
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal ion exchange in Prussian blue analogues: Cu(II)-exchanged Zn–Co PBAs as highly selective catalysts for A3 coupling†
Carlos
Marquez
 ,
Francisco G.
Cirujano
,
Simon
Smolders
,
Cédric
Van Goethem
,
Ivo
Vankelecom
,
Dirk
De Vos
* and
Trees
De Baerdemaeker
*
,
Francisco G.
Cirujano
,
Simon
Smolders
,
Cédric
Van Goethem
,
Ivo
Vankelecom
,
Dirk
De Vos
* and
Trees
De Baerdemaeker
*
Centre for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be; trees.debaerdemaeker@kuleuven.be
First published on 4th March 2019
Abstract
The occurrence of metal ion exchange in Zn3[Co(CN)6]2 and Cu3[Co(CN)6]2 Prussian blue analogues (Zn–Co and Cu–Co PBAs) was demonstrated for the first time. While Cu(II) ion exchange easily occurs in Zn–Co PBA, the exchange of Cu(II) atoms in Cu–Co PBA by Zn(II) proved to be more difficult. At low to medium Cu(II) loadings, the catalytic activity of the exchanged PBAs for the A3 coupling reaction of benzaldehyde, piperidine and phenylacetylene was higher than that of the bimetallic PBAs and that of multi metal PBAs of similar composition prepared by co–precipitation. This result showcases the benefits of the ion exchange process as a preparation method of PBA catalysts, since it is believed to lead to the incorporation of the desired metal in a more accessible position for reactant molecules. At higher Cu(II) loadings, ion exchange with Cu(CH3COO)2·H2O also resulted in co-incorporation of CH3COO−. This incorporation considerably boosted the catalytic activity of the PBAs by providing a basic function that facilitates the C–H activation of phenylacetylene. The most active of the studied PBAs, catallytically outperforms other Cu(II) based A3 coupling catalysts and completely suppresses the activity for the homocoupling of phenylacetylene, even under oxidative conditions. Furthermore, the basicity of the PBAs was investigated in the nitroaldol (Henry) reaction, where a clear effect of the presence of CH3COO− was observed. The CH3COO− containing PBAs exhibited an activity three times higher than the rest of the PBAs. The presence of the basic CH3COO− groups represents the first case of basic functionalization of PBAs.
Introduction
Prussian blue analogues (PBAs) are porous coordination polymers with a structural formula of M1u[M2(CN)6]v (M1-M2 PBAs). M1 can be, for example, a divalent metal, such as Zn(II), Fe(II), Cu(II), Co(II), and M2 a trivalent metal such as Co(III), Fe(III) or Cr(III). The structure of PBAs typically consists of a rock salt type arrangement of [M1]v+ and [M2(CN)6]u−. For v = 2 and u = 3, one out of three [M2(CN)6]3− sites is vacant to obtain charge neutrality. PBAs – and especially Prussian blue – are among the oldest known synthetic coordination compounds, as their use as dyes and pigments dates back to the 18th century.1–3 Since then, they have received substantial attention due to their interesting magnetic,4–6 textural,7–9 electrochemical,10–12 ion exchange13–15 and catalytic properties.1,16–19 PBAs can be easily synthesized by a fast precipitation reaction using aqueous solutions of a M1 salt (M1Zv, where Z is usually Cl−, NO3− or CH3COO−) and a M2 cyanide salt (Yu[M2(CN)n], where Y is usually K+ or Na+). The stoichiometry of the final product depends on the oxidation states of the metals and the possible incorporation of other cations.20 One of the advantages of PBAs is their high tunability due to the numerous potential variations in M1. This leads to the preparation of complexes with different compositions and properties, even allowing the incorporation of two different divalent metals on the M1 position in the PBA.21 For example, Liu et al.14 prepared a series of FexZn1−x–Co PBAs for the capture of Cs+ that showed an improved performance as the Zn/Fe ratio increased. Likewise, the group of Reguera studied mixed composition PBAs and metal nitroprussides for H2 storage and found that the adsorption potential can be modulated by combining different metals.21,22 For electrochemical applications, varying the composition of PBA electrode materials by element doping can enhance their electrochemical performance, as shown by studies of NixFe1−x–Fe, NixMn1−x–Fe, NixCo1−x–Fe, NixCu1−x–Fe and CoxMn1−x–Fe PBAs as cathode materials.11,23–26 In catalysis, García-Ortiz et al.27 employed FexCu1−x–Co PBAs as solid catalysts for the aerobic oxidation of oximes to the corresponding ketone and recently, our group reported the use of a series of CuxZn1−x–Co PBAs as heterogeneous catalyst for the A3 coupling of phenylacetylene, benzaldehyde and piperidine.28However, in all these examples, the respective multi metal PBA was synthesized by the standard co-precipitation procedure, and while this is an easy approach, the final solid most likely consists of a random arrangement of the different divalent metals on the M1 position all throughout the crystals.21 For some applications, especially in catalysis on the outer surface and where two different M1 cooperate in the catalytic cycle, there is no need to introduce a second M1 in the bulk of the crystals. Therefore, the selective incorporation of the second M1 closer to the external surface would be beneficial for the catalytic activity of PBAs. Since M1 atoms at the outer surface are, on average, coordinated to fewer N atoms, they are more likely to be released from the structure first,14 and post-synthetic metal ion exchange (PSE) of these atoms represents an interesting preparation method of catalytically active multi metal PBAs. PSE of metal ions has already been observed for several complexes, like CdSe ionic nanocrystals,29 octanuclear Cu(II) wheels30 and MOFs31–33 but to the best of our knowledge, observation of such phenomena in PBAs has not been reported.
Herein, PSE of metal ions in Zn–Co and Cu–Co PBAs is studied for the first time based on the quantitative correlation between the concentration of metal ions released from the PBA framework and the metal loading in the solid. We chose to work with these two PBAs because of the reported high activity of CuxZn1−x–Co PBA catalysts for A3 coupling reactions by combining the high conversion obtained with Cu–Co PBA with the excellent selectivity obtained with Zn–Co PBA.28 The ion exchanged PBAs were characterized by powder X–ray diffraction (PXRD), thermogravimetric analysis coupled with mass spectroscopy (TGA–MS), N2 physisorption, transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR) and FTIR with pyridine as probe molecule. Their catalytic performance for the A3 coupling of phenylacetylene, benzaldehyde and piperidine was evaluated. Furthermore, the basic character of selected samples was studied for the nitroaldol (Henry) reaction of nitromethane and benzaldehyde.
