
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium(I) complexes with carborane-substituted P,N ligands: investigations of electronic structure and dynamic behaviour†
Peter
Coburger
 a,
Gizem
Kahraman
ab,
Axel
Straube
a and
Evamarie
Hey-Hawkins
*a
a,
Gizem
Kahraman
ab,
Axel
Straube
a and
Evamarie
Hey-Hawkins
*a
aLeipzig University, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bChemistry Department, Yildiz Technical University, 34220, Istanbul, Turkey
First published on 4th March 2019
Abstract
Two distorted square-planar RhI complexes (1 and 2) were obtained from [{RhCl(η4-cod)}2] and the respective P,N ligands. The metal–ligand interaction was probed with density functional theory (DFT) and ab initio (CASSCF-NEVPT2) calculations. NMR spectroscopy proved dynamic binding behaviour of the ligands in solution. The tetradenticity of the ligands also affects the electrochemical behaviour of complexes 1 and 2 significantly. Finally, preliminary catalytic studies, namely the dehydrocoupling of dimethylamine-borane, are presented.
Introduction
In homogeneous catalysis, ligands, which combine soft and hard donor atoms, according to the HSAB principle, can act as hemilabile ligands and are, therefore, often superior to ligands with either hard or soft donor atoms.1 Often encountered are combinations of phosphorus with nitrogen or oxygen donor atoms.1b,c Alkyl and aryl groups have been predominantly used as substituents at phosphorus. In contrast, icosahedral carborane moieties have been rarely used as substituents in these ligands, despite their appealing properties.2 Especially the 1,2-dicarba-closo-dodecaborane (ortho-carborane) displays some properties which make it very attractive for ligand design such as a flexible Ccluster–Ccluster bond, its electron-withdrawing effect, the possibility of the hydridic B–H hydrogen atoms to participate in metal bonding, and the possibility of converting the closo cluster into an anionic nido carborane.2d–f Indeed, only eight different structural motifs which could be classified as potential hemilabile P,N or P,O ligands have been published (Fig. 1).3 Among those, only I and II have been employed in homogeneous catalysis so far.3a,d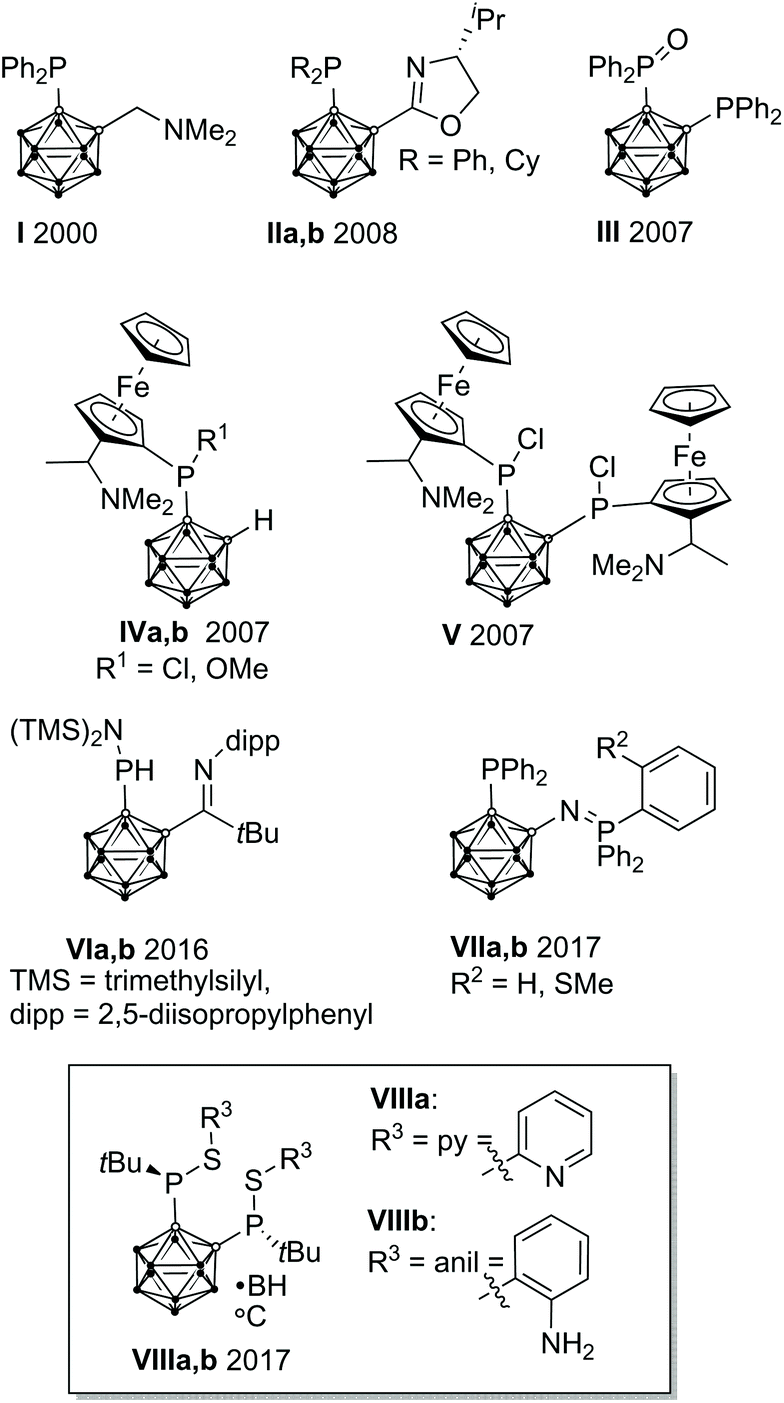 | ||
| Fig. 1 (Potentially) hemilabile ligand structures containing the ortho-carborane backbone. | ||
Previously, we reported the tetradentate ligands VIIIa,b and their coordination chemistry with CuI and AgI. Both complexes showed a tetrahedral coordination environment.3g We reasoned that the combination of VIIIa,b with transition metals which prefer square-planar coordination geometries, such as RhI, might lead to complexes with interesting structural properties and reactivity.
Results and discussion
The reaction of VIIIa and VIIIb with [{RhCl(η4-cod)}2] (cod = 1,5-cyclooctadiene) yielded the respective monomeric RhI complexes 1 and 2. When the reaction is carried out in toluene, the complexes precipitate from the reaction mixture and are obtained in moderate to good yields (Scheme 1). Single crystals of 1 and 2 suitable for X-ray crystallography were obtained from THF/hexane (1) or toluene (2) at r.t. 1 crystallises in the Sohncke group P21 and 2 crystallises in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In both complexes, the rhodium atom exhibits a distorted square-planar coordination geometry with VIIIa,b acting as tridentate P,P,N ligands (Fig. 2).
