
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
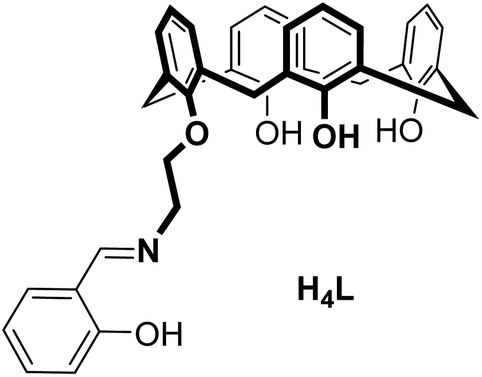
Dinuclear lanthanide complexes supported by a hybrid salicylaldiminato/calix[4]arene-ligand: synthesis, structure, and magnetic and luminescence properties of (HNEt3)[Ln2(HL)(L)] (Ln = SmIII, EuIII, GdIII, TbIII)†
Steve
Ullmann
a,
Peter
Hahn
a,
Laura
Blömer
a,
Anne
Mehnert
a,
Christian
Laube
b,
Bernd
Abel
cb and
Berthold
Kersting
 *a
*a
aInstitut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: b.kersting@uni-leipzig.de; Fax: +49(0)341-97-36199
bWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstraße 2, D-04103 Leipzig, Germany
cLeibniz Institute for Surface Engineering (IOM), Department Functional Surfaces, D-04318 Leipzig, Germany
First published on 22nd February 2019
Abstract
The synthesis, structures, and properties of a new calix[4]arene ligand with an appended salicylaldimine unit (H4L = 25-[2-((2-methylphenol)imino)ethoxy]-26,27,28-trihydroxy-calix[4]arene) and four lanthanide complexes (HNEt3)[Ln2(HL)(L)] (Ln = SmIII (4), EuIII (5), GdIII (6), and TbIII (7)) are reported. X-ray crystallographic analysis (for 4 and 6) reveals an isostructural series of dimeric complexes with a triply-bridged NO3Ln(μ-O)2(OH⋯O)LnO3N core and two seven coordinated lanthanide ions. According to UV-vis spectrometric titrations in MeCN and ESI-MS the dimeric nature is maintained in solution. The apparent stability constants range between log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 5.8 and 6.3. The appended salicylaldimines sensitize EuIII and TbIII emission (λexc 311 nm) in the solid state or immersed in a polycarbonate glass at 77 K (for 5, 7) and at 295 K (for 7).
K = 5.8 and 6.3. The appended salicylaldimines sensitize EuIII and TbIII emission (λexc 311 nm) in the solid state or immersed in a polycarbonate glass at 77 K (for 5, 7) and at 295 K (for 7).
Introduction
Calix[4]arenes1 have turned out to be versatile backbones for multidentate supporting ligands,2,3 and a large number of donor groups have been appended at the lower and upper rim in order to control the properties of the resulting complexes.4–17 Lanthanide complexes of such ligands have also been well investigated,18–24 particularly for their potential in liquid–liquid extraction.25–34 Recently, some research in this area has been directed towards the development of lanthanide-based single-molecule magnets35 and luminescent probes and materials.36–40 The calixarenes are typically designed to saturate the metal's coordination sphere, and several luminescent complexes have been investigated.41–46 Despite the maturity of the field, not many lanthanide complexes of pendant calix[4]arenes were structurally characterized. Most structures are derived from calix[4]arene diamides47 or tetraamides.48 Only a handful of structures with triply appended calix[4]arenes have been reported,49 and as far as we are concerned no structures exist with one-armed calix[4]arenes.50 To fill this gap, we decided to prepare a mono-substituted calix[4]arene-Schiff base ligand H4L and investigate its coordination chemistry towards some lanthanide ions. Hybrid ligands of this sort are known to complex first-row transition metals readily, but their lanthanide chemistry remains largely unexplored.51–57This study demonstrates that H4L supports dinuclear lanthanide complexes (for Ln = Sm, Gd, Eu, and Tb) – a property which contrasts the mononucleating behavior of the double and fourfold functionalized calix[4]arene amides. Their synthesis and characterization along with the investigation of photophysical, magnetic and structural properties are presented herein.
Results and discussion
Synthesis and characterization of the ligand
The salicylaldimine-appended calix[4]arene H4L was readily prepared according to a procedure reported by Zhang et al. for related bis(salicylaldimine)-p-tert-butylcalix[4]arenes (Scheme 1).58 Alkylation of the parent calix[4]arene 1 with bromoacetonitrile followed by reduction of the nitrile 2 provided the amine 3, which was condensed with salicylaldehyde in the presence of MgSO4, to provide the title compound as a pale-yellow solid in 21% overall yield. The IR spectrum of H4L reveals two sharp (3635 and 3500 cm−1) and one broad OH band (3320 cm−1) indicative of hydrogen bonding interactions.59 The CN stretch appears at 1635 cm−1, a typical value for salicylaldimines.60 The calixarene adopts a cone conformation in CH2Cl2 as evidenced by NMR (two characteristic AB systems for the Ar–CH2–Ar groups).61–63 The free ligand displays intense absorption bands in the UV (Table 1), attributed to π → π* transitions of aromatic rings of the calix[4]arene (254, 286 nm)64 and the salicylaldimine (311 nm).65,66 A weak band around 403 nm (ε = 117 M−1 cm−1) can be assigned to the n → π* transition of the imine group.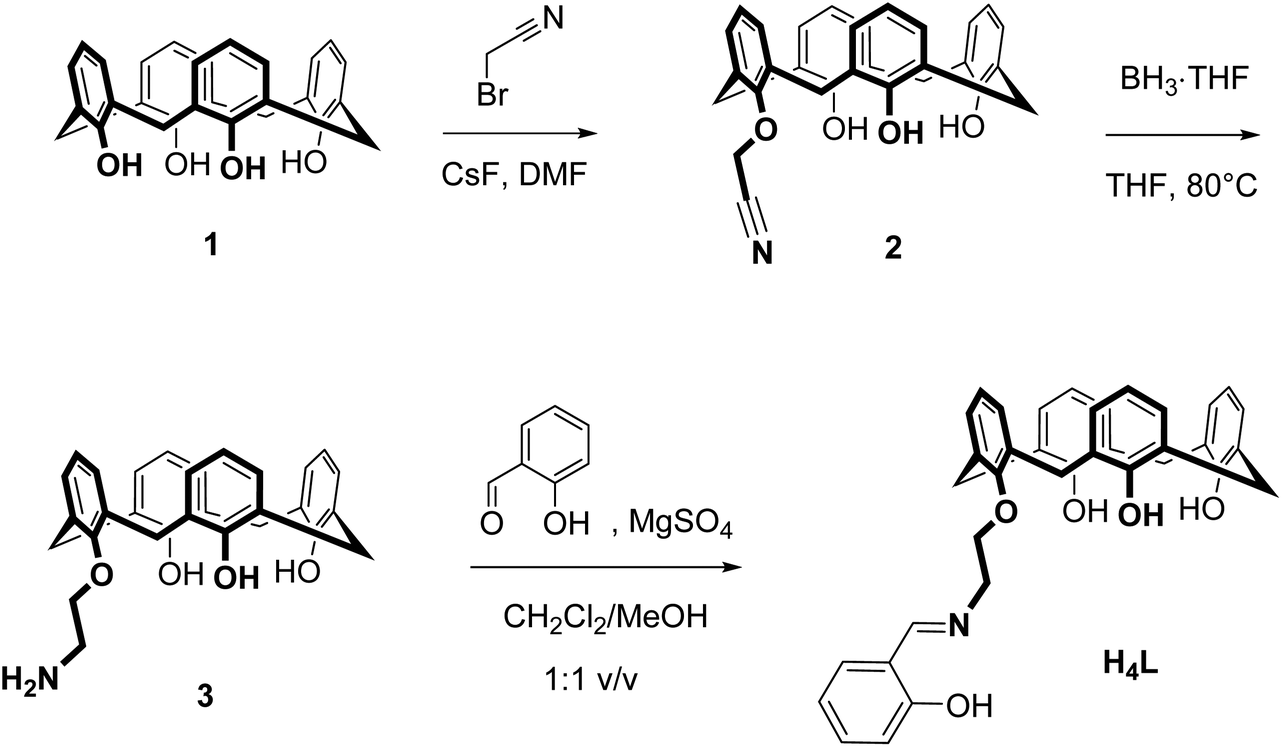 | ||
| Scheme 1 Synthesis of the ligand H4L. | ||
| Compound | ESI-MS(−) | IR/cm−1 | λ max/nm (ε/M−1 cm−1)b |
|---|---|---|---|
| a Concentration of solutions were ∼1.0 × 10−5 M, T = 298 K. b MeCN solution. | |||
| H4L | 570.3 | 1635, 1338 | 254 (13397), 286 (8446), 311 (6321), 403 (117) |
| 4 (Sm) | 1439.2, 719.1 | 1635, 1316 | 299 (9874), 346 (4417) |
| 5 (Eu) | 1441.3, 720.1 | 1636, 1317 | 300 (9845), 346 (4662) |
| 6 (Gd) | 1451.3, 725.1 | 1636, 1325 | 300 (9742), 346 (4513) |
| 7 (Tb) | 1453.3, 726.1 | 1637, 1303 | 300 (9941), 344 (4367) |
The crystal structure of the free ligand (Fig. 1) shows a cone conformation stabilized by three intramolecular OH⋯O hydrogen bonds (O1⋯O2, O2⋯O3, O3⋯O4). The pendant Schiff-base is almost perfectly planar forming an intramolecular OH⋯N hydrogen bond, as in other o-hydroxyaryl Schiff bases.67 An intramolecular CH⋯π interaction manifests itself by a short H8b⋯X1centroid distance of 2.84 Å. Self-inclusion mediated by intermolecular CH2⋯π interactions of length 2.90 Å occurs. This leads to one-dimensional chains as illustrated in Fig. 1.
