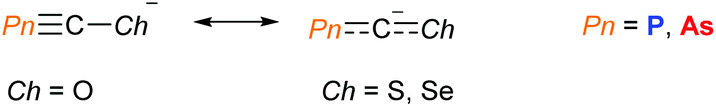DOI:
10.1039/C9DT00574A
(Frontier)
Dalton Trans., 2019,
48, 4460-4466
Heavier pnictogens – treasures for optical electronic and reactivity tuning
Received
7th February 2019
, Accepted 19th February 2019
First published on 20th February 2019
Abstract
We highlight recent advances in organopnictogen chemistry contrasting the properties of lighter and heavier pnictogens. Exploring new bonding situations, discovering unprecedented reactivities and producing fascinating opto-electronic materials are some of the most prominent directions of current organopnicogen research. Expanding the chemical toolbox towards the heavier group 15 elements will continue to create new opportunities to tailor molecular properties for small molecule activation/reactivity and materials applications alike. This frontier article illustrates the elemental substitution approach in selected literature examples.
Introduction
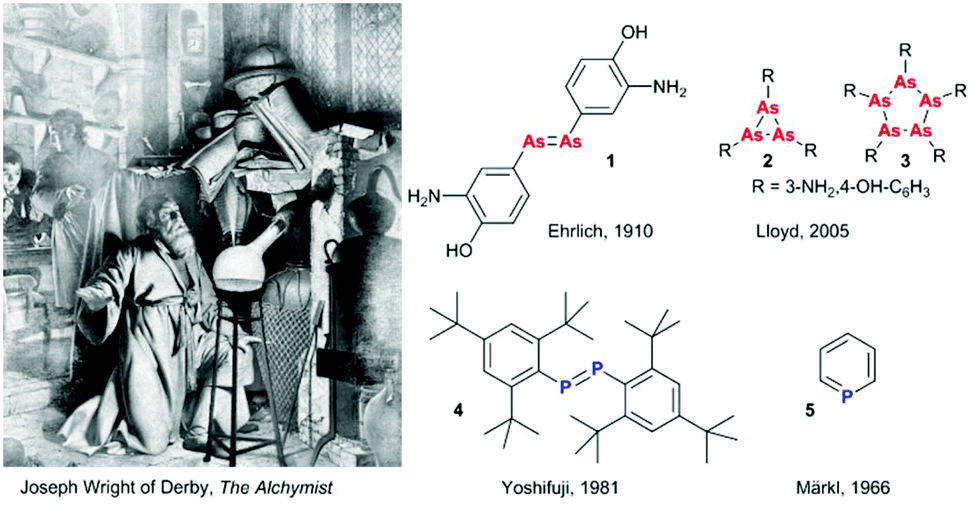
The discovery of phosphorus by Hennig Brand in 1669 pictured in the famous painting by Joseph Wright of Derby was the beginning of pnictogen (Pn) chemistry (Fig. 1). Discoveries that followed the incidental P4 preparation included the development of salvarsan (1), 3-amino-4-hydroxiphenyl arsenic(I), the first drug for syphilis treatment (Fig. 1).1 Originally proposed to feature an arsenic–arsenic double bond by Ehrlich in the beginning of the 20th century, it was later argued to have an oligo- or polymeric structure with a saturated (cyclo-)polyarsine core since such unsaturated motifs would require kinetic stabilisation. In 2005, Lloyd et al. ultimately resolved the debate concerning the composition of “compound 606” as a mixture of various small ring structures (R–As)n, where n = three (2) and five (3) dominate the original composition (Fig. 1).2 In the second half of the 20th century, fascinating examples of stabilised unsaturated low-valent and low-coordinate organopnictogens have been reported, challenging the empirical double bond rule. These include the first kinetically stabilised diphosphene (4) reported by Yoshifuji in 1981 (Fig. 1),3 the first complete series of homologous dipnictenes R–Pn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Pn–R (Pn = P, As, Sb, or Bi, where R is a meta-terphenyl substituent), and the thermodynamically (aromatically) stabilised phosphabenzene 5 (λ3-phosphinine).4 Early unsaturated pnictogen chemistry was dominated by the discovery of new bonding motifs and fundamental reactivity studies, but despite all these efforts, not all of the possible unsaturated pnictogen–heteroelement combinations have been synthetically realised and fundamental work has continued to fascinate researchers around the world. More recently, exciting news of arsenic thriving species of bacteria5 have then been disproven to incorporate arsenate.6 On the other hand, newly developed synthetic routes have made previously scarce or difficult to handle building blocks readily available, with broader applications of these systems now being studied.
Pn–R (Pn = P, As, Sb, or Bi, where R is a meta-terphenyl substituent), and the thermodynamically (aromatically) stabilised phosphabenzene 5 (λ3-phosphinine).4 Early unsaturated pnictogen chemistry was dominated by the discovery of new bonding motifs and fundamental reactivity studies, but despite all these efforts, not all of the possible unsaturated pnictogen–heteroelement combinations have been synthetically realised and fundamental work has continued to fascinate researchers around the world. More recently, exciting news of arsenic thriving species of bacteria5 have then been disproven to incorporate arsenate.6 On the other hand, newly developed synthetic routes have made previously scarce or difficult to handle building blocks readily available, with broader applications of these systems now being studied.