Experimental
Catalysts preparation
 | (1) |
 | (2) |
 | ||
| Scheme 1 Post-synthetic metal ion exchange procedure. | ||
| Entry | PBA | Cu(II) salt | Concentration (mM) | Time (h) | Cu (wt%) | Zn (wt%) | Co (wt%) | M1/Coa | Znloss![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (wt%) b (wt%) |
IEDCuc (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a (Cu + Zn)/Co molar ratio in the final PBA multi metal complex. b Zn wt% loss of the sample, based on the concentration of Zn released into solution during the exchange process. c Atomic ion exchange degree defined as atoms of Zn exchanged per atoms of Zn present in Zn–Co PBA. d This sample was synthesized by mixing of an aqueous solution of K3[Co(CN)6] and an aqueous solution of CuCl2·2H2O. e This sample was synthesized by mixing of an aqueous solution of K3[Co(CN)6] and an aqueous solution of Cu(CH3COO)2·H2O. | ||||||||||
| 1 | Zn–Co | — | — | — | — | 32.0 | 18.7 | 1.54 | — | — |
| 2 | [Cu(ClO4)2]0.10@Zn0.90–Co | Cu(ClO4)2 | 5 | 16 | 3.02 | 28.2 | 18.7 | 1.55 | 3.03 | 11.9 |
| 3 | [Cu(ClO4)2]0.33@Zn0.67–Co | Cu(ClO4)2 | 25 | 16 | 10.2 | 20.4 | 18.9 | 1.50 | 10.2 | 36.3 |
| 4 | [Cu(OAc)2]0.11@Zn0.89–Co(a) | Cu(OAc)2 | 2.5 | 8 | 3.45 | 28.1 | 18.5 | 1.58 | 9.69 | |
| 5 | [Cu(OAc)2]0.11@Zn0.89–Co(b) | Cu(OAc)2 | 2.5 | 16 | 3.51 | 28.3 | 18.5 | 1.60 | 9.06 | |
| 6 | [Cu(OAc)2]0.15@Zn0.85–Co | Cu(OAc)2 | 5 | 4 | 4.69 | 27.0 | 18.5 | 1.59 | 4.18 | 13.4 |
| 7 | [Cu(OAc)2]0.19@Zn0.81–Co | Cu(OAc)2 | 5 | 16 | 6.05 | 26.1 | 18.4 | 1.62 | 14.4 | |
| 8 | [Cu(OAc)2]0.20@Zn0.80–Co | Cu(OAc)2 | 5 | 8 | 6.46 | 25.4 | 18.5 | 1.60 | 18.4 | |
| 9 | [Cu(OAc)2]0.21@Zn0.79–Co | Cu(OAc)2 | 7 | 16 | 6.90 | 25.6 | 18.3 | 1.65 | 6.63 | 20.0 |
| 10 | [Cu(OAc)2]0.45@Zn0.55–Co | Cu(OAc)2 | 12.5 | 8 | 15.3 | 18.5 | 18.0 | 1.74 | 40.6 | |
| 11 | [Cu(OAc)2]0.53@Zn0.47–Co | Cu(OAc)2 | 12.5 | 16 | 17.8 | 16.1 | 18.0 | 1.75 | 48.4 | |
| 12 | [Cu(OAc)2]0.67@Zn0.33–Co | Cu(OAc)2 | 25 | 8 | 24.0 | 11.8 | 17.5 | 1.89 | 62.2 | |
| 13 | [Cu(OAc)2]0.70@Zn0.30–Co | Cu(OAc)2 | 25 | 16 | 24.8 | 10.7 | 17.6 | 1.87 | 65.9 | |
| 14 | [Cu(OAc)2]0.77@Zn0.23–Co | Cu(OAc)2 | 50 | 4 | 25.2 | 7.56 | 18.4 | 1.65 | 24.1 | 75.8 |
| 15 | [Cu(OAc)2]0.90@Zn0.10–Co | Cu(OAc)2 | 50 | 16 | 30.5 | 3.23 | 18.1 | 1.72 | 29.5 | 89.9 |
| 16 | [CuCl2]0.20@Zn0.80–Co | CuCl2 | 5 | 16 | 6.67 | 26.1 | 18.4 | 1.62 | 5.79 | 18.4 |
| 17 | [CuCl2]0.42@Zn0.58–Co | CuCl2 | 50 | 8 | 14.9 | 20.5 | 17.7 | 1.80 | 11.2 | 35.9 |
| 18 | [CuCl2]0.55@Zn0.45–Co | CuCl2 | 200 | 16 | 18.1 | 14.6 | 18.3 | 1.66 | 54.4 | |
| 19 | [CuCl2]0.83@Zn0.17–Co | CuCl2 | 300 | 16 | 18.2 | 5.70 | 18.2 | 1.71 | 82.2 | |
| 20 | CuCl2–Cod | CuCl2 | — | — | 35.8 | — | 17.6 | 1.83 | — | — |
| 21 | Cu(OAc)2–Coe | Cu(OAc)2 | — | — | 38.0 | — | 17.0 | 2.01 | — | — |
Characterization
The metal content of the PBAs was determined by ICP-OES analysis using a Varian 720-ES equipped with a double-pass glass cyclonic spray chamber, a Sea Spray concentric glass nebulizer and a high solids torch. The digestion of the samples was done following a previously reported procedure.35 PXRD patterns were collected on a Malvern PANalytical Empyrean diffractometer (in transmission mode) over a 1.3–50° 2θ range, using a PIXcel3D solid state detector and Cu anode (Cu Kα1: 1.5406 Å; Cu Kα2: 1.5444 Å). Lattice parameters were refined with TOPAS-Academic V5 using the Le Bail method in space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (or Pmm for CuCl2–Co PBA).36,37 The textural properties of selected samples were studied by N2 physisorption. The N2 isotherms were collected at −196.15 °C on a Micromeritics 3Flex Surface Analyzer. The specific surface area (SBET) was obtained using the BET method (0.05–0.3 p/p0 range), and the micropore volume (Vmicro) and external surface area (Sext) were determined using t-plot analysis. Before analyses, the samples were evacuated at 110 °C for 16 h. FTIR spectra of KBr wafers (containing ∼1 wt% of sample) were collected on a Bruker IFS 66 v/S Vacuum FTIR spectrometer. The acid nature and acid site density were determined by pyridine adsorption followed by FTIR spectroscopy using a Nicolet 6700 FTIR spectrometer. For this, a self-supported wafer (∼10 mg cm−2) was placed in a cell under vacuum and activated at 250 °C for 1 h. After this, a reference spectrum was recorded at 150 °C. The cell was then cooled down further and pyridine (25 mbar) was adsorbed onto the wafer at 50 °C until the sample was saturated. The weakly coordinated pyridine was removed by evacuation for 30 min before reheating to 150 °C to record the IR spectrum. The Lewis acid site density was calculated from the area of the absorption band at 1450 cm−1 in the difference spectrum using the integrated molar extinction coefficient from Emeis.38 High angle annular dark field (HAADF) images and EDX maps were obtained in a JEOL ARM-200F TEM with a probe Cs corrector operated at 200 kV. Prior to imaging, the samples were suspended in ethanol and dropped onto a Cu grid (300 Mesh, Pacific Grid Tech, USA) coated with a Lacey carbon layer. Thermogravimetric analysis (TGA) measurements were carried out on a TGA Q500 of TA Instruments with a heating rate of 10 °C min−1 under compressed air atmosphere. Additional TGA–MS analyses were performed under similar conditions on a NETZSCH STA 449 F3 Jupiter® thermal analyser coupled with a Hiden HPR-20 EGA gas analysis system.