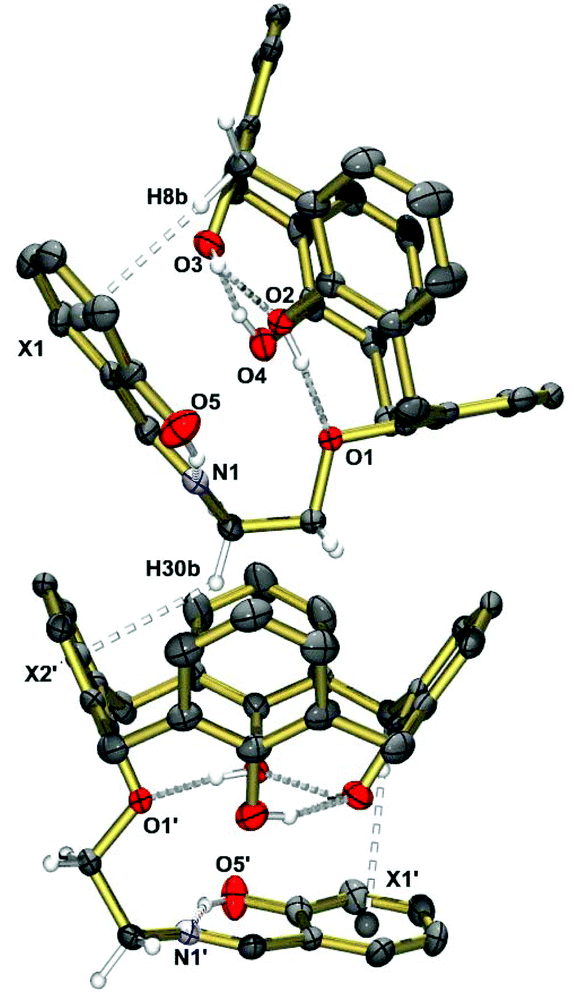 | ||
| Fig. 1 Solid-state structure of compound H4L as determined by single-crystal X-ray diffraction. Selected distances [Å]: O1⋯O2 2.668(3), O2⋯O3 2.628(3), O3⋯O4 2.693(3), O4⋯O1 2.981(3), O5⋯N1 2.609(3); H8b⋯X1 2.84, H30b⋯X2′ 2.90. Symmetry code used to generate equivalent atoms: −x, 0.5 + y, 0.5 − z (′). | ||
The new ligand and all intermediates were characterized by IR, UV-vis, 1H and 13C NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS). 2D NMR experiments (NOESY, HSQC, HMBC) were used to correctly assign the chemical shifts of hydrogen and carbon atoms (ESI†).
Synthesis and characterization of complexes
The reaction of H4L with samarium(III) nitrate hexahydrate was performed with NEt3 as a base (pKa 18.82, MeCN)68 to deprotonate the phenol functions. At a ∼1:1:4.5 molar ratio in a mixed CH2Cl2/MeOH solution at room temperature a pale-yellow solution forms, from which a dinuclear compound of composition (NHEt3)[Sm2(HL)(L)] (4, where L and HL represent the fourfold and threefold deprotonated versions of H4L) could be reproducibly obtained in 82% yield (Scheme 2).
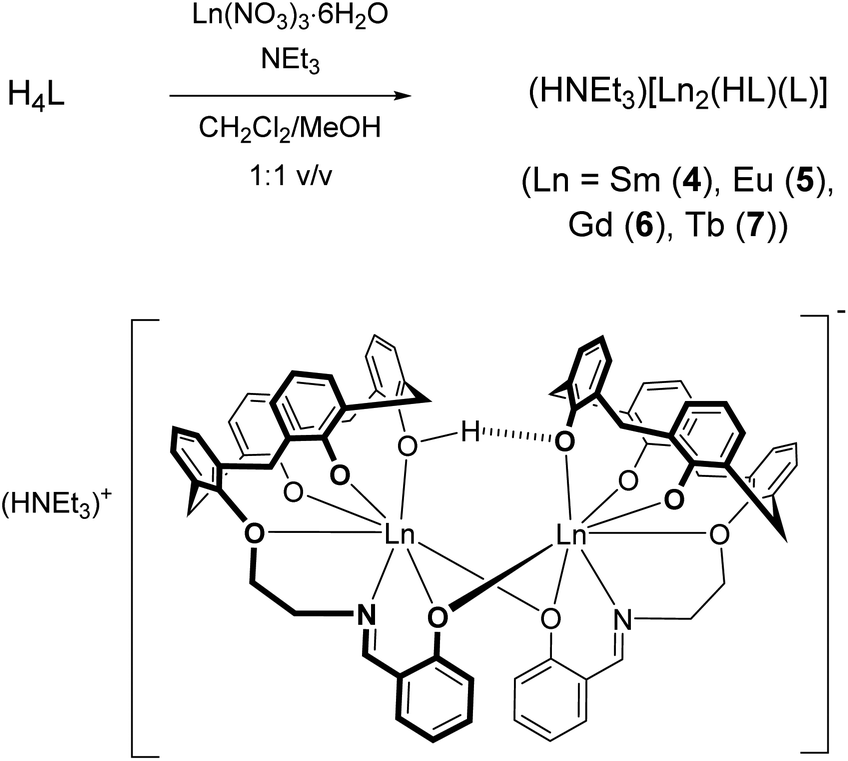 | ||
| Scheme 2 Synthesis of the complexes 4–7. | ||
Analogous europium(III) (5), gadolinium(III) (6) and terbium(III) complexes (7) were also synthesized in this manner. According to ESI-MS, mononuclear complexes of composition [LnL]− are also present (Table 1), but all attempts to isolate these entities failed. The exclusive formation of the [Sm2(HL)(L)]− dimers may be due to a lower solubility although other factors such as packing or specific intermolecular interactions cannot be ruled out. All complexes are air-stable but hygroscopic, and exhibit good solubility in methylene chloride, chloroform and THF. They are moderately soluble in toluene, and only sparingly soluble in protic solvents.