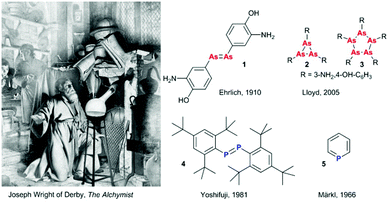 |
| | Fig. 1 Depiction of Hennig Brand's discovery of phosphorus. Painted in 1771 (redone in 1795) by Joseph Wright of Derby: “The Alchymist, in Search of the Philosopher's Stone, Discovers Phosphorus, and prays for the successful Conclusion of his operation, as was the custom of the Ancient Chymical Astrologers”. Originally proposed structure of salvarsan (compound 606) by Ehrlich,1 and the revised cycloarsine structures.2 First isolated diphosphene (4) by Yoshifuji and coworkers,3 and the first isolated phosphinine (5) by Märkl.4 | |
Research into replacing group 14 and 16 elements such as C, O, or S with their heavier analogues in small molecules and polymeric materials for optoelectronics applications has been investigated for a number of years. For example, the elemental substitution of S by Se or Te in unsaturated five membered rings can lead to alterations in energy levels and band gaps, solid-state packing and phosphorescence, and improved singlet–triplet energy conversion, making them of interest for a number of applications including organic light-emitting diodes (OLEDs), organic photovoltaics (OPVs), organic field-effect transistors (OFETs),7 and sensors.8 Similarly, the recent expansion in the use of heavier pnictogens has clearly demonstrated their potential to tailor opto-electronic properties and reactivity.
Exploration of the diverse reactivity of elemental pnictogens and unsaturated organopnictogens
Exploring the reactivity and properties of simple models is key to expanding our knowledge and discovering new areas where pnictogen-containing systems could have applications. The following sections describe important advances in molecular pnictogen chemistry, with a particular focus on contrasting the reactivity arising from pnictogen substitution. Demonstrative examples are given from seminal work and more recent contributions within the field.
Heavier cyanates, ChCPn−
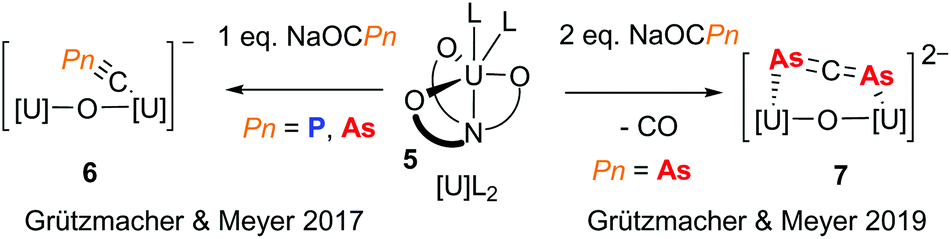
The pnictaethynolates Na(OCP) and Na(OCAs) have been known for many years and recent publications outlining simplified syntheses for these reagents has resulted in a wealth of interesting chemistry, including pnictogen-containing small molecules,9 larger π-delocalised organic molecules,10 and metal complexes spanning the p-, d- and f-blocks.11 While the chemistries of P and As are often assumed to be similar, the reactivity of pnictaethynolates has shown that subtle differences can influence reaction outcomes. This is well exemplified by a recent contribution from the groups of Meyer and Grützmacher, who investigated the reactivity of Na(OCPn) (Pn = P and As) with a highly reducing uranium(III) complex (5).12 When exposed to a single equivalent of pnictaethynolate, a new complex with a terminal cyapnictide ([C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Pn]−) is obtained in both cases (Fig. 2, 6). On the other hand, gas evolution was observed upon reaction with two equivalents of Na(OCAs) and a new complex with a bridging diarsaallenediide ([AsCAs]2−) ligand was obtained (Fig. 2, 7). Notably, the formation of the phosphorus analogue was not observed even with prolonged reaction times. This is likely due to the tendency of the OCAs ion to act as an As atom transfer reagent by decarbonylation.13 With two previously unseen arsenic-based ligands ([CAs]− and [AsCAs]2−), this contribution from Meyer, Grützmacher and co-workers highlights the usefulness of the pnictaethynolates for discovering new pnictogen chemistry.
Pn]−) is obtained in both cases (Fig. 2, 6). On the other hand, gas evolution was observed upon reaction with two equivalents of Na(OCAs) and a new complex with a bridging diarsaallenediide ([AsCAs]2−) ligand was obtained (Fig. 2, 7). Notably, the formation of the phosphorus analogue was not observed even with prolonged reaction times. This is likely due to the tendency of the OCAs ion to act as an As atom transfer reagent by decarbonylation.13 With two previously unseen arsenic-based ligands ([CAs]− and [AsCAs]2−), this contribution from Meyer, Grützmacher and co-workers highlights the usefulness of the pnictaethynolates for discovering new pnictogen chemistry.
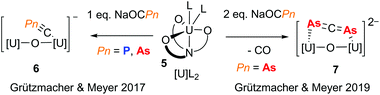 |
| | Fig. 2 Reactivity of a uranium(III) complex with phosphaethynolate and arsaethynolate. | |
The Goicoechea group have reported the synthesis and characterisation of the heavier chalcogen analogues Na(PnCS) and Na(PnCSe) (Pn = P or As), based on a modified procedure for Na(OCPn).14 These were found to have higher stability towards air and moisture due the greater degree of electronic delocalisation across the molecules (Fig. 3). Unlike the O-containing derivatives, the negative charge was found to reside primarily on the central carbon atom. An earlier study by Chen, Cummins, Borden and Wang found that the electron density in PCS was distributed over the P and S centres. Additionally, the negative ion photoelectron spectra of the OCP, OCAs and PCS radicals revealed that the electron affinities of these radicals are markedly lower than those of the NCO and NCS radicals, suggesting them to be softer pseudohalide ligands than their N-containing congeners.15 These significant differences between the oxygen and heavier chalcogen analogues will most likely lead to new and interesting reactivity/chemistry.
