---|---|---|
| [H-ZSM-5300] | 69 | 45 | 114 |
| [Ti/ZSM-5] 1 | 146 | 40 | 186 |
However, the Lewis acid site (LAS) density of the grafted catalysts [Ti/ZSM-5] is very close compared to the pristine [H-ZSM-5300]. This result shows that the grafting of Ti on the surface of [H-ZSM-5300] by the SOMC strategy enhances the Brønsted of the catalysts but does not change the Lewis acidity.
Fig. 2A and B show the high-resolution XPS spectra of the Ti 2p core level for the fresh ([Ti/ZSM-5]) and the treated [Ti–H/ZSM-5] catalysts. It is worth mentioning here that the Ti 2p3/2 core level is fitted with three components for the case of fresh [Ti/ZSM-5]. Whereas, it is fitted with almost one component in the case of the treated catalyst [Ti–H/ZSM-5] 2. This means that titanium was present in three oxidation states (Ti4+, Ti3+, and Ti2+) in the fresh catalyst and then it is transformed to one oxidation state after the treatment of this catalyst under H2 at 550 °C. As mentioned above, the Ti 2p3/2 core level of the fresh catalyst is fitted using three components. The first component centered at 458.9 eV is associated with Ti ions with a formal valence 4 + (Ti4+) while the remaining components exist in a very small amount at a lower binding energy of ∼456.6 eV and ∼455.5 eV which are associated with Ti ions with a reduced charge state (Ti3+) and (Ti2+), respectively.43
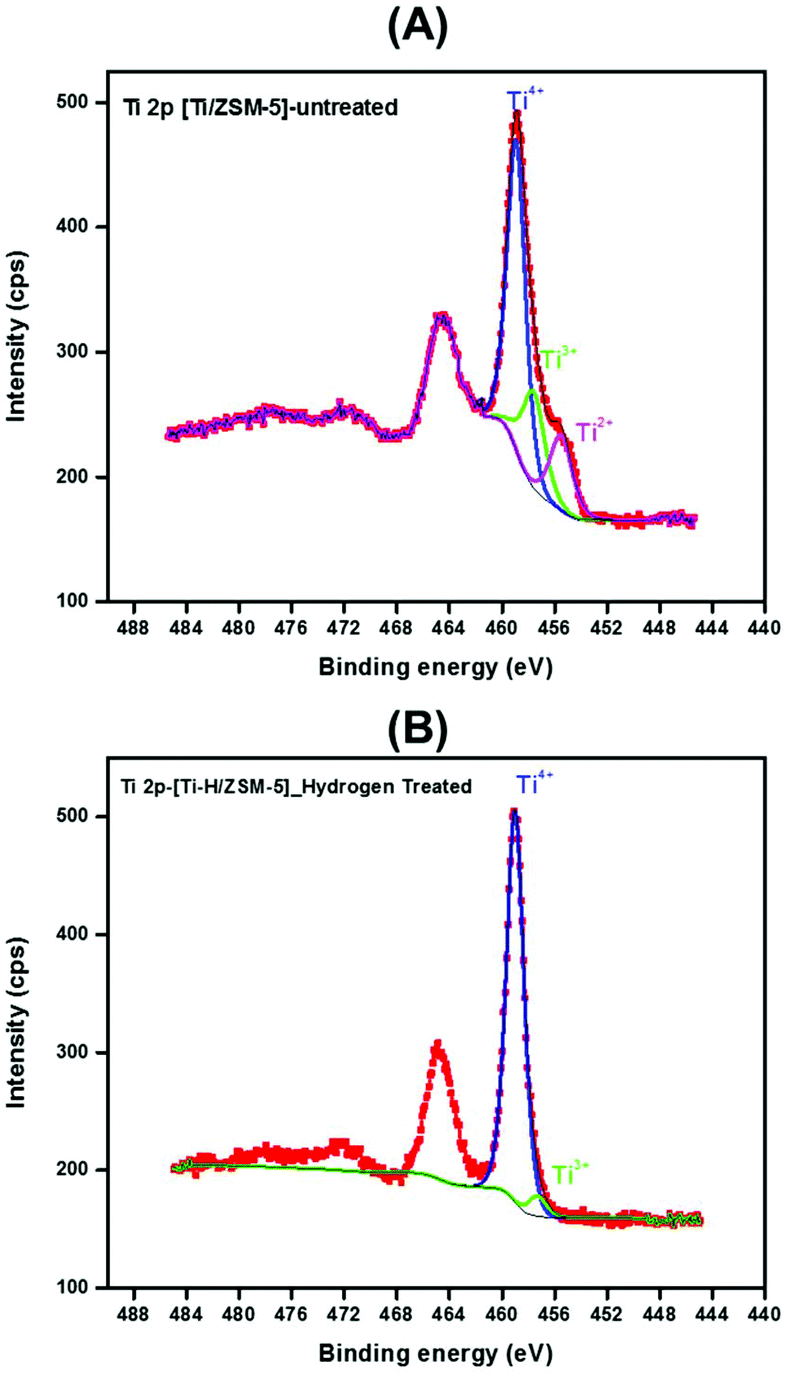 | ||
| Fig. 2 XPS spectra of fresh and treated catalysts: (A) Ti 2p XPS spectra of fresh catalyst [Ti/ZSM-5] 1, (B) Ti 2p XPS spectra of [Ti–H/ZSM-5] 2. | ||
Furthermore, the Si/Ti ratio determined from XPS spectra was changed from 6.5 to 8.6 determined for the fresh [Ti/ZSM-5] 1 and [Ti–H/ZSM-5] 2, respectively. This minor change in the Si/Ti ratio implies that there is no titanium diffusion from the Zeolite surface to its bulk.44,45 The survey spectra for the fresh and treated catalysts are shown in Fig. 4-ESI A, and B.† Titanium, Si, Al, O, and C elements are detected.