. In both complexes, the rhodium atom exhibits a distorted square-planar coordination geometry with VIIIa,b acting as tridentate P,P,N ligands (Fig. 2).
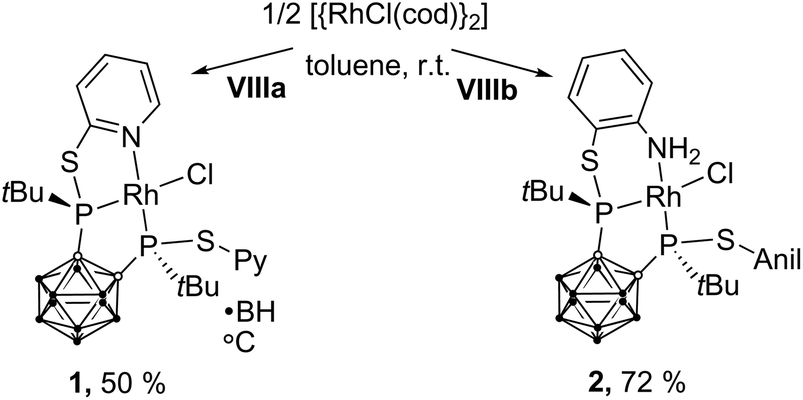 | ||
| Scheme 1 Synthesis of 1 and 2. 1 and 2 were obtained as a racemic mixture. For clarity, in this and all following figures and schemes only one enantiomer is shown. | ||
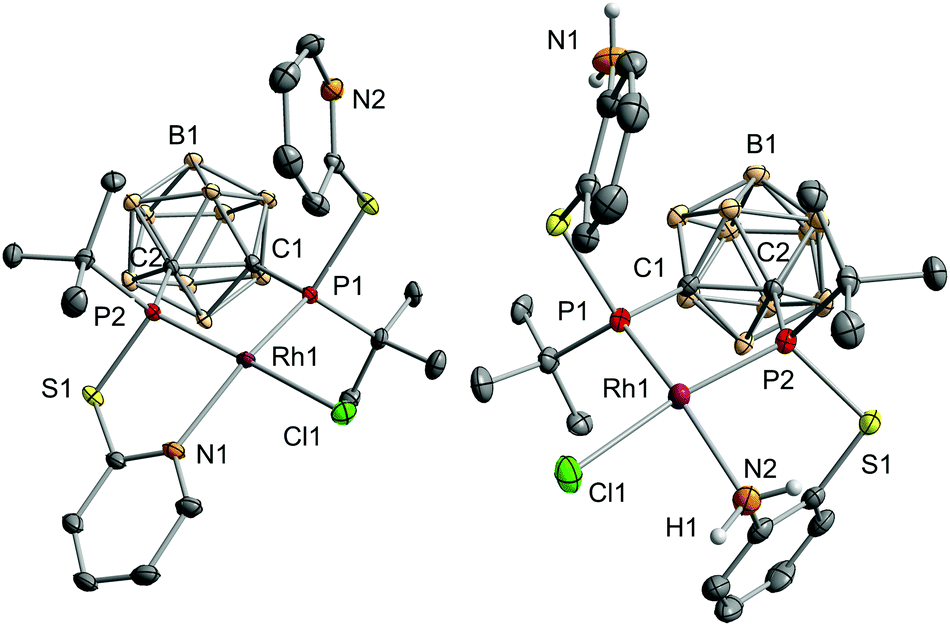 | ||
| Fig. 2 Molecular structures of (RP, RP)-1 (left) and (SP, SP)-2 (right) in the solid state with ellipsoids drawn at the 50% probability level. All hydrogen atoms, except for those at the nitrogen atoms in 2, are omitted for clarity. Selected bond lengths [Å] and angles [°]: 1: Rh1–P1: 2.2028(6), Rh1–P2: 2.1417(6), Rh1–N1: 2.160(2), Rh1–Cl1: 2.3930(6), C1–C2: 1.702(3), P1–Rh1–P2: 90.77(2), P1–Rh1–N1: 156.06(5), P1–Rh1–Cl1: 99.45(2), P2–Rh1–N2: 84.43(6), P2–Rh1–Cl1: 165.09(2). 2: Rh1–P1: 2.1958(8), Rh1–P2: 2.1605(9), Rh1–N2: 2.155(3), Rh1–Cl1: 2.396(1), C1–C2: 1.701(4), P1–Rh1–P2: 91.20(3), P1–Rh1–N2: 160.48(8), P1–Rh1–Cl1: 101.68(3), P2–Rh1–N2: 88.09(8), P2–Rh1–Cl1: 166.21(3). | ||
Especially the P–Rh–N and P–Rh–Cl bond angles deviate significantly from linearity (e.g.1: P1–Rh1–N1: 156.06(5)°, P2–Rh1–Cl1: 165.09(2)°, Fig. 2) leading to a geometry index τ4 of 0.27 for 1 and 0.23 for 2.4 In fact, these values indicate that 1 and 2 adopt the largest deviation from the ideal square-planar geometry among all RhI complexes featuring two phosphorus donor atoms in a cis arrangement and one nitrogen and one chlorine atom (see the ESI† for references). In contrast to the bond angles, the Rh–P, Rh–N and Rh–Cl bond lengths in both complexes are within the range of standard values.5 The phosphorus atoms trans to the chlorine atoms form significantly shorter Rh–P bonds than the phosphorus atoms cis to chlorine (e.g.1: Rh1–P1: 2.2028(6), Rh1–P2: 2.1417(6), Fig. 2). This could be due to the trans effect or, as reasoned for a structurally related complex, [RhCl(L)] (L = 2-(2-{[3-(di-tert-butylphosphanyl)propyl]-(phenyl)phosphanyl}ethyl)pyridine, partly due to the steric repulsion between Cl1 and the tert-butyl group at P1.5a
We probed the metal–ligand interactions in 1 and 2 further by ab initio ligand field theory (AILFT as implemented in the ORCA program package)6 calculations and energy decomposition analyses at the DFT level (see the ESI† for details). Despite their distorted structures, 1 and 2 exhibit d orbital splittings similar to an ideal square-planar complex (Fig. 3, left), however the dxz and dyz orbitals are not degenerated in our complexes. Thereby, the splitting is slightly larger in 1 as indicated by the higher dx2−y2–dz2 separation of 3.58 eV (2: 3.53 eV). These values indicate a slightly stronger metal–ligand interaction in 1 as underpinned by the smaller Racah parameter B (649 cm−1vs. 657 cm−1 in 2). Additionally, our energy decomposition analyses revealed that the interaction energy of the ligand with the rhodium centre is higher by 2.7 kcal mol−1 in 1.
