
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of a gold(I)/platinum(0) frustrated Lewis pair with germanium and tin dihalides†
Nereida
Hidalgo
 a,
Sonia
Bajo
a,
Juan José
Moreno
a,
Carlos
Navarro-Gilabert
a,
Brandon Q.
Mercado
b and
Jesús
Campos
*a
a,
Sonia
Bajo
a,
Juan José
Moreno
a,
Carlos
Navarro-Gilabert
a,
Brandon Q.
Mercado
b and
Jesús
Campos
*a
aInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA). Universidad de Sevilla and Consejo Superior de Investigaciones Científicas (CSIC), Avenida Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: jesus.campos@iiq.csic.es
bDepartment of Chemistry, Yale University, 225 Prospect St, New Haven, CT 06511, USA
First published on 24th May 2019
Abstract
The reactivity of germanium and tin dichlorides with a transition metal-only frustrated Lewis pair based on Au(I) and Pt(0) compounds bearing bulky phosphine ligands is described in this work. We have examined both the reactivity of tetrylene dihalides towards the individual components of the metallic pair, as well as under metal/metal cooperative conditions. These studies allowed us to isolate several uncommon homo- and heterometallic structures. Computational methods have been employed to investigate the bonding scheme of one of these highly-reduced metallic aggregates. In addition, we have developed a tin-promoted strategy to access heteroleptic diphosphine platinum(0) compounds.
Introduction
The use of molecular donor–acceptor pairs has served as a fruitful tool to stabilize or intercept reactive inorganic species with ambiphilic character.1 The strategy has been particularly successful in the study of heavier tetrylenes, :EX2 (E = Si, Ge, Sn, Pb), compounds based on a divalent heavier group 14 element, which possess relatively reduced HOMO–LUMO gaps and dual nucleophilic (lone electron pair) and electrophilic (empty p orbital) nature. The cooperative stabilization conferred by a donor and an acceptor that mutually bind an ambiphilic molecule is understood in terms of the electronic push–pull bonding scheme that emerges. Representative examples of otherwise highly unstable tetrylene fragments include E(CH3)2,2 EH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 or SiCl2,4 which have been characterized by this approach, providing fundamental understanding of their bonding and reactivity. Stabilizing heavier tetrylenes by intra- or intermolecular donors has also been exploited in their use as more robust ligands in coordination chemistry.5
3 or SiCl2,4 which have been characterized by this approach, providing fundamental understanding of their bonding and reactivity. Stabilizing heavier tetrylenes by intra- or intermolecular donors has also been exploited in their use as more robust ligands in coordination chemistry.5
From a related perspective, this electronic push–pull stabilization highly resembles the chemistry of frustrated Lewis pairs (FLPs). These systems have been widely employed to capture an ample range of small molecules by the synergistic combination of an acid and a base for which adduct formation has been quenched.6 However, the presence of heavier group 14 elements within the field of FLPs mostly focuses on their use as acidic partners,7 while reactivity studies of traditional FLP systems towards the tetrel series finds little precedent.8 We recently entered the FLP arena by describing the first transition metal-only FLP (TMOFLP) in which the two constituents were based on transition metals, more precisely Au(I) and Pt(0) as the acidic and basic counterparts respectively (Fig. 1).9 Somehow related metal-only donor–acceptor pairs (Rh/W and Pt/W) have been recently employed by Rivard to stabilize low-valent group 14 species.10 Encouraged by these results we decided to explore the reactivity of our Au(I)/Pt(0) FLP towards simple forms of low-valent group 14 compounds, particularly GeCl2 and SnCl2. It is pertinent to note that after push–pull stabilization, germanium and tin dihalides could serve as suitable precursors towards their corresponding dialkyl2 or dihydride3 derivatives, which in turn can be the source of functional nanomaterials.11 We will firstly present the reactivity of germanium and tin dihalides with the gold and platinum single components of the FLP. The discussion will then be continued by describing their combined reactivity. In addition, the present studies reveal the key role of tin dichloride in promoting phosphine exchange reactions for the platinum component of the metallic FLP.
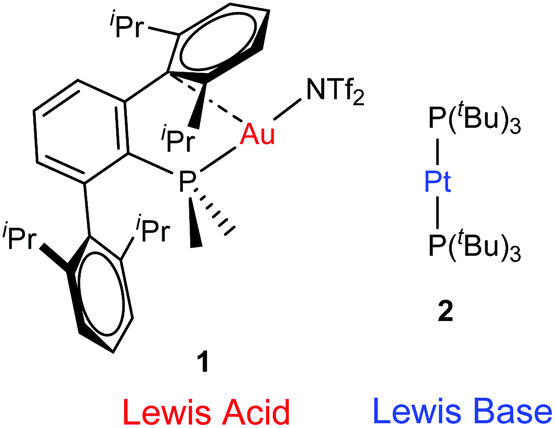 | ||
| Fig. 1 Transition metal-only frustrated Lewis pair (TMOFLP) studied in this work, where the weakly coordinating triflimide anion ([N(SO2CF3)2]−) is represented by NTf2−. | ||
Results and discussion
Reactivity of GeCl2 and SnCl2 towards gold compound 1
We began our studies by exploring the reactivity of germanium and tin dihalides towards gold compound (PMe2ArDipp2)Au(NTf2), 1. In both cases reactions proceed readily to yield compounds 3 and 4 after the respective insertion of GeCl2 or SnCl2 into the Au-N(SO2CF3)2 bond of 1 (Scheme 1) in quantitative spectroscopic yield. These species were isolated as white powders and their purity confirmed by microanalysis. While 3 features a broad 31P{1H} NMR resonance in CD2Cl2 at 4.8 ppm, shifted to higher frequency by about 16 ppm relative to 1 (δ = −11.5 ppm), the analogous broad signal due to 4 appears at −9.3 ppm. These resonances become sharp upon cooling the NMR probe to −40 °C suggesting fluxional behaviour for both compounds likely due to the lability of the triflimide anion. Similarly, all the resonances observed in their 1H NMR spectra become sharper when recorded at low temperatures and do not exhibit any relevant features that differ from those of precursor 1. Fluxional behaviour seems to be hampered in THF-d8 solution, where the 31P{1H} NMR resonances of the gold germyl and stannyl compounds shift to higher frequencies (3·THF, 6.7 ppm; 4·THF, −3.1 ppm) likely due to the displacement of the weakly coordinating triflimide anion by a solvent molecule (Scheme 1). As introduced above, the bonding scheme in these cationic complexes may be understood in terms of the push–pull interactions provided by the Au/THF pair to the :ECl2 moiety. However, while 4·THF remains stable in solution for at least one day, its germanium analogue is acidic enough to readily promote the electrophilic ring-opening polymerization of THF.12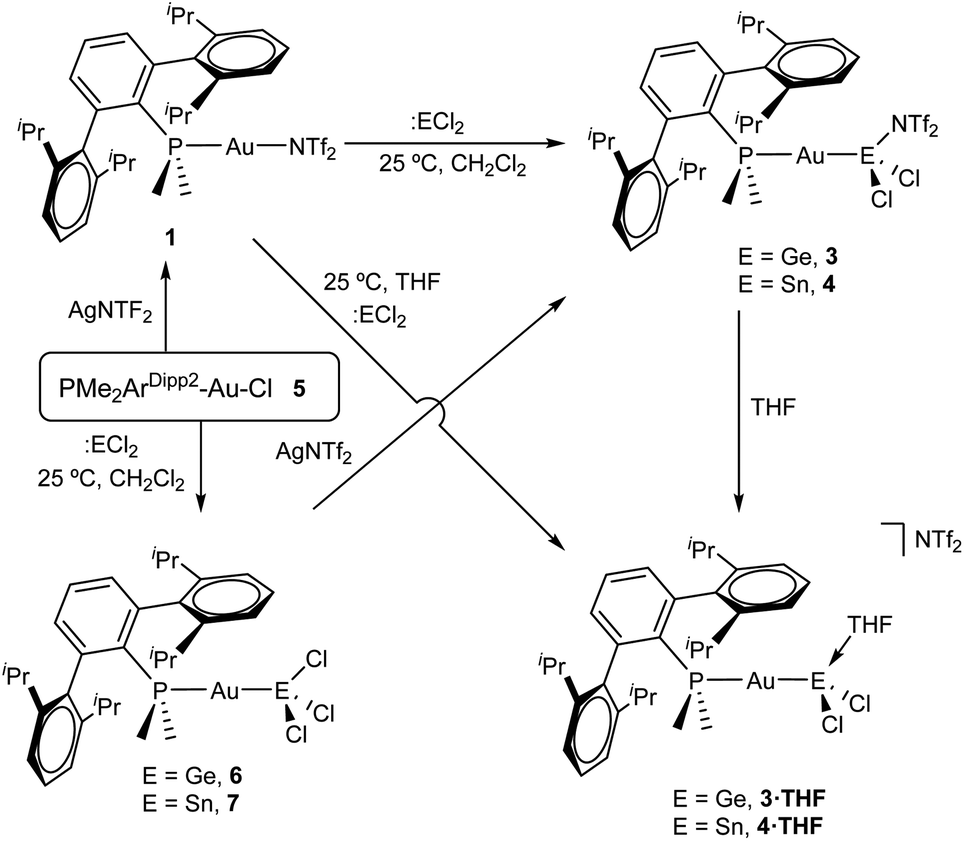 | ||
| Scheme 1 Reactivity of tetrylene dihalides with gold compounds bearing a terphenyl phosphine. | ||
Despite our efforts, we were unable to grow single crystals of enough quality to authenticate the proposed formulation for compounds 3 and 4. Nevertheless, the insertion of germylenes and stannylenes into gold-halide and other related bonds is well-documented. In fact, the same reactivity is observed when GeCl2·dioxane or SnCl2 are added to dichloromethane solutions of the gold chloride compound (PMe2ArDipp2)AuCl (5),13 precursor of 1via salt metathesis with AgNTf2 (Scheme 1). The resulting gold-tetryl species are characterized by 31P{1H} NMR resonances at 5.0 and −2.2 ppm due to the germyl (6) and stannyl (7) insertion products respectively, while their 1H NMR spectra match with those of their precursor 5, as well as with other gold derivatives previously described by some of us.13 Subsequent chloride abstraction by silver triflimide results in quantitative formation of compounds 3 and 4, respectively, as expected for the proposed molecular formulations collected in Scheme 1. The insertion of tetrylenes into gold-halide bonds has provided complexes with Au–E (E = Ge, Sn) bonds with a variety of geometries and coordination environments,14 as well as interesting photophysical properties.15 Most examples rely on the use of sterically unhindered phosphines that permit the formation of supramolecular aggregates by aurophilic and other non-covalent interactions. The former interactions have indeed been suggested as key for the reported photoluminiscent properties of these species. The solid-state structure of complex 6 is depicted in Fig. 2, revealing that no gold aggregates are formed. At variance with prior examples, gold–gold and gold-chloride contacts are replaced by a weak Au⋯Carene interaction with the ipso carbon of a lateral terphenyl ring (dAu–Cipso = 2.95(4) Å), a common feature for gold complexes of biaryl phosphines.13,16 This forces the coordination geometry around gold to bend from linearity (P–Au–Ge 171.30(4)°), while other distances and angles lie within normal values. The two flanking aryl rings of the terphenyl fragment are equivalent by NMR, while the 13C{1H} NMR resonance of the interacting ipso-carbon (138.1 ppm, 3JCP = 6 Hz) lies close to the analogous one in the free phosphine (142.5 ppm, 3JCP = 5 Hz). This data, along with the long Au⋯CArene distance, suggests that the secondary interaction is weak. For the sake of comparison, we aimed to examine the supramolecular structure of a compound analogous to 6 but constructed around the less hindered terphenyl phosphine PMe2ArXyl2 (where ArXyl2 = C6H3-2,6-(C6H3-2,6-Me2)2), in which the isopropyl groups of the lateral aryl rings were replaced by methyl groups. The related gold germyl compound was prepared in good yields (ca. 90%) by the same strategy followed to access its bulkier counterpart (see ESI† for details). Its solid-state structure was almost identical to 6 and exhibits a similar secondary Au–arene interaction characterized by a dAu–Cipso of 3.05(4) Å and a reduced P–Au–Ge angle of 165.77(2)° (Fig. S1†).