The microscopic observations of [Ti–H/ZSM-5] 2 after treatment under H2 at 550 °C for 2 h were performed using scanning transmission electron microscopy (HAADF-STEM) and are shown in Fig. 3(A). The [Ti–H/ZSM-5] catalyst 2 is made of large zeolite crystals (100–400 nm). There is no apparent formation of Ti oxide-based nanoparticles lying on the surface of the [ZSM-5300] crystals. This observation was strongly supported by the Raman spectroscopy analysis for the fresh and hydrogen treated catalysts. Furthermore, no features attributed to TiO2 were observed at 145, 440 or 805 cm−1 suggesting the absence of oligomeric Ti species (Fig. 7-ESI†).46,47 Subsequently, the location of Ti metals could be revealed by elemental mapping carried out by STEM-EDS.
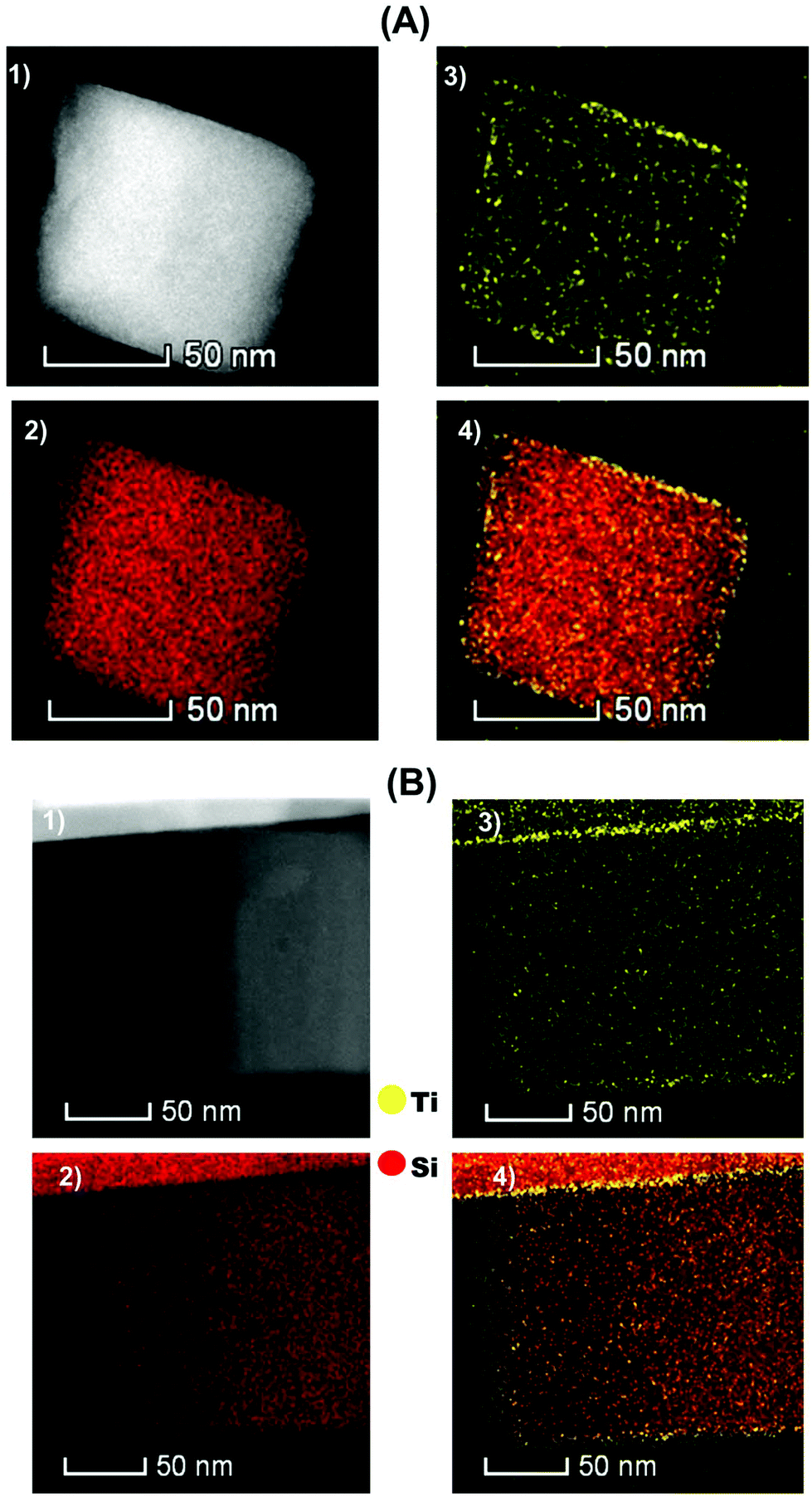 | ||
| Fig. 3 (A): (1) HAADF-STEM image for [Ti–H/ZSM-5] after treatment at 550 °C for 2 h under H2. EDX elemental mapping images of [Ti–H/ZSM-5]; (2) Si red, (3) Ti (yellow) and (4) Ti are displayed on the surface of Si. (B): (1) HAADF-STEM image for [Ti–H/ZSM-5] after the propane conversion reaction at 550 °C for 5 h. EDX elemental mapping images of [Ti–H/ZSM-5]; (2) Si (red), (3) Ti (yellow) and (4) Ti is displayed on the surface of Si. | ||
Integration of the ensemble of EDS spectra acquired for representative Ti mapping is shown in Fig. 5-ESI.† The latter was used to compute the Ti loading: 0.7 ± 0.1 wt% for Ti which is in close agreement with elemental analysis performed by ICP (0.8 ± 0.1). Titanium is uniformly distributed mostly on the external surface of [H-ZSM-5300] crystals (Fig. 3(A); Ti: Yellow, Si: Red) as it was shown previously by XPS spectroscopy. The bonding of Ti–H, after the treatment of the fresh catalyst under H2 at 550 °C to the surface of [H-ZSM-5300] seems to be very strong. This would explain why the Brønsted acidity inherent to the zeolite is not affected by Ti grafting.