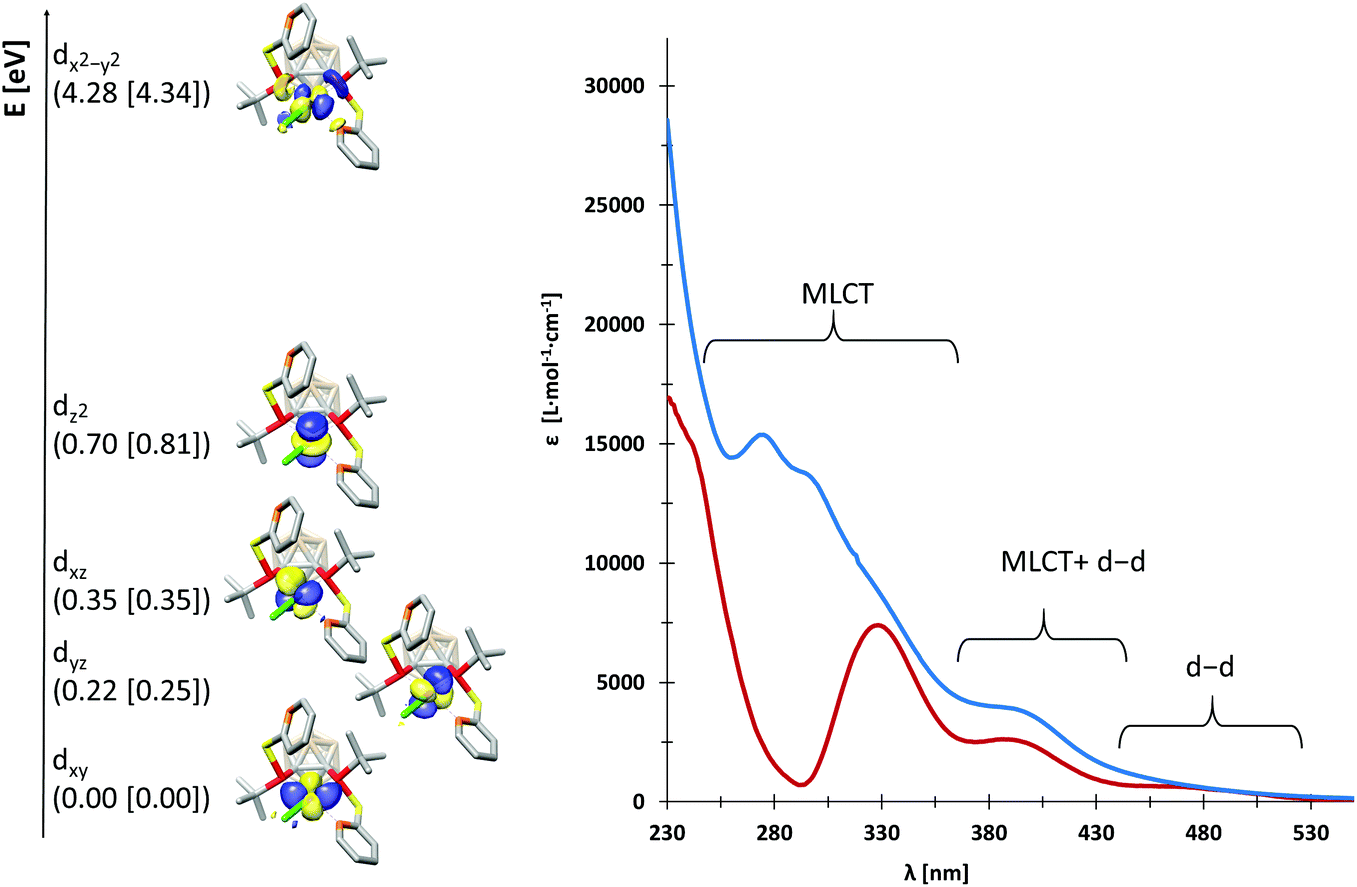 | ||
| Fig. 3 Left: Orbital scheme for the d orbitals in 1 and 2 (energies for 2 are given in brackets). Exemplarily, the d orbitals for 1 are shown with an isosurface value of 0.05. Right: UV-vis spectra of 1 (blue) and 2 (red) in diethyl ether. | ||
Due to their different nitrogen donor groups, the overall electronic structure of 1 and 2 is rather different as shown by our combined UV-vis spectroscopy and TDDFT investigations (see the ESI†). In diethyl ether, the visible part of the spectra (390 nm to 700 nm) is rather similar for both complexes, exhibiting one metal-to-ligand charge transfer (MLCT) band and several d–d transitions. Both spectra predominantly exhibit MLCT bands between 230 and 390 nm, however, their overall shape is significantly different. In particular, the nature of the MLCT bands in 1 and 2 highlights their different electronic structure: in 1, the MLCT bands largely consist of d–π* transitions from d orbitals on rhodium to antibonding orbitals of the aromatic systems, while in 2, d–σ* transitions from d orbitals on rhodium to P–C and P–S antibonding orbitals dominate. These findings are in accordance with our previous theoretical investigations on the ligands VIIIa,b which showed that the energetically lowest acceptor orbitals of VIIIa are located at the pyridine rings, whereas the acceptor orbitals of VIIIb reside at the phosphorus atoms.3g
Having investigated the electronic structure of 1 and 2, we moved on to probe their behaviour in solution. Based on their temperature-dependent NMR spectra, both complexes undergo dynamic processes, which involve the phosphorus atoms and nitrogen donor groups. At low temperatures, the 31P{1H} NMR spectrum consists of two doublets of doublets (1 (−40 °C): 150.5 (1JPRh = 219 Hz) and 158.8 (1JPRh = 186 Hz) ppm, 2JPP = 38 Hz; 2 (−5 °C): 162.4 (1JPRh = 230 Hz) and 195.7 (1JPRh = 216 Hz) ppm, 2JPP = 40 Hz). Due to the higher P–Rh coupling constants, the multiplets at 158.8 and 195.7 ppm are tentatively assigned to P2, which forms a shorter P–Rh bond than P1 in both complexes (Fig. 2). For 1, a broad singlet is observed at 154.4 ppm (νFWHM = 530 Hz) at 60 °C (see the ESI†); in contrast, the signals in the 31P{1H} NMR spectrum of 2 just broaden at higher temperature. Together with an analysis of the temperature-dependent 1H NMR spectra of 1 and 2, this observation proves a higher activation barrier for the dynamic processes in 2 compared to 1.