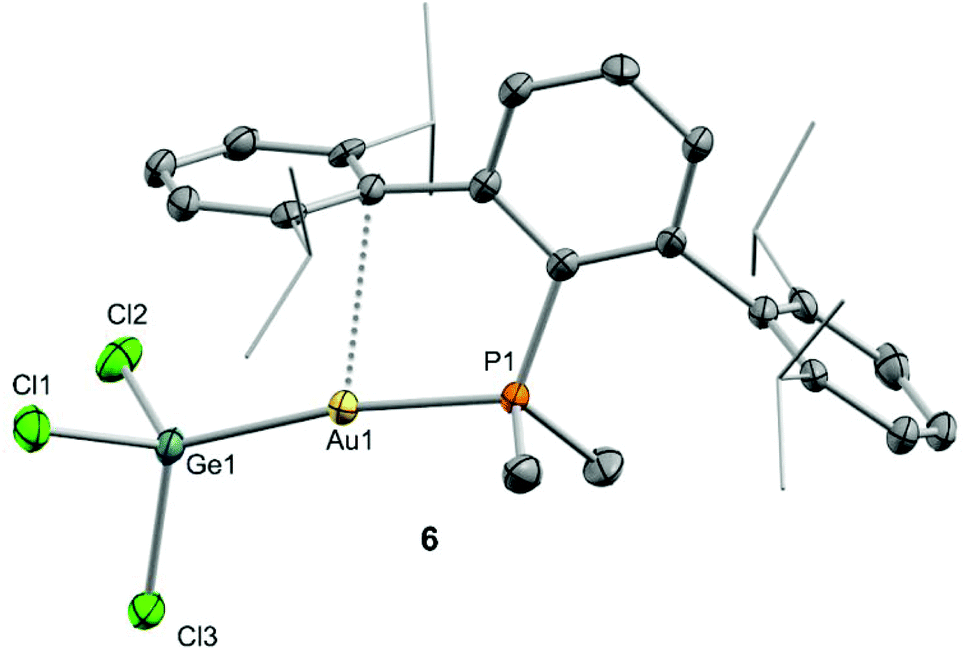 | ||
| Fig. 2 ORTEP diagram of compound 6; for the sake of clarity hydrogen atoms are excluded and some substituents have been represented in wireframe format, while thermal ellipsoids are set at 50% probability. | ||
Drawing on the same theme, we wondered if the steric properties of terphenyl phosphines would still permit the insertion of bulkier tetrylenes across the gold-chloride/triflimide bond.17 We chose stannylene Sn[N(SiMe3)]2 to carry out these experiments since its insertion into Au–Cl bonds was recently documented.17b Its addition to benzene solutions of 1 and 5 indeed resulted in almost quantitative formation of compounds 9 and 8, respectively (Scheme 2). Trace amounts of another gold complex were detected, as discussed below. The new Au–Sn heterobimetallic compounds are characterized by a higher-frequency shift of their 31P{1H} NMR resonances (8, 15.4 ppm; 9, 13.8 ppm) of around 23 ppm with respect to their precursors. The 31P{1H} NMR signal of compound 8 exhibits a strong two-bond coupling due to the trans tin centre (2JPSn = 3201 Hz). A new intense signal in the 1H NMR spectrum is collected at around 0.47 ppm due to the trimethylsilyl groups for both 8 and 9, while the rest of their 1H NMR spectra is comparable to other related samples described herein. Authenticating the proposed molecular structures proved challenging due to the poor quality of the crystals grown with the selected phosphine system. However, we succeeded in growing crystals with a related bulkier phosphine, namely PCyp2ArXyl2 (Cyp = C5H9). Thus, compound 8Cyp was isolated in moderate yield as a white crystalline material following the same synthetic procedure described to access 8 (see Experimental section). Its 31P{1H} NMR resonance displays a 2JPSn coupling constant of 2846 Hz, somewhat smaller relative to compound 8. This may result from the steric pressure exerted by the cyclopentyl substituents of the phosphorus centre onto the bulky bis(trimethylsilyl)amido fragments, which could weaken the metal–metal bond. In fact, its X-ray diffraction structure (Fig. 3) reveals a Au–Sn bond distance that accounts for 2.65(1) Å, relatively elongated compared to previous linear gold–tin complexes (ca. 2.57 Å).18
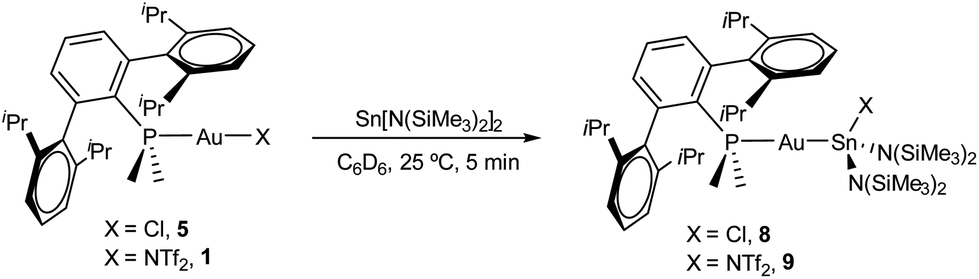 | ||
| Scheme 2 Reaction of stannylene Sn[N(SiMe3)]2 with gold precursors 1 and 5. | ||
 | ||
| Fig. 3 ORTEP diagram of compound 8Cyp; for the sake of clarity hydrogen atoms are excluded and some substituents have been represented in wireframe format, while thermal ellipsoids are set at 50% probability. | ||
In addition, we could isolate the main side product resulting from the reactions represented in Scheme 2 (<5% by 31P{1H} NMR spectroscopy), which consists of an amido-bridged cationic digold complex of formula [Au2(μ-N(SiMe3)2)(PMe2ArDipp2)2] (see ESI† for details) due to the transfer of a trimethylsilylamido substituent from stannylene to a gold centre. This compound could be independently synthesized by mixing equimolar amounts of gold-triflimide 1 and [PMe2ArDipp2]Au[N(SiMe3)2], prepared by reaction of gold-chloride 5 and Li[N(SiMe3)2]. The molecular formulation of the amido-bridged digold compound based on PMe2ArXyl2 phosphine was further confirmed by single-crystal X-ray diffraction studies (Fig. S2†) and represents an uncommon example of this motif in the context of gold chemistry.19
Reactivity of GeCl2 and SnCl2 towards platinum compound 2
The reaction of tetrel dihalides,20 as well as aluminum trichloride,21 with linear platinum(0) compounds has been investigated by Braunschweig and co-workers. In those studies the reactions of :GeCl2 and :SnCl2 with [Pt(PCy3)2] (Cy = cyclohexyl) yielded the corresponding mononuclear dihalogermylene and -stannylene compounds (PCy3)2Pt![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ECl2 (E = Ge, Sn). In stark contrast, reactions of equimolar amounts of :ECl2 and [Pt(PtBu3)2] (2) did not result in major alterations of the resonances recorded by 1H and 31P{1H} NMR spectroscopy relative to reactant 2, although a rapid colour change from colourless to dark red was noticeable in both cases. The dissimilar reactivity of compound 2 and its PCy3 analogue towards :ECl2 is reminiscent to their reaction with H2, which is rapid for [Pt(PCy3)2]22 but impracticable for 2 unless gold complex 1 is present, in which case an FLP-like H2 splitting takes place.9 We ascribe the lack of reactivity of 2 to the high steric shielding provided by the tert-butyl phosphines.
ECl2 (E = Ge, Sn). In stark contrast, reactions of equimolar amounts of :ECl2 and [Pt(PtBu3)2] (2) did not result in major alterations of the resonances recorded by 1H and 31P{1H} NMR spectroscopy relative to reactant 2, although a rapid colour change from colourless to dark red was noticeable in both cases. The dissimilar reactivity of compound 2 and its PCy3 analogue towards :ECl2 is reminiscent to their reaction with H2, which is rapid for [Pt(PCy3)2]22 but impracticable for 2 unless gold complex 1 is present, in which case an FLP-like H2 splitting takes place.9 We ascribe the lack of reactivity of 2 to the high steric shielding provided by the tert-butyl phosphines.
Although the Pt(0) compound 2 remained practically unchanged, we observed a new 31P{1H} NMR signal at 94.5 ppm after the addition of one equivalent of :GeCl2·dioxane to its CD2Cl2 solution, but it accounted for only around 5% of the overall phosphorus content, preventing the observation of a 195Pt–31P coupling constant. Based on its chemical shift and in comparison with prior studies by Braunschweig this signal could be tentatively assigned to a Pt germylene compound analogous to (PCy3)2PtGeCl2.20b However, addition of excess :GeCl2 did not lead to a major increase in the proportion of this species, which remained as the minor product (<10%) under all attempted conditions. We decided to examine whether an equilibrium towards the formation of a Pt germylene could be observed at variable temperature. Low-temperature multinuclear NMR spectroscopic studies revealed dynamic behaviour in solution, although the proportion of the suggested Pt germylene remained practically unaltered. However, an additional broad 31P{1H} NMR resonance at 119.4 ppm exhibiting a large 1JPPt coupling constant of 4670 Hz became discernible below −20 °C and reached a proportion of around 20% at −60 °C. Although we are unsure of the nature of this new species, it seems to result from the dissociation of a phosphine ligand, as evinced by a 31P{1H} NMR signal at 59.9 ppm of intensity equal to the newly formed compound and corresponding to PtBu3. Based on the analogous reactivity with :SnCl2 (vide infra) we tentatively suggest the formation of a dinuclear platinum compound stabilized by bridging germanium halides.