The formulation of the complexes was ascertained in all cases by elemental analysis, mass spectrometry, IR and UV-vis spectroscopy, and in case of the SmIII and GdIII complexes also by X-ray crystallography. The negative ESI-MS spectra of dilute (10−3 M) MeCN/CH2Cl2 solutions exhibit molecular ion peaks at m/z = 1439.2 (4), 1441.3 (5), 1451.3 (6), and 1453.3 (7), respectively, with the correct isotopic peak pattern for dimeric [Ln2(HL)(L)]− anions (ESI). Under these conditions, signals at m/z = 719.1 (4), 720.1 (5), 725.1 (6) and 726.1 (7) for monomeric [LnL]− species are also observed. The IR spectra of all complexes reveal a band at 1635–1636 cm−1 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching frequency, a typical value for imine functions coordinated to LnIII ions.66 O–H stretching bands were absent indicative of LnIII bound phenolate groups. The C–O stretching frequency observed for H4L at 1338 cm−1 is shifted to lower frequencies in the complexes (1327–1316 cm−1), indicative of the coordination of the phenol ether moiety as well.69
N stretching frequency, a typical value for imine functions coordinated to LnIII ions.66 O–H stretching bands were absent indicative of LnIII bound phenolate groups. The C–O stretching frequency observed for H4L at 1338 cm−1 is shifted to lower frequencies in the complexes (1327–1316 cm−1), indicative of the coordination of the phenol ether moiety as well.69
Crystallographic characterization
Single crystals of (HNEt3)[Sm2(HL)L(MeCN)2]·MeCN (4·3MeCN) obtained from MeCN were analyzed by X-ray diffraction. The structure comprises dinuclear [Sm2(HL)(L)(MeCN)2]− anions (Fig. 2), HNEt3+ cations and MeCN molecules. The latter occupy voids in the structure and the calixarene cavities, as in other structures.70 The HNEt3+ ion is located in a cleft generated by three phenyl rings of the dimer and71 hydrogen bonds to a MeCN solvate molecule N3⋯N6 2.90 Å (see Fig. S1†). Significant interactions between the HNEt3+ ion and phenolate O atoms are not observed (N3⋯O7 4.33 Å), presumably due to the fact that the latter are buried by the organic residues of the supporting ligand.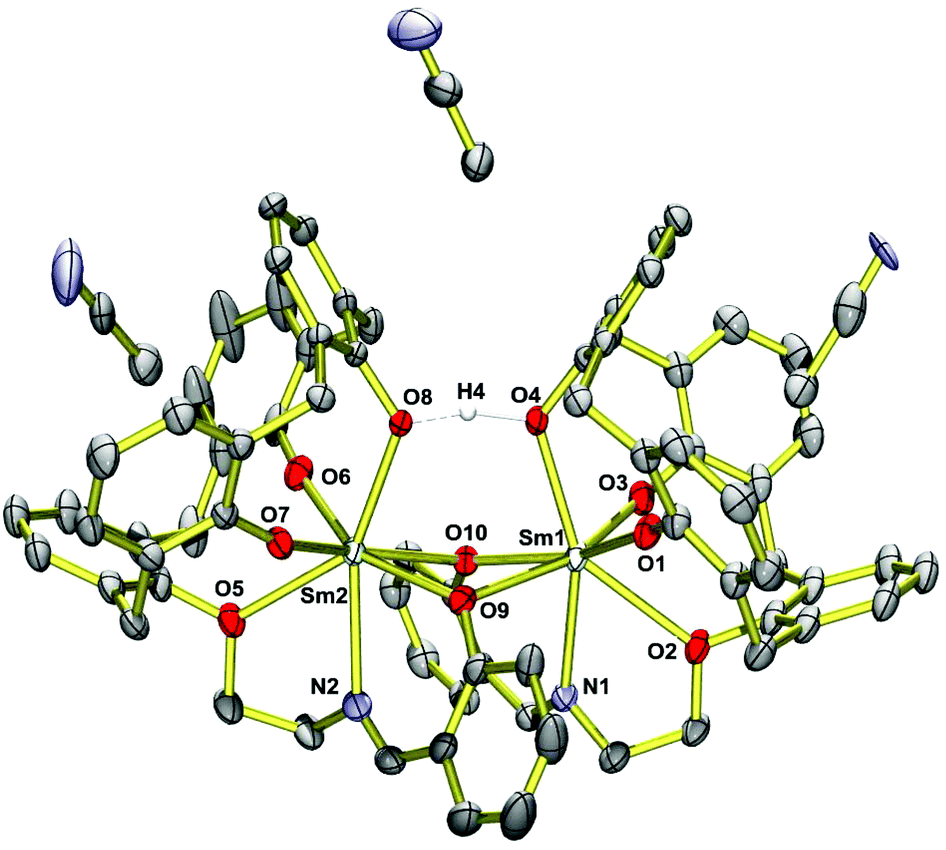 | ||
| Fig. 2 Single-crystal X-ray diffraction structure of the [Sm2(HL)(L)(MeCN)2]− anion in crystals of (HNEt3)[Sm2(HL)(L)(MeCN)2]·MeCN (4·3MeCN). The HNEt3+ ion is omitted for clarity. Thermal ellipsoids are shown at the 30% probability level. | ||
The complex has idealized C2 symmetry comprising two mononuclear SmIIIL units joined by two phenolato bridges to give a four-membered Sm2O2 ring, a motif quite common in lanthanide calixarene structures but herein realized from phenol groups of the salicylidene moieties.72–74 This assembly is reinforced by a hydrogen bond between O4 and O8 of the calixarene bowls (O4⋯O8 2.40 Å, O4–H4⋯O8 164°).75–77 Each samarium atom is further bonded to four calix[4]arene O atoms and to the imine N atom of the Schiff base unit, giving rise to coordination number seven (Fig. 3).
 | ||
| Fig. 3 Ball-and-stick representation of the immediate coordination environments of the Ln atoms in the [Ln2(HL)(L)(MeCN)2]− complex anions. | ||
The calix[4]arene adopts a distorted cone conformation with an “elliptical” rather than a “circular” cross section very similar to that observed in [Eu2(HL)2(dmf)4]·7dmf (where HL represents triply deprotonated p-t-butyl-calix[4]arene).74 The Sm–O bond lengths vary significantly from 2.18–2.59 Å. Four Sm–O bonds are very short (Sm1–O1,O3; Sm2–O6,O7) ca. 2.18 Å. The Sm–O bonds involving the bridging phenolate oxygen donors are significantly longer at 2.38–2.48 Å (Sm1–O9,O10, Sm2–O9,O10), which is not unusual for such bridges. The phenol ether O atoms (O2,O5) are weakly coordinating and form the longest Sm–O bonds (2.58 Å). They compare well with those in samarium complexes [(pic-O)Sm{(L–H)(EtOH)0.5(CH2Cl2)0.5}](pic)·EtOH·2H2O and [Sm(L-2H)(pic)], where pic = picrate anion and L is a bis- or tris-substituted calixarene.78 The Sm–N bonds in 4 are also quite long at 2.56 Å. The presence of the hydrogen bonding interaction is supported by the relatively long Sm1–O4 and Sm2–O8 distances of 2.372 Å and 2.375 Å.
The structure of the gadolinium compound (HNEt3)[Gd2(HL)L(MeCN)2]·MeCN (6·3MeCN) is isomorphous with 4·3MeCN, having slightly shorter Gd–O and Gd–N distances (Table 2), in agreement with its smaller ionic radius.79 The Ln⋯Ln distance is 3.9067(3) Å in 4 and 3.8965(4) in 6. In essence the NO5 donor set of H4L cannot saturate the coordination sphere of the lanthanide ions and so dimerization occurs to share some of the O donors.74 There are no significant intermolecular bonding interactions between the [Ln2(L)(HL)]− complexes. The shortest intermolecular Ln⋯Ln distances are 10.725 Å in 4 and 10.696 Å in 6.
| M | 4·3MeCN (M = Sm) | 6·3MeCN (M = Gd) |
|---|---|---|
| M1–O1 | 2.186(2) | 2.175(3) |
| M1–O2 | 2.586(3) | 2.572(4) |
| M1–O3 | 2.163(3) | 2.158(4) |
| M1–O4 | 2.375(2) | 2.344(3) |
| M1–O9 | 2.474(2) | 2.451(3) |
| M1–O10 | 2.406(2) | 2.385(3) |
| M1–N1 | 2.571(3) | 2.542(4) |
| M2–O5 | 2.576(3) | 2.561(4) |
| M2–O6 | 2.179(3) | 2.162(4) |
| M2–O7 | 2.181(3) | 2.168(4) |
| M2–O8 | 2.372(2) | 2.342(3) |
| M2–O9 | 2.380(2) | 2.367(3) |
| M2–O10 | 2.467(2) | 2.452(3) |
| M2–N2 | 2.554(3) | 2.525(5) |
| M1⋯M2 | 3.9067(3) | 3.8965(4) |
Magnetic properties
The lanthanide complexes were further studied by temperature-dependent magnetic susceptibility measurements using a SQUID-Magnetometer (MPMS Quantum Design) in applied magnetic fields of 0.5 T over a temperature range 2–300 K. Plots of χMT versus T for 4–7 are shown in Fig. 4.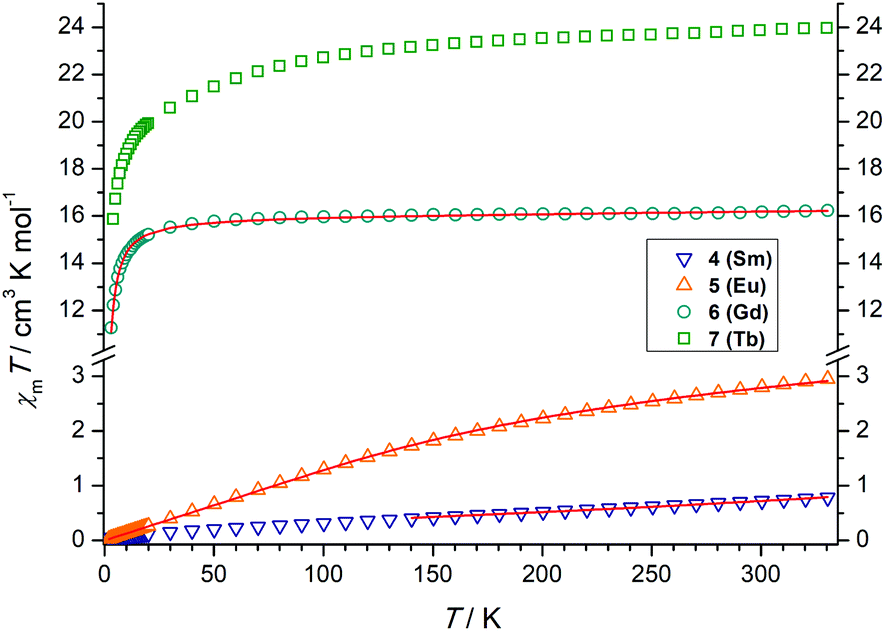 | ||
| Fig. 4 Temperature dependence of the χMT product (at 5000 G) for the dinuclear complexes 4–7, χm being the molar susceptibility per dinuclear complex defined as M/H. The solid lines correspond to the best fits. | ||
For the SmIII2 complex 4 the χMT value is 0.72 cm3 K mol−1 at room temperature, slightly larger than the expected value of 0.64 cm3 K mol−1 for two non-interacting SmIII ions.81 On lowering the temperature, χMT decreases and tends to a value of ca. 0.01 cm3 K mol−1 at 2 K. For Sm3+, with a 6H5/2 (gJ = 2/7) ground state, the multiplet spacing is on the order of kBT and thermal population of excited 6HJ/2 states (J = 7, 9, 11, 13, 15) contributes significantly to the susceptibility. The crystal field, which partially lifts the degeneracy of the J states in zero field, may also affect the susceptibility.