In order to get further insight, we calculated the mechanism of the dynamic process for both complexes at the DFT level (see the ESI† for details), which is depicted for 1 in Scheme 2. A similar mechanism was proposed for PdII and PtII complexes of a structurally related ligand, 1,3-bis{methyl[2-(pyridin-2-yl)ethyl]phosphanyl}propane.5a The mechanism involves the formation of a distorted square-pyramidal intermediate 1-qpym, in which VIIIa acts as a tetradentate ligand. 1-qpym isomerises to 1-qpym′via a rocking vibration involving the chlorine atom. Finally, one nitrogen donor group in 1-qpym′ dissociates to form 1′. This process equilibrates both the phosphorus atoms and pyridyl groups in 1. A very similar mechanism was determined for complex 2 (see the ESI† for details). The first step (a, Scheme 2) is rate determining for both complexes. In agreement with our NMR spectroscopy experiments, the activation energy for the formation of the square-pyramidal complexes 1-qpym or 2-qpym, respectively, is considerably higher in the case of 2 (19.4 kcal mol−1vs. 11.5 kcal mol−1). Our calculations show that the higher energy barrier in 2 can be traced back to steric effects and is caused by the structural rearrangements necessary to adopt the tetra-coordinating geometry of the ligand in 2-qpym (see the ESI†).
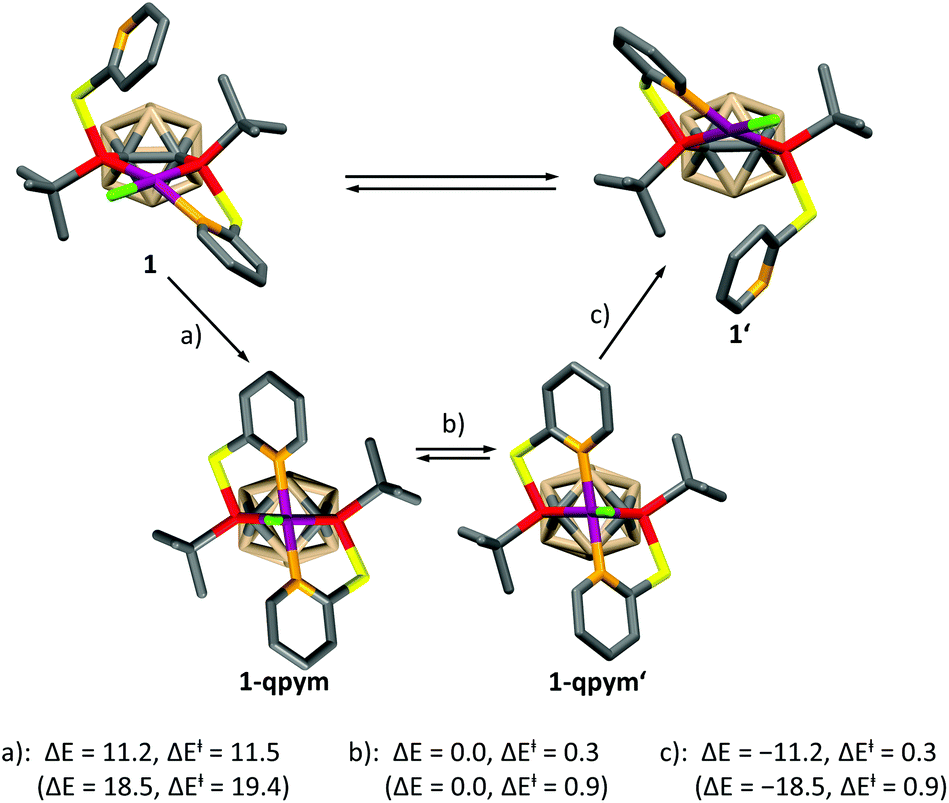 | ||
| Scheme 2 Calculated mechanism of the dynamic processes in 1. Energy differences are given in kcal mol−1. Energy differences are given relative to 1 (or 2) in step (a), relative to 1-qpym (or 2-qpym) in step (b) and relative to 1-qpym′ in step (c). Values in parentheses correspond to the values calculated for the respective structures with the ligand VIIIb. Hydrogen atoms have been omitted for clarity. | ||
The tetra-coordination of the ligands in 1 and 2 also has a great influence on their electrochemistry. Both complexes undergo an irreversible reduction in the typical range of carborane derivatives (1−/1: Epeak(cathodic) = −1.88 V, 2−/2: Epeak(cathodic) = −1.95 V, vs. FcH/[FcH]+),7 and our calculations confirm that the reduction of 1 and 2 occurs at the carborane backbone based on the spin densities of the monoanions 1− and 2− (Fig. 4). The reduction is accompanied by a change to a square-pyramidal structure in 1−, whereas the distorted square-planar structure is retained in 2− (see the ESI† for details). For both 1 and 2 the calculated reduction potentials agree well with the measured cathodic peak potentials (1−/1: −2.14 V, 2−/2: −2.07 V, vs. FcH/[FcH]+, see the ESI† for details).
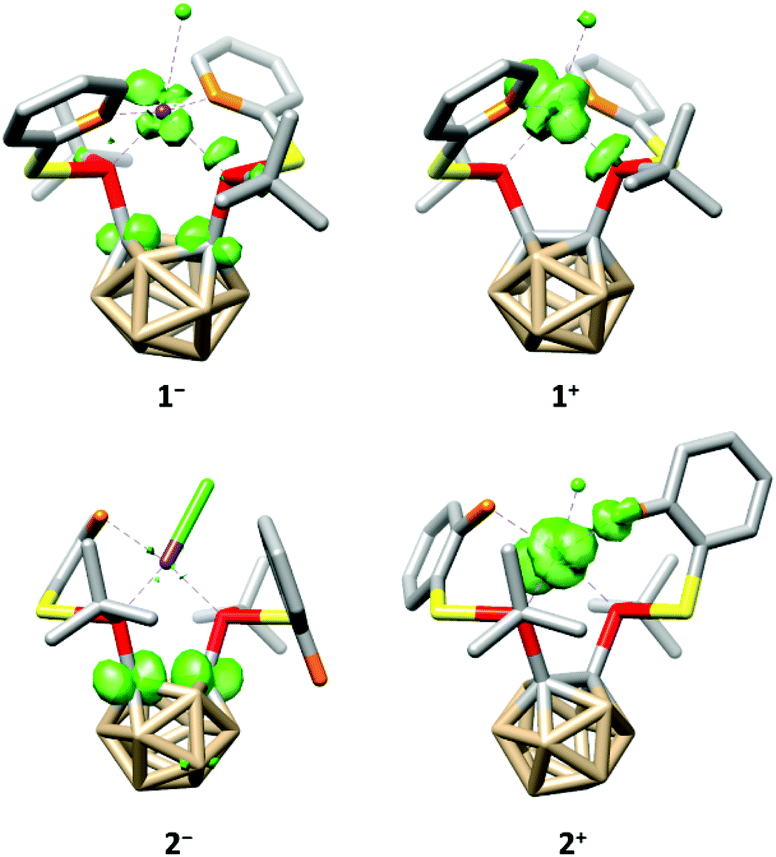 | ||
| Fig. 4 Calculated spin densities (green, surface isovalue = 0.05) and structures of 1−, 1+, 2− and 2+. Hydrogen atoms have been omitted for clarity. | ||
At higher potentials, 1 is oxidised in two steps. While the first event appears to be quasi-reversible, the second is irreversible (1/1+: E1/2 = −588 mV, 1+/12+: Epeak(anodic) = −296 mV, vs. FcH/[FcH]+). Complex 2 behaves similarly; however, both oxidation events are irreversible and appear at significantly higher potentials (2/2+: Epeak(anodic) = −27 mV, 2+/22+: Epeak(anodic) = 149 mV, vs. FcH/[FcH]+). As for the reductions, our calculated oxidation potentials for the first oxidation events of 1 and 2 are in good agreement8 with the experimental values (1/1+: −800 mV, 2/2+: −150 mV, vs. FcH/[FcH]+, see the ESI† for details) and reveal that these oxidations take place at the rhodium centre (Fig. 4). Both oxidation events in 1 and 2 are accompanied by a structural change to a distorted square-pyramidal structure, again underpinning the influence of the tetra-coordination of the ligands on the properties of 1 and 2.