Although treatment of CD2Cl2 solutions of 2 with equimolar amounts of :SnCl2 led to an immediate colour change to dark red, we could not observe the formation of Pt stannylene or the existence of an equilibrium with such a species by low-temperature 1H and 31P{1H} NMR monitoring. Identical results were derived from reactions in tetrahydrofuran where tin dichloride exhibits better solubility. In contrast, the addition of a second equivalent of :SnCl2 in chlorinated solutions drastically changed the reaction outcome. Complete disappearance of Pt(0) compound 2 is immediately recorded upon addition of the second equivalent of tin dichloride at room temperature to yield a complex mixture of species, in which we could unambiguously identify several platinum compounds (Scheme 3). A 31P{1H} NMR resonance recorded at 52.8 ppm and exhibiting a 1JPSn coupling-constant of 1855 Hz accounts for the formation of the tin-phosphine adduct 11, confirmed by the independent reaction between :SnCl2 and PtBu3. It is worth mentioning that in all experiments described herein variable amounts of [PtClH(PtBu3)2] (12) were formed, likely due to reaction of 2 with hydrochloric acid produced by adventitious traces of water in the presence of :SnCl2. Phosphonium cation [H(PtBu3)]+ was produced by the same reason and displays a distinctive 1H NMR doublet at 6.02 ppm (1JHP = 408 Hz). More interestingly, the higher-frequency region of the 31P{1H} NMR spectra reveals the formation of a major compound (10) that resonates at 128.3 ppm and is accompanied by both 119Sn (2JPSn = 110 Hz) and 195Pt (1JPPt = 4874 Hz) satellites. We managed to grow crystals from the crude dichloromethane reaction mixtures that exhibit an intense dark red colour by slow diffusion of pentane at −30 °C. X-ray diffraction studies proved the formation of a dinuclear Pt(0) compound 10 in which each metal bears a single tri(tert-butyl)phosphine ligand. The capacity of tin chloride to promote phosphine dissociation has been examined in more detail and will be discussed later. The dinuclear platinum fragment in 10 is held together by three tin chloride units, one of which formally appears as an anionic bridging stannyl fragment. A phosphonium cation [H(PtBu3)]+ linked to the Pt-cluster by P–H+⋯Cl interactions (average dH–Cl 3.0 Å) compensates the anionic character of the Pt2Sn3 cluster. The anionic part of the molecular structure depicted in Fig. 4 can be described as a distorted trigonal bipyramid with a missing Pt–Sn edge and characterized by a Pt–Pt distance of 2.706(1) Å. The average Pt–Sn bond distances accounts for 2.58 Å, except for the SnCl3− termini, for which one of the two Pt–Sn contacts is elongated to 2.998(2) Å. The P–Pt–Pt–P escapes from linearity due to the presence of the stannyl-bridged group, which distorts the ideal symmetry. Thus, one of the phosphine ligands tilts to accommodate the SnCl3− group resulting in a Pt–Pt–P bond angle of 171.2 Å. A somehow related structure has been previously described in which the bridging divalent tin nuclei are stabilized by acetylacetonate ligands.23 As in prior cases, the highly reduced character of the heteropolymetallic cluster is likely responsible for its high instability.24
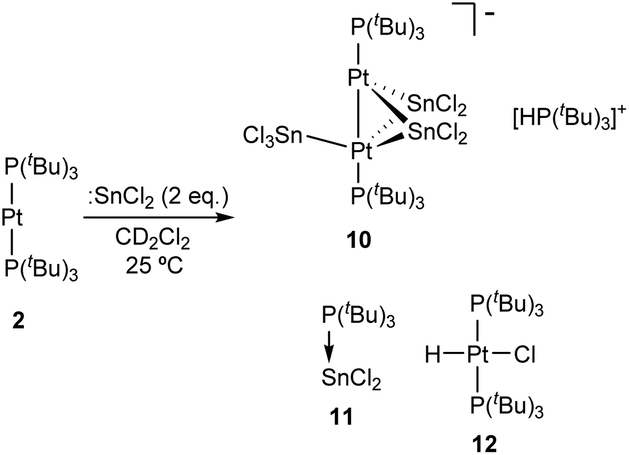 | ||
| Scheme 3 Reactivity of Pt(0) compound 2 with 2 equivalents of :SnCl2. | ||
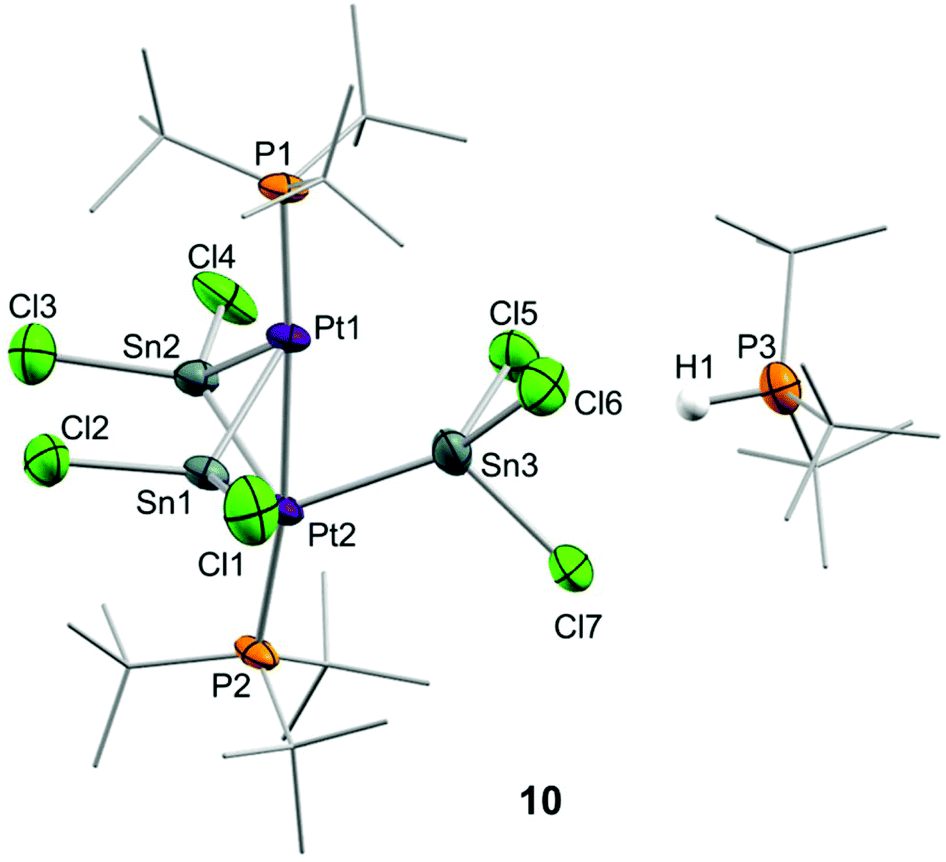 | ||
| Fig. 4 ORTEP diagram of compound 10; for the sake of clarity most hydrogens have been omitted and tert-butyl substituents have been represented in wireframe format. Thermal ellipsoids are set at 50% probability. | ||
We found of interest to interrogate the bonding scheme in diplatinum 10 by computational methods. Optimization of its molecular geometry at the ωB97XD/6-31G(d,p) level of theory was in agreement with the solid state structure, with Pt–Pt and Pt–Sn bond distances of 2.77 and 2.63–2.69 Å, respectively, except for the SnCl3− group, for which one of the two Pt–Sn contacts is elongated to 3.21 Å. Analysis of the computed electron density (QTAIM) performed at the same level of theory disclosed bond critical points (BCPs) and the corresponding bond paths (BPs) connecting each SnCl2 fragment to both Pt atoms, while the SnCl3− group binds to a single Pt centre (Pt(2)) (Fig. 5). Additionally, one BCP was located at the path between the Pt atoms, supporting the bonding interaction suggested by the short solid state Pt–Pt distance.
 | ||
| Fig. 5 Plot of the laplacian of the electron density, ∇2ρ, of complex 10 in the Pt(1)–Pt(2)–SnCl3 (left) and the Pt(1)–Pt(2)–SnCl2 (right) planes calculated with the ωB97X-D functional. The solid and dashed lines correspond to positive and negative values of ∇2ρ, respectively. In plane BCPs and BPs of the electron density are superimposed. | ||
The analysis of the electron density was complemented with an analysis of localized molecular orbitals to rationalize the interactions between the [Pt(PtBu3)], SnCl2 and SnCl3− fragments, following the Pipek–Mezey25 and NBO criteria. Both localization schemes provide similar information revealing that the three SnCln fragments donate electron density to one of the platinum atoms, Pt(2), whereas Pt(1) acts as a donor by delocalizing d-electron density onto empty p orbitals of the two SnCl2 fragments (Fig. 6a). Besides, Pt(1) also behaves as an acceptor, since the Pt–Pt interaction arises from electron delocalization from one d orbital on Pt(2) onto Pt(1) as seen in Fig. 6b. Overall, the bonding in compound 10 could be rationalize by the schematic representation depicted in Fig. 6a, where each metal atom (except for SnCl3−) exhibits ambiphilic donor–acceptor character.