m (or Pmm for CuCl2–Co PBA).36,37 The textural properties of selected samples were studied by N2 physisorption. The N2 isotherms were collected at −196.15 °C on a Micromeritics 3Flex Surface Analyzer. The specific surface area (SBET) was obtained using the BET method (0.05–0.3 p/p0 range), and the micropore volume (Vmicro) and external surface area (Sext) were determined using t-plot analysis. Before analyses, the samples were evacuated at 110 °C for 16 h. FTIR spectra of KBr wafers (containing ∼1 wt% of sample) were collected on a Bruker IFS 66 v/S Vacuum FTIR spectrometer. The acid nature and acid site density were determined by pyridine adsorption followed by FTIR spectroscopy using a Nicolet 6700 FTIR spectrometer. For this, a self-supported wafer (∼10 mg cm−2) was placed in a cell under vacuum and activated at 250 °C for 1 h. After this, a reference spectrum was recorded at 150 °C. The cell was then cooled down further and pyridine (25 mbar) was adsorbed onto the wafer at 50 °C until the sample was saturated. The weakly coordinated pyridine was removed by evacuation for 30 min before reheating to 150 °C to record the IR spectrum. The Lewis acid site density was calculated from the area of the absorption band at 1450 cm−1 in the difference spectrum using the integrated molar extinction coefficient from Emeis.38 High angle annular dark field (HAADF) images and EDX maps were obtained in a JEOL ARM-200F TEM with a probe Cs corrector operated at 200 kV. Prior to imaging, the samples were suspended in ethanol and dropped onto a Cu grid (300 Mesh, Pacific Grid Tech, USA) coated with a Lacey carbon layer. Thermogravimetric analysis (TGA) measurements were carried out on a TGA Q500 of TA Instruments with a heating rate of 10 °C min−1 under compressed air atmosphere. Additional TGA–MS analyses were performed under similar conditions on a NETZSCH STA 449 F3 Jupiter® thermal analyser coupled with a Hiden HPR-20 EGA gas analysis system.
A3 coupling
The catalysts (10 mg) were activated at 80 °C under vacuum for 16 h and then loaded into glass reaction vials with phenylacetylene (0.05 mmol), piperidine (0.1 mmol), benzaldehyde (0.1 mmol), 2-butanol (0.5 mL, solvent) and dodecane (0.1 mmol, internal standard). The vials were then placed in a preheated aluminum block at 110 °C and stirred at 300 rpm using a magnetic stirring bar. After reaction, the catalyst was removed by centrifugation and the liquid supernatant was analyzed by GC (Shimadzu 2014 GC equipped with a FID detector and a CP-Sil 5 CB column) and GC-MS (Agilent 6890 gas chromatograph, equipped with a HP-5MS column, coupled to a 5973 MSD mass spectrometer). Recycling test reactions were carried out after recovery and re-activation of the sample before each run.Results and discussion
Ion exchange
The amount of Cu(II) incorporated in the Zn–Co PBA via ion exchange was quantified by ICP-OES (Table 1). The Cu(II) loading was found to be dependent on the metal salt used in the ion exchange process. The lowest level of exchange was achieved with Cu(ClO4)2·6H2O (ion exchange degree, IEDCu, 12%; entry 2) in comparison to the other salts (IEDCu = 14% for Cu(CH3COO)2·H2O, entry 7; and IEDCu = 18% for CuCl2·2H2O, entry 16). At higher Cu concentrations an IEDCu of 90% could be achieved when Cu(CH3COO)2·H2O was used as copper source, with Cu constituting 30.5 wt% of the PBA structure (entry 15). The Cu(II) salt anion may influence the exchange process in several ways: changes in the pH of the ion exchange solution (Fig. S1†) and the stability constants of the Zn salts resulting from the exchange process,39,40 may account for the notable differences in IEDCu. Furthermore, there are clear differences between these salts regarding dissociation: while dissociation is complete for Cu(ClO4)2·6H2O, Cu(CH3COO)2·H2O is known to produce [Cu(CH3COO)]+ in solution.41 This suggests that the lattice-terminating Zn(II) cation is more readily replaced by a soft [Cu(CH3COO)]+ cation with charge +1, than by a fully hydrated cation with charge +2.The effect of time on the ion exchange process is presented in Fig. 1. The Cu(II) content increases with the Cu(II) salt concentration in the solution. Nevertheless, for the [Cu(OAc)2]x@Zn1−x–Co series, increasing ion exchange time from 8 to 16 h hardly affected the final Cu loading, especially at low Cu(CH3COO)2·H2O concentrations (Fig. 1 and Table 1, entries 4 and 5, and entries 7 and 8). High solid yields (>90%, mass based) were obtained at all studied exchange conditions and no Co was detected in solution after exchange, confirming that Zn is the only metal ion extracted from the structure and that there is no further dissolution of the PBA framework during the post synthetic exchange treatment (Co loss <1 wt% leached from the PBA).