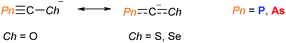 |
| | Fig. 3 Electron localisation differences in [PnCCh]−. Resonance structures with localised charges on the carbon atom and chalcogens are seen for the S/Se and oxygen containing derivatives, respectively. | |
Pn4 activation
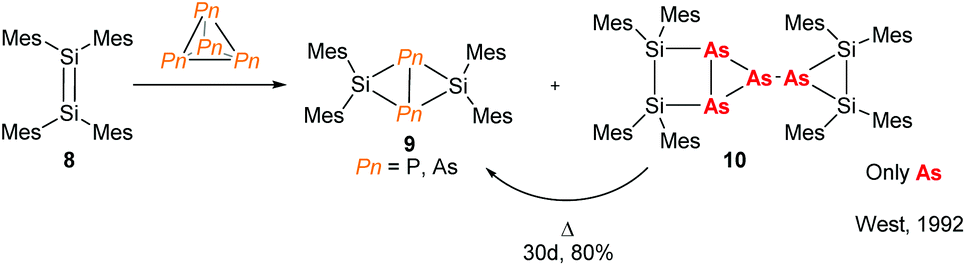
The need for high value phosphorus-containing fertilisers from inexpensive sources means that research into the chemistry of P4 has been incentivised in both academic and industrial circles. This has resulted in many examples of P4 activation, using compounds from across the periodic table.16 The chemistry of As4, on the other hand, remains relatively unexplored. For main-group elements, there are a few reports of disilenes and silylenes reacting with As4 which demonstrate differing reactivity when compared to P4.17 For instance, early work published by West and co-workers shows that reaction of tetramesityldisilene (8) with As4 not only forms the butterfly complex (9) seen for P4 activation but also an arrested dimeric species 10 which forms the butterfly complex 9 by thermal decomposition (Fig. 4).
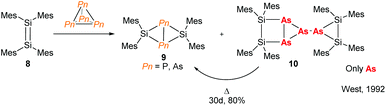 |
| | Fig. 4 Reaction of a disilene with Pn4 illustrating the different reactivities of phosphorus and arsenic.17 | |
The scarce reports of As4 activation can be attributed to the challenges associated with its preparation and handling (high light- and air-sensitivity). These challenges have recently been addressed by the Scheer group, with a report detailing the preparation of activated carbon-supported As4 and P4 (As4@C and P4@C) which are air- and light-stable and which were employed as synthons in P4 and As4 activation chemistry.18 While the synthesis of As4@C still requires the preparation of in situ As4 solutions, the increased stability of As4@C is a marked improvement and has potential for the production of preparative-scale quantities.
Organopnictogen radicals
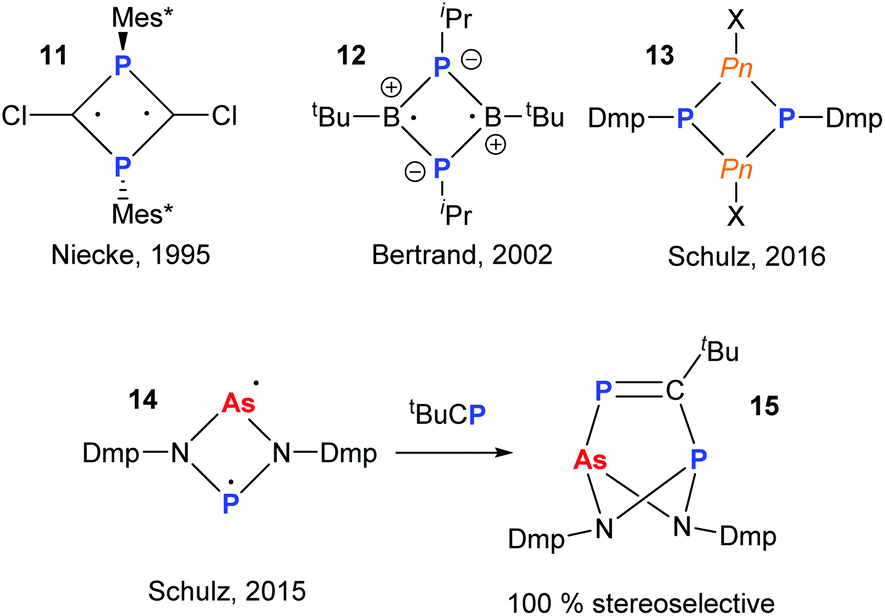
The radical chemistry of organopnictogen compounds is particularly important as it provides a fundamental insight into their chemical bonding and reactivity. In particular, the chemistry of main group biradicaloids has been stimulated by the work of Niecke and coworkers on 1,3-diphosphacyclobutane-2,4-yls (11).19 Significant work has been carried out on cyclo-P2B2-radicals (12)20 and, more recently, heavier cyclo-P2Pn2 (Pn = P, As, Sb, Bi; 13) potential biradical precursors (Fig. 5).21 In 2015, Schulz and co-workers reported a very interesting mixed As and P diradicaloid 14, which was shown to have non-equivalent radical character on the As- and P-centres by theoretical and experimental techniques.22 For instance, upon reaction with tBuCP a single product 15 was obtained stereoselectively, indicating the P-centre to be more nucleophilic than the As.