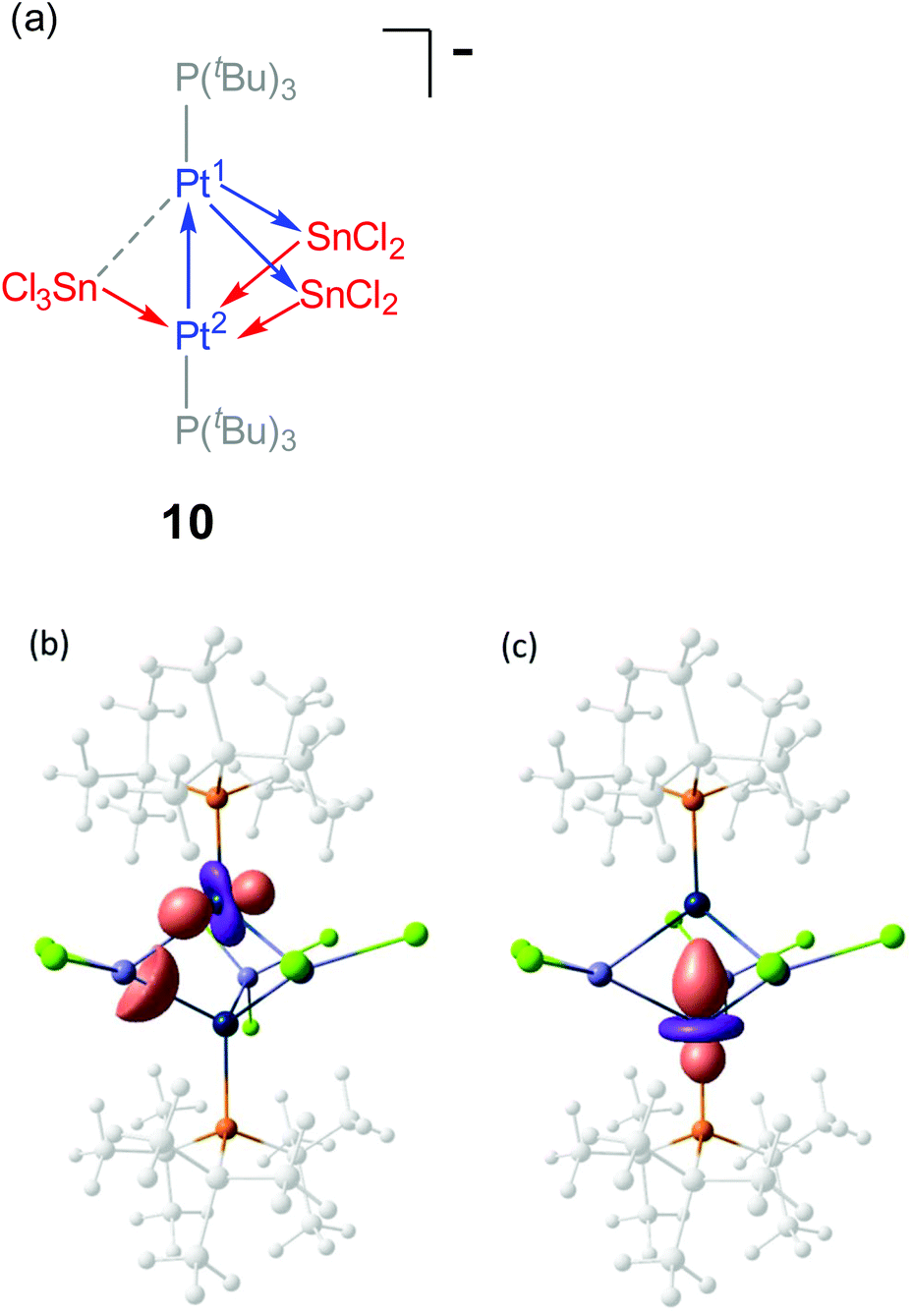 | ||
| Fig. 6 (a) Simplified bonding scheme representation of compound 10, where arrows describe electron donation between metal centres; (b, c) Representative localized molecular orbitals (Pipek–Mezey) involved in the Pt–Sn (b) and the Pt–Pt bonding (c). | ||
Reactivity of GeCl2 and SnCl2 towards a Au/Pt frustrated Lewis pair
After examining the reactivity of tetrylene dihalides with compounds 1 and 2 we moved to investigate their chemistry with the two metallic fragments in cooperation. Before describing the details of these experiments it must be noted that the reaction outcomes were independent of the order in which the three components were mixed together. In other words, the reaction of platinum compound 2 with pre-formed germyl or stannyl derivatives 3 and 4, respectively, led to identical product distributions than those detected after the addition of gold compound 1 to dichloromethane solutions of 2 and the corresponding tetrylene. We previously showed that TMOFLP 1/2 exists in solution as an equilibrium between the independent metallic fragments and a metal-only Lewis pair (MOLP)26 in which the electron-rich platinum forms a dative bond with the electrophilic gold nucleus. Although we could not detect the metallic Pt→Au adduct by spectroscopic methods, NMR line-broadening upon mixing 1 and 2 supported this assumption, with the prevalence of the individual components relative to the metallic adduct being ascribed to steric frustration.To our surprise, treatment of dichloromethane solutions of the 1/2 pair with one equivalent of :GeCl2·dioxane cleanly generated the metallic adduct whose existence we had previously proposed9 (compound 13 in Scheme 4). The tetrylene seems to be key in withdrawing the triflimide anion from gold, likely by formation of NTf2→GeCl2. The formulation of the unsupported heterobimetallic compound 13 was ascertained by NMR spectroscopy and validated by microanalysis. In the 31P{1H} NMR spectrum a doublet at 94.5 ppm (1JPPt = 3159 Hz) with a small 3JPP coupling constant of 2 Hz was accompanied by a triplet at −34.2 ppm, highly shifted to lower frequencies with respect to gold compound 1 (δ = −11.5 ppm) and also exhibiting an identical coupling constant of 2 Hz. Besides, the latter signal arises from the phosphine directly bound to the gold centre but features a relatively strong coupling to platinum (2JPPt = 1984 Hz). This 31P{1H} NMR pattern supports the fact that a new Pt→Au dative bond is present in compound 13. The proposed molecular structure was also authenticated by single-crystal X-ray diffraction studies. The triflimide salt of compound 13 co-crystallized with half a molecule of [(PMe2ArDipp2)2Au]+ cation per asymmetric unit, as well as with another half a molecule of triflimide as counteranion. This is not surprising since cationic diphosphine gold species have been described as recurrent side-products in gold chemistry,27 although its presence in solution was minimal (ca. 5%) as monitored by NMR spectroscopic techniques. The molecular structure of compound 13 is represented in Fig. 7. The platinum centre exhibits a slightly distorted T-shaped coordination environment, with a relatively reduced P–Pt–P bond of 167.59(5)° likely due to the bulkiness of the Au(PMe2ArDipp2) unit bound to the Pt(0) centre. The Pt–Au distance amounts to 2.575(1) Å, significantly shortened when compared to its related heterobimetallic dihydride compound,9 but marginally longer than in compound [(PCy3)2Pt]→Au(PCy3) (dAu–Pt = 2.54 Å), the only other unsupported Pt(0)–Au(I) species structurally characterized to date.28
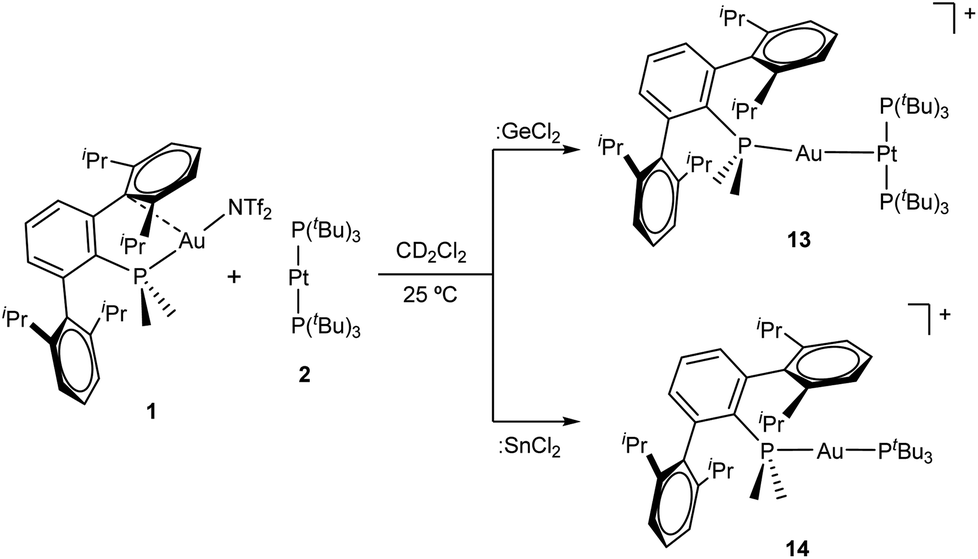 | ||
| Scheme 4 Combined reactivity of compounds 1 and 2 towards germanium and tin dihalides. | ||
 | ||
| Fig. 7 ORTEP diagram of the cation of complex 13; hydrogen atoms, half molecule of [(PMe2ArDipp2)2Au]+ and triflimide anions are excluded for clarity and thermal ellipsoids are set at 50% probability. tert-Butyl and iso-propyl substituents have been drawn in wireframe format. | ||
As briefly noted earlier, compound 13 was alternatively synthesized by treatment of gold germyl 3 with [Pt(PtBu3)2] 2, which reflects the lability of the Au–Ge bond in these species. However, the reactivity of :SnCl2 with Au/Pt 1/2 pair markedly differs and no metal-only Lewis adduct 13 was detected in any of our experiments. Instead, tin dichloride promoted an interesting phosphine exchange to yield the heteroleptic compound [(PMe2ArDipp2)Au(PtBu3)]+ (14) as the only gold-containing species. 31P{1H} NMR spectroscopy revealed the immediate formation of compound 14 upon mixing the three reaction components, as evidenced by two set of doublets at 100.6 and 14.6 ppm, characterized by a two-bond coupling-constant of 312 Hz, analogous to other cationic and heteroleptic diphosphine gold derivatives.29 We could not observe, however, any other signal by 31P{1H} NMR corresponding to the remaining Pt-bound tri-tert-butylphosphine.
Tin-promoted phosphine exchange reactions
The ability of :SnCl2 to mediate the transfer of a phosphine ligand from Pt(0) to Au(I) prompted us to investigate the possibility of accessing heteroleptic diphosphine Pt(0) compounds, of which there are very few reported examples.30 Prior studies have shown that ligand-exchange reactions between [Pt(PCy3)2] and several N-heterocyclic carbenes (NHCs) provide access to heteroleptic NHC-Pt-PCy3 derivatives,31 but the analogous substitution reaction to incorporate bulky phosphines into P–Pt(0)–P′ structures remains unknown. To carry out these studies we chose three bulky phosphines, more precisely PCy3, whose stannylene-platinum chemistry has already been outlined,31 as well as PMeXyl2 (Xyl = 2,6-Me2C6H3) and PMe2ArDtbp2 (ArDtbp2 = C6H3-2,6-(C6H3-3,5-(CMe3)2)), whose coordination chemistry and reactivity has been reported by our group.32 The progress of the exchange reactions can be easily monitored by 31P{1H} NMR spectroscopy. Heating equimolar solutions of Pt(0) compound 2 and each of the three aforementioned phosphine ligands at 80 °C for several days did not result in any apparent transformation in view of the resulting NMR spectra, except for PCy3, where minor amounts of unknown species were detected. Likewise, the inertness of 2 towards ligand substitution stands unaltered under excess of the free phosphine (up to 10 equivalents). In stark contrast, the addition of one equivalent of :SnCl2 to the previous solutions led to rapid phosphine-exchange reactions that had in common the appearance of free PtBu3 as the main side-product. Best yields were obtained by the use of 1.5 equivalents of :SnCl2. In the case of PCy3, instead of the aimed heteroleptic Pt(0) compound, immediate formation of (PCy3)2PtSnCl2 at 25 °C was evinced by 31P{1H} NMR spectroscopy (Scheme 5). A characteristic broad singlet at 51.4 ppm flanked by 195Pt satellites (1JPPt = 3525 Hz), as previously described by Braunschweig,20 demonstrated its formation, which became quantitative when performing the reaction with 2.1 equivalents of PCy3. The formation of (PCy3)2PtSnCl2 was accompanied by the presence of unbound PtBu3 in a 1:2 ratio, with a 31P{1H} NMR signal at 59.9 ppm.