 | ||
| Fig. 1 Effect of time of the exchange in the final Cu(II) loading in [Cu(OAc)2]x@Zn1−x–Co PBAs as a function of the solution concentration. | ||
The insertion of monovalent, divalent and even trivalent cations into the interstitial sites of PBA frameworks has been the prime focus of several studies in diverse fields, like energy storage10–12,23–26,42–45 and radionuclide sorption.13–15,46–48 In electrochemical insertions – charging and discharging of cations – it is believed that the intercalation of these cations in the so-called A sites occurs concomitantly with the redox reaction of the central anionic group.43,44,49 However, as no reduction of Co(III) takes place during the ion exchange process (Fig. S2†), a mechanism similar to that observed in electrochemical insertions is, in all likelihood, not the main pathway of the Cu(II) exchange. On the other hand, the mechanism of radionuclide sorption in PB and PBAs is still under debate. Cs+ is widely believed to adsorb on PBAs by K+ or H+ exchange.15,48 Of these two possible processes, only proton exchange is relevant in this research, as elemental analysis (ICP) of the parent Zn–Co PBA material showed no presence of K in its structure. In order to study the possibility of a H+ exchange mechanism, we investigated the pH of the supernatant solutions during the ion exchange process. The negligible changes in pH observed during the ion exchange process (Fig. S1†) prove that the Cu(II) loading does not induce dissociation of adsorbed water molecules and subsequent release of protons into solution. Additionally, the amount of Zn released into the solution (Znloss) corresponds with the amount of copper introduced in the sample after ion exchange (Cu wt%) and with the decrease in Zn content compared to the parent Zn–Co PBA (Table 1). This implies that the Zn(II) atoms in the PBA framework have solely been replaced by Cu(II) atoms.
To study if Cu(II) atoms in Cu–Co PBAs could also be exchanged with Zn(II), CuCl2–Co PBA was dispersed in aqueous solutions of Zn(CH3COO)2·2H2O or ZnCl2 (Table S1†). In this case, the metal ion exchange process was less effective, leading only to an IEDZn of ∼47% at high salt concentrations, which suggests that Cu(II) ions bind preferentially — in comparison to Zn(II) — to the cyanide group. This has also been observed in the synthesis of multi metal CuxZn1−x–Co PBAs by co-precipitation.28 However, when the ion exchange was carried out at higher temperatures, a greater Zn loading could also be achieved (IEDZn = 79 for the sample [Zn(OAc)2]0.78@Cu0.22–Co PBA, prepared at 80 °C).
Characterization
The crystallinity of the samples was confirmed by PXRD (Fig. 2, S3, S4†). The patterns of the [Cu(Z)v]x@Zn1−x–Co PBAs show no prominent differences when compared to that of Zn–Co PBA, with all samples exhibiting reflections corresponding to the cubic space group Fmm, typical of PBAs.50–52 However, the peak positions are slightly shifted to higher angles as the Cu content is increased, which is expected given the smaller ionic radius of Cu(II) compared to that of Zn(II).53 Moreover, using Le Bail refinements, the cell parameter a was refined for the exchanged samples. A linear correlation between the value of a and the Cu/(Cu + Zn) ratio was obtained (Fig. 3), which is in accordance with Vegard's law and again implies partial replacement of Zn(II) by Cu(II) in the framework.21,54–56 Furthermore, from HAADF-STEM images (Fig. 4) no segregated Cu-rich or Zn-rich phases could be observed.
 | ||
| Fig. 2 XRD patterns of selected [Cu(OAc)2]x@Zn1−x–Co PBAs. | ||
 | ||
| Fig. 3 Variation of the cell parameter (a) with the Cu(II) content for [Cu(OAc)2]x@Zn1−x–Co PBAs. Empty circles (○) correspond to a obtained from the Le Bail refinement of the PXRD data. | ||
 | ||
| Fig. 4 HAADF-STEM images and EDX composition mapping for Cu and Zn of the samples: (a) [Cu(OAc)2]0.45@Zn0.55–Co PBA, (b) [Cu(OAc)2]0.67@Zn0.33–Co PBA and (c) [Cu(OAc)2]0.90@Zn0.10–Co PBA. Scale bar: 200 nm. | ||
The physicochemical properties of the ion exchanged samples, namely textural properties, Lewis acid site density and thermal stability, were found to be intermediate between those of the bimetallic PBAs (Zn–Co, CuCl2–Co and Cu(OAc)2–Co). For all studied samples, a type I isotherm was obtained, characteristic of microporous PBAs (Fig. S5†). Furthermore, there was no decrease in the SBET and Vmicro of the Cu(II) exchanged samples, compared to the SBET and Vmicro of Zn–Co PBA (Table S2†). This indicates that the Cu(II) atoms were incorporated into the PBA framework and that no Cu-rich particles are blocking the pores. The difference FTIR spectra of adsorbed pyridine obtained for selected PBA samples are presented in Fig. S6.† The amount of pyridine adsorbed on Lewis acid sites per mass of PBA decreases in the order Zn–Co PBA (0.10 mmol g−1) > [Cu(OAc)2]0.90@Zn0.10–Co PBA (0.072 mmol g−1) > Cu–Co PBA (0.044 mmol g−1). The TGA profiles of all studied samples (Fig. S7†) exhibit a first mass decay before 100 °C attributed to the loss of water and a sharp mass decay between 300 and 370 °C, corresponding to the decomposition of the framework. However, the TGA profile of sample [Cu(OAc)2]0.90@Zn0.10–Co PBA presents an additional mass loss between 200 and 300 °C. In order to identify the compounds removed at certain temperatures, TGA-MS studies were performed. Results show (Fig. S8,†m/z 60 curve) that this additional, intermediate step in the TGA curve corresponds to the release of acetic acid from the PBA structure, which was incorporated during ion exchange with Cu(CH3COO)2·H2O.
FTIR spectra of selected samples (Fig. S9†) show all the typical bands of PBAs: a band around 1600 cm−1 corresponding to the bending vibration of water molecules, a wide band observed around 3500 cm−1, due to the symmetric and asymmetric vibration of water molecules, a band around 470 cm−1 attributed to the bending vibrations of the Co–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N chain and a band at ∼2190 cm−1 ascribed to the stretching vibrations of the CN bond.57 However, the FTIR spectra of [Cu(OAc)2]0.70@Zn0.30–Co, [Cu(OAc)2]0.77@Zn0.23–Co and [Cu(OAc)2]0.90@Zn0.10–Co present additional absorption bands in the range 1500–1300 cm−1, which corresponds to the –COO− stretching region.58–60 Such bands were also observed in the FTIR spectrum of Cu(CH3COO)2·H2O. This observation is in agreement with the TGA-MS analyses that revealed the presence of acetate in the structure of [Cu(OAc)2]0.90@Zn0.10–Co PBA. In contrast, these bands were not observed in the FTIR spectra of the sample Cu(OAc)2–Co PBA. Moreover, the frequency of the CN stretching vibration allows a qualitative characterization of the distribution of Cu–N and Zn–N bonds in the samples. Indeed, a closer inspection of the CN stretching region in the FTIR spectra of the [Cu(OAc)2]x@Zn1−x–Co PBAs (Fig. 5) reveals a continuous variation in the ν(CN) as a function of the Cu(II) content.