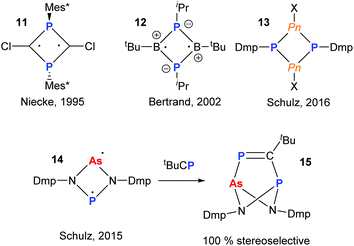 |
| | Fig. 5 Main group hetero-cyclobutane based biradicals. Mixed As/P diradicaloid and reactivity with phosphaalkyne. | |
Stabilising low coordinate pnictogen centres
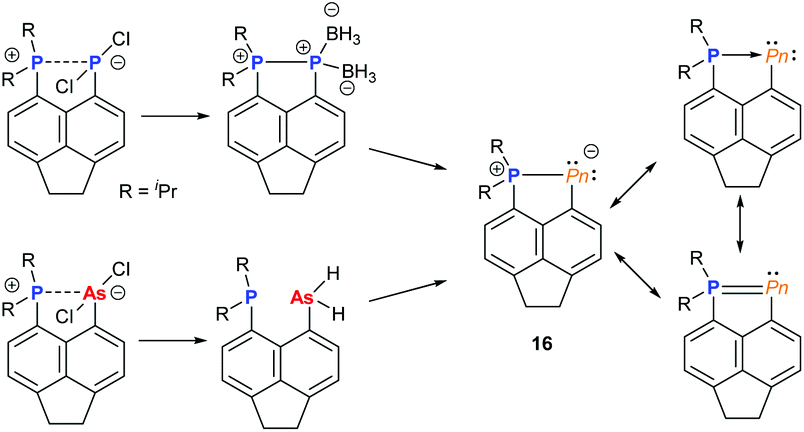
Over the past decade, the group of Kilian has reported a fascinating array of peri-substituted acenaphthene complexes. The P–Pn (Pn = P, As) donor–acceptor stabilised system can be converted in a two-step procedure into cyclic pnictinylidene-λ4-phosphoranes (16) with significant electronic resonance (Fig. 6). These dark-red coloured systems have a low coordinate (λ2) pnictogen site formed via a borane reduction–deborylation or hydride transfer with LiAlH4 followed by reductive H2 elimination for phosphorus and arsenic, respectively.23 Other examples of low-coordinate NHC-stabilised dipnictogens have been reported by Robinson and co-workers.24
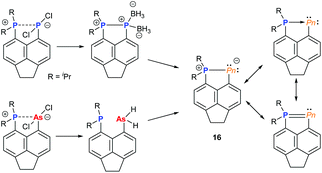 |
| | Fig. 6
peri-substituted acenaphthene dipnictogen complexes. | |
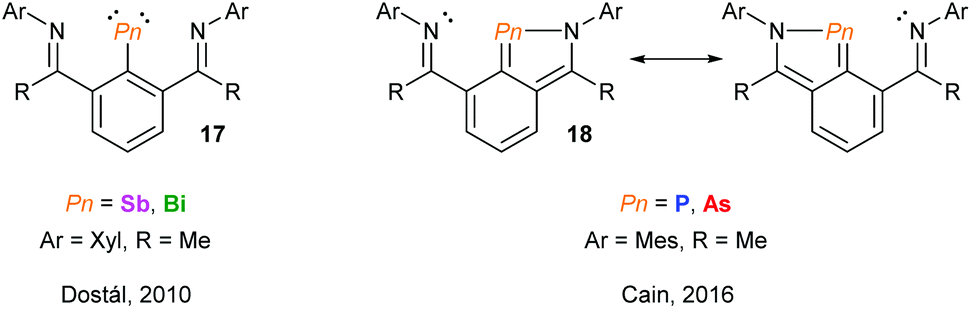
Using NCN pincer ligands, a series of pnictinidene complexes were reported separately by the Dostál and Cain groups (Fig. 7).25 All complexes were found to exhibit C2V symmetry in solution by NMR spectroscopy. However, unlike the Sb and Bi systems (17) reported by Dostál and co-workers, the Cain group found that the As and P analogues (18) were unsymmetrical in the solid-state with non-equivalent Pn–N bond distances. The high symmetry observed in solution, even at low temperatures, suggests that the Pn atom is hopping from one N centre to the other like a “bell clapper”, which is supported by computational experiments. The significant degree of Pn–C double bond character for As and P results in the masking of the pnictinidene fragment, and the pnictaalkene was computationally determined to be a lower energy intermediate than the pnictinidene in the oscillation pathway. Conversely, the pnictaalkene is more energetically demanding for Sb and Bi, resulting in a ground state pnictinidene.
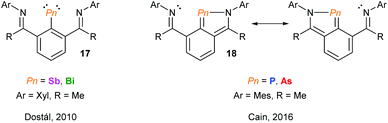 |
| | Fig. 7 NCN pincer ligand supported pnictinidene and masked pnictinidene complexes reported by the groups of Dostál and Cain, respectively. | |
Optoelectronic materials containing heavy pnictogens
As mentioned above, the use of heavier pnictogens in optoelectronics applications has been a growing field over the last few years. The properties seen in conjugated systems containing heavier group 15 elements, for example phospholes and arsoles, are largely attributed to interactions between the σ* orbital of the exocyclic Pn–C bond and the surrounding π* system, which lowers the LUMO level of the conjugated material far more than it affects the HOMO level.26 In addition, the accessibility of both the +3 and +5 valence states of group 15 elements allows for interactions with Lewis acids or transition metals, with such coordination leading to further changes in the optoelectronics of the systems. This gives these materials potential as sensors as well as providing further opportunities for catalysis or other applications, potentially mirroring the development of compounds such as phospholes, pioneered by the groups of Réau and Hissler as active materials for opto-electronic devices. Detailed reviews of their contributions and those of others in this area already exist,27 while the purpose of this Frontier article is to focus on materials containing higher pnictogens.