 | ||
| Scheme 5 Tin-mediated phosphine exchange reactions at Pt(0) 2. | ||
The reaction of PMeXyl2 and Pt(0) 2 in the presence of :SnCl2 (1.5 equiv.) proceeds rapidly towards compound 15 in quantitative spectroscopic yield (Scheme 5). At variance with PCy3, the use of PMeXyl2 permitted the formation of the desired heteroleptic Pt(0) species in which only one of the two PtBu3 ligands was substituted by the incoming phosphine. In fact, using an excess of PMeXyl2 did not lead to the homoleptic Pt(0) compound analogous to (PCy3)2PtSnCl2 even under moderate heating. The use of the bulkier phosphine PMe2ArDtbp2 bearing a terphenyl group led to the formation of heteroleptic platinum stannylene 16 (Scheme 5), though it required longer reaction times. The high-resolution mass spectra of 15 and 16 fit exactly to their proposed formulation (see Experimental section and ESI†), albeit without the bound SnCl2 fragment, not surprisingly given the lability of the Pt→Sn bond. Both compounds feature similar 31P{1H} NMR spectra characterized by two doublets exhibiting 2JPP of around 300 Hz, indicating the trans disposition of the two phosphines. Compound 15 leads to resonances at 94.6 and 6.3 ppm due to PtBu3 and PMeXyl2, respectively, while the analogous signals appear at 97.3 and 12.6 ppm due to PtBu3 and PMe2ArDtbp2 in compound 16. These resonances are flanked by 195Pt satellites with strong coupling constants (15: 1JPPt = 3776 and 3244 Hz; 16: 1JPPt = 3788 and 3504 Hz). The presence of a tin centre bound to platinum was inferred in the two compounds from the satellites that escort the PtBu3 doublet (2JPSnca. 250 Hz). In 195Pt NMR spectra, their platinum centres resonate at about −5000 ppm as double doublets and, in the case of 15, we could detect a large 1JPtSn coupling constant of 3210 that further corroborates the coordination of tin. Our attempts to record 119Sn{1H} NMR resonances were unsuccessful, though this is not unexpected due to the high asymmetry of the 119Sn centres in these compounds, which results in an increased effect of chemical shift anisotropy in the relaxation of their NMR signals.33 Coupling to the variety of neighbouring NMR-active nuclei adds to the difficulty of observing 119Sn{1H} NMR signals for 15 and 16.
As a side note, we observed that the methyl group directly bound to the phosphorus centre in compound 15 resonates at 2.93 ppm (dd, 3JHPt = 50.7, 2JHP = 9.0, 4JHP = 2.5 Hz) in the 1H NMR spectrum, shifted to surprisingly higher frequency compared to free phosphine (1.63 ppm)34 or to other Pt-PMeXyl2 compounds previously reported by us (ca. 1.5–1.7 ppm).33 However, its corresponding 13C{1H} resonance appears at 21.0 ppm (1JCP = 37 Hz), that is, within the expected range for a Ar2PMe group. The unexpected 1H NMR chemical shift served though to validate the proposed molecular structure of 15 by means of computational studies. A conformational analysis was calculated at the ωB97XD/6-31G(d,p) level of theory and disclosed no close contacts between the P–CH3 moiety and the Sn or Pt centres. The geometric parameters of the minimum energy conformer of complex 15 (see Fig. S3†) are also comparable to previous platinum(0) diphosphine stannylene systems.20 With this model in hand, we calculated the theoretical 1H NMR chemical shifts of 15 by means of the GIAO method (ωB97XD/6-311+G(2d,p)//ωB97XD/6-31G(d,p)).35 To calibrate these results 1H NMR data of compounds 2, [Pt(PCy3)2(SnCl2)]20a and [Pt(IMes)(PCy3)(SnCl2)]20a (IMes = 1,3-dimesitylimidazol-2-ylidene) were also evaluated. The linear relationship found between calculated and experimental proton chemical shifts (R2 = 0.996, Fig. 8) gives an expected δ of 2.71 ppm for the PMe moiety in complex 15, in reasonable agreement with the experimental value (2.93 ppm).
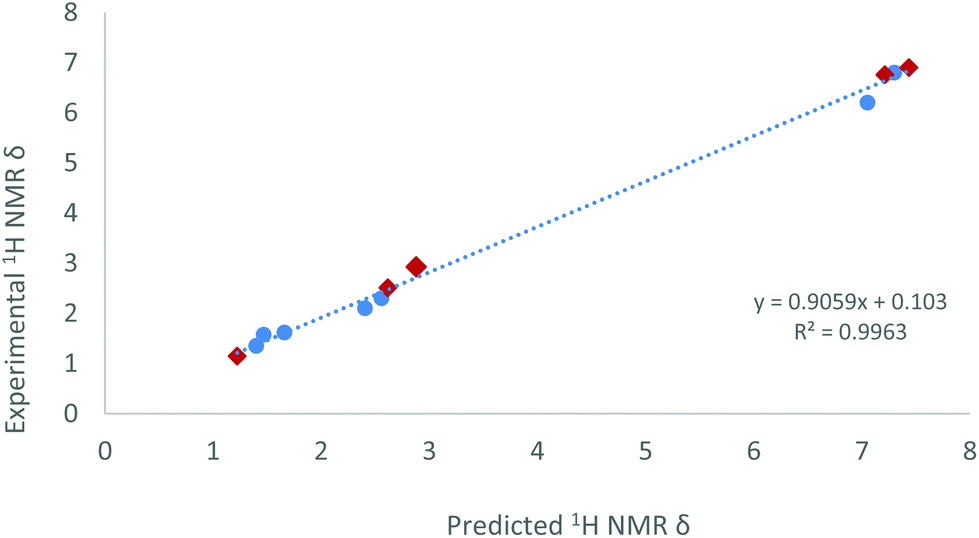 | ||
| Fig. 8 Predicted and experimental 1H NMR data relative to complexes 15 (red diamond), benchmarked against 2, [Pt(PCy3)2(SnCl2)]20a and [Pt(IMes)(PCy3)(SnCl2)].20a | ||
Experimental section
General considerations
All preparations and manipulations were carried out using standard Schlenk and glove-box techniques, under an atmosphere of argon and of high purity nitrogen, respectively. All solvents were dried, stored over 4 Å molecular sieves, and degassed prior to use. Toluene (C7H8), n-pentane (C5H12) and n-hexane (C6H14) were distilled under nitrogen over sodium. Tetrahydrofuran (THF) and diethyl ether were distilled under nitrogen over sodium/benzophenone. [D6]Benzene and [D8]Toluene were distilled under argon over sodium/benzophenone, THF was dried over molecular sieves (4 Å) and CD2Cl2 over CaH2 and distilled under argon. [AuCl(THT)] (THT = tetrahydrotiophene),36 phosphine ligands PMe2ArDipp2,13 PCyp2ArXyl2,32c PMe2ArXyl2,33b PMe2ArDtbp232c and compounds 1,13237 and 513 were prepared as described previously. Other chemicals were commercially available and used as received. Solution NMR spectra were recorded on Bruker AMX-300, DRX-400 and DRX-500 spectrometers. Spectra were referenced to external SiMe4 (δ: 0 ppm) using the residual proton solvent peaks as internal standards (1H NMR experiments), or the characteristic resonances of the solvent nuclei (13C NMR experiments), while 31P and 195Pt were referenced to H3PO4 and Na[PtCl6], respectively. Spectral assignments were made by routine one- and two-dimensional NMR experiments where appropriate (Fig. 9). For elemental analyses a LECO TruSpec CHN elementary analyzer, was utilized. HRMS data were obtained on a JEOL JMS-SX 102A mass spectrometer by the Mass Spectrometry Services of the University of Seville (CITIUS).CCDC 1897306–1897311† (compounds 6, 6Xyl, 8Cyp, [Au2(μ-N(SiMe3)2)(PMe2ArXyl2)2], 13, 10) contain the supplementary crystallographic data for this paper.