Finally, we explored the use of 1 and 2 as homogeneous catalysts. As a model reaction, we investigated the dehydrocoupling of dimethylamine-borane (3) to give the diazadiboretidine derivative 4 and molecular hydrogen (Scheme 3) in toluene-d8. This reaction was initially conducted with [{RhCl(cod)}2] as catalyst.9 Since then, a variety of transition metal complexes, including zirconium, iridium and cobalt, was shown to efficiently catalyse this reaction.10 Our results indicate that both 1 and 2 convert 3 completely and at a comparable rate like [{RhCl(cod)}2] (Scheme 3, see the ESI† for details). However, they are much less selective, especially in the case of 1, producing a significant amount of the aminoborane HB(NMe2)2 and an unidentified side product. However, this side product decomposes slowly under the reaction conditions, whereas the desired cyclic product 4 is stable, leading to 4 as the only product detectable by 11B NMR spectroscopy (1: after 72 h; 2: after 48 h). Due to the formation of this side product, the yield of 4 is reduced compared to [{RhCl(cod)}2]. The different performance of the catalysts 1 and 2 might be explained by the different nitrogen-donor groups present in both complexes. In 2, the aniline groups present a much stronger Lewis base than the pyridine groups in 1. Therefore, the aniline groups in 2 might interact more strongly with the Lewis-acidic substrate (dimethylamine-borane) leading to a more selective reaction. However, further kinetic and computational studies would be required for definite conclusions.
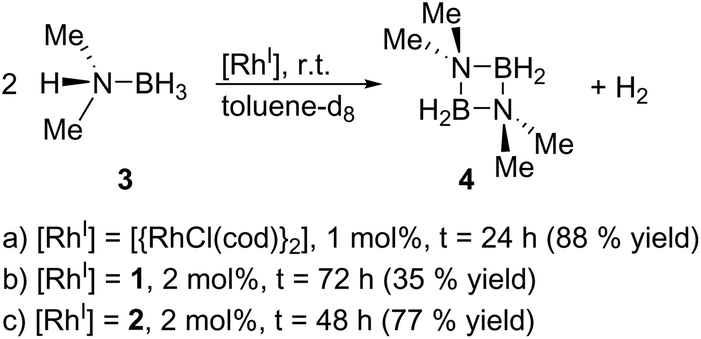 | ||
| Scheme 3 Performance of [{RhCl(cod)}2], 1 and 2 in the dehydrocoupling of dimethylamine-borane (3). | ||
Conclusions
In summary, we have prepared and thoroughly characterised two distorted square-planar RhI complexes with tetradentate, hemilabile carborane-substituted P,N ligands. The coordination mode of these ligands strongly influences the dynamic behaviour in solution as well as the redox properties of 1 and 2. Both complexes catalyse the dehydrocoupling reactions of amine-boranes, but are less active and selective than [{RhCl(cod)}2]. However, due to their significantly distorted structure and hemilabile ligands, they could be more promising in other reactions. Respective investigations are currently underway in our group.Experimental
Materials and methods
All reactions and manipulations were carried out under a nitrogen atmosphere by using standard Schlenk techniques unless stated otherwise. Solvents were either obtained from an MBraun Solvent Purification System, or dried and stored according to common procedures.11 Dimethylamine-borane is commercially available and was used as obtained. [{RhCl(cod)}2] and ligands VIIIa and VIIIb were synthesised according to literature procedures.3g,12 NMR spectra were recorded with a Bruker AVANCE DRX 400 MHz NMR spectrometer at room temperature. All isolated compounds were measured in deuterated tetrahydrofuran (THF-d8). Tetramethylsilane was used as internal standard for 1H NMR spectra. 85% H3PO4 was used as external standard for 31P{1H} NMR spectra, and BF3·OEt2 was used as external standard for 11B{1H} NMR spectra. NMR spectra were recorded at the following frequencies: 1H: 400.13 MHz, 11B: 128.38 MHz, 31P: 161.99 MHz. IR spectra were recorded as ATR spectra in the range of 4000–640 cm−1 with a Bruker TENSOR 27 IR spectrometer.Crystallography
X-ray diffraction studies on 1 were performed with an Agilent Technologies SuperNova diffractometer using Cu-Kα radiation and ω-scan rotation. X-ray diffraction studies on 2 were performed with an Oxford Diffraction CCD Xcalibur-S diffractometer using Mo-Kα radiation and ω-scan rotation. Data were collected using ω steps accumulating area detector frames (data reduction was carried out with CrysAlis Pro).13 All data were corrected for Lorentz polarisation and long-term intensity fluctuations. Absorption effects were corrected based on analytical corrections for 1 and on the basis of multiple equivalent reflections for 2. Structures were solved by direct methods and refined by full-matrix least-squares techniques against F2 using the SHELX program package.14 Hydrogen atoms were assigned riding isotropic displacement parameters and constrained to idealised geometries. Structure figures were generated with DIAMOND (Table 1).15 The crystal structures of 1 and 2 have been deposited in the CCDC (1: 1896568, 2: 1896567).†| Compound | 1 | 2 |
|---|---|---|
| Empirical formula | C20H36B10ClN2P2RhS2 | C22H40B10ClN2P2RhS2 |
| Formula weight | 677.03 | 705.08 |
| T [K] | 123 | 130 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21 |