We analyzed the temperature dependence of the magnetic susceptibility of the SmIII complex by utilizing the analytical expression given by Kahn (eqn (S1)†).80 This model considers only the effect of spin–orbit coupling, which is appropriate given that magnetic exchange interactions are weak as suggested by the results for the analogous Gd complex (see below). Indeed, a reasonable fit was possible (excluding the low temperature data). The value of the spin–orbit coupling constant was determined to be λ = 254 cm−1. This value is comparable to that of the free Sm3+ ion (284 cm−1).81
The χMT value of the GdIII2 complex 6 amounts to 16.16 cm3 K mol−1 at 300 K, somewhat larger than the expected value 15.77 cm3 K mol−1 of two uncoupled 8S7/2 centers. Upon cooling, χMT slowly decreases to 15.5 cm3 K mol−1 at 23.3 K and drops to 11.2 cm3 K mol−1 at 3 K, indicative of a very weak antiferromagnetic exchange interaction as in other phenolato-bridged GdIII2 complexes.82,83 For GdIII ions, first-order spin–orbit coupling is absent (L = 0). Therefore, the exchange interaction can be analyzed by using the isotropic spin Hamiltonian H = −J·SGd1·SGd2 with SGd1 = SGd2 = 7/2. The magnetic susceptibility for a dinuclear GdIII complex is given by eqn (1), where g is the Landé factor, μB the Bohr magneton, Na the Avogadro number, kB the Boltzmann constant, and x = J/kBT.84
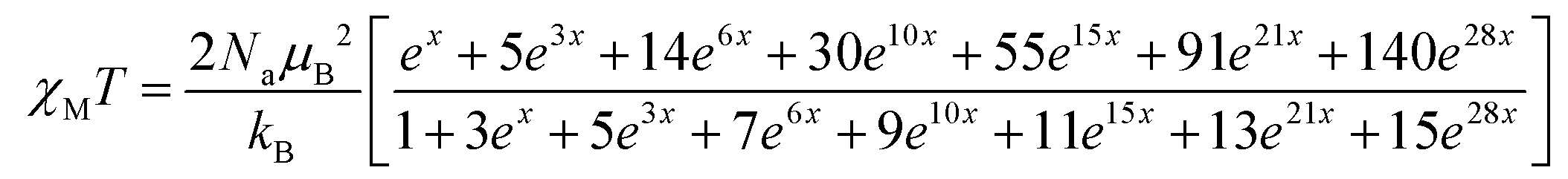 | (1) |
A good fit of the experimental data is possible applying J = −0.065 cm−1 and g = 2.01. Such a weak antiferromagnetic coupling constant is a typical value for phenolato-bridged GdIII systems.82,84–87
The χMT value of the EuIII2 complex 5 is 2.80 cm3 K mol−1 at 300 K, a value which is close to that expected for two non-interacting EuIII ions (2.65 cm3 K mol−1), with non-negligible population of excited 7F1–7F6 levels. The deviation from the Hund-Landé expectation value (0μB) is also attributable to contributions from the second order Zeeman effect in the ground 7F0 multiplet.88 Upon cooling, the χMT values decrease steadily, reaching 0.03 cm3 K mol−1 at 2 K, as in other dinuclear EuIII complexes.89 The magnetic susceptibility of the EuIII2 complex can be fitted to the formula derived by Kahn (eqn (S2)†) by considering only λ (multiplet spacing) as a parameter as also done for the SmIII complex. Again, the magnetic interaction between the EuIII ions are assumed negligible.
Indeed, an excellent fit was possible over the whole temperature range to give λ = 324 cm−1. The multiplet spacing is within the range of kBT, and significant population of the first excited state at 300 K explains the deviation from the Curie law. The λ parameter for 6 agrees with other dinuclear EuIII complexes. In [Eu2(L′)2], for example, where L′ is derived from a calixarene ligand with two hydroxyquinolinolato arms, and the EuIII ions in an N4O4 environment λ = 325 cm−1.90
The χMT value of the dinuclear TbIII complex at 300 K with 23.87 cm3 K mol−1 is slightly higher than the expected value of 23.60 cm3 K mol−1 for a 7F6 ground state. Upon cooling the χMT values decrease first slowly to 22.71 cm3 K mol−1 at 100 K and then more rapidly to 15.87 cm3 K mol−1 at 4 K. TbIII complexes are known to exhibit significant magnetic anisotropy, and fitting of susceptibility data is therefore difficult.84 The field dependence of the magnetization for complex 7 was determined in order to see whether magnetic anisotropy is present in this complex. Indeed, at 2 K the magnetization values increase rapidly at low fields and then linearly but without a clear saturation, reflecting the presence of a significant magnetic anisotropy (Fig. 5). Moreover, a M vs. H/T plot (Fig. 6) illustrates that the curves are not really superimposed on each other as expected for an isotropic system with a well-defined ground state. Nevertheless, hysteresis effects were not observed in M vs. H data at 2 K. Alternative current (ac) measurements were also undertaken to determine potential SMM behavior. However, neither maxima nor imaginary components of the ac susceptibility were observed in the χ′′Mvs. T plots, ruling out an SMM behavior for 7. This may be attributed to the low local symmetry of the TbIII ions.91
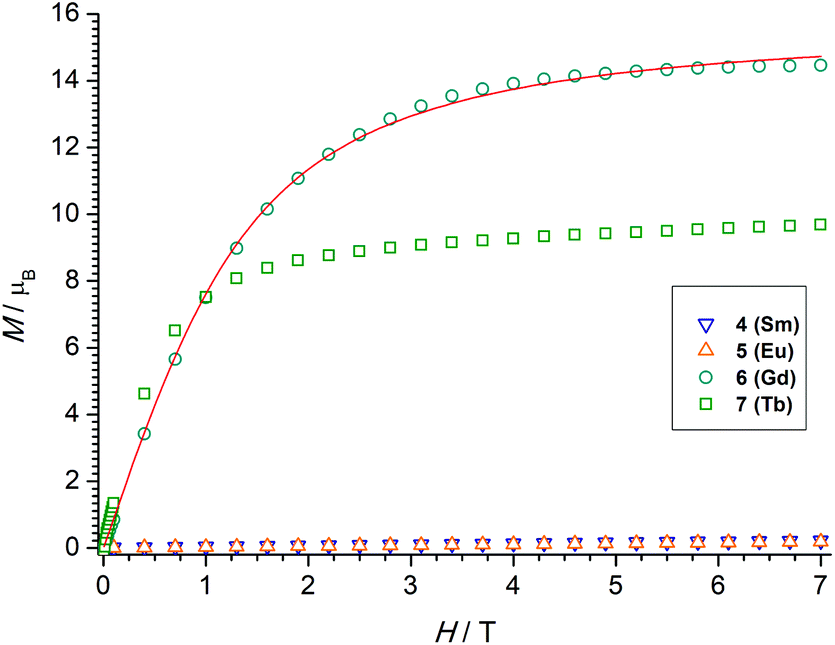 | ||
| Fig. 5 Field dependence of the magnetization for powdered samples of 4–7 at 2 K. | ||
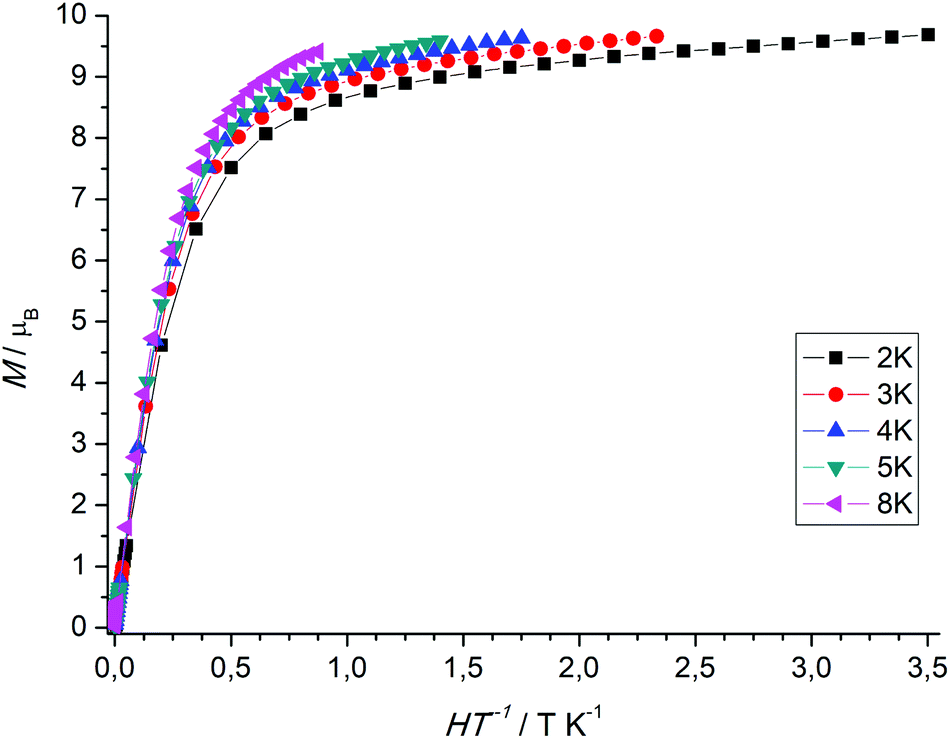 | ||
| Fig. 6 Plot of M versus H/T for the TbIII complex 7 at various temperatures. | ||
Spectrophotometric titrations