N chain and a band at ∼2190 cm−1 ascribed to the stretching vibrations of the CN bond.57 However, the FTIR spectra of [Cu(OAc)2]0.70@Zn0.30–Co, [Cu(OAc)2]0.77@Zn0.23–Co and [Cu(OAc)2]0.90@Zn0.10–Co present additional absorption bands in the range 1500–1300 cm−1, which corresponds to the –COO− stretching region.58–60 Such bands were also observed in the FTIR spectrum of Cu(CH3COO)2·H2O. This observation is in agreement with the TGA-MS analyses that revealed the presence of acetate in the structure of [Cu(OAc)2]0.90@Zn0.10–Co PBA. In contrast, these bands were not observed in the FTIR spectra of the sample Cu(OAc)2–Co PBA. Moreover, the frequency of the CN stretching vibration allows a qualitative characterization of the distribution of Cu–N and Zn–N bonds in the samples. Indeed, a closer inspection of the CN stretching region in the FTIR spectra of the [Cu(OAc)2]x@Zn1−x–Co PBAs (Fig. 5) reveals a continuous variation in the ν(CN) as a function of the Cu(II) content.
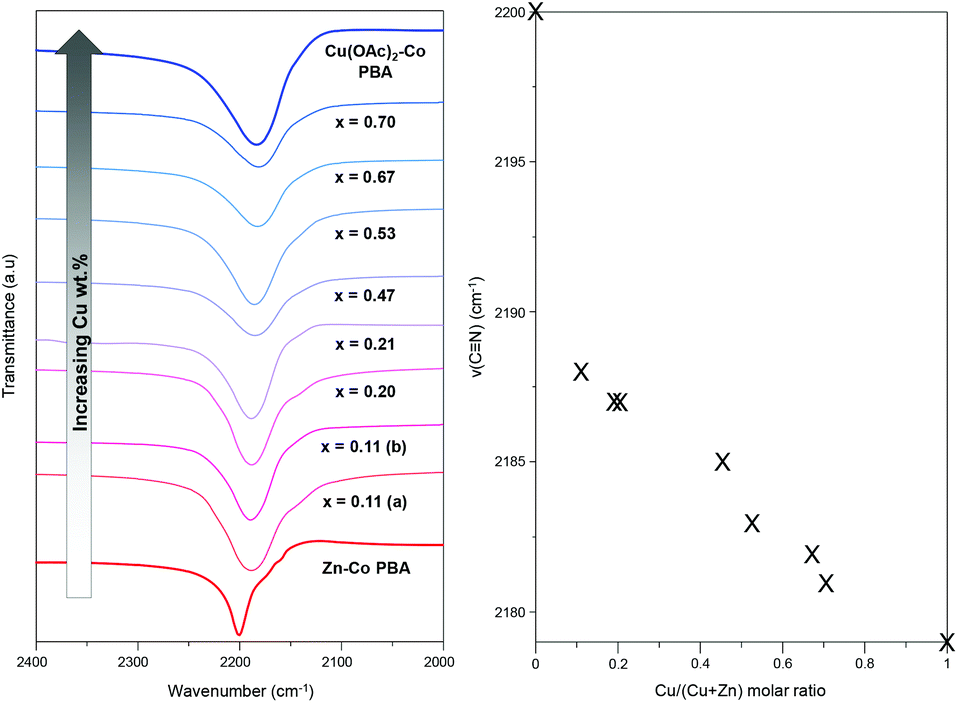 | ||
| Fig. 5 CN stretching region of the FTIR spectra (left) of [Cu(OAc)2]x@Zn1−x–Co PBAs starting from the parent material (bottom spectrum) to the reference PBA (top spectrum) and variation of the ν(CN) with the Cu(II) content (right). | ||
A3 coupling
The catalytic activity of the [Cu(Z)v]x@Zn1−x–Co PBAs was investigated in the synthesis of propargylamines via the A3 coupling of phenylacetylene, benzaldehyde and piperidine. Propargylamines are versatile intermediates in the production of N-containing compounds, and their motif is part of pharmaceutical products used for treatment of chronic neurodegenerative diseases.61–64 The yield of A3 product obtained with the studied samples is presented in Fig. 6 and Table S3.† The incorporation of Cu into the structure increases the catalytic activity of the material in comparison to Zn–Co PBA, as the Cu(II) atoms are necessary for the rapid activation of phenylacetylene via generation of a metal acetylide intermediate.65–67 Furthermore, a higher selectivity to the A3 product was obtained when the reaction was catalyzed by multi metal PBAs, in comparison to that obtained with bimetallic Cu–Co PBAs. Zn(II) atoms can facilitate the formation of the iminium ion intermediate and aid in the coupling between the iminium ion and the metal acetylide,28,68 thus increasing the yield of the A3 product over phenylacetylene-derived side products. | ||
Fig. 6 Yield of A3 product after 6 h reaction time for the coupling of phenylacetylene (0.05 mmol), piperidine (0.1 mmol) and benzaldehyde (0.1 mmol) at 383 K over 10 mg of Zn–Co ( ), CuCl2–Co ( ), CuCl2–Co ( ), Cu(OAc)2–Co ( ), Cu(OAc)2–Co ( ), [Cu(OAc)2]x@Zn1−x–Co ( ), [Cu(OAc)2]x@Zn1−x–Co ( ), [CuCl2]x@Zn1−x–Co ( ), [CuCl2]x@Zn1−x–Co ( ) and CuxZn1−x–Co PBAs (□) with different Cu/(Cu + Zn). ) and CuxZn1−x–Co PBAs (□) with different Cu/(Cu + Zn). | ||
At low to medium Cu(II) loadings (up to Cu/(Cu + Zn) 0.67), the [Cu(Z)v]x@Zn1−x–Co PBAs exhibit a higher activity than PBAs of similar composition synthesized by co-precipitation (CuxZn1−x–Co PBAs, ESI†). Although no Cu(II) concentration gradient could be observed in the EDX composition mapping of the [Cu(Z)v]x@Zn1−x–Co PBAs (Fig. 4), it is likely that the Cu(II) content at the crystal outer edges is higher – and closer to an optimal Zn(II)/Cu(II) ratio – than in the bulk. Based on Zn K–edge EXAFS measurements, Liu et al.14 concluded that Zn(II) atoms coordinated by fewer N atoms – for instance, at defects or surrounded by a higher than average number of [Co(CN)6]3− vacancies, but also at the crystal outer surface – are more likely to be released from the structure. Consequently, these highly accessible sites would be occupied more easily by Cu(II) atoms as a result of the ion exchange procedure. In contrast, via a co–precipitation procedure both divalent metals (Zn and Cu) are arbitrarily arranged in the M1 position, so a higher bulk Cu(II) content is required for an optimal Zn(II)/Cu(II) ratio near the accessible outer surface.21 Remarkably, at high Cu(II) loading and especially with the catalysts prepared using copper acetate for PSE ([Cu(OAc)2]x@Zn1−x–Co series), the yield of A3 product is even more increased. In fact, with the sample [Cu(OAc)2]0.77@Zn0.23–Co PBA, full conversion is achieved after only 6 h