P |
| a [Å] | 9.5859(2) | 9.4717(2) |
| b [Å] | 13.2274(2) | 10.4441(3) |
| c [Å] | 11.9819(2) | 16.3936(4) |
| α | 90 | 88.649(2) |
| β | 103.849(2) | 75.070(2) |
| γ | 90 | 89.069(2) |
| V [Å3] | 1475.10(5) | 1566.44(7) |
| Z | 2 | 2 |
| ρ calcd [g cm−3] | 1.524 | 1.495 |
| θ max | 30.679 | 28.281 |
| F(000) | 688 | 720 |
| Reflns collected | 9062 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 286 286 |
| Independent reflns | 4139 | 7337 |
| R 1/wR2 [I > 2σ(I)] | 0.0150/0.0387 | 0.0415/0.0763 |
| R 1/wR2 (all data) | 0.0150/0.0387 | 0.0615/0.0823 |
| Largest diff. peak/hole/(e Å−3) | 0.289/−0.350 | 0.775/−0.496 |
Synthetic procedures
[{RhCl(cod)}2] (46 mg, 0.09 mmol, 0.500 eq.) and the respective ligand (1: ligand VIIIa: 101 mg, 0.19 mmol, 1.06 eq.; 2: ligand VIIIb: 100 mg, 0.18 mmol, 1.00 eq.) were dissolved in toluene and stirred for 2 d. The resulting suspensions were filtered, and the residues were washed with toluene (2 × 2 mL) to afford 1 (64 mg, 50% yield) or 2 (90 mg, 72% yield) as orange solids. Single crystals of 1 suitable for X-ray diffraction studies were obtained by layering a saturated THF solution with n-hexane. In the case of 2, single crystals were obtained directly from the reaction mixture when stirring was discontinued after the reactants had dissolved. 1: IR:![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3063 (m, νC–H), 3043 (m, νC–H), 3018 (m, νC–H), 3002 (m, νC–H), 2981 (m, νC–H), 2958 (m, νC–H), 2957 (m, νC–H), 2902 (m, νC–H), 2863 (m, νC–H), 2648 (s, νB–H), 2620 (s, νB–H), 2611 (s, νB–H), 2594 (s, νB–H), 2569 (vs, νB–H), 2008 (w), 1981 (w), 1960 (w), 1892 (w), 1872 (w), 1847 (w), 1587 (m), 1582 (m), 1576 (m), 1449 (s), 1415 (m), 1394 (m), 1364 (m), 1302 (w), 1262 (w), 1167 (m), 1164 (m), 1111 (m), 1070 (m), 1009 (m), 987 (m), 904 (w), 888 (w), 863 (w), 826 (w), 801 (s), 776 (s), 733 (w), 720 (w), 649 (w) cm−1. UV-vis (Et2O): λmax = 398 (ε = 3713 cm L mol−1), 298 (shoulder, ε = 13497 cm L mol−1), 273 (ε = 15375 cm L mol−1) nm. Anal. calcd for C20H36B10ClN2P2RhS2: C 35.48, H 5.36, N 4.14. Found: C 35.56, H 5.10, N 3.91. 1H NMR: δ = 1.33 (bs, 18H, CH3), 1.28–3.60 (m, 10H, BH), 7.25 (ddd, 3JHH = 7.3 Hz, 3JHH = 5.5 Hz, 4JHH = 1.2 Hz, 2H, arom. CH), 7.75 (td, 3JHH = 7.8 Hz, 4JHP = 1.9 Hz, 2H, arom. CH), 8.36 (bs, 2H, arom. CH), 8.96 (bs, 2H, arom. CH) ppm. 1H{11B} NMR δ = 1.31 (bs, 18H, CH3), 2.41– 2.45 (m, 3H, BH), 2.54 (s, 2H, BH), 2.63 (s, 3H, BH), 3.45 (s 2H, BH), 7.25–7.28 (m, 2H, arom. CH), 7.76 (td, 3JHH = 7.8 Hz, 4JHP = 1.9 Hz, 2H, arom. CH), 8.39 (bs, 2H, arom. CH), 8.98 (bs, 2H, arom. CH) ppm.11B{1H} NMR: δ = −9.3 (bs, 6B), −4.6 (bs, 2B), −1.3 (bs, 2B) ppm. 11B NMR: δ = −10.0 to −8.7 (m, 6B), −4.6 (d, 1JBH = 144 Hz, 2B), −1.3 (d, 1JBH = 153 Hz, 2B) ppm. 31P{1H} NMR (r.t.): δ = 151.8 (bs), 159.9 (bs) ppm. 31P{1H} NMR (−40 °C): δ = 150.5 (dd, 1JPRh = 219 Hz, 2JPP = 38 Hz), 158.8 (dd, 1JPRh = 186 Hz, 2JPP = 38 Hz) ppm. 2: IR: = 3484 (s, νN–H), 3385 (m, νN–H), 3313 (m, νN–H), 3245 (m, νN–H), 3015 (m, νC–H), 3022 (m, νC–H), 3001 (m, νC–H), 2947 (m, νC–H), 2927 (m, νC–H), 2866 (m, νC–H), 2631 (s, νB–H), 2557 (vs, νB–H), 1609 (s), 1588 (m), 1545 (s), 1477 (s), 1446 (m), 1395 (m), 1257 (m), 1171 (m), 1158 (m), 1141 (w), 1068 (s), 1015 (s), 970 (w), 931 (m), 904 (w), 858 (w), 802 (m), 745 (vs), 676 (w) cm−1. UV-vis (Et2O): λmax = 471 (ε = 622 cm L mol−1), 389 (ε = 2611 cm L mol−1), 328 (ε = 7393 cm L mol−1), 243 (shoulder, ε = 14637 cm L mol−1) nm. Anal. calcd for C22H40B10ClN2P2RhS2: C 37.48, H 5.72, N 3.97. Found: C 37.86, H 5.63, N 3.91. 1H NMR: δ = 1.20 (bd, 3JHP = 15.4 Hz, 18H, CH3), 1.28–3.44 (m, 10H, BH), 5.01 (bs, 2H, NH), 5.46 (bs, 1H, NH), 5.75 (bs, 1H, NH), 6.59–6.64 (m, 2H, arom. CH), 7.01 (bs, 2H, arom. CH), 7.15 (bd, 3JHH = 8.1 Hz, 1H, arom. CH), 7.25–7.27 (m, 1H, arom. CH), 7.48 (bd, 3JHH = 7.7 Hz, 1H, arom. CH), 8.54 (bd, 3JHH = 7.8 Hz, 1H, arom. CH) ppm. 1H{11B} NMR: δ = 1.22 (bd, 3JHP = 15.4 Hz, 18H, CH3), 2.16 (bs, 1H, BH), 2.29–2.35 (m, 3H, BH), 2.46 (bs, 2H, BH), 2.68 (bs, 3H, BH), 2.88 (bs, 1H, BH), 5.01 (bs, 2H, NH), 5.47 (bs, 1H, NH), 5.76 (bs, 1H, NH), 6.62 (bs, 2H, arom. CH), 7.02 (bs, 2H, arom. CH), 7.16 (bs, 1H, arom. CH), 7.28 (bs, 1H, arom. CH), 7.49 (bs, 1H, arom. CH), 8.58 (bs, 1H, arom. CH) ppm. 11B{1H} NMR: δ = −11.3 (bs, 4B), −8.3 (bs, 2B), −4.8 (bs, 1B), −2.3 (bs, 3B) ppm. 11B NMR: δ = −14.0 to −8.2 (m, 6B), −5.4 to −1.8 (m, 4B) ppm. 31P{1H} NMR (r.t.): δ = 161.9 (dd, 1JPRh = 230 Hz, 2JPP = 39 Hz), 195.5 (dd, 1JPRh = 218 Hz, 2JPP = 44 Hz) ppm. 31P{1H} NMR (−5 °C): δ = 162.4 (dd, 1JPRh = 230 Hz, 2JPP = 40 Hz), 195.7 (dd, 1JPRh = 216 Hz, 2JPP = 41 Hz) ppm.