While this Frontier article aims to highlights some of the recent progress in heavier organopnictogens and point out the differences within the pnictogen family, readers are also referred to some excellent recent reviews discussing the history of the topic: Gates and coworkers examined p-block elements in polymers,28 Rivard and coworkers discussed heavy main group elements in π-conjugated systems,29 and a third from the Orthaber group predominantly focused on P-containing materials for use in organic electronics.30 Readers interested in a more in-depth review of the history of organoarsenic π-conjugated materials are also encouraged to read Imoto and Naka's thorough overview of the topic.31
As-Containing heterocycles
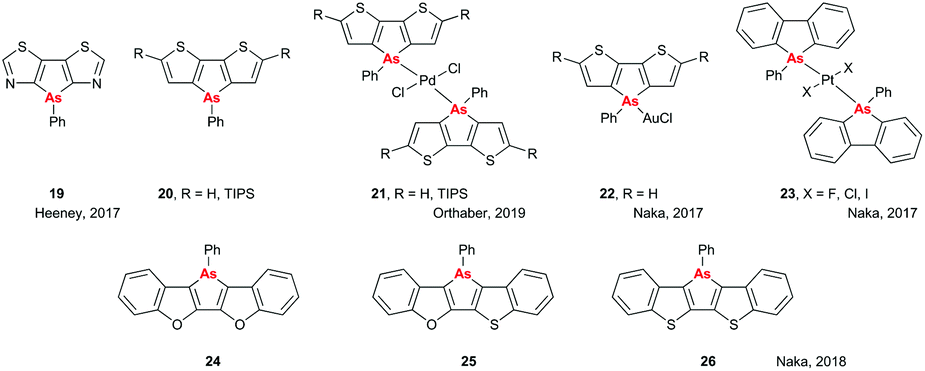
There have been a number of interesting developments in the area of small molecules containing heavy pnictogens in recent years. Both the Heeney and Orthaber groups have investigated the optical properties and aromaticities of arsenic-containing heterocycles. In the first paper on this subject, a dithiazoloarsole compound (Fig. 8, 19) was prepared,32 and its crystal structure and optical properties were compared to those of its dithiazolophosphole analogue, previously prepared and studied by Baumgartner et al.33 Only minor changes were observed in the optical properties on replacing P with As, with λab and λem red-shifting and blue-shifting by 5 nm, respectively, although there was a noticeable decrease in the photoluminescent quantum yield (PLQY; 4% for As vs. 22% for P). Subsequent work by the Orthaber group investigated the impact of changing As from the +3 to +5 valence state by coordination to Pd on the optical and structural properties of dithienoarsole (Fig. 8, 20). Coordination to Pd had a more subtle effect on the optical properties of the silylated arsole (21, R = TIPS) than was seen for the analogous phosphole, with λab shifting by only 5 nm and emission still taking place on photoexcitation of the ligand absorption band rather than from the charge-transfer band, as was seen for the phosphole.34 Interestingly, work by Naka and coworkers found that coordination of the non-silylated dithienoarsole (R = H) to AuCl led to a significant red shift (93 nm) in the emission spectrum of 22.35 However, this was accompanied by a substantial decrease in PLQY (<1% compared to 7% for the uncoordinated material), whereas for the analogous phosphole material the PLQY increased from 49% to 64% on coordination to AuCl.36
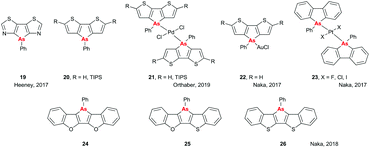 |
| | Fig. 8 Structures of arsole-containing systems. | |
In another unexpected result, it was found that altering the halide co-ligands in trans-[PtX2L2] complexes 23 led to significant alterations in both the geometry of the resulting crystals and the emissive properties of the complexes, with red shifts of up to 20 nm and 50-fold increases in PLQY being observed when replacing Cl with I.37 Finally, work by Naka et al. in 2018 showed that the central arsole rings in heteropentacenes 24–26 (Fig. 8) showed weak anti-aromaticity as determined by NICS, which was attributed to the further expansion of the conjugated systems. Additionally, it was found that altering the flanking bridging elements could change the packing structure and hence the solid state optical properties and charge carrier mobility, with As–S interactions in 26 leading to enhanced charge carrier mobility but quenching of emission in the solid state.38 Overall, the variation in optical properties obtainable by making minor modifications shows the diverse characteristics available when using arsole-containing moieties, and demonstrate that there is still much we do not understand about the optoelectronics of arsenic-containing heterocycles; hopefully this will stimulate further study in the future.
Sb- and Bi-containing heterocycles
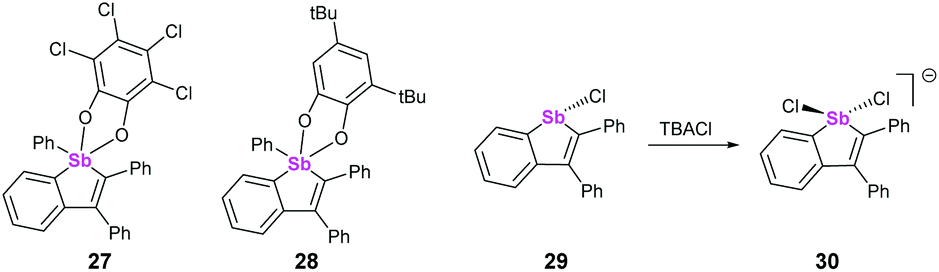
Both λ5 and λ3 stibaindoles (Fig. 9, 27/28 and 29, respectively) have been investigated by Gabbaï et al. as potential sensing materials capable of binding anions at their Sb sites. The pentacoordinated derivatives 27 and 28 showed strong σ*–π* conjugation, and were capable of binding to fluoride ions via the σ* orbital of the Ph–Sb bond due to the Lewis acidity of the Sb atom. Coordination of F− caused significant changes to the absorption spectra of 27 and 28, as this prevented the σ*–π* conjugation from taking place; the group subsequently tested 27 as an anion sensor for fluoride in aqueous solutions, achieving promising results.39 Compound 29 also showed strong σ*–π* conjugation, which again led to Lewis acidic behaviour at the Sb atom. Chloride anions were found to bind to Sb opposite the existing Cl, with the Cl–Sb–Cl angle being almost 180° (Fig. 9, 30).40
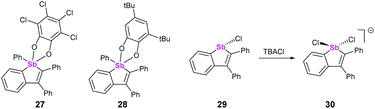 |
| | Fig. 9 Structures of stibole-containing systems. | |
This behaviour is highly similar to the pnictogen bonding recently described by Matile and coworkers, who found that tri(pentafluorophenyl)antimony was capable of binding to chloride ions through σ-hole interactions opposite to the strongly electron-withdrawing pentafluorophenyl substituents.41 Interestingly, they subsequently used this Sb–Cl interaction to catalyse the Reissert-type substitutions of isoquinoline and 1-chloroisochromane, which when combined with the findings of Gabbaï suggests that carefully designed heterocycles containing heavy pnictogens may also be suitable for catalysis.