The catalytic tests realized on the successive reactions of dehydrogenation, oligomerization and aromatization were carried out in a continuous flow reactor (PID® unit) at 550 °C with a space velocity = 16.08 h−1, and a total flow rate (100% C3H8) of 20 mL min−1. All reactions were performed at atmospheric pressure (P = 1 bar). During the reaction, various products have been obtained including methane, ethane, ethylene, propylene, n-butane, iso-butane, toluene, benzene and xylenes (para, ortho, meta-xylene). The recorded conversion of propane, the selectivity and the yield of aromatics, and the selectivity of propylene as a first intermediate produced during the reaction are presented in Fig. 4.
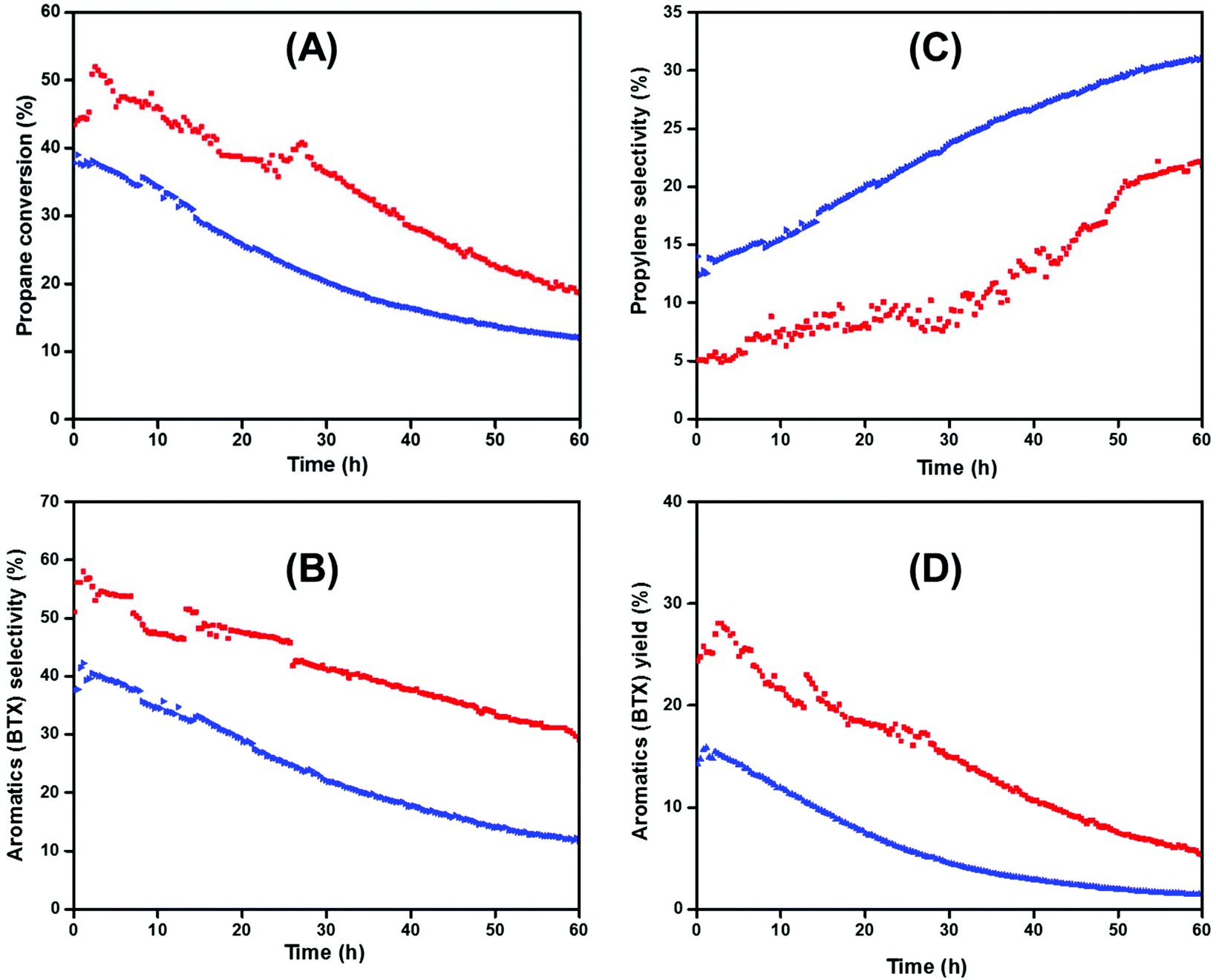 | ||
| Fig. 4 (A) Conversion of propane catalyzed over [Ti–H/ZSM-5] (red), and [H-ZSM-5300] (blue). (B) Propylene selectivity obtained over [Ti–H/ZSM-5] (red), and [H-ZSM-5300] (blue). (C) Aromatics (BTX) selectivity obtained over [Ti–H/ZSM-5] (red), and [H-ZSM-5300] (blue). (D) Aromatics (BTX) Yield (%) obtained over [Ti–H/ZSM-5] (red), and [H-ZSM-5300] (blue). Reaction conditions: m (catalyst) = 150 mg, P = 1 bar, T = 550 °C, total flow rate (100% C3H8) = 20 mL min−1. The error of measurements for the GC is around ±2%. | ||
The pristine [H-ZSM-5300] gave the lowest initial conversion of propane (ca. 37%), compared to catalyst 2. This propane conversion slowly decreased with time on stream to reach 10% after 60 h (Fig. 4A). However, the addition of Ti on the [H-ZSM-5300] improved the initial conversion of propane significantly. Under the same conditions, high conversion of propane (ca. 52%) was recorded over [Ti–H/ZSM-5] 2 especially at the beginning of the reaction. As time on stream goes on, this conversion decreased slightly over time, and the conversion of propane remained significant even after 60 h time on stream (25%) compared to [H-ZSM-5300] (ca.13%) (Fig. 4A). It should be noted that the deactivation is very similar for the catalysts, in agreement with the fact that Ti is outside the pores and does not modify the internal Brønsted acidity responsible for coke formation.56
As observed in Fig. 4B, the selectivity for aromatics (BTX) increased significantly in the presence of 2 compared to [H-ZSM-5300]. The [Ti–H/ZSM-5] catalyst showed a high selectivity for aromatics (60%) associated with good stability maintaining at 30% after 60 hours in time on stream. Otherwise, it is worth highlighting that 2 ([Ti–H/ZSM-5]) exhibited an induction period (2 h) to reach the highest activity toward the aromatic selectivity. This phenomenon was less observed with the pristine [H-ZSM-5300] (the induction period = 30 min).
In the presence of pristine [H-ZSM-5300] and [Ti–H/ZSM-5], the first chemical intermediate produced initially during the time on stream was the propylene which was obtained with a low initial selectivity (ca. = 5 and 12%, respectively). This selectivity increased slightly and reached 19 and 30%, respectively, with time on stream (Fig. 4C).48,49 This fact is maybe correlated to the catalytic deactivation of catalysts by blocking of active sites (with the formation of coke inside and outside of the zeolite pores).