reaction time. Furthermore the turnover frequency (TOF), determined at initial reaction rates, was calculated to be 6.48 h−1 (compared to TOF = 2.1 h−1 with Cu0.86Zn0.14–Co PBA). The series [Cu(OAc)2]x@Zn1−x–Co not only exhibits a higher activity than that of CuxZn1−x–Co PBAs, but is also more active than the [CuCl2]x@Zn1−x–Co PBAs of similar composition. This outstanding activity is attributed to the presence of CH3COO− in the PBA framework, as evidenced by FTIR and TGA-MS. The acetate ion has been found to participate in other coupling reactions;69,70 it can provide a basic function to the PBA, facilitating the C–H activation of phenylacetylene (generation of the metal acetylide intermediate).71–74
To test the basicity of the CH3COO− containing PBAs, selected samples were studied as catalysts for the nitroaldol or Henry reaction of nitromethane and benzaldehyde. This organic transformation is an interesting route towards the production of nitroaldol intermediates.75,76 Since this reaction usually requires the presence of a base to proceed, it can serve as a tool to probe the basic nature of the samples.77–79 As can be observed in Fig. S10,† the benzaldehyde conversion obtained with the sample [Cu(OAc)2]0.90@Zn0.10–Co PBA was more than three times higher than the one obtained with the rest of the PBAs. This is a clear effect of the presence of CH3COO− moieties in the structure of [Cu(OAc)2]0.90@Zn0.10–Co PBA, which provide the basic function to facilitate the nitroaldol reaction. This represents, to the best of our knowledge, the first report of basic functionalization of PBAs.
The catalytic activity for the A3 coupling reaction of [Cu(OAc)2]0.77@Zn0.23–Co PBA was further compared to that of homogeneous catalysts, like CuCl2·2H2O, Cu(ClO4)2·6H2O or Cu(CH3COO)2·H2O (Table 2). Although a high phenylacetylene conversion is obtained with all the copper salts (entries 2–4), only 1,4–diphenylbuta-1,3-diyne (formed by oxidative homocoupling of phenylacetylene) was detected in the product mixture. Considering that Zn has been found to increase the selectivity to the A3 product,28 the reaction was repeated using a mixture of ZnCl2 and CuCl2·2H2O (entry 5), and ZnCl2 and Cu(CH3COO)2·H2O (entry 6) in the same molar Cu:Zn ratio of the most active PBA (Cu:Zn 77:23). Once again, high phenylacetylene conversions are achieved after 6 h reaction time. However, in this case, the presence of Zn as a homogenous salt in the reaction mixture has a negligible effect on the selectivity to the A3 product. This indicates that the coordination of Cu and Zn in the PBA framework is effective in suppressing the homocoupling of phenylacetylene, even in the presence of air, by stabilization of the oxidation state of Cu(II).28,66 Additionally, we compared the activity of the ion exchanged PBAs with that of other heterogeneous Cu(II) catalysts (ESI†), namely Cu(II)/MCM-41 (Cu wt% = 19.4), Cu(II)/BEA (Cu wt% = 23.6), Cu(II)/SiO2 (Cu wt% = 19.8) and Cu(II)-BTC (Cu wt% = 26.2%). These materials also exhibited a lower phenylacetylene conversion and selectivity to the A3 product than [Cu(OAc)2]0.77@Zn0.23–Co PBA (Table 2, entries 7–10), which highlights the advantages of the ion exchanged PBAs over other heterogeneous A3 coupling catalysts. Moreover, the heterogeneity of the PBA catalyst was studied by a hot filtration test (Fig. S11†). No appreciable activity is observed after removal of the PBA, indicating that no active species leach from the solid. Finally, recycling tests show that the sample [Cu(OAc)2]0.90@Zn0.10–Co PBA maintains its activity after 10 runs (Fig. S12†), with no notable loss of crystallinity or phase change observed after reaction (Fig. S13†).
| Entry | Catalyst | Xa (%) | Sb (%) | Y (%) |
|---|---|---|---|---|
|
a Conversion of phenylacetylene.
b Selectivity to the A3 product based on phenylacetylene.
c Acetophenone was the only phenylacetylene-derived side-product detected.
d Mixture of salts in a Cu:Zn 77:23 molar ratio.
e 1,4-Diphenylbuta-1,3-diyne (from the homocoupling of phenylacetylene) was the only phenylacetylene-derived side-product detected.
f Both acetophenone and 1,4-diphenylbuta-1,3-diyne were detected. Reaction conditions: Phenylacetylene (0.05 mmol), piperidine (0.1 mmol) and benzaldehyde (0.1 mmol) at 383 K over 10 mg of catalyst for 6 h.