= 3063 (m, νC–H), 3043 (m, νC–H), 3018 (m, νC–H), 3002 (m, νC–H), 2981 (m, νC–H), 2958 (m, νC–H), 2957 (m, νC–H), 2902 (m, νC–H), 2863 (m, νC–H), 2648 (s, νB–H), 2620 (s, νB–H), 2611 (s, νB–H), 2594 (s, νB–H), 2569 (vs, νB–H), 2008 (w), 1981 (w), 1960 (w), 1892 (w), 1872 (w), 1847 (w), 1587 (m), 1582 (m), 1576 (m), 1449 (s), 1415 (m), 1394 (m), 1364 (m), 1302 (w), 1262 (w), 1167 (m), 1164 (m), 1111 (m), 1070 (m), 1009 (m), 987 (m), 904 (w), 888 (w), 863 (w), 826 (w), 801 (s), 776 (s), 733 (w), 720 (w), 649 (w) cm−1. UV-vis (Et2O): λmax = 398 (ε = 3713 cm L mol−1), 298 (shoulder, ε = 13497 cm L mol−1), 273 (ε = 15375 cm L mol−1) nm. Anal. calcd for C20H36B10ClN2P2RhS2: C 35.48, H 5.36, N 4.14. Found: C 35.56, H 5.10, N 3.91. 1H NMR: δ = 1.33 (bs, 18H, CH3), 1.28–3.60 (m, 10H, BH), 7.25 (ddd, 3JHH = 7.3 Hz, 3JHH = 5.5 Hz, 4JHH = 1.2 Hz, 2H, arom. CH), 7.75 (td, 3JHH = 7.8 Hz, 4JHP = 1.9 Hz, 2H, arom. CH), 8.36 (bs, 2H, arom. CH), 8.96 (bs, 2H, arom. CH) ppm. 1H{11B} NMR δ = 1.31 (bs, 18H, CH3), 2.41– 2.45 (m, 3H, BH), 2.54 (s, 2H, BH), 2.63 (s, 3H, BH), 3.45 (s 2H, BH), 7.25–7.28 (m, 2H, arom. CH), 7.76 (td, 3JHH = 7.8 Hz, 4JHP = 1.9 Hz, 2H, arom. CH), 8.39 (bs, 2H, arom. CH), 8.98 (bs, 2H, arom. CH) ppm.11B{1H} NMR: δ = −9.3 (bs, 6B), −4.6 (bs, 2B), −1.3 (bs, 2B) ppm. 11B NMR: δ = −10.0 to −8.7 (m, 6B), −4.6 (d, 1JBH = 144 Hz, 2B), −1.3 (d, 1JBH = 153 Hz, 2B) ppm. 31P{1H} NMR (r.t.): δ = 151.8 (bs), 159.9 (bs) ppm. 31P{1H} NMR (−40 °C): δ = 150.5 (dd, 1JPRh = 219 Hz, 2JPP = 38 Hz), 158.8 (dd, 1JPRh = 186 Hz, 2JPP = 38 Hz) ppm. 2: IR: = 3484 (s, νN–H), 3385 (m, νN–H), 3313 (m, νN–H), 3245 (m, νN–H), 3015 (m, νC–H), 3022 (m, νC–H), 3001 (m, νC–H), 2947 (m, νC–H), 2927 (m, νC–H), 2866 (m, νC–H), 2631 (s, νB–H), 2557 (vs, νB–H), 1609 (s), 1588 (m), 1545 (s), 1477 (s), 1446 (m), 1395 (m), 1257 (m), 1171 (m), 1158 (m), 1141 (w), 1068 (s), 1015 (s), 970 (w), 931 (m), 904 (w), 858 (w), 802 (m), 745 (vs), 676 (w) cm−1. UV-vis (Et2O): λmax = 471 (ε = 622 cm L mol−1), 389 (ε = 2611 cm L mol−1), 328 (ε = 7393 cm L mol−1), 243 (shoulder, ε = 14637 cm L mol−1) nm. Anal. calcd for C22H40B10ClN2P2RhS2: C 37.48, H 5.72, N 3.97. Found: C 37.86, H 5.63, N 3.91. 1H NMR: δ = 1.20 (bd, 3JHP = 15.4 Hz, 18H, CH3), 1.28–3.44 (m, 10H, BH), 5.01 (bs, 2H, NH), 5.46 (bs, 1H, NH), 5.75 (bs, 1H, NH), 6.59–6.64 (m, 2H, arom. CH), 7.01 (bs, 2H, arom. CH), 7.15 (bd, 3JHH = 8.1 Hz, 1H, arom. CH), 7.25–7.27 (m, 1H, arom. CH), 7.48 (bd, 3JHH = 7.7 Hz, 1H, arom. CH), 8.54 (bd, 3JHH = 7.8 Hz, 1H, arom. CH) ppm. 1H{11B} NMR: δ = 1.22 (bd, 3JHP = 15.4 Hz, 18H, CH3), 2.16 (bs, 1H, BH), 2.29–2.35 (m, 3H, BH), 2.46 (bs, 2H, BH), 2.68 (bs, 3H, BH), 2.88 (bs, 1H, BH), 5.01 (bs, 2H, NH), 5.47 (bs, 1H, NH), 5.76 (bs, 1H, NH), 6.62 (bs, 2H, arom. CH), 7.02 (bs, 2H, arom. CH), 7.16 (bs, 1H, arom. CH), 7.28 (bs, 1H, arom. CH), 7.49 (bs, 1H, arom. CH), 8.58 (bs, 1H, arom. CH) ppm. 11B{1H} NMR: δ = −11.3 (bs, 4B), −8.3 (bs, 2B), −4.8 (bs, 1B), −2.3 (bs, 3B) ppm. 11B NMR: δ = −14.0 to −8.2 (m, 6B), −5.4 to −1.8 (m, 4B) ppm. 31P{1H} NMR (r.t.): δ = 161.9 (dd, 1JPRh = 230 Hz, 2JPP = 39 Hz), 195.5 (dd, 1JPRh = 218 Hz, 2JPP = 44 Hz) ppm. 31P{1H} NMR (−5 °C): δ = 162.4 (dd, 1JPRh = 230 Hz, 2JPP = 40 Hz), 195.7 (dd, 1JPRh = 216 Hz, 2JPP = 41 Hz) ppm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Studienstiftung des deutschen Volkes (doctoral fellowships to P. C. and A. S.), TUBITAK 2214-A (research fellowship to G. K.) and the Graduate School BuildMoNa is gratefully acknowledged. We thank Prof. R. Wolf (Regensburg University) for X-ray measurement time.Notes and references
- (a) P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193–195, 499–556 CrossRef CAS; (b) P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 ( Angew. Chem. , 2001 , 113 , 702–722 ) CrossRef CAS; (c) V. V. Grushin, Chem. Rev., 2004, 104, 1629–1662 CrossRef CAS PubMed.