Consequently, the 2 reported the highest yield of aromatics at the initial stage (30%). Furthermore, this yield dropped slightly to 10% after 60 h of time on stream without any regeneration. In the case of pristine [H-ZSM-5300], the yield for aromatics dropped drastically from 15 at the beginning of the reaction to less than 3% after 60 h of time on stream (Fig. 4D).
We should note that, in terms of propane conversion and selectivity to aromatics (BTX), the catalyst 2 yields better catalytic performances with time on stream compared to the hierarchical [Ga/HZSM-5] catalysts prepared by ionic exchange or by physical mixture (ca. 40% selectivity for a duration of 1 h versus 50% selectivity for 60 h in our case).50 Moreover, the catalyst 2 is much active on propane conversion using harsh reaction conditions (100% propane) compared to the other catalysts (Ga, Zn, Mo, and Re impregnated [ZSM-5]) reported by Haijun Wan et al. (1 wt% of metals load on ZSM-5) where 1 g of catalyst having 1 wt% of loading was used instead of 0.15 g and 0.8 wt% of loading in our case.51
In parallel, we reported in Fig. 8-ESI† the selectivity of other chemical products produced (methane, ethane, ethylene, n-butane iso-butane, and butane) during the catalytic reaction of propane aromatization. The selectivity for methane, ethane, and ethylene, formed by the protolytic cracking reaction occurred via a pentacoordinate carbonium ion,52 was more pronounced by using the pristine [H-ZSM-5300] as a catalyst (55 to 60%) compared to 2.53 Note that for pristine [H-ZSM-5300] and 2, the ethane and ethylene selectivity is equal to methane selectivity, which is a logical result corresponding to the cracking of propane to C1 and C2, with various degrees of dehydrogenations with time on stream (Fig. 8-ESI (A and B)†).30,54
The catalysts were subjected, after the reaction, to various analyses: N2-physisorption (BET), thermal gravimetric analysis (TGA) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM).55–57 The obtained results are shown in Table 3. After 60 h of the reaction, H-ZSM-5300 showed the smallest amount of coke. 2 exhibited a value of 7 wt% coke (Fig. 9-ESI†). A similar tendency was observed after 5 hours of the reaction (Fig. 10-ESI†). The N2-physisorption analysis (BET) of the spent catalysts confirmed the decreases of the specific surface area (350 m2 g−1, and 296 m2 g−1, respectively, for [H-ZSM-5300] + coke and [Ti–H/ZSM-5] + coke.
| Spent catalysts | Cokea (wt%) | Cokeb (wt%) | Surface areac (m2 g−1) |
|---|---|---|---|
| a TGA analysis for the spent catalysts collected after 60 h of conversion of propane for all catalysts. b TGA of the spent catalysts collected after 5 h of reaction over [Ti–H/ZSM-5]. c BET, according to Rouquerol criteria. Reaction conditions: m (catalyst) = 150 mg, P = 1 bar, T = 550 °C, total flow rate (100% C3H8) = 20 mL min−1. | |||
| [H-ZSM-5300] + coke | 2 | — | 350 |
| [Ti–H/ZSM-5] + coke | 7 | 2 | 296 |
The microscopic observations of 2 ([Ti–H/ZSM-5] after 5 h of the reaction at 550 °C without removing coke were performed using scanning transmission electron microscopy (HAADF-STEM) and are shown in Fig. 3(B) for [Ti/ZSM-5]. The obtained results showed that the distribution of titanium is mostly not changed after 5 h of reaction, and the titanium was uniformly distributed mainly on the surface of ZSM-5 crystals.