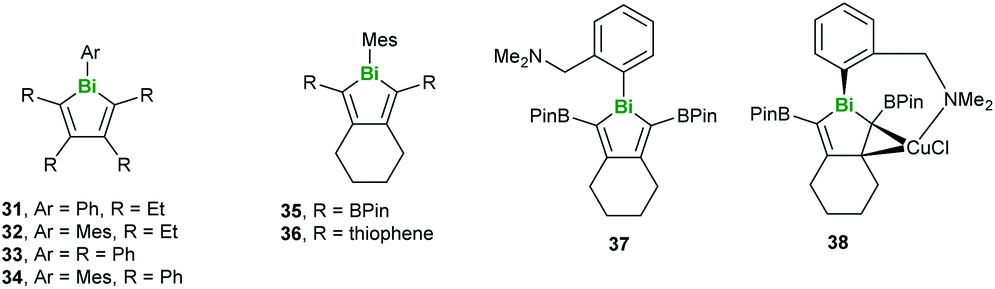
Regarding the heaviest pnictogen, Bi, there have been two studies investigating conjugated molecules containing bismuth published recently. Rivard and coworkers synthesised a series of bismole-containing systems via metallacycle transfer from related zirconacycles, noting that while 31 decomposed under ambient conditions, compounds 32–36 could be stored in air without degradation (Fig. 10). The photoluminescent properties of 36–38 were studied in depth, with 36 being found to fluoresce at low temperatures only due to the limited participation of the Bi orbitals (Fig. 10). On the other hand, 37 and 38 exhibited both fluorescence and phosphorescence due to increased singlet–triplet mixing in the lower-energy excited states.42
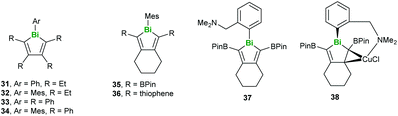 |
| | Fig. 10 Structures of bismole-containing systems. | |
The same group have also produced a series of Bi-containing polymers (both homopolymers P1–2 and random and block copolymers P3–4 with alkylarene comonomers) by ring-opening metathesis polymerisation (ROMP, Fig. 11). After polymerisation, there was a 60 nm red shift in the luminescence and a decrease in intensity, attributed to increased free volume around the bismoles as well as stabilisation of singlet and triplet states.43
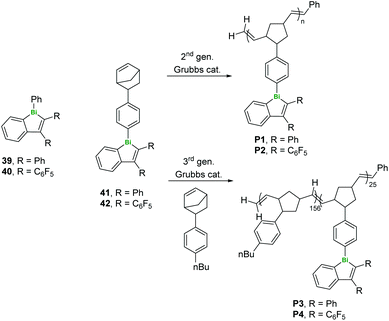 |
| | Fig. 11 Structures of bismole-containing polymers. | |
Heteroalkenes
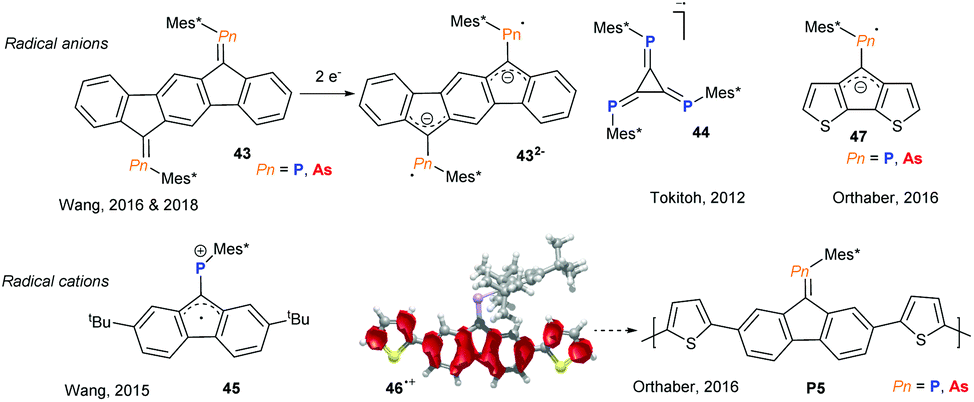
Incorporating group 15 heteroalkenes into conjugated frameworks has been a long lasting effort, providing fascinating materials that exhibit pronounced acceptor character and offer an orthogonal tuning site via the heteroelement lone pair, allowing the opto-electronic properties to be altered. However, these efforts have been mainly limited to phosphaalkenes and diphosphenes, with arsaalkenes recently becoming attractive alternatives. The Wang group has studied a series of fused aromatic systems with exocyclic phosphaalkenes, which upon one and two electron reductions display persistent phosphorus centred radical and diradical character, respectively (Fig. 12, 43).44 Interestingly, the corresponding arsenic based diradical systems have thermally accessible triplet states due to the reduced singlet–triplet energy gap.45 Tokitoh and coworkers described the facile consecutive reduction of 4,5,6-triphospha-[3]-radialene 44 to the mono- and dianions as pnictogen analogs of deltate anions.46 An inverse radical character was described for the corresponding radical cation of alkyl substituted fluorenyl-phosphaalkene by Wang and coworkers (Fig. 12, 45).47 By thienyl extension of the conjugated fluorenyl core, Orthaber and coworkers were able to spread the spin density towards the α-thienyl carbon atoms (46˙+), allowing for the cationic radical polymerisation of both arsa- and phosphaalkene derivatives (Fig. 12, P5). The resulting polymer films display reversible electrochromic switching between the oxidised and neutral states, which were dark blue and orange in colour, respectively.48 The Orthaber group also exemplified the LUMO-lowering effects of P- and As-alkene derivatives of cyclopentadithiophenes (47) based on optical spectroscopy and the electrochemically reversible formation of radical anions. Remarkably, excited state lifetimes are extended by a factor of 104 to ca. 18 ns upon Au(I)Cl coordination to the phosphaalkene. Interestingly, the increased s-character of the lone pair in arsaalkenes precludes such metal coordination in this system.49
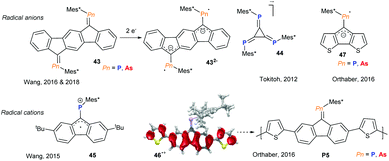 |
| | Fig. 12 Phospha- and arsaalkene-based radicals and radical reactions. | |
Conclusions