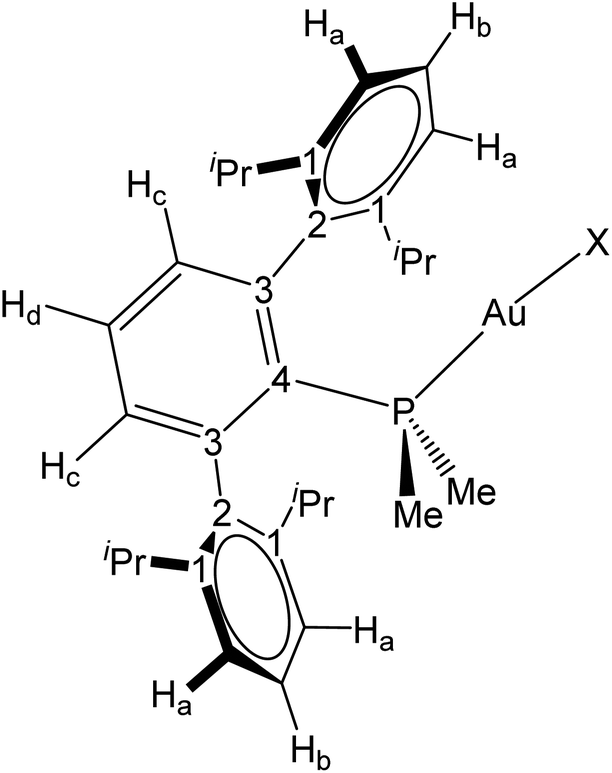 | ||
| Fig. 9 Labeling scheme used for 1H and 13C{1H} NMR assignments. | ||
Compound 3
A solution of 1 (30 mg, 0.03 mmol) in CD2Cl2 (0.5 mL) was treated with :GeCl2·dioxane (7.4 mg, 0.03 mmol) in a J. Young NMR tube. The tube was shaken resulting in the immediate formation of compound 3, (15 mg, 46%). Anal. calcd for C34H43AuCl2F6GeNO4PS2: C, 37.8; H, 4.0; N, 1.3; S, 5.9. Found: C, 38.2; H, 4.2; N, 1.5; S, 5.5. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.60 (t, 1 H, 3JHH = 7.6 Hz, Hd), 7.47 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.4 (d, 4 H, 3JHH = 7.6 Hz, Ha), 7.26 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.7 Hz, Hc), 2.48 (sept, 4 H, 3JHH = 6.8 Hz, iPr(CH)), 1.37 (d, 6 H, 2JHP = 10 Hz, PMe2), 1.36 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)), 1.06 (d, 12 H, 3JHH = 6.7 Hz, iPr(CH3)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 147.2 (C1), 146.2 (d, 2JCP = 12 Hz, C3), 138.2 (d, 3JCP = 6 Hz, C2), 133.2 (d, 3JCP = 8 Hz, CHc), 131.1 (CHd), 130.4 (CHb), 129.0 (d, 1JCP = 82 Hz, C4), 124.8 (CHa), 119.7 (q, 1JCF = 323 Hz, CF3), 31.7 (iPr(CH)), 25.5 (iPr(CH3)), 23.1 (iPr(CH3)), 16.2 (d, 1JCP = 36 Hz, PMe2). 31P{1H} NMR (160 MHz, CD2Cl2, 25 °C) δ: 4.8.Compound 4
In an NMR tube, a solution of 1 (30 mg, 0.03 mmol) in CD2Cl2 (0.5 mL) was treated with tin(II) chloride (6.0 mg, 0.03 mmol). The tube was shaken resulting in the immediate formation of compound 3, (17 mg, 48%). Anal. calcd for C34H43AuCl2F6NO4PS2Sn: C, 36.3; H, 3.9; N, 1.2; S, 5.7. Found: C, 36.3; H, 3.9; N, 1.5; S, 5.5. Spectroscopic data for compound 4·THF: 1H NMR (400 MHz, THF-d8, 25 °C) δ: 7.58 (td, 1 H, 3JHH = 7.6 Hz, 5JHP = 1.9 Hz, Hd), 7.42 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.29 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.4 Hz, Hc), 7.25 (d, 4 H, 3JHH = 7.6 Hz, Ha), 2.62 (sept, 4 H, 3JHH = 6.7 Hz, iPr(CH)), 1.35 (d, 12 H, 3JHH = 7.0 Hz, iPr(CH3)), 1.27 (d, 6 H, 2JHP = 10.4 Hz, PMe2), 1.03 (d, 12 H, 3JHH = 7.0 Hz, iPr(CH3)). 13C{1H} NMR (100 MHz, THF-d8, 25 °C) δ: 147.1 (C1), 145.7 (d, 2JCP = 11 Hz, C3), 139.4 (d, 3JCP = 5 Hz, C2), 133.7 (d, 3JCP = 8 Hz, CHc), 130.2 (CHd), 130.0 (CHb), 129.3 (d, 1JCP = 56 Hz, C4), 124.0 (CHa), 121.0 (q, 1JCF = 320 Hz, CF3), 31.9 (iPr(CH)), 25.6 (iPr(CH3)), 23.1 (iPr(CH3)), 17.9 (d, 1JCP = 38 Hz, PMe2). 31P{1H} NMR (160 MHz, THF-d8, 25 °C) δ: −3.1.Compound 6
A THF (5 mL) solution of 5 (150 mg, 0.22 mmol) was added under argon atmosphere over a solution of :GeCl2·dioxane (50 mg, 0.22 mmol) in THF (5 mL), then stirred for 30 minutes at room temperature. The solvent was then removed under vacuum to give compound 6 as a fine white powder (161 mg, 88%). Crystals suitable for X-ray studies were grown by slow diffusion of pentane into a dichloromethane solution of 6. Anal. calcd for C32H43AuCl3GeP: C, 46.1; H, 5.2. Found: C, 45.8; H, 5.5. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.58 (td, 1 H, 3JHH = 7.6 Hz, 5JHP = 1.6 Hz, Hd), 7.47 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.38 (d, 4 H, 3JHH = 7.6 Hz, Ha), 7.24 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.5 Hz, Hc), 2.48 (sept, 4 H, 3JHH = 6.8 Hz, iPr(CH)), 1.36 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)), 1.33 (d, 6 H, 2JHP = 10 Hz, PMe2), 1.05 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 146.9 (C1), 146.4 (d, 2JCP = 11 Hz, C3), 138.1 (d, 3JCP = 6 Hz, C2), 133.1 (d, 3JCP = 7 Hz, CHc), 130.8 (CHd), 130.4 (CHb), 127.5 (d, 1JCP = 52 Hz, C4), 124.6 (CHa), 31.7 (iPr(CH)), 25.6 (iPr(CH3)), 23.1 (iPr(CH3)), 16.5 (d, 1JCP = 33 Hz, PMe2). 31P{1H} NMR (160 MHz, CD2Cl2, 25 °C) δ: 5.0.Compound 7
A THF (5 mL) solution of 5 (150 mg, 0.22 mmol) was added under argon atmosphere over a solution of tin(II) chloride (41 mg, 0.22 mmol) in THF (5 mL), then stirred for 30 minutes at room temperature. The solvent was then removed under vacuum to give compound 7 as a fine white powder (172 mg, 89%). Anal. calcd for C32H43AuCl3PSn: C, 43.6; H, 4.9. Found: C, 43.5; H, 5.1. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.56 (td, 1 H, 3JHH = 7.6 Hz, 5JHP = 1.8 Hz, Hd), 7.47 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.33 (d, 4 H, 3JHH = 7.6 Hz, Ha), 7.24 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.5 Hz, Hc), 2.52 (sept, 4 H, 3JHH = 6.8 Hz, iPr(CH)), 1.35 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)), 1.33 (d, 6 H, 2JHP = 10 Hz, PMe2), 1.05 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 147.0 (C1), 146.0 (d, 2JCP = 11 Hz, C3), 138.7 (d, 3JCP = 4 Hz, C2), 133.2 (d, 3JCP = 7 Hz, CHc), 130.5 (CHd), 130.0 (CHb), 124.2 (CHa), 31.7 (iPr(CH)), 25.6 (iPr(CH3)), 23.1 (iPr(CH3)), 17.4 (d, 1JCP = 38 Hz, PMe2). The quaternary carbon C4 could not be located neither in the 13C{1H} NMR spectrum or by two-dimensional 1H–13C correlations. 31P{1H} NMR (160 MHz, CD2Cl2, 25 °C) δ: −2.2.Compound 8
In an NMR tube, a solution of 5 (30 mg, 0.04 mmol) in C6D6 (0.5 mL) was treated with tin(II) bis(trimethylsilyl)amide (19 mg, 0.04 mmol). The tube was shaken resulting in the formation of compound 8 after 5 minutes. The compound could be isolated as a white powder after removing the solvent under reduced pressure (22 mg, 48%).1H NMR (400 MHz, C6D6, 25 °C) δ: 7.45 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.24 (d, 4 H, 3JHH = 7.6 Hz, Ha), 7.15 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.5 Hz, Hc), 6.98 (td, 1 H, 3JHH = 7.6 Hz, 5JHP = 1.8 Hz, Hd), 2.52 (sept, 4 H, 3JHH = 6.8 Hz, iPr(CH)), 1.30 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)) 1.10 (d, 6 H, 2JHP = 10 Hz, PMe2), 0.90 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)), 0.47 (s, 2JHSi = 15.3 Hz, 4JHSn = 6.5 Hz, SiMe3). 13C{1H} NMR (100 MHz, C6D6, 25 °C) δ: 146.9 (C1), 146.3 (C3), 137.8 (d, 3JCP = 6 Hz, C2), 132.6 (d, 3JCP = 7 Hz, CHc), 130.7 (CHd), 129.6 (CHb), 124.3 (CHa), 123.2 (d, 1JCP = 60 Hz, C4), 31.6 (iPr(CH)), 25.6 (iPr(CH3)), 23.2 (iPr(CH3)), 17.1 (d, 1JCP = 30 Hz, PMe2), 7.27 (1JCSi = 55 Hz, 3JCSn = 16 Hz, SiMe3). 31P{1H} NMR (160 MHz, C6D6, 25 °C) δ: 15.4 (2JPSn = 3201 Hz).
Compound 9
In an NMR tube, a solution of 1 (30 mg, 0.03 mmol) in C6D6 (0.5 mL) was treated with tin(II) bis(trimethylsilyl)amide (14 mg, 0.03 mmol). The tube was shaken resulting in the formation of 9 after 5 minutes. The compound could be isolated as a white powder after removing the solvent under reduced pressure (20 mg, 48%).1H NMR (400 MHz, C6D6, 25 °C) δ: 7.26 (m, 3 H, Hd, Hb), 7.14 (d, 4 H, 3JHH = 7.6 Hz, Ha), 6.94 (m, 2 H, Hc), 2.56 (sept, 4 H, 3JHH = 6.8 Hz, iPr(CH)), 1.58 (d, 6 H, 2JHP = 10 Hz, PMe2), 1.28 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)), 0.89 (d, 12 H, 3JHH = 6.8 Hz, iPr(CH3)), 0.47 (s, SiMe3). 13C{1H} NMR (100 MHz, C6D6, 25 °C) δ: 146.6 (C1), 145.9 (d, 2JCP = 10 Hz, C3), 138.3 (d, 3JCP = 5 Hz, C2), 132.9 (d, 3JCP = 7 Hz, CHc), 130.0 (CHd), 129.6 (CHb), 124.3 (CHa), 120.0 (q, 1JCF3 = 322 Hz, CF3), 31.6 (iPr(CH)), 25.6 (iPr(CH3)), 23.2 (iPr(CH3)), 16.6 (d, 1JCP = 33 Hz, PMe2), 6.7 (1JCSi = 55 Hz, 3JCSn = 16 Hz, SiMe3). 31P{1H} NMR (160 MHz, C6D6, 25 °C) δ: 13.8.
Compound 10
A CH2Cl2 (3 mL) solution of 2 (90 mg, 0.15 mmol) was added under argon atmosphere over tin(II) chloride (56 mg, 0.30 mmol) and the resulting red solution was stirred for 5 minutes at room temperature. Compound 10 could be crystallized by slow diffusion of hexane at −30 °C (26 mg, 11%).1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 6.15 (d, 1 H, 1JHP = 452 Hz, H–P(C(CH3)3), 1.69 (d, 27 H, 3JHP = 15.0 Hz, H–P(C(CH3)), 1.55 (d, 54 H, 3JHP = 13.0 Hz, Pt–P(C(CH3)3)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 40.5 (vt, 1JCP = 7 Hz, Pt–P(C(CH3)3), 38.0 (d, 1JCP = 28 Hz, H–P(C(CH3)3), 33.6 (Pt–P(C(CH3)3), 30.6 (H–P(C(CH3)3). 31P{1H} NMR (160 MHz, CD2Cl2, 25 °C) δ: 128.3 (2JPSn = 110 Hz, 1JPPt = 4874 Hz, Pt–P(C(CH3)3), 51.9 (H–P(tBu)3). UV-vis (CH2Cl2) λmax (ε [cm−1 M−1]): 572 nm (102).