In Scheme 2, we summarize the mechanism of this reaction, assuming that Ti is playing a significant role in the dehydrogenation of propane to propylene by the well-known mechanism of Sigma bond metathesis and β-H elimination,27,30 followed by the oligomerization cyclization and aromatization by the carbocationic mechanism.58
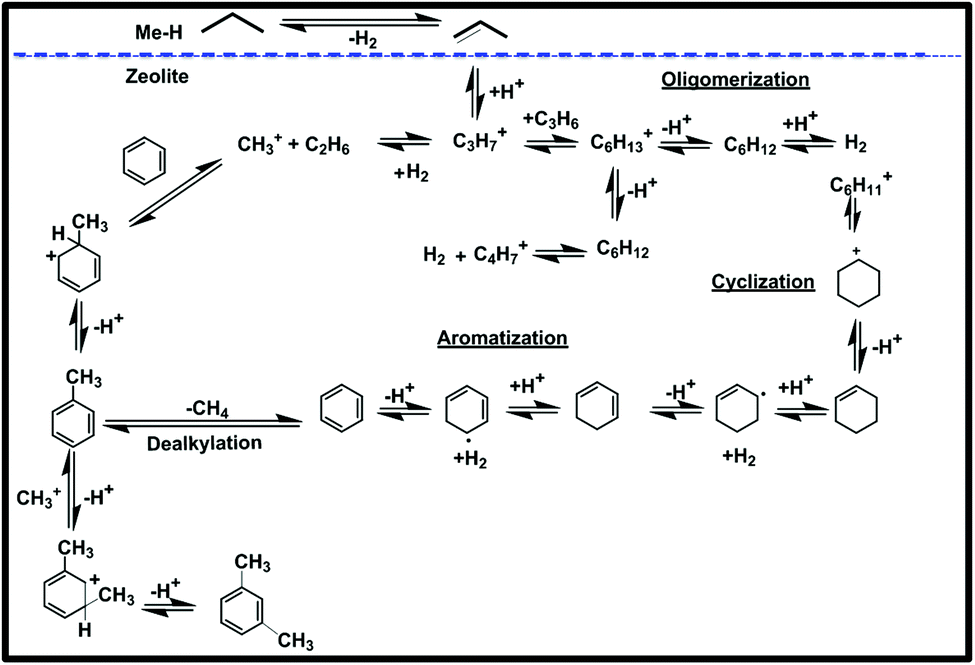 | ||
| Scheme 2 Mechanism of aromatization taking into account Norsic et al. and Guisnet et al.27,30,58 | ||
Conclusions
Our strategy of catalysis by design seems to be based on the catalyst 2 where Ti is selectively grafted at the periphery of [H-ZSM-5300] which gives good and relatively stable activities on the conversion of propane to aromatics (BTX) compared to the pristine [H-ZSM-5300]. All the analytical tools and catalytic tests indicate that the sequence of reactions, dehydrogenation, oligomerization, cyclisation, and aromatization can be partly controlled by assuming that at the periphery of [H-ZSM-5300], the Ti promotes a selective dehydrogenation of propane to propylene which was effectively observed. This was expected from our previous work on the role of Ti-hydride for C–H bond activation in the hydrogenolysis of alkanes. Interestingly, Ti remains mostly outside of the cavity at least after 5 hours of the reaction.24,27,30Experimental section
General procedures
All experiments were carried out under an inert atmosphere (Ar and N2), using Schlenk and glovebox techniques for organometallic syntheses. For the synthesis and treatment of supported species, reactions were carried out using high-vacuum lines (ca. 1 MPa) and gloveboxes. All solvents were distilled from Na-benzophenone and degassed using freeze–pump–thaw cycles. The TiNp4 organometallic complex was synthesized according to a published method.59,60Preparation of [H-ZSM-5300]
[H-ZSM-5] was obtained after the calcination of commercial [NH4-ZSM-5] purchased from Alpha-Aesar at 550 °C under air for 6 h. The dehydration of [H-ZSM-5] at 300 °C under vacuum (∼5 × 10−5 atm) affords the [H-ZSM-5300]. The concentration of isolated silanols (Si–OH), used further for grafting of tetrakis(neopentyl)titanium (TiNp4), after dehydration at 300 °C of [H-ZSM-5300] was calculated by the quantity of MeLi reacting with Si–OH. 0.4 ± 0.1 mmol of Si–OH per gram of [H-ZSM-5300] are found.
Synthesis of [Ti/ZSM-5] (catalyst 1)
A mixture of (150 mg, 0.80 mmol) TiNp4 and [H-ZSM-5300] (1 g) in pentane (15 mL) was stirred at room temperature for 8 h. After filtration, the solid was washed 3 times with pentane. The resulting powder was dried under vacuum (10−5 bar) at 80 °C. Elemental analysis: Ti = 0.8 wt%, C = 3.9 wt%.The catalyst 2 [Ti–H/ZSM-5] was prepared after heat treatment under H2 of catalyst 1 freshly prepared at 550 °C for 2 h. Elemental analysis: Ti = 0.87 wt%, C ∼0.2 wt%.