To study the complexation reactions of H4L with SmIII, EuIII, GdIII, and TbIII in solution UV-vis spectrophotometric batch titrations were carried out. The experiments were performed at room temperature in acetonitrile at constant ionic strength (10−2 M nBu4NPF6) and pH value (5 × 10−4 M NEt3 buffer). Data were recorded in the 200–600 nm range. The titration of H4L with Sm(NO3)3·6H2O is representative for all complexes (Fig. 7). For the other compounds, see ESI Fig. S29–S35.† Upon addition of aliquots of Sm(NO3)3·6H2O (0–5 equiv.) clear changes occur in the UV-vis ligand spectra. The bands at 254, 286, and 311 nm for H4L vanish with increasing SmIII concentration, while new bands (for [Sm2(HL)(L)]−) develop with maxima at 300 and 345 nm. The final spectra match the recorded spectra of the isolated metal complexes. An isosbestic point at 325 nm indicates that SmIII binding occurs to a single equilibrium.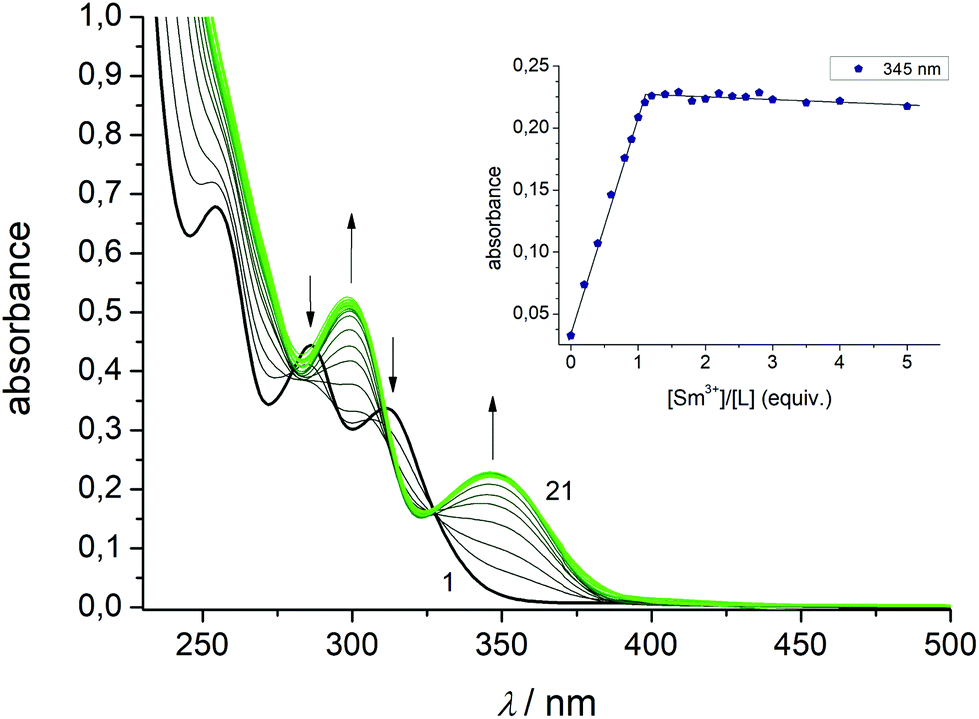 | ||
| Fig. 7 Spectrophotometric titration of H4L with Sm(NO3)3·6H2O in CH3CN (10−5 M concentration) at constant ionic strength (10−2 M NnBu4PF6, T = 298 K) in the presence of 5 × 10−4 M NEt3. The green curve refers to a final molar ratio of M/H4L = 5.0. The inset shows the evolution of absorbance values at 345 nm versus the [SmIII]/[H4L] molar ratio. | ||
To determine the stoichiometry of the resulting species the mole ratio method was applied.92 The inset of Fig. 7 shows a plot of absorbance values at 345 nm versus molar ratio [SmIII]/[H4L]. The values increase steadily up to a molar ratio of about unity and then remains constant. No further changes were observed for up to five-fold excess of lanthanide salt, signifying the formation of a complex species with 1:1 ligand/metal stoichiometry. The other lanthanide ions behave in a very similar fashion. Irrespective of the type of the lanthanide ion, only 1:1 compounds were systematically detected. Nonlinear least-squares refinements of the titration data converged for a speciation model involving the ligand and its 1:1 complexes with apparent stability constants of 6.08(4) (SmIII), 6.21(7) (EuIII), 5.81(4) (GdIII), and 6.34(6) (TbIII). The stability constants show a strong affinity of L4− towards lanthanides and decrease with decreasing ionic radii with the strongest interaction observed with TbIII and the weakest interaction observed for SmIII. There are only very few studies reporting thermodynamic data for f-element calixarene complexes in non-aqueous solvents.93–96 Danil de Namor and Jafour have studied the complexation of trivalent cations by p-tert-butylcalix[4]arene tetraethanoate, p-tert-butylcalix[4]arene tetramethyl ketone, and p-tert-butylcalix[4]arene tetraacetamide in acetonitrile.97 Borisova and co-workers have determined stability constants for lanthanide complexes supported by 2,2′-bipyridyl-6,6′-dicarboxylic acid diamide and 2,6-pyridinedicarboxylic acid diamide ligands in the same solvent. The binding constants of these complexes were found to lie in a similar range (logK ∼ 4–6).98
Spectroscopic and photophysical properties
The new compounds were further characterized by UV-vis absorption and emission spectroscopy. The electronic absorption spectra were measured in acetonitrile (complexes) at room temperature. Table 1 lists the data. All complexes show three intense absorption bands around 220 nm, 300 nm, and 350 nm, respectively. The first two high-energy bands are associated with 1(π–π)* transitions centered on the phenol ether and phenolate groups of the calix[4]arene backbone. The transition at 350 nm can be attributed to the phenyl ring of the salicylaldimine unit. Deprotonation and coordination of the lanthanides red-shifts these features by 15 and 40 nm relative to those of the free ligand. The change of the lanthanide ion appears to have little if any impact on the spectrophotometric properties. Hence, upon going from the Sm to the Tb complex a slight blue-shift of the lowest energy band of ca. 2 nm can be detected.The luminescence properties of the Eu and Tb complexes were investigated in view of literature reports that calix[4]arenes can act as an antenna for the sensitization of lanthanide luminescence.37,41 The free ligand shows a single emission band with a maximum at 455 nm when excited at 285 nm. The two complexes are not emissive in solution (CH3CN, CH2Cl2). However, when embedded in a polymer Eu complex 5 displays four relatively broad and intense transitions (Fig. 8), attributed to 5D0 → 7FJ transitions (J = 1–2) when excited at 311 nm at 77 K.99 Both, the 5D0 → 7F1 (580 nm, 595 nm,) and the 5D0 → 7F2 transitions (620, 630 nm) appear as doublets. In view of the low local symmetry of the coordination polyhedron (C1 in this case), this may be related to crystal-field splitting of the 7F1 and 7F2 levels. Splitting of these levels is not unusual for Eu(III) complexes with such a low site symmetry.100 The 5D0 → 7F0 transition (expected in the 570–585 nm range), is a strictly forbidden transition in site symmetries other than Cnv, Cn or Cs.100 It is also not observed for the present compound.