- (a) S. P. Fisher, A. W. Tomich, S. O. Lovera, J. F. Kleinsasser, J. Guo, M. J. Asay, H. M. Nelson and V. Lavallo, Chem. Rev., 2019 DOI:10.1021/acs.chemrev.8b00551; (b) B. J. Eleazer, M. D. Smith, A. A. Popov and D. V. Peryshkov, Chem. Sci., 2018, 9, 2601–2608 RSC; (c) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed; (d) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (e) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (f) A. R. Popescu, F. Teixidor and C. Viñas, Coord. Chem. Rev., 2014, 269, 54–84 CrossRef CAS.
- (a) H.-S. Lee, J.-Y. Bae, J. Ko, Y. S. Kang, H. S. Kim, S.-J. Kim, J.-H. Chung and S. O. Kang, J. Organomet. Chem., 2000, 614–615, 83–91 CrossRef CAS; (b) J. Dou, D. Zhang, D. Li and D. Wang, Eur. J. Inorg. Chem., 2007, 2007, 53–59 CrossRef; (c) S. Tschirschwitz, P. Lönnecke and E. Hey-Hawkins, Organometallics, 2007, 26, 4715–4724 CrossRef CAS; (d) J.-D. Lee, T. T. Co, T.-J. Kim and S. O. Kang, Synlett, 2009, 771–774 CAS; (e) A. Herrera, A. Briceño, T. Gonzalez, A. Linden, F. W. Heinemann, G. Agrifoglio, J. Pastrán and R. Dorta, Tetrahedron: Asymmetry, 2016, 27, 759–767 CrossRef CAS; (f) P. Crujeiras, J. L. Rodríguez-Rey and A. Sousa-Pedrares, Eur. J. Inorg. Chem., 2017, 2017, 4653–4667 CrossRef CAS; (g) P. Coburger, J. Schulz, J. Klose, B. Schwarze, M. B. Sárosi and E. Hey-Hawkins, Inorg. Chem., 2017, 56, 292–304 CrossRef CAS PubMed.
- τ 4 = 0 represents an ideal square-planar complex, and τ4 = 1 represents the case of an ideal tetrahedral complex: L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) A. Heßler, J. Fischer, S. Kucken and O. Stelzer, Chem. Ber., 1994, 127, 481–488 CrossRef; (b) F. W. B. Einstein, R. H. Jones, Y. Zhang and D. Sutton, Inorg. Chem., 1988, 27, 1004–1010 CrossRef CAS; (c) C. Hahn, J. Sieler and R. Taube, Polyhedron, 1998, 17, 1183–1193 CrossRef CAS; (d) F. Lorenzini, B. O. Patrick and B. R. James, Inorg. Chim. Acta, 2008, 361, 3199–3204 CrossRef CAS; (e) Y. Wang, B. Zheng, Y. Pan, C. Pan, L. He and K.-W. Huang, Dalton Trans., 2015, 15111–15115 RSC; (f) H.-C. Böttcher and P. Mayer, Z. Anorg. Allg. Chem., 2016, 642, 299–301 CrossRef.
- (a) M. Atanasov, D. Ganyushin, D. A. Pantazis, K. Sivalingam and F. Neese, Inorg. Chem., 2011, 50, 7460–7477 CrossRef CAS PubMed; (b) S. K. Singh, J. Eng, M. Atanasov and F. Neese, Coord. Chem. Rev., 2017, 344, 2–25 CrossRef CAS; (c) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (d) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- (a) M.-H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407–7412 CrossRef CAS; (b) Y. Shimodaira, T. Miura, A. Kudo and H. Kobayashi, J. Chem. Theory Comput., 2007, 3, 789–795 CrossRef CAS PubMed; (c) L. Yan, Y. Lu and X. Li, Phys. Chem. Chem. Phys., 2016, 18, 5529–5536 RSC.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, Chem. Commun., 2001, 962–963 RSC.
- (a) O. J. Metters, S. R. Flynn, C. K. Dowds, H. A. Sparkes, I. Manners and D. F. Wass, ACS Catal., 2016, 6, 6601–6611 CrossRef CAS; (b) E. M. Titova, E. S. Osipova, A. A. Pavlov, O. A. Filippov, S. V. Safronov, E. S. Shubina and N. V. Belkova, ACS Catal., 2017, 7, 2325–2333 CrossRef CAS; (c) T. M. Maier, S. Sandl, I. G. Shenderovich, A. Jacobi von Wangelin, J. J. Weigand and R. Wolf, Chem. – Eur. J., 2019, 25, 238–245 CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 2009 Search PubMed.
- G. Giordano, R. H. Crabtree, R. M. Heintz, D. Forster and D. E. Morris, Inorg. Synth., 2007, 28, 88–90 CrossRef.
- CrysAlis Pro: Data Collection and Data Reduction Software Package, Agilent Technologies.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- K. Brandenburg and H. Putz, DIAMOND, Crystal Impact GbR, Bonn, Germany, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR investigations, computational details. CCDC 1896567 and 1896568. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt00628a |
| This journal is © The Royal Society of Chemistry 2019 |