Characterization of catalysts
IR spectroscopy: IR spectra of solid samples were recorded with a Nicolet 6700 FT-IR spectrometer by using a Diffuse Reflectance Infrared Fourier Transformation (DRIFT) cell equipped with CaF2 windows. The samples were prepared in an argon-filled glove-box. Typically, 64 scans were accumulated for each spectrum (resolution 4 cm−1).Raman spectroscopy was performed on a Horiba Yvon LabRAM Aramis with a CCD-camera as a detector using a 50× objective, an 1800 gr mm−1 grating, a 100 μm slit and a 473 nm cobalt laser. The Raman spectra were collected on the samples sealed under an Ar atmosphere, which were packed in a closed cell fitted with a rubber O ring and a Quartz window.
Fourier transform infrared (FTIR) spectroscopy of the adsorbed pyridine was carried out using a Bruker IFS 66 spectrometer. Self-supporting zeolite wafers were first degassed at 10−3 mbar and 673 K for 4 h. After saturation with pyridine, weakly bound molecules were evacuated at room temperature for 15 min and subsequently at 473 K for 2 h. The concentrations of Brønsted and Lewis acid sites were determined using the reported extinction coefficients of 1.67 and 2.94 cm μmol−1, respectively.39
Elemental analyses for Ti were performed at the Core Laboratory Facilities (KAUST) using inductively coupled plasma atomic emission spectroscopy (ICP–AES) on a Thermo-Electron 3580 instrument. XRD patterns were collected using a Bruker D8 Advanced diffractometer in Bragg–Brentano geometry fitted with a copper tube operating at 40 kV and 40 mA and a linear position sensitive detector (opening 2.9°). The diffractometer was configured with a 0.36° diverging slit, 2.9° anti-scattering slit, 2.5° Soller slits, and a Ni filter. The datasets were acquired in a continuous scanning mode (0.008° s−1) over the 2θ range of 15–120°, using a step interval of 0.04° and a counting time of 5 s per step. The gas-phase analysis of alkanes was performed using an Agilent 6850 gas chromatography column with a split injector coupled with a flame ionization detector. An HP-PLOT Al2O3 KCl capillary column (30 m × 0.53 mm; 20.00 mm) coated with a stationary phase of aluminum oxide deactivated with KCl was used with helium as the carrier gas at 32.1 kPa. The coke content in the samples after the catalytic tests was quantified from the weight loss observed between 200 and 800 °C during thermogravimetric analysis (TGA) in air using a Mettler Toledo TGA/DSC 1 system.
One-dimensional 1H MAS and 13C CP-MAS solid-state NMR
Spectra were recorded on a Bruker AVANCE III spectrometer operating at 400 and 100 MHz resonance frequencies for 1H and 13C, respectively, with a conventional double resonance 4 mm CPMAS probe. The samples were introduced under argon into zirconia rotors, which were then tightly closed. The spinning frequency was set to 14 and 10 kHz for 1H, 13C spectra, respectively. NMR chemical shifts are reported concerning TMS as an external reference.Electron microscopy and elemental mapping
Transmission electron microscopy (TEM) of the samples was performed with a Titan Themis-Z microscope from Thermo-Fisher Scientific by operating it at an accelerating voltage of 300 kV. Prior to the analysis, the microscope was set to the scanning TEM (STEM) mode to acquire atomic number (Z) sensitive STEM images with an attached a high-angle annular dark-field (HAADF) detector. Furthermore, a high throughput X-ray energy dispersive spectrometer (EDS) was also utilized in conjunction with DF-STEM imaging to acquire STEM-EDS spectrum-imaging datasets. During the acquisition of these datasets, at every image-pixel, a corresponding EDS spectrum was also obtained for generating the elemental maps of Si, Al, O, and Ti, simultaneously. It is also pertinent to note herein that Spectrum-imaging datasets were acquired in the so-called frame mode in which the electron beam was allowed to dwell at each pixel for the only time of few microseconds in order to keep a total frame time to merely one second or less. However, each spectrum-imaging dataset was collected until more than 200 frames were completed. This mode of operation allowed us to have a high signal to noise ratio in the acquired STEM-EDS spectrum-imaging datasets while causing a little or no damage to beam-sensitive zeolite samples by the electron beam. Both imaging and spectroscopy datasets for each sample were both acquired as well as analysed with a newly developed software package called Velox from Thermo Fisher Scientific.XPS spectroscopy
XPS studies were carried out in a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) operating at 150 W, a multi-channel plate and delay line detector under a vacuum of ∼10−9 mbar. All spectra were recorded using an aperture slot of 300 μm × 700 μm. Survey spectra were collected using a pass energy of 160 eV and a step size of 1 eV. A pass energy of 20 eV and a step size of 0.1 eV were used for the high-resolution spectra; for XPS analysis, samples were mounted in a floating mode in order to avoid differential charging. Charge neutralization was required for all samples. Binding energies were referenced to the sp3 hybridized (C–C) carbon for the C 1s peak set at 284.8 eV. The sample was mounted on the holder in a glovebox under controlled environment (argon) and then transferred to the XPS instrument using a transfer vessel for air-sensitive samples.Catalytic test
The catalytic performance of the catalysts toward propane conversion into aromatics (BTX) was studied in a stainless steel continuous flow reactor unit (PID® unit) = 1 bar, T = 550 °C, total flow rate = 20 mL min−1 (100 vol% propane), with (2 mL) of N2 as standard and catalyst = (150 mg). The gases were purified by passing through a filter (Cu2O/Al2O3), with flow rates controlled by Brooks mass flow controllers. Catalysts were loaded into the reactor inside the glovebox. The lines were purged before starting the reaction. The produced gases were analyzed by Gas Chromatography in a Varian CG-450 with three channels: 1 TCD and 2 FID as described below:• 1 TCD connected to Molsieve 13× column for N2 detection.