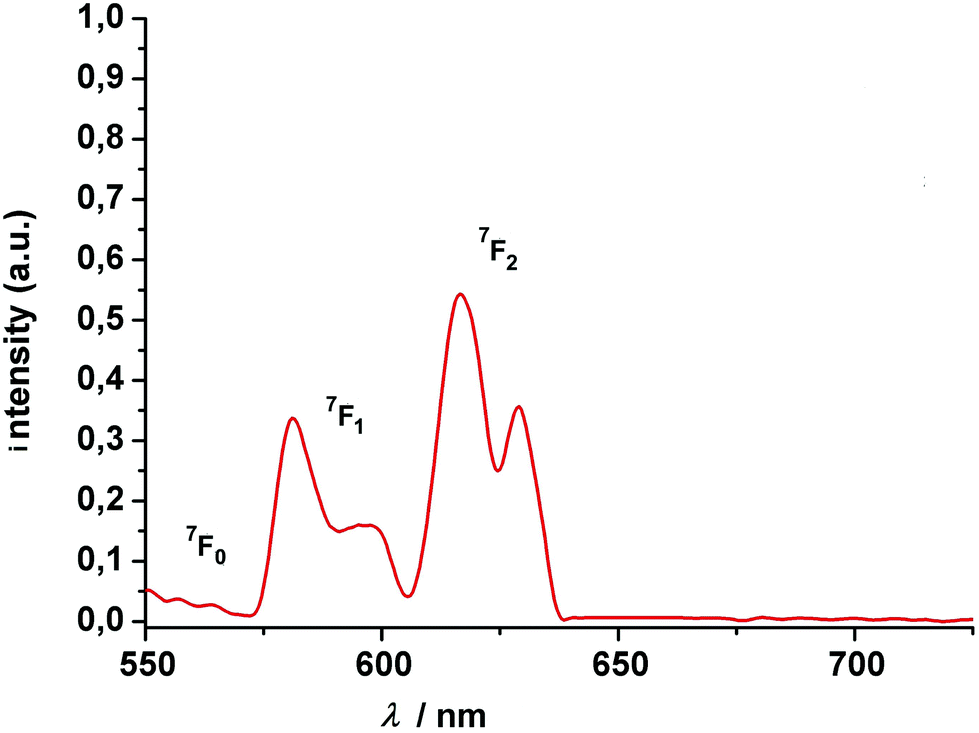 | ||
| Fig. 8 Luminescence spectrum of (HNEt3)[Eu2(HL)(L)] (5) at 77 K (polycarbonate thin films doped with 4 wt% Eu). The excitation wavelength is 311 nm. The transitions above 575 nm start from the 5D0 state. | ||
The intensity of the hypersensitive 5D0 → 7F2 transition (or the ratio R of the intensities I(5D0 → 7F2)/I(5D0 → 7F1) is also often used as a measure for the asymmetry of the Eu3+ site, since the 5D0 → 7F2 signal is strictly forbidden for a Eu3+ at a site with inversion symmetry. In our case, there is no inversion symmetry about the Eu3+ ion. The 5D0 → 7F2 is observed and is 1.6 times more intense than the 5D0 → 7F1 transition, in good agreement with the theoretical predictions.101–103
The Tb complex gives rise to four transitions at 490, 545, 584 and 619 nm, assigned to the 5D4 → 7FJ (J = 6, 5, 4, 3) transitions, again split by crystal-field effects. Of these, the “green” 5D4 → 7F5 transition at 545 nm has the highest intensity. Note that the intensity decreases with increasing temperature, which might be traced to quenching via enhanced vibrational relaxation (energy transfer to the O–H⋯O vibration modes).104,105
The excited state luminescence decay of the immobilized Tb complex is biexponential, although the first exponential term is dominating (99% of the initializing luminescence intensity, I0) with a lifetime of about τ1 = 81 ± 2.5 μs. A small contribution of a second term with a time constant of τ2 = 305 ± 3 μs was determined. The origin of the second term may be a different conformation of the complex due to the imbedding into the polymer matrix, as often observed for imbedded dyes.106 This will be further investigated in a subsequent work. The features of the lifetimes are comparable to values reported for other luminescent Tb calixarene complexes (Fig. 9 and 10).37,107
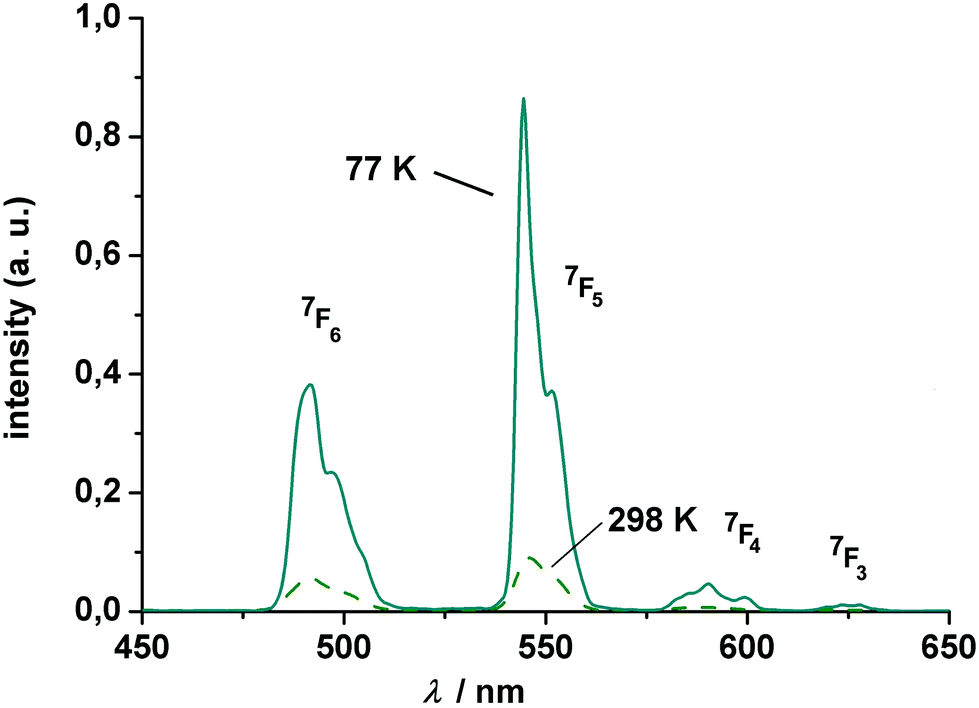 | ||
| Fig. 9 Luminescence spectrum of (HNEt3)[Tb2(HL)(L)] (7) at 77 and 298 K (polycarbonate thin film doped with 4 wt% Tb). The excitation wavelength is 311 nm. All transitions start from the 5D4 state. | ||
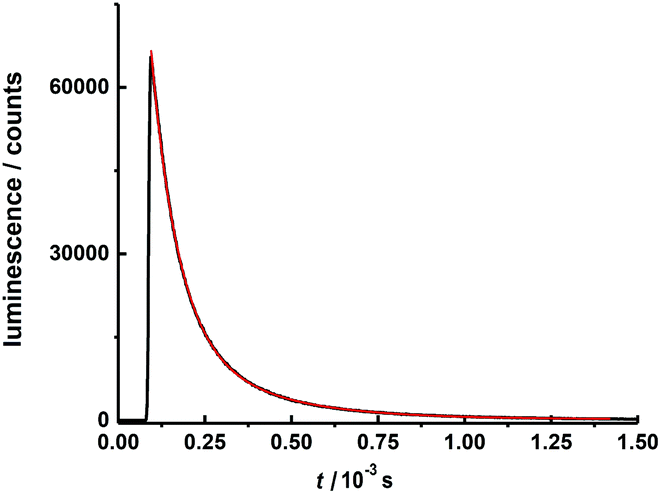 | ||
| Fig. 10 Luminescence decay curve for 4 wt% (HNEt3)[Tb2(HL)(L)] in polycarbonate matrix at 298 K after excitation at λex = 310 nm (pulse width = 12.6 μs) detected at 545 nm within the time range of 1.5 ms. | ||
The luminescence properties of the Gd compound 6 were examined in order to determine the triplet state energy of the Schiff base ligand. The emission spectrum of compound 6 embedded in polycarbonate at 77 K is strong (Fig. S38†), with the shortest wavelength 0–0 transition of the ligand peaking at ca. 457 nm (21882 cm−1). This triplet-state energy compares well with those of other Schiff base ligands66 and lies well above the resonance energy levels of the Eu(III) and Tb(III) ions. These results suggest that a ligand triplet state is indeed involved in the energy transfer mechanism to the resonance state of the Ln(III) ions, from which emission occurs.