|
||||
| 1 | [Cu(OAc)2]0.77@Zn0.23–Co | >99 | 98c | 98 |
| 2 | Cu(OAc)2 | 97 | 4e | 4 |
| 3 | Cu(ClO4)2 | 69 | <1e | <1 |
| 4 | CuCl2 | 94 | <1e | <1 |
| 5 | (CuCl2 + ZnCl2)d | 90 | <1e | <1 |
| 6 | (Cu(OAc)2 + ZnCl2)d | 95 | 7e | 7 |
| 7 | Cu(II)/BEA | 63 | 68f | 43 |
| 8 | Cu(II)/MCM-41 | 42 | 73f | 31 |
| 9 | Cu(II)/SiO2 | 61 | 69f | 42 |
| 10 | Cu(II)-BTC | 87 | 76f | 66 |
Conclusions
We have demonstrated the occurrence of PSE of metal ions in PBAs for the first time, based on the quantification of the amount of atoms released from the structure and the amount incorporated in the PBA, as well as, on the characterization of the PBA after the PSE. While high temperatures were necessary to achieve a high Zn(II) ion exchange degree (IEDZn), the metathesis between Zn(II) and Cu(II) in Zn–Co PBA occurred easily at room temperature. At low to medium Cu(II) loadings, the Cu(II) exchanged PBAs exhibited a higher activity in the A3 coupling reaction than the bimetallic PBAs and the multi metal samples prepared by co-precipitation. At high Cu(II) loadings, the presence of the CH3COO− (confirmed by FTIR and TGA-MS) provided a basic function resulting in a superior catalytic activity for the series [Cu(OAc)2]x@Zn1−x–Co. The sample [Cu(OAc)2]0.77@Zn0.23–Co was also more active than other commonly used A3 coupling catalysts, effectively suppressing the activity of the oxidative homocoupling of phenylacetylene, even in the presence of air. The basicity of PBAs was further studied in the nitroaldol (Henry) reaction, where a higher conversion was obtained with the CH3COO− containing PBAs. All in all, we believe that these findings can have several implications in the way these materials are synthesized, as the ion exchange procedure allows the incorporation of the desired metal in a more accessible site, thus increasing the activity of PBAs for applications in catalysis on the outer surface. Additionally, the possibility to incorporate a basic function in the PBAs can expand their already interesting catalytic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 641887 (project acronym: DEFNET). F. G. C. would like to thank the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 750391 (Project acronym SINMOF) for financial support. F. W. O. Vlaanderen (Research Foundation Flanders) is also acknowledged for project funding (D. D. V.: research projects), aspiring funding (S. S.) and a Postdoctoral Fellowship (T. D. B.). D. D. V. thanks KU Leuven for the Metusalem grant CASAS. The Hercules project AKUL/13/19 is kindly acknowledged for TEM funding. C. V. G. and I. V. thank prof. Jin Won Seo (KU Leuven) for TEM support.Notes and references
- P. Valvekens and D. De Vos, in New Materials for Catalytic Applications, ed. V. I. Parvulescu and E. Kemnitz, Elsevier, Amsterdam, 2016, ch. 1, pp. 1–12 Search PubMed.
- A. Ludi, J. Chem. Educ., 1981, 58, 1013–1013 CrossRef CAS.
- M. Verdaguer and G. Girolami, in Magnetism: Molecules to Materials V, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2005, ch. 9, pp. 283–346 Search PubMed.
- S. Ferlay, T. Mallah, R. Ouahès, P. Veillet and M. Verdaguer, Nature, 1995, 378, 701–703 CrossRef CAS.
- S.-I. Ohkoshi, K. Imoto, Y. Tsunobuchi, S. Takano and H. Tokoro, Nat. Chem., 2011, 3, 564–569 CrossRef CAS PubMed.
- N. Hoshino, F. Iijima, G. N. Newton, N. Yoshida, T. Shiga, H. Nojiri, A. Nakao, R. Kumai, Y. Murakami and H. Oshio, Nat. Chem., 2012, 4, 921–926 CrossRef CAS PubMed.
- A. Takahashi, H. Tanaka, D. Parajuli, T. Nakamura, K. Minami, Y. Sugiyama, Y. Hakuta, S.-I. Ohkoshi and T. Kawamoto, J. Am. Chem. Soc., 2016, 138, 6376–6379 CrossRef CAS PubMed.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 CrossRef CAS PubMed.
- M. R. Hartman, V. K. Peterson, Y. Liu, S. S. Kaye and J. R. Long, Chem. Mater., 2006, 18, 3221–3224 CrossRef CAS.
- W. Zhang, Y. Zhao, V. Malgras, Q. Ji, D. Jiang, R. Qi, K. Ariga, Y. Yamauchi, J. Liu, J.-S. Jiang and M. Hu, Angew. Chem., 2016, 128, 8368–8374 CrossRef.
- H. Fu, C. Liu, C. Zhang, W. Ma, K. Wang, Z. Li, X. Lu and G. Cao, J. Mater. Chem. A, 2017, 5, 9604–9610 RSC.
- S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2015, 3, 959–962 RSC.
- T. Vincent, C. Vincent, Y. Barre, Y. Guari, G. Le Saout and E. Guibal, J. Mater. Chem. A, 2014, 2, 10007–10021 RSC.
- J. Liu, X. Li, A. I. Rykov, Q. Fan, W. Xu, W. Cong, C. Jin, H. Tang, K. Zhu, A. Sundaram Ganeshraja, R. Ge, X. Wang and J. Wang, J. Mater. Chem. A, 2017, 5, 3284–3292 RSC.
- M. Ishizaki, S. Akiba, A. Ohtani, Y. Hoshi, K. Ono, M. Matsuba, T. Togashi, K. Kananizuka, M. Sakamoto, A. Takahashi, T. Kawamoto, H. Tanaka, M. Watanabe, M. Arisaka, T. Nankawa and M. Kurihara, Dalton Trans., 2013, 42, 16049–16055 RSC.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- A. Peeters, P. Valvekens, R. Ameloot, G. Sankar, C. E. Kirschhock and D. E. De Vos, ACS Catal., 2013, 3, 597–607 CrossRef CAS.
- S. Lee, S. T. Baek, K. Anas, C.-S. Ha, D.-W. Park, J. W. Lee and I. Kim, Polymer, 2007, 48, 4361–4367 CrossRef CAS.
- C. Marquez, M. Rivera-Torrente, P. P. Paalanen, B. M. Weckhuysen, F. G. Cirujano, D. De Vos and T. De Baerdemaeker, J. Catal., 2017, 354, 92–99 CrossRef CAS.
- M. B. Zakaria and T. Chikyow, Coord. Chem. Rev., 2017, 352, 328–345 CrossRef CAS.
- C. P. Krap, J. Balmaseda, L. F. del Castillo, B. Zamora and E. Reguera, Energy Fuels, 2010, 24, 581–589 CrossRef CAS.
- L. Reguera, J. Balmaseda, C. P. Krap and E. Reguera, J. Phys. Chem. C, 2008, 112, 10490–10501 CrossRef CAS.
- D. Z. Yang, J. Xu, X. Z. Liao, Y. S. He, H. M. Liu and Z. F. Ma, Chem. Commun., 2014, 50, 13377–13380 RSC.
- M. Xie, M. Xu, Y. Huang, R. Chen, X. Zhang, L. Li and F. Wu, Electrochem. Commun., 2015, 59, 91–94 CrossRef CAS.
- T.-F. Hung, H.-L. Chou, Y.-W. Yeh, W.-S. Chang and C.-C. Yang, Chem. – Eur. J., 2015, 21, 15686–15691 CrossRef CAS PubMed.
- X. Jiang, H. Liu, J. Song, C. Yin and H. Xu, J. Mater. Chem. A, 2016, 4, 16205–16212 RSC.
- A. García-Ortiz, A. Grirrane, E. Reguera and H. García, J. Catal., 2014, 311, 386–392 CrossRef.