• 1 FID connected to Alumina column for alkanes and olefins from C1 to C4.
• 1 FID connected to Rxi-624 Sil MS column (10 m × 0.53 mm × 3 μm) for aromatics.
The conversion of propane was calculated concerning the carbon numbers from FID.
Conflicts of interest
No conflicts of interest on this workAcknowledgements
This work received support from the King Abdullah University of Science and Technology (KAUST) in collaboration with SABIC Corporate Research and Development. The authors acknowledge the KAUST Nuclear Magnetic Resonance Core Lab and the analytical core lab (ACL). The authors acknowledge Dr. Sandeep Mishra as well for the Raman spectroscopy analysis.Notes and references
- H. Xiao, J. Zhang, P. Wang, Z. Zhang, Q. Zhang, H. Xie, G. Yang, Y. Han and Y. Tan, RSC Adv., 2015, 5, 92222–92233 RSC.
- M. Corbetta, F. Manenti, C. Pirola, M. V. Tsodikov and A. V. Chistyakov, Comput. Chem. Eng., 2014, 71, 457–466 CrossRef CAS.
- H. A. Zaidi and K. K. Pant, Catal. Today, 2004, 96, 155–160 CrossRef CAS.
- A. A. Rownaghi and J. Hedlund, Ind. Eng. Chem. Res., 2011, 50, 11872–11878 CrossRef CAS.
- I. Vollmer, I. Yarulina, F. Kapteijn and J. Gascon, ChemCatChem, 2019, 11, 39–52 CrossRef CAS.
- W. Wannapakdee, C. Wattanakit, V. Paluka, T. Yutthalekha and J. Limtrakul, RSC Adv., 2016, 6, 2875–2881 RSC.
- W. J. H. Dehertog and G. F. Fromen, Appl. Catal., A, 1999, 189, 63–75 CrossRef CAS.
- R.-l. Liu, H.-q. Zhu, Z.-w. Wu, Z.-f. Qin, W.-b. Fan and J.-g. Wang, J. Fuel Chem. Technol., 2015, 43, 961–969 CrossRef CAS.
- H. Xiao, J. Zhang, X. Wang, Q. Zhang, H. Xie, Y. Han and Y. Tan, Catal. Sci. Technol., 2015, 5, 4081–4090 RSC.
- T. C. Hoff, R. Thilakaratne, D. W. Gardner, R. C. Brown and J.-P. Tessonnier, J. Phys. Chem. C, 2016, 120, 20103–20113 CrossRef CAS.
- I. Petrovic, A. Navrotsky, M. E. Davis and S. I. Zones, Chem. Mater., 1993, 5, 1805–1813 CrossRef CAS.
- K. L. Yeung and W. Han, Zeolites and Catalysis: Synthesis, Reactions and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, vol. 2, pp. 827–861 Search PubMed.
- P. Schulz and M. Baerns, Appl. Catal., 1991, 78, 15–29 CrossRef CAS.
- A. Bhan and W. Nicholas Delgass, Catal. Rev., 2008, 50, 19–151 CrossRef CAS.
- S. Yuan, S. B. Derouane-Abd Hamid, Y. Li, P. Ying, Q. Xin, E. G. Derouane and C. Li, J. Mol. Catal. A: Chem., 2002, 184, 257–266 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71–83 CrossRef CAS.
- G. Giannetto, R. Monque and R. Galiasso, Catal. Rev. – Sci. Eng., 1994, 36, 271–304 CrossRef CAS.
- E. A. Pidko, V. B. Kazansky, E. J. M. Hensen and R. A. Van Santen, J. Catal., 2006, 240, 73–84 CrossRef CAS.
- E. J. M. Hensen, E. A. Pidko, N. Rane and R. A. van Santen, Stud. Surf. Sci. Catal., 2007, 170, 1182–1189 CrossRef.
- V. B. Kazansky, I. R. Subbotina, R. A. van Santen and E. J. M. Hensen, J. Catal., 2005, 233, 351–358 CrossRef CAS.
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS.
- J. C. Mohandas, E. Abou-Hamad, E. Callens, M. K. Samantaray, D. Gajan, A. Gurinov, T. Ma, S. Ould-Chikh, A. S. Hoffman, B. C. Gates and J.-M. Basset, Chem. Sci., 2017, 8, 5650–5661 RSC.
- N. Maity, S. Barman, E. Callens, M. K. Samantaray, E. Abou-Hamad, Y. Minenkov, V. D'Elia, A. S. Hoffman, C. M. Widdifield, L. Cavallo, B. C. Gates and J.-M. Basset, Chem. Sci., 2016, 7, 1558–1568 RSC.
- C. Rosier, G. P. Niccolai and J.-M. Basset, J. Am. Chem. Soc., 1997, 119, 12408–12409 CrossRef CAS.
- P. L. Watson and D. C. Roe, J. Am. Chem. Soc., 1982, 104, 6471–6473 CrossRef CAS.
- M. K. Samantaray, S. Kavitake, N. Morlanes, E. Abou-Hamad, A. Hamieh, R. Dey and J.-M. Basset, J. Am. Chem. Soc., 2017, 139, 3522–3527 CrossRef CAS.