Conclusions
A new monofunctionalized calix[4]arene-Schiff base ligand has been synthesized and its coordination chemistry towards selected lanthanide ions (Sm, Eu, Gd, Tb) investigated in solution and solid state. The chemistry of this ligand system is distinct from that of the well-studied bis- and tetrakis-lower rim functionalized calix[4]arenes, which tend to support only monomeric structures. Dimerization occurs via the salicylidene's phenolate groups, not via bridging O atoms from the calix[4]arene, as seen for some heteroleptic complexes involving the parent calix[4]arenes to give coordination number 7 with an highly irregular coordination geometry. The assembly is further stabilized by an intramolecular OH⋯O–hydrogen bond established in second sphere of the calixarene bowls. The dimeric units are also present in MeCN solution as suggested by ESI MS. There are little – if any – magnetic exchange interactions in the dimers, and the absence of SMM behavior may be associated with the low local symmetry of the lanthanide ions. The present study enlarges the database, may contribute to current knowledge of structure–property relationship in Ln calixarene containing SMMs, luminescent materials, and chemosensors.Experimental section
Materials and methods
The calix[4]arene 1 was prepared as described in the literature.108 All reagents and solvents were commercial grade and used without further purification. Melting points were determined with an Electrothermal IA9000 series instrument using open glass capillaries and are uncorrected. Elemental analyses were carried out on a VARIO EL elemental analyzer (Elementar Analysensysteme GmbH, Hanau). NMR spectra were recorded on a Bruker FT 300 spectrometer or AVANCE DRX 400 spectrometer at 298 K. Chemical shifts refer to solvent signals. Mass spectra were obtained using the negative ion electrospray ionization modus (ESI) on a Bruker Daltronics ESQUIRE 3000 Plus ITMS or Impact II UHR Qq-TOF instrument. Infrared spectra (4000–400 cm−1) were recorded at 1 cm−1 resolution on a Bruker TENSOR 27 (equipped with a MIRacle ZnSe ATR accessory from PIKE Technologies) FT-IR spectrometer. Solution absorption spectra were collected on a Jasco V-670 UV–vis–NIR device. Steady state fluorescence absorption and emission spectra were recorded on a PerkinElmer LS 50B luminescence spectrometer using 1 cm quartz cells (Hellma). The magnetic susceptibility measurements were performed with the use of a MPMS 7XL SQUID magnetometer (Quantum Design) working between 1.8 and 330 K for applied dc fields ranging from −7 to 7 T. Measurements were performed on polycrystalline samples over the temperature range 2–330 K at applied magnetic field of 0.1, 0.5, 1.0 T. The observed susceptibility data were corrected for the underlying diamagnetism.Synthesis and analysis of compounds
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, C30), 121.5 (p-CAr c[4]a, C5/17), 122.6 (p-CAr c[4]a, C11), 127.8 (p-CAr c[4]a, C23), 128.1 (o-CAr c[4]a, C7/15), 128.6 (o-CAr c[4]a, C3/19), 129.05 (o-CAr c[4]a, C9/13), 129.1 (m-CAr c[4]a, C4/18), 129.4 (m-CAr c[4]a, C6/16), 129.6 (m-CAr c[4]a, C10/12), 130.5 (m-CAr c[4]a, C22/24), 134.1 (o-CAr c[4]a, C1/21), 149.3 (ipso CAr–OH c[4]a, C27), 150.9 (ipso CAr–O–CH2 c[4]a, C25), 151.5 (ipso CAr–OH c[4]a, C26/28). ATR-IR (ZnSe): ν/cm−1 = 3290 (s, br, ν O–H), 3271 (s, br, ν O–H), 3040 (w), 2931 (w), 2866 (w), 1593 (w, ν CC), 1467 (s, ν CC), 1454 (s, ν CC), 1430 (m), 1377 (m), 1349 (m), 1297 (w), 1272 (m), 1260 (m), 1243 (m), 1226 (m), 1210 (m), 1180 (m), 1156 (w), 1146 (w), 1085 (w), 1034 (m), 1029 (w), 976 (w), 959 (w), 949 (w), 908 (w), 895 (w), 841 (w), 806 (w), 796 (w), 778 (w), 753 (s), 742 (m), 705 (w), 692 (w), 686 (w).
C), 1461 (s, ν CC), 1445 (s, ν CC), 1399 (m), 1364 (w), 1251 (m), 1212 (m), 1181 (w), 1149 (w), 1090 (m), 1079 (m), 1008 (w), 971 (w), 913 (w), 885 (w), 840 (w), 828 (w), 802 (w), 755 (s), 700 (w).
:1, v/v) was added MgSO4 (100 mg). The resulting mixture was stirred at r.t. for 12 h, filtered and evaporated in vacuum to ∼1/3 of its original volume. The resulting yellow precipitate was filtered, washed with methanol (20 mL) and dried at 60 °C to give 495 mg (81% based on 1) of pure H2L1 as a yellow powder. M.p. 184 °C. Elemental analysis for C37H33NO5·1/3MeOH·1/3CH2Cl2 (571.67 + 10.68 + 28.32) calcd: C 74.09, H 5.78, N 2.29%; found C 74.24, H 5.43, N 2.17%. m/z (ESI−, CH2Cl2/MeCN): C37H33NO5 (571.24) [M − H+]− calcd: 570.23; found 570.3. 1H-NMR (400 MHz, CH2Cl2-d2, for atom labels see Fig. S13†): δ = 3.38 (d, 2J = 13.8 Hz, 2H, ArCHeqHAr, C8/14), 3.51 (d, 2J = 13.1 Hz, 2H, ArCHeqHAr, C2/20), 3.98 (d, 2J = 13.8 Hz, 2H, ArCHHaxAr, C8/14), 4.31–4.40 (m, 4H, Ar–CHHax–Ar, NCH2, C2/20, C30), 4.48 (t, 3J = 4.9 Hz, 2H, OCH2, C29), 6.66 (t, 3J = 7.5 Hz, 2H, p-ArH, C5/17), 6.67 (t, 3J = 7.5 Hz, 1H, C11), 6.87–7.02 (m, 7H, p-ArH, m-ArH, ArH sal, C23, C10/12, C6/16, C34, C36), 7.07 (dd, 3J = 7.6 Hz, 4J = 1.7 Hz, 2H, m-ArH, C4/18), 7.11 (d, 3J = 7.6 Hz, 2H, m-ArH, C22/24), 7.35 (ddd, 3J = 8.2 Hz, 7.4 Hz, 4J = 1.7 Hz, 1H, ArH sal, C35), 7.57 (dd, 3J = 7.6 Hz, 4J = 1.7 Hz, 1H, ArH sal, C33), 8.81 (s, 1H, CHN, C31). 13C{1H} NMR (100 MHz, CH2Cl2-d2, for atom labels see Fig. S13†): δ = 31.8 (ArCH2Ar, C2/20), 32.0 (ArCH2Ar, C8/14), 60.5 (CH2N, C30), 76.5 (OCH2, C29), 117.2 (CAr sal, C36), 119.2 (CAr sal, C34), 119.7 (CAr sal, C32), 121.3 (p-CAr c[4]a, C5/17), 122.4 (p-CAr c[4]a, C11), 126.6 (p-CAr c[4]a, C23), 128.6 (o-CAr c[4]a, C7/15), 128.7 (o-CAr c[4]a, C3/19), 129.0 (m-CAr c[4]a, C4/18), 129.2 (m-CAr c[4]a, C6/16), 129.25 (m-CAr c[4]a, C10/12), 129.3 (o-CAr c[4]a, C9/13), 130.0 (m-CAr c[4]a, C22/24), 132.6 (CAr sal, C33), 133.1 (CAr sal, C35), 134.7 (o-CAr c[4]a, C1/21), 149.6 (ipso CAr–OH c[4]a, C27), 151.5 (ipso CAr–OH c[4]a, C26/28), 151.7 (ipso CAr–O–CH2 c[4]a, C25), 161.7 (CAr sal, C37), 168.7 (CHN, C31). ATR-IR (ZnSe): ν/cm−1 = 3635 (w, νOH), 3500 (w, νOH), 3320 (m, νOH), 3152 (m, νOH), 2952 (w), 2925 (w), 2890 (w), 1635 (s, ν CN), 1591 (w, νC=C), 1583 (w), 1496 (w), 1464 (s, νCC), 1448 (s, νCC), 1429 (m), 1406 (m), 1367 (w), 1337 (w), 1280 (m), 1267 (m), 1246 (m), 1212 (m), 1195 (m), 1150 (m), 1124 (m), 1087 (w), 1058 (w), 1028 (m), 923 (w), 913 (w), 884 (w), 840 (w), 752 (s), 734 (m). UV–vis (MeCN): λmax/nm (ε/M−1 cm−1) = 202sh (39953), 217 (49380), 254 (13567), 286 (8882), 311 (6749). This compound was additionally characterized by X-ray crystallography. Crystals suitable for X-ray crystallography were obtained from a solution of H4L in MeOH.
:v/v, 30 mL) was added a solution of Sm(NO3)3·6H2O (128 mg, 0.289 mmol) at room temperature. The reaction mixture was stirred for 12 h and evaporated to ca. 10 mL to give a pale-yellow solid which was isolated by filtration, washed with MeOH (5 mL), and dried at 60 °C. The crude product was further purified by recrystallization from a mixed CH2Cl2/EtOH (1:1) mixture. Yield: 166 mg (82% based on H4L). mp > 245 °C (decomposes without melting). m/z (ESI−, CH2Cl2): C80H75N3O10Sm2 (1541.385) [2M − HNEt3+]− calcd: 1439.256; found 1439.2; [M]− calcd 719.124; found 719.1. Found: C 59.57, H 5.12, N 2.70; C80H75N3O10Sm2·4H2O (1539.21 + 72.06) requires: C 59.63, H 5.19, N 2.61. AT-IR (ZnSe): ν/cm−1 = 3580 (w), 3052 (w), 3025 (w), 2911 (w), 1635 (m, ν CN), 1593 (w, ν CC), 1548 (w), 1450 (s, ν CC), 1427 (m), 1399 (w), 1379 (w), 1316 (w), 1300 (m), 1288 (m), 1266 (w), 1246 (w), 1224 (w), 1186 (m), 1152 (m), 1124 (w), 1087 (m), 1072 (w), 1032 (m), 932 (w), 889 (m), 867 (m), 833 (w), 812 (w), 802 (w), 784 (w), 756 (s), 745 (s), 714 (m), 694 (w). Magnetic moment: μeff,dim = 2.40μB (per binuclear unit, 300 K), μeff = 1.70μB (per Sm3+). Single crystals of [HNEt3][Sm2(HL)(L)(MeCN)2]·MeCN were grown by diethyl ether vapor diffusion into a acetonitrile solution and analyzed by X-ray diffraction.