- C. Marquez, F. G. Cirujano, C. Van Goethem, I. Vankelecom, D. De Vos and T. De Baerdemaeker, Catal. Sci. Technol., 2018, 8, 2061–2065 RSC.
- D. H. Son, S. M. Hughes, Y. D. Yin and A. P. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- J. Zhao, L. Mi, J. Hu, H. Hou and Y. Fan, J. Am. Chem. Soc., 2008, 130, 15222–15223 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- C. K. Brozek and M. Dinca, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- C. K. Brozek and M. Dinca, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- J. Kuyper and G. Boxhoorn, J. Catal., 1987, 105, 163–174 CrossRef CAS.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- A. Le Bail, H. Duroy and J. L. Fourquet, Mater. Res. Bull., 1988, 23, 447–452 CrossRef CAS.
- A. Coelho, TOPAS-Academic: General Profile and Structure Analysis Software for Powder Diffraction Data, version 4.1, Coelho Software, Brisbane, Australia, 2007 Search PubMed.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- A. Krężel and W. Maret, Arch. Biochem. Biophys., 2016, 611, 3–19 CrossRef PubMed.
- R. Bruce Martin, Inorg. Chim. Acta, 2002, 339, 27–33 CrossRef.
- S. Sattar and D. Eden, J. Phys. Chem., 1982, 86, 140–144 CrossRef CAS.
- L. D. Reed, S. N. Ortiz, M. Xiong and E. J. Menke, Chem. Commun., 2015, 51, 14397–14400 RSC.
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef PubMed.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Yi Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed.
- T. Shiga, H. Kondo, Y. Kato and M. Inoue, J. Phys. Chem. C, 2015, 119, 27946–27953 CrossRef CAS.
- N. L. Torad, M. Hu, M. Imura, M. Naito and Y. Yamauchi, J. Mater. Chem., 2012, 22, 18261–18267 RSC.
- S.-C. Jang, Y. Haldorai, G.-W. Lee, S.-K. Hwang, Y.-K. Han, C. Roh and Y. S. Huh, Sci. Rep., 2015, 5, 17510 CrossRef CAS PubMed.
- H. Yang, L. Sun, J. Zhai, H. Li, Y. Zhaoa and H. Yu, J. Mater. Chem. A, 2014, 2, 326–332 RSC.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS PubMed.
- D. F. Mullica, W. O. Milligan, G. W. Beall and W. L. Reeves, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3558–3561 CrossRef.
- A. Ludi, H. U. Guedel and M. Ruegg, Inorg. Chem., 1970, 9, 2224–2227 CrossRef CAS.
- G. W. Beall, W. O. Milligan, J. Korp and I. Bernal, Inorg. Chem., 1997, 16, 2715–2718 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- A. R. Denton and N. W. Ashcroft, Phys. Rev. A, 1991, 43, 3161–3164 CrossRef CAS.
- O. Halbherr and R. A. Fischer, in The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications, ed. S. Kaskel, Wiley-VCH, Weinheim, 2016, ch. 26, pp. 795–822 Search PubMed.
- M. Lammert, C. Glißmann and N. Stock, Dalton Trans., 2017, 46, 2425–2429 RSC.
- C. P. Krap, B. Zamora, L. Reguera and E. Reguera, Microporous Mesoporous Mater., 2009, 120, 414–420 CrossRef CAS.
- E. A. Secco and G. G. Worth, Can. J. Chem., 1987, 65, 2504–2508 CrossRef CAS.
- N. Masciocchi, E. Corradi, A. Sironi, G. Moretti, G. Minelli and P. J. Porta, Solid State Chem., 1997, 131, 252–262 CrossRef CAS.
- D. C. Pereira, D. L. A. de Faria and V. R. L. Constantino, J. Braz. Chem. Soc., 2006, 17, 1651–1657 CrossRef.
- C. Wei and C.-J. Li, J. Am. Chem. Soc., 2003, 125, 9584–9485 CrossRef CAS PubMed.
- J. Dulle, K. Thirunavukkarasu, M. C. Mittelmeijer-Hazeleger, D. V. Andreeva, N. Raveendran Shiju and G. Rothenberg, Green Chem., 2013, 15, 1238–1243 RSC.
- V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790–3807 RSC.
- W.-J. Yoo, L. Zhao and C.-J. Li, Aldrichimica Acta, 2011, 44, 43–54 CAS.
- M. J. Albaladejo, F. Alonso and M. J. González-Soria, ACS Catal., 2015, 5, 3446–3456 CrossRef CAS.
- I. Luz, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2012, 285, 285–291 CrossRef CAS.
- G. H. Dang, H. Q. Lam, A. T. Nguyen, D. T. Le, T. Truong and N. T. S. Phan, J. Catal., 2016, 337, 167–176 CrossRef CAS.
- R. R. Mondal, S. Khamarui and D. K. Maiti, ACS Omega, 2016, 1, 251–263 CrossRef CAS.
- B. Li, S. Bi, Y. Liu, B. Ling and P. Li, Organometallics, 2014, 33, 3453–3463 CrossRef CAS.
- B. Wang, H.-X. Sun and Z.-H. Sun, Eur. J. Org. Chem., 2009, 3688–3692 CrossRef CAS.
- D. E. Frantz, R. Fässler, C. S. Tomooka and E. M. Carreira, Acc. Chem. Res., 2000, 33, 373–381 CrossRef CAS PubMed.
- R. J. Rahaim Jr. and J. T. Shaw, J. Org. Chem., 2008, 73, 2912–2915 CrossRef PubMed.
- T. Sugiishi and H. Nakamura, J. Am. Chem. Soc., 2012, 134, 2504–2507 CrossRef CAS PubMed.
- V. F. Brameld, M. T. Clark and A. P. Seyfang, J. Chem. Technol. Biotechnol., 1947, 66, 346–353 CrossRef CAS.
- F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS.
- C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442–5444 CrossRef CAS PubMed.
- Y.-M. Chung, Res. Chem. Intermed., 2018, 44, 3673–3685 CrossRef CAS.
- D. Markad and S. K. Mandal, Dalton Trans., 2018, 47, 5928–5932 RSC.
- J.-M. Gu, W.-S. Kim and S. Huh, Dalton Trans., 2011, 40, 10826–10829 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Elemental analysis of the Zn(II) exchanged samples, synthesis procedure of multi metal PBAs (co–precipitation) and Cu(II)-supported materials, experimental details of nitroaldol reaction, additional characterization (PXRD, TGA-MS, N2 sorption, FTIR spectra) and additional catalytic data. See DOI: 10.1039/c9dt00388f |
| This journal is © The Royal Society of Chemistry 2019 |