- S. Norsic, C. Larabi, M. Delgado, A. Garron, A. de Mallmann, C. Santini, K. C. Szeto, J.-M. Basset and M. Taoufik, Catal. Sci. Technol., 2012, 2, 215–219 RSC.
- J. Corker, F. Lefebvre, C. Lécuyer, V. Dufaud, F. Quignard, A. Choplin, J. Evans and J.-M. Basset, Science, 1996, 271, 966–969 CrossRef CAS.
- M. Sato, T. Kanbayashi, N. Kobayashi and Y. Shima, J. Catal., 1967, 7, 342–351 CrossRef CAS.
- C. Larabi, N. Merle, S. Norsic, M. Taoufik, A. Baudouin, C. Lucas, J. Thivolle-Cazat, A. de Mallmann and J.-M. Basset, Organometallics, 2009, 28, 5647–5655 CrossRef CAS.
- K. A. Tarach, K. Gora-Marek, J. Martinez-Triguero and I. Melian-Cabrera, Catal. Sci. Technol., 2017, 7, 858–873 RSC.
- R. Fricke, H. Kosslick, G. Lischke and M. Richter, Chem. Rev., 2000, 100, 2303–2405 CrossRef CAS.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Petrini, G. Leofanti, M. Padovan and C. Otero Arean, J. Chem. Soc., Faraday Trans., 1992, 88, 2959–2969 RSC.
- E. Loeffler, U. Lohse, C. Peuker, G. Oehlmann, L. M. Kustov, V. L. Zholobenko and V. B. Kazansky, Zeolites, 1990, 10, 266–271 CrossRef CAS.
- S. E. Ashbrook and M. E. Smith, Chem. Soc. Rev., 2006, 35, 718–735 RSC.
- M. Hunger, Catal. Rev. Sci. Eng., 1997, 39, 345–393 CrossRef CAS.
- V. d. O. Rodrigues, J.-G. Eon and A. C. Faro, J. Phys. Chem. C, 2010, 114, 4557–4567 CrossRef CAS.
- S. Mitchell, M. Boltz, J. Liu and J. Perez-Ramirez, Catal. Sci. Technol., 2017, 7, 64–74 RSC.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- T. Barzetti, E. Selli, D. Moscotti and L. Forni, J. Chem. Soc., Faraday Trans., 1996, 92, 1401–1407 RSC.
- H. Xiao, J. Zhang, P. Wang, Z. Zhang, Q. Zhang, H. Xie, G. Yang, Y. Han and Y. Tan, RSC Adv., 2015, 5, 92222–92233 RSC.
- V. B. Kazansky, I. R. Subbotina, R. A. van Santen and E. J. M. Hensen, J. Catal., 2004, 227, 263–269 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- F. Langerame, A. M. Salvi, M. Silletti and G. Moretti, Surf. Interface Anal., 2008, 40, 695–699 CrossRef CAS.
- A. C. Alba-Rubio, J. L. G. Fierro, L. Leon-Reina, R. Mariscal, J. A. Dumesic and M. Lopez Granados, Appl. Catal., B, 2017, 202, 269–280 CrossRef CAS.
- E. L. Lee and I. E. Wachs, J. Phys. Chem. C, 2008, 112, 6487–6498 CrossRef CAS.
- C. Jin, B. Liu, Z. Lei and J. Sun, Nanoscale Res. Lett., 2015, 10, 95 CrossRef PubMed.
- T. Blasco, Chem. Soc. Rev., 2010, 39, 4685–4702 RSC.
- A. A. Gabrienko, S. S. Arzumanov, D. Freude and A. G. Stepanov, J. Phys. Chem. C, 2010, 114, 12681–12688 CrossRef CAS.
- M. Raad, S. Hamieh, J. Toufaily, T. Hamieh and L. Pinard, J. Catal., 2018, 366, 223–236 CrossRef CAS.
- H. Wan and P. Chitta, J. Anal. Appl. Pyrolysis, 2016, 121, 369–375 CrossRef CAS.
- Y. V. Kissin, Catal. Rev., 2001, 43, 85–146 CrossRef CAS.
- J. A. Lercher, R. A. van Santen and H. Vinek, Catal. Lett., 1994, 27, 91–96 CrossRef CAS.
- M. K. Samantaray, R. Dey, S. Kavitake, E. Abou-Hamad, A. Bendjeriou-Sedjerari, A. Hamieh and J.-M. Basset, J. Am. Chem. Soc., 2016, 138, 8595–8602 CrossRef CAS PubMed.
- C.-T. Shao, W.-Z. Lang, X. Yan and Y.-J. Guo, RSC Adv., 2017, 7, 4710–4723 RSC.
- B. K. Vu, M. B. Song, I. Y. Ahn, Y.-W. Suh, D. J. Suh, J. S. Kim and E. W. Shin, J. Ind. Eng. Chem., 2011, 17, 71–76 CrossRef CAS.
- Y. Shan, Z. Sui, Y. Zhu, D. Chen and X. Zhou, Chem. Eng. J., 2015, 278, 240–248 CrossRef CAS.
- M. Guisnet and N. S. Gnep, Catal. Today, 1996, 31, 275–292 CrossRef CAS.
- R. A. Kovar, H. Derr, D. Brandau and J. O. Callaway, Inorg. Chem., 1975, 14, 2809–2814 CrossRef CAS.
- J. Cheon, L. H. Dubois and G. S. Girolami, J. Am. Chem. Soc., 1997, 119, 6814–6820 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt00905a |
| This journal is © The Royal Society of Chemistry 2019 |