Organopnictogen chemistry is a flourishing field of chemical research. Exploring the heavier congeners for the discovery of new optical, electronic and bonding phenomena has stimulated a wealth of research over the past two decades, the history of which is summarised in reviews of main-group elements in polymeric systems by Jäkle and Vidal and small molecules by Quin.50 Additionally, the continuing fundamental research into new reagents and the reactivity and properties of model systems provides a thriving environment to develop new motifs and discover new applications for heavier pnictogens. This context will allow this newly regenerated field to move in novel and unexpected directions while building on the knowledge of the past.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the EU-COST action on Smart Inorganic Polymers (CM1302, SIPs), the Swedish Research Council (Vetenskapsrådet), the Swedish Energy Agency (Energimyndigheten), the Wenner Gren Foundation, Carl-Trygger foundation, Byggmästare-Olle-Engkvist Foundation.
Notes and references
- P. Ehrlich and A. Bertheim, Ber. Dtsch. Chem. Ges., 1912, 45, 756–766 CrossRef CAS.
- N. C. Lloyd, H. W. Morgan, B. K. Nicholson and R. S. Ronimus, Angew. Chem., Int. Ed., 2005, 44, 941–944 CrossRef CAS PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- G. Märkl, Angew. Chem., 1966, 78, 907–908 CrossRef.
- F. Wolfe-Simon, J. S. Blum, T. R. Kulp, G. W. Gordon, S. E. Hoeft, J. Pett-Ridge, J. F. Stolz, S. M. Webb, P. K. Weber, P. C. W. Davies, A. D. Anbar and R. S. Oremland, Science, 2011, 332, 1163–1166 CrossRef CAS PubMed.
- M. L. Reaves, S. Sinha, J. D. Rabinowitz, L. Kruglyak and R. J. Redfield, Science, 2012, 337, 470–473 CrossRef CAS PubMed.
-
(a) B. D. Datko, A. K. Thomas, Z. Fei, M. Heeney and J. K. Grey, Phys. Chem. Chem. Phys., 2017, 19, 28239–28248 RSC;
(b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed;
(c) S. M. Parke and E. Rivard, Isr. J. Chem., 2018, 58, 915–926 CrossRef CAS.
- X. He and T. Baumgartner, RSC Adv., 2013, 3, 11334–11350 RSC.
-
(a) A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131–19134 CrossRef CAS PubMed;
(b) K. M. Szkop, A. R. Jupp and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 12751–12755 CrossRef CAS PubMed.
- Y. Mei, D.-J. Wu, J. E. Borger and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 5512–5515 CrossRef CAS PubMed.
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
-
(a) C. J. Hoerger, F. W. Heinemann, E. Louyriac, L. Maron, H. Grützmacher and K. Meyer, Organometallics, 2017, 36, 4351–4354 CrossRef CAS;
(b) C. J. Hoerger, F. W. Heinemann, E. Louyriac, M. Rigo, L. Maron, H. Grützmacher, M. Driess and K. Meyer, Angew. Chem., Int. Ed., 2019, 58, 1679–1683 CrossRef CAS PubMed.
- A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 8536–8541 CrossRef CAS PubMed.
- F. Tambornino, A. Hinz, R. Köppe and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 8230–8234 CrossRef CAS PubMed.
- G.-L. Hou, B. Chen, W. J. Transue, Z. Yang, H. Grützmacher, M. Driess, C. C. Cummins, W. T. Borden and X.-B. Wang, J. Am. Chem. Soc., 2017, 139, 8922–8930 CrossRef CAS PubMed.
-
(a) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed;
(b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed;
(c) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed;
(d) Z. R. Turner, Inorganics, 2015, 3, 597 CrossRef CAS.
-
(a) R. P. Tan, N. M. Comerlato, D. R. Powell and R. West, Angew. Chem., Int. Ed. Engl., 1992, 31, 1217–1218 CrossRef;
(b) A. E. Seitz, M. Eckhardt, S. S. Sen, A. Erlebach, E. V. Peresypkina, H. W. Roesky, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2017, 56, 6655–6659 CrossRef CAS PubMed.
- A. E. Seitz, F. Hippauf, W. Kremer, S. Kaskel and M. Scheer, Nat. Commun., 2018, 9, 361 CrossRef PubMed.