Compound 13
A solid mixture of compounds 1 (100 mg, 0.106 mmol), 2 (64 mg, 0.106 mmol) and :GeCl2·dioxane (25 mg, 0.106 mmol) was placed in a Schlenk flask inside a dry box, dissolved in CH2Cl2 (5 mL) and stirred at room temperature for 15 minutes. Addition of pentane (10 mL) caused precipitation of 13 as an orange solid that was washed with pentane (150 mg, 92%). This compound can be recrystallized by slow diffusion of pentane into a toluene solution (2:1 by vol.) at −20 °C. Anal. calcd for C58H97AuF6NO4P3PtS2: C, 45.4; H, 6.4; N, 0.9; S, 4.2. Found: C, 45.8; H, 6.2; N, 0.8; S, 4.0. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.52 (td, 1 H, 3JHH = 7.6 Hz, 5JHP = 2.0 Hz, Hd), 7.42 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.26 (d, 4 H, 3JHH = 7.6 Hz, Ha), 7.14 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.6 Hz, Hc), 2.56 (sept, 4 H, 3JHH = 6.5 Hz, iPr(CH)), 1.50 (vt, 54 H, 3JHP = 6.4 Hz, tBu), 1.30 (d, 12 H, 3JHH = 6.7 Hz, iPr(CH3)), 1.19 (d, 6 H, 2JHP = 10 Hz, PMe2), 1.00 (d, 12 H, 3JHH = 6.7 Hz, iPr(CH3)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 146.8 (C1), 144.4 (d, 2JCP = 10 Hz, C3), 139.2 (d, 5JCP = 3 Hz, C2), 134.8 (d, 3JCP = 9 Hz, CHc), 130.0 (CHb), 129.2 (CHd), 127.4 (d, 1JCP = 41 Hz, C4), 124.1 (CHa), 120.5 (q, 1JCF = 323 Hz, CF3), 39.5 (vt, 1JCP = 8 Hz, 2JCPt = 20 Hz, Pt–P(C(CH3)3), 33.8 (Pt–P(C(CH3)3), 31.5 (iPr(CH), 25.9 (iPr(CH3), 23.8 (iPr(CH3), 19.7 (d, 1JCP = 35 Hz, PMe2). 31P{1H} NMR (160 MHz, CD2Cl2, 25 °C) δ: 94.5 (d, 3JPP = 2, 1JPPt = 3159 Hz), −34.2 (t, 3JPP = 2, 2JPPt = 1984 Hz).
Compound 14
A dichloromethane (5 mL) solution of compound 1 (50 mg, 0.05 mmol) was treated with PtBu3 (10.7 mg, 0.05 mmol) under argon atmosphere. The solution was stirred at −80 °C for 5 min and the temperature was slowly raised to 25 °C. Compound 14 was precipitated by the addition of pentane as a white solid that was further washed with the same solvent (49 mg, 86%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.60 (td, 1 H, 3JHH = 7.6 Hz, 5JHP = 1.7 Hz, Hd), 7.44 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.31 (d, 4 H, 3JHH = 7.6 Hz, Ha), 7.23 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.4 Hz, Hc), 2.53 (sept, 4 H, 3JHH = 6.8 Hz, iPr(CH)), 1.57 (d, 6 H, 2JHP = 10.4 Hz, PMe2), 1.35 (d, 27 H, 3JHP = 15 Hz, tBu), 1.23 (d, 12 H, 3JHH = 7.0 Hz, iPr(CH3)), 1.06 (d, 12 H, 3JHH = 7.0 Hz, iPr(CH3)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 146.6 (C1), 146.1 (d, 2JCP = 10 Hz, C3), 137.7 (d, 3JCP = 3 Hz, C2), 132.9 (d, 3JCP = 7 Hz, CHc), 129.9 (CHd), 125.2 (CHb), 123.8 (CHa), 119.7 (q, 1JCF = 323 Hz, CF3), 39.9 (d, 1JCP = 16 Hz, P(C(CH3)3), 32.3 (P(C(CH3)3), 31.3 (iPr(CH)), 25.0 (iPr(CH3)), 22.9 (iPr(CH3)), 16.0 (d, 1JCP = 34 Hz, PMe2). 31P{1H} NMR (160 MHz, CD2Cl2, 25 °C) δ: 100.6 (2JPP = 312 Hz), 14.6 (2JPP = 312 Hz).Compound 15
An NMR tube was charged with PMeXyl2 (18 mg, 0.075 mmol), Pt(PtBu3)2 (30 mg, 0.05 mmol), tin(II) dichloride (14.4 mg, 0.075 mmol) and deuterated benzene or toluene (0.5 mL). The initial white suspension became a red solution after several hours and was stirred for an overall period of 8 hours (85% NMR yield). HRMS (electrospray, m/z): calcd for C29H49P2Pt: [M − SnCl2 + H]+ 654.7249, found 654.2952. 1H NMR (400 MHz, tol-d8, 25 °C) δ: 6.90 (t, 2 H, 3JHH = 7.4 Hz, p-C6H3), 6.76 (dd, 4 H, 3JHH = 7.4 Hz, 4JHP = 3.6 Hz, m-C6H3), 2.93 (dd, 3 H, 3JHPt = 50.7 Hz, 2JHP = 9.0 Hz, 4JHP = 2.5 Hz, PMe), 2.51 (s, 12 H, MeXyl), 1.15 (d, 27 H, 3JHP = 12.6 Hz, tBu). 13C{1H} NMR (100.6 MHz, C6D6, 25 °C) δ: 141.6 (d, 2JCP = 9 Hz, o-C6H3), 134.0 (d, 1JCP = 48 Hz, ipso-C6H3), 130.0 (d, 3JCP = 8 Hz, meta-C6H3), 129.1 (d, 4JCP = 2 Hz, para-C6H3), 39.2 (d, 1JCP = 13, 2JCPt = 55 Hz, Pt–P(C(CH3)3), 32.2 (Pt–P(C(CH3)3), 25.1 (d, 3JCP = 7 Hz, MeXyl), 21.0 (d, 1JCP = 37 Hz, PMe). 31P{1H} NMR (161.98 MHz, tol-d8, 25 °C) δ: 94.6 (d, 1JPPt = 3776 Hz, 2JPP = 299 Hz, 2JPSn = 250 Hz, PtBu3), 6.3 (d, 1JPPt = 3244 Hz, 2JPP = 299 Hz, PMeXyl). 195Pt{1H} NMR (86.16 MHz, tol-d8, 25 °C) δ: −4947 (dd, 1JPPt = 3776 Hz, 1JPtP = 3244 Hz, 1JPtSn = 3210 Hz).Compound 16
An NMR tube was charged with PMe2ArDtbp2 (38.7 mg, 0.075 mmol), compound 2 (30 mg, 0.05 mmol), tin(II) dichloride (14.4 mg, 0.075 mmol) and deuterated benzene or toluene (0.5 mL). The initial white suspension became a red solution after several hours and was stirred for an overall period of one day after which we determined a spectroscopic yield of ca. 50%. HRMS (electrospray, m/z): calcd for C48H78P2Pt: [M − SnCl2] 912.1584, found 912.5291.1H NMR (400 MHz, C6D6, 25 °C) δ: 7.27–7.23 (m, 8H, Ar), 7.07 (td, 1H, 3JHH = 7.5 Hz, 5JHP = 1.5 Hz, p-C6H3), 1.98 (dd, 6 H, 2JHP = 10.0 Hz, 4JHP = 1.9 Hz, PMe2), 1.49 (br, 36 H, tBu (PMe2ArDtbp2)), 1.34 (d, 27 H, 3JHP = 12.6 Hz, tBu (PtBu3)). 13C{1H} NMR (100.6 MHz, C6D6, 25 °C) δ: 151.7 (s, m-Dtbp), 149.9 (d, 2JCP = 10 Hz, o-C6H3), 141.7 (d, 3JCP = 4 Hz, ipso-Dtbp), 131.7 (d, 1JCP = 8 Hz, ipso-C6H3), 130.7 (d, 4JCP = 7 Hz, m-C6H3), 127.8 (p-C6H3), 124.6 (s, o-Dtbp), 121.6 (s, p-Dtbp), 39.6 (d, 1JCP = 12, 2JCPt = 42 Hz, Pt–P(C(CH3)3), 34.8 (s, C(CH3)), 32.4 (Pt–P(C(CH3)3), 31.9 (s, C(CH3)), 19.5 (d, 1JCP = 36 Hz, PMe2). 31P{1H} NMR (161.98 MHz, C6D6, 25 °C) δ: 97.3 (d, 1JPPt = 3788 Hz, 2JPP = 307 Hz, 2JPSn = 255 Hz, PtBu3), 12.6 (d, 1JPPt = 3504 Hz, 2JPP = 307 Hz, PArtBu). 195Pt{1H} NMR (86.16 MHz, C6D6, 25 °C) δ: −5067 (dd, 1JPPt = 3788 Hz, 1JPtP = 3504 Hz).
Conclusion
In summary, we have analyzed the reactivity of tin and germanium dihalides with a transition metal-only frustrated Lewis pair based on Pt(0) and Au(I) fragments. Our results reveal a dissimilar reactivity of the tetrylenes in the presence of the two metals compared to the reactions displayed with the individual Au(I) and Pt(0) monometallic species. While the insertion chemistry of :GeCl2 and :SnCl2 into Au–X bonds is analogous to prior studies, their reactivity with [Pt(PtBu3)2] (2) contrasts with previous work based on less hindered phosphines. As such, we have demonstrated that :SnCl2 promotes phosphine exchange reactions at Pt(0) centres to access uncommon heteroleptic diphosphine platinum(0) compounds. In addition, an unusual highly-reduced heteropolymetallic aggregate containing a Pt2Sn3 core has been isolated and characterized by X-ray diffraction techniques, while its bonding scheme has been analyzed by computational methods. The different reactivity exhibited by :GeCl2 compared to :SnCl2 is also apparent by their addition to the Au(I)/Pt(0) pair. In the former case a metal-only Pt → Au Lewis adduct is readily produced, while in the latter experiment a cationic heteroleptic diphosphine gold compound is the major species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, Project 756575). J. J. M. thanks the Universidad de Sevilla for a research grant. The use of computational facilities at the Supercomputing Centre of Galicia (CESGA) is acknowledged. We are grateful to J. López-Serrano, M. Roselló, F. Molina, Francisco F. de Córdova and Eleuterio Alvarez for valuable discussions.References
- E. Rivard, Dalton Trans., 2014, 43, 8577 RSC.
- (a) T. J. Marks, J. Am. Chem. Soc., 1971, 93, 7090 CrossRef CAS; (b) T. J. Marks and A. R. Newman, J. Am. Chem. Soc., 1973, 95, 769 CrossRef CAS; (c) C. Eisenhut, T. Szilvasi, G. Dübek, N. C. Breit and S. Inoue, Inorg. Chem., 2017, 56, 10061 CrossRef CAS PubMed; (d) S. K. Grumbine, D. A. Straus, T. D. Tilley and A. L. Rheingold, Polyhedron, 1995, 14, 127 CrossRef CAS.
- (a) M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2011, 133, 8874 CrossRef CAS PubMed; (b) K. C. Thimer, S. M. I. Al-Rafia, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2009, 7119 RSC; (c) A. K. Swarnakar, S. M. McDonald, K. C. Deutsch, P. Choi, M. J. Ferguson, R. McDonald and E. Rivard, Inorg. Chem., 2014, 53, 8662 CrossRef CAS PubMed; (d) S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson and E. Rivard, J. Am. Chem. Soc., 2011, 133, 777 CrossRef CAS PubMed; (e) S. M. I. Al-Rafia, O. Shynkaruk, S. M. McDonald, S. K. Liew, M. J. Ferguson, R. McDonald, R. H. Herber and E. Rivard, Inorg. Chem., 2013, 52, 5581 CrossRef CAS PubMed.
- (a) R. S. Ghadwal, H. W. Roesky, S. Merkel and D. Stalke, Chem. – Eur. J., 2010, 16, 85 CrossRef CAS PubMed; (b) R. Azhakar, G. Tavcar, H. W. Roesky, J. Hey and D. Stalke, Eur. J. Inorg. Chem., 2011, 475 CrossRef CAS; (c) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Chem. Commun., 2012, 48, 1308 RSC; (d) R. S. Ghadwal, R. Azhakar, K. Pröpper, J. J. Holstein, B. Dittrich and H. W. Roesky, Inorg. Chem., 2011, 50, 8502 CrossRef CAS PubMed.
- (a) J. A. Cabeza, P. García-Álvarez and D. Polo, Eur. J. Inorg. Chem., 2016, 10 CrossRef CAS; (b) M. F. Lappert and R. S. Rowe, Coord. Chem. Rev., 1990, 100, 267 CrossRef CAS; (c) W. Petz, Chem. Rev., 1986, 86, 1019 CrossRef CAS.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 334, 1 CrossRef PubMed; (c) D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 332, 1 CrossRef CAS PubMed; (d) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (e) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018 CrossRef CAS PubMed; (f) D. W. Stephan, Science, 2016, 354, 1248 CrossRef CAS PubMed.
- (a) B. Michelet, C. Bour and V. Gandon, Chem. – Eur. J., 2014, 20, 14488 CrossRef CAS PubMed; (b) J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098 CrossRef CAS PubMed; (c) J. Backs, M. Lange, J. Possart, A. Wollschlager, C. Muck-Lichtenfeld and W. Uhl, Angew. Chem., Int. Ed., 2017, 56, 3094 CrossRef CAS PubMed; (d) Y. Yu, J. Li, W. Liu, O. Yeb and H. Zhu, Dalton Trans., 2016, 45, 6259 RSC.
- (a) A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Inorg. Chem., 2009, 48, 7645 CrossRef CAS PubMed; (b) A. Jana, G. Tavčar, H. W. Roesky and C. Schulzke, Dalton Trans., 2010, 39, 6217 RSC.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944 CrossRef CAS PubMed.
- A. K. Swarnakar, M. J. Ferguson, R. McDonald and E. Rivard, Dalton Trans., 2016, 45, 6071 RSC.
- (a) H. Yang, J. Zhao, M. Qiu, P. Sun, D. Han, L. Niu and G. Cui, Biosens. Bioelectron., 2019, 124–125, 191 CrossRef CAS PubMed; (b) L. Wang, E. Guan, J. Zhang, J. Yang, Y. Zhu, Y. Han, M. Yang, C. Cen, G. Fu, B. C. Gates and F.-S. Xiao, Nat. Commun., 2018, 9, 1362 CrossRef PubMed.
- (a) A. Aouissi, S. S. Al-Deyab and H. Al-Shahri, Molecules, 2010, 15, 1398 CrossRef CAS PubMed; (b) B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564 CrossRef CAS PubMed.
- M. F. Espada, J. Campos, J. López-Serrano, M. L. Poveda and E. Carmona, Angew. Chem., Int. Ed., 2015, 54, 15379 CrossRef CAS PubMed.
- (a) A. Bauer, A. Schier and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 1995, 2919 RSC; (b) A. Bauer and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 2919 Search PubMed; (c) J. A. Dilts and M. P. Johnson, Inorg. Chem., 1966, 5, 2079 CrossRef CAS; (d) D. M. Mingos, H. R. Powell and T. L. Stolberg, Transition Met. Chem., 1992, 17, 334 CrossRef CAS.
- (a) R. V. Bojan, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, R. Echeverría, O. Lehtonen and D. Sundholm, ChemPlusChem, 2016, 81, 176 CrossRef CAS; (b) R. V. Bojan, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, R. Echeverria, O. Lehtonen and D. Sundholm, ChemPlusChem, 2014, 79, 67 CrossRef.
- P. Pérez-Galán, N. Delpont, E. Herrero- Gómez, F. Maseras and A. M. Echavarren, Chem. – Eur. J., 2010, 16, 5324 CrossRef PubMed.
- (a) U. Anandhi and P. R. Sharp, Inorg. Chim. Acta, 2006, 359, 3521 CrossRef CAS; (b) J. A. Cabeza, J. M. Fernández-Colinas, P. García-álvarez and D. Polo, Inorg. Chem., 2012, 51, 3896 CrossRef CAS PubMed; (c) M. Walewska, J. Hlina, W. Gaderbauer, H. Wagner, J. Baumgartner and C. Marschner, Z. Anorg. Allg. Chem., 2016, 642, 1304 CrossRef CAS.
- (a) J. Hlina, H. Arp, M. Walewska, U. Florke, K. Zangger, C. Marschner and J. Baumgartner, Organometallics, 2014, 33, 7069 CrossRef CAS PubMed; (b) B. Findeis, M. Contel, L. H. Gade, M. Laguna, M. C. Gimeno, I. J. Scowen and M. McPartlin, Inorg. Chem., 1997, 36, 2386 CrossRef CAS; (c) C. Marschner, J. Baumgartner, H. Arp, K. Rasmussen, N. Siraj, P. Zark and T. Muller, J. Am. Chem. Soc., 2013, 135, 7949 CrossRef PubMed.
- (a) S. D. Bunge, O. Just and W. S. Rees Jr., Angew. Chem., Int. Ed., 2000, 39, 3082 CrossRef CAS; (b) K. Angermaier and H. Schmidbaur, Chem. Ber., 1995, 128, 817 CrossRef CAS; (c) A. Shiotani and H. Schmidbaur, J. Am. Chem. Soc., 1970, 92, 7003 CrossRef; (d) S. D. Bunge and J. L. Steele, Inorg. Chem., 2009, 48, 2701 CrossRef CAS PubMed.
- (a) F. Hupp, M. Ma, F. Kroll, J. O. C. Jimenez-Halla, R. D. Dewhurst, K. Radacki, A. Stasch, C. Jones and H. Braunschweig, Chem. – Eur. J., 2014, 20, 16888 CrossRef CAS PubMed; (b) H. Braunschweig, M. A. Celik, R. D. Dewhurst, M. Heid, F. Huppa and S. S. Sen, Chem. Sci., 2015, 6, 425 RSC.
- (a) H. Braunschweig, K. Gruss and K. Radacki, Angew. Chem., Int. Ed., 2007, 46, 7782 CrossRef CAS PubMed; (b) J. Bauer, H. Braunschweig, P. Brenner, K. Kraft, K. Radacki and K. Schwab, Chem. – Eur. J., 2010, 16, 11985 CrossRef CAS PubMed.
- T. Yoshida and S. Otsuka, J. Am. Chem. Soc., 1977, 99, 2134 CrossRef CAS.
- G. W. Bushnell, D. T. Eadie, A. Pidcock, A. R. Sam, R. D. Holmes-Smith and S. R. Stobart, J. Am. Chem. Soc., 1982, 104, 5837 CrossRef CAS.
- Z. Béni, R. Scopelliti and R. Roulet, Inorg. Chem. Commun., 2005, 8, 99 CrossRef.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916 CrossRef CAS.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed.
- See for example: (a) Y. Liu, F. Song and S. Guo, J. Am. Chem. Soc., 2006, 128, 11332 CrossRef CAS PubMed; (b) F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 818 CrossRef CAS PubMed; (c) K. J. Kilpin, W. Henderson and B. K. Nicholson, Dalton Trans., 2008, 3899 RSC; (d) J. Zhang, C.-G. Yang and C. He, J. Am. Chem. Soc., 2006, 128, 1798 CrossRef CAS PubMed.
- J. Bauer, H. Braunschweig, A. Damme and K. Radacki, Angew. Chem., Int. Ed., 2012, 51, 10030 CrossRef CAS PubMed.
- (a) S. Arndt, M. M. Hansmann, P. Motloch, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem. – Eur. J., 2017, 23, 2542 CrossRef CAS PubMed; (b) R. Uson, J. Gimeno, J. Fornies, F. Martinez and C. Fernandez, Inorg. Chim. Acta, 1982, 63, 91 CrossRef CAS; (c) H. El-Amouri, A. A. Bahsoun, J. Fischer, J. A. Osborn and M.-T. Youinou, Organometallics, 1991, 10, 3582 CrossRef.
- (a) M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917 CrossRef CAS PubMed; (b) B. E. Cowie, F. A. Tsao and D. J. H. Emslie, Angew. Chem., Int. Ed., 2015, 54, 2165 CrossRef CAS PubMed.
- J. Bauer, H. Braunschweig, P. Brenner, K. Kraft, K. Radacki and K. Schwab, Chem. – Eur. J., 2010, 16, 11985 CrossRef CAS PubMed.
- (a) J. Campos, R. Peloso and E. Carmona, Angew. Chem., Int. Ed., 2012, 51, 8255 CrossRef CAS PubMed; (b) J. Campos, L. Ortega-Moreno, S. Conejero, R. Peloso, J. López-Serrano, C. Maya and E. Carmona, Chem. – Eur. J., 2015, 21, 8883 CrossRef CAS; (c) M. Marín, J. J. Moreno, C. Navarro-Gilabert, E. Álvarez, C. Maya, R. Peloso, M. C. Nicasio and E. Carmona, Chem. – Eur. J., 2019, 25, 260 CrossRef.
- (a) R. R. Sharp and J. W. Tolan, J. Chem. Phys., 1976, 65, 522 CrossRef CAS; (b) R. R. Sharp, J. Chem. Phys., 1972, 57, 5321 CrossRef CAS.
- J. Campos, M. F. Espada, J. López-Serrano and E. Carmona, Inorg. Chem., 2013, 52, 6694 CrossRef CAS.
- (a) K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS; (b) M. Häser, R. Ahlrichs, H. P. Baron, P. Weiss and H. Horn, Theor. Chim. Acta, 1992, 83, 455 CrossRef.
- A. Uson, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, Inorg. Synth., 2007, 26, 85 CrossRef.
- H.-R. C. Jaw and W. R. Mason, Inorg. Chem., 1989, 28, 4310 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization of new compounds, crystallographic and computational details and NMR and HRMS spectra. CCDC 1897306–1897311. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt00702d |
| This journal is © The Royal Society of Chemistry 2019 |