- E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1995, 34, 555–557 CrossRef CAS.
- D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1881 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Inorg. Chem., 2016, 55, 3692–3699 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 668–672 CrossRef CAS PubMed.
-
(a) B. A. Chalmers, M. Bühl, K. S. Athukorala Arachchige, A. M. Z. Slawin and P. Kilian, J. Am. Chem. Soc., 2014, 136, 6247–6250 CrossRef CAS PubMed;
(b) B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Angew. Chem., Int. Ed., 2012, 51, 10150–10153 CrossRef CAS PubMed.
-
(a) M. Y. Abraham, Y. Wang, Y. Xie, R. J. Gilliard, P. Wei, B. J. Vaccaro, M. K. Johnson, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2013, 135, 2486–2488 CrossRef CAS PubMed;
(b) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed.
-
(a) P. Šimon, F. de Proft, R. Jambor, A. Růžička and L. Dostál, Angew. Chem., Int. Ed., 2010, 49, 5468–5471 CrossRef PubMed;
(b) J. Hyvl, W. Y. Yoshida, A. L. Rheingold, R. P. Hughes and M. F. Cain, Chem. – Eur. J., 2016, 22, 17562–17565 CrossRef CAS PubMed;
(c) V. Kremláček, J. Hyvl, W. Y. Yoshida, A. Růžička, A. L. Rheingold, J. Turek, R. P. Hughes, L. Dostál and M. F. Cain, Organometallics, 2018, 37, 2481–2490 CrossRef.
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 613–1622 CrossRef PubMed.
- M. P. Duffy, W. Delaunay, P. A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296–5310 RSC.
- A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Chem. Soc. Rev., 2016, 45, 922–953 RSC.
- S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485–9505 RSC.
- M. A. Shameem and A. Orthaber, Chem. – Eur. J., 2016, 22, 10718–10735 CrossRef CAS PubMed.
- H. Imoto and K. Naka, Chem. – Eur. J., 2019, 25, 1883–1894 CrossRef CAS PubMed.
- J. P. Green, S. J. Cryer, J. Marafie, A. J. P. White and M. Heeney, Organometallics, 2017, 36, 2632–2636 CrossRef CAS.
- X. He, A. Y. Y. Woo, J. Borau-Garcia and T. Baumgartner, Chem. – Eur. J., 2013, 19, 7620–7630 CrossRef CAS PubMed.
- J. P. Green, A. K. Gupta and A. Orthaber, Eur. J. Inorg. Chem., 2019 DOI:10.1002/ejic.201801169.
- H. Imoto, I. Kawashima, C. Yamazawa, S. Tanaka and K. Naka, J. Mater. Chem. C, 2017, 5, 6697–6703 RSC.
- Y. Dienes, S. Durben, T. Kárpáti, T. Neumann, U. Englert, L. Nyulászi and T. Baumgartner, Chem. – Eur. J., 2007, 13, 7487–7500 CrossRef CAS PubMed.
- H. Imoto, H. Sasaki, S. Tanaka, T. Yumura and K. Naka, Organometallics, 2017, 36, 2605–2611 CrossRef CAS.
- H. Imoto, T. Fujii, S. Tanaka, S. Yamamoto, M. Mitsuishi, T. Yumura and K. Naka, Org. Lett., 2018, 20, 5952–5955 CrossRef CAS PubMed.
- A. M. Christianson, E. Rivard and F. P. Gabbaï, Organometallics, 2017, 36, 2670–2676 CrossRef CAS.
- A. M. Christianson and F. P. Gabbaï, Organometallics, 2017, 36, 3013–3015 CrossRef CAS.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- S. M. Parke, M. A. B. Narreto, E. Hupf, R. McDonald, M. J. Ferguson, F. A. Hegmann and E. Rivard, Inorg. Chem., 2018, 57, 7536–7549 CrossRef CAS PubMed.
- S. M. Parke, E. Hupf, G. K. Matharu, I. de Aguiar, L. Xu, H. Yu, M. P. Boone, G. L. C. de Souza, R. McDonald, M. J. Ferguson, G. He, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2018, 57, 14841–14846 CrossRef CAS PubMed.
- G. Tan, S. Li, S. Chen, Y. Sui, Y. Zhao and X. Wang, J. Am. Chem. Soc., 2016, 138, 6735–6738 CrossRef CAS PubMed.
- Y. Fang, L. Zhang, C. Cheng, Y. Zhao, M. Abe, G. Tan and X. Wang, Chem. – Eur. J., 2018, 24, 3156–3160 CrossRef CAS PubMed.
-
(a) H. Miyake, T. Sasamori, J. I. C. Wu, P. v. R. Schleyer and N. Tokitoh, Chem. Commun., 2012, 48, 11440–11442 RSC;
(b) H. Miyake, T. Sasamori and N. Tokitoh, Angew. Chem., 2012, 124, 3514–3517 CrossRef.
- X. Pan, X. Wang, Z. Zhang and X. Wang, Dalton Trans., 2015, 44, 15099–15102 RSC.
- D. Morales Salazar, E. Mijangos, S. Pullen, M. Gao and A. Orthaber, Chem. Commun., 2017, 53, 1120–1123 RSC.
- A. El Nahhas, M. A. Shameem, P. Chabera, J. Uhlig and A. Orthaber, Chem. – Eur. J., 2017, 23, 5673–5677 CrossRef CAS PubMed.
-
(a) F. Jäkle and F. Vidal, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201810611;
(b)
L. D. Quin, A guide to orgaophosphorus chemistry, John Wiley & Sons, Inc., New York, 2000 Search PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Jordann A. L.
Wells†
,
Jordann A. L.
Wells†