N), 1594 (w, ν CC), 1548 (w), 1465 (s, ν CC), 1450 (s, ν CC), 1427 (m), 1399 (w), 1317 (w), 1300 (m), 1288 (m), 1266 (w), 1246 (w), 1223 (w), 1188 (w), 1154 (w), 1123 (w), 1086 (w), 1071 (w), 1033 (w), 930 (w), 911 (w), 889 (m), 867 (m), 834 (w), 813 (w), 801 (w), 754 (s), 745 (s), 731 (w), 713 (w), 695 (w), 677 (w). Magnetic moment: μeff,dim = 4.73μB (per binuclear unit, 300 K), μeff = 3.34μB (per Eu3+).
N), 1595 (w, ν CC) 1582 (m), 1547 (w), 1496 (s, ν CC), 1454 (s, ν CC), 1425 (m), 1402 (w), 1325 (m), 1290 (m), 1245 (w), 1222 (w), 1189 (w), 1152 (w), 1085 (w), 1073 (w), 1035 (w), 934 (w), 900 (w), 868 (w), 849 (w), 834 (w), 814 (w), 803 (w), 757 (m), 732 (m), 715 (w). Magnetic moment: μeff,dim = 11.37μB (per binuclear unit, 300 K), μeff = 8.04μB (per Gd3+). Single crystals of [HNEt3][Gd2(HL)(L)(MeCN)2]·MeCN were grown by diethyl ether vapor diffusion into a acetonitrile solution and analyzed by X-ray diffraction.
N), 1596 (w, ν CC), 1589 (w, ν CC), 1548 (w), 1466 (s, ν CC), 1456 (s, ν CC), 1427 (m), 1440 (w), 1400 (w), 1385 (w), 1326 (w), 1303 (m), 1292 (m), 1265 (w), 1246 (w), 1224 (w), 1190 (w), 1152 (w), 1125 (w), 1085 (w), 1074 (w), 1049 (w), 1036 (w), 935 (w), 911 (w), 902 (w), 869 (m), 848 (w), 835 (w), 815 (w), 802 (w), 786 (w), 758 (m), 715 (w), 693 (w), 677 (w), 626 (w), 592 (w), 572 (w), 555 (w), 515 (w), 507 (w). Magnetic moment: μeff,dim = 13.82μB (per binuclear unit, 300 K), μeff = 9.77μB (per Tb3+).
N, C30), 121.5 (p-CAr c[4]a, C5/17), 122.6 (p-CAr c[4]a, C11), 127.8 (p-CAr c[4]a, C23), 128.1 (o-CAr c[4]a, C7/15), 128.6 (o-CAr c[4]a, C3/19), 129.05 (o-CAr c[4]a, C9/13), 129.1 (m-CAr c[4]a, C4/18), 129.4 (m-CAr c[4]a, C6/16), 129.6 (m-CAr c[4]a, C10/12), 130.5 (m-CAr c[4]a, C22/24), 134.1 (o-CAr c[4]a, C1/21), 149.3 (ipso CAr–OH c[4]a, C27), 150.9 (ipso CAr–O–CH2 c[4]a, C25), 151.5 (ipso CAr–OH c[4]a, C26/28). ATR-IR (ZnSe): ν/cm−1 = 3290 (s, br, ν O–H), 3271 (s, br, ν O–H), 3040 (w), 2931 (w), 2866 (w), 1593 (w, ν CC), 1467 (s, ν CC), 1454 (s, ν CC), 1430 (m), 1377 (m), 1349 (m), 1297 (w), 1272 (m), 1260 (m), 1243 (m), 1226 (m), 1210 (m), 1180 (m), 1156 (w), 1146 (w), 1085 (w), 1034 (m), 1029 (w), 976 (w), 959 (w), 949 (w), 908 (w), 895 (w), 841 (w), 806 (w), 796 (w), 778 (w), 753 (s), 742 (m), 705 (w), 692 (w), 686 (w).
C), 1461 (s, ν CC), 1445 (s, ν CC), 1399 (m), 1364 (w), 1251 (m), 1212 (m), 1181 (w), 1149 (w), 1090 (m), 1079 (m), 1008 (w), 971 (w), 913 (w), 885 (w), 840 (w), 828 (w), 802 (w), 755 (s), 700 (w).
:1, v/v) was added MgSO4 (100 mg). The resulting mixture was stirred at r.t. for 12 h, filtered and evaporated in vacuum to ∼1/3 of its original volume. The resulting yellow precipitate was filtered, washed with methanol (20 mL) and dried at 60 °C to give 495 mg (81% based on 1) of pure H2L1 as a yellow powder. M.p. 184 °C. Elemental analysis for C37H33NO5·1/3MeOH·1/3CH2Cl2 (571.67 + 10.68 + 28.32) calcd: C 74.09, H 5.78, N 2.29%; found C 74.24, H 5.43, N 2.17%. m/z (ESI−, CH2Cl2/MeCN): C37H33NO5 (571.24) [M − H+]− calcd: 570.23; found 570.3. 1H-NMR (400 MHz, CH2Cl2-d2, for atom labels see Fig. S13†): δ = 3.38 (d, 2J = 13.8 Hz, 2H, ArCHeqHAr, C8/14), 3.51 (d, 2J = 13.1 Hz, 2H, ArCHeqHAr, C2/20), 3.98 (d, 2J = 13.8 Hz, 2H, ArCHHaxAr, C8/14), 4.31–4.40 (m, 4H, Ar–CHHax–Ar, NCH2, C2/20, C30), 4.48 (t, 3J = 4.9 Hz, 2H, OCH2, C29), 6.66 (t, 3J = 7.5 Hz, 2H, p-ArH, C5/17), 6.67 (t, 3J = 7.5 Hz, 1H, C11), 6.87–7.02 (m, 7H, p-ArH, m-ArH, ArH sal, C23, C10/12, C6/16, C34, C36), 7.07 (dd, 3J = 7.6 Hz, 4J = 1.7 Hz, 2H, m-ArH, C4/18), 7.11 (d, 3J = 7.6 Hz, 2H, m-ArH, C22/24), 7.35 (ddd, 3J = 8.2 Hz, 7.4 Hz, 4J = 1.7 Hz, 1H, ArH sal, C35), 7.57 (dd, 3J = 7.6 Hz, 4J = 1.7 Hz, 1H, ArH sal, C33), 8.81 (s, 1H, CHN, C31). 13C{1H} NMR (100 MHz, CH2Cl2-d2, for atom labels see Fig. S13†): δ = 31.8 (ArCH2Ar, C2/20), 32.0 (ArCH2Ar, C8/14), 60.5 (CH2N, C30), 76.5 (OCH2, C29), 117.2 (CAr sal, C36), 119.2 (CAr sal, C34), 119.7 (CAr sal, C32), 121.3 (p-CAr c[4]a, C5/17), 122.4 (p-CAr c[4]a, C11), 126.6 (p-CAr c[4]a, C23), 128.6 (o-CAr c[4]a, C7/15), 128.7 (o-CAr c[4]a, C3/19), 129.0 (m-CAr c[4]a, C4/18), 129.2 (m-CAr c[4]a, C6/16), 129.25 (m-CAr c[4]a, C10/12), 129.3 (o-CAr c[4]a, C9/13), 130.0 (m-CAr c[4]a, C22/24), 132.6 (CAr sal, C33), 133.1 (CAr sal, C35), 134.7 (o-CAr c[4]a, C1/21), 149.6 (ipso CAr–OH c[4]a, C27), 151.5 (ipso CAr–OH c[4]a, C26/28), 151.7 (ipso CAr–O–CH2 c[4]a, C25), 161.7 (CAr sal, C37), 168.7 (CHN, C31). ATR-IR (ZnSe): ν/cm−1 = 3635 (w, νOH), 3500 (w, νOH), 3320 (m, νOH), 3152 (m, νOH), 2952 (w), 2925 (w), 2890 (w), 1635 (s, ν CN), 1591 (w, νC=C), 1583 (w), 1496 (w), 1464 (s, νCC), 1448 (s, νCC), 1429 (m), 1406 (m), 1367 (w), 1337 (w), 1280 (m), 1267 (m), 1246 (m), 1212 (m), 1195 (m), 1150 (m), 1124 (m), 1087 (w), 1058 (w), 1028 (m), 923 (w), 913 (w), 884 (w), 840 (w), 752 (s), 734 (m). UV–vis (MeCN): λmax/nm (ε/M−1 cm−1) = 202sh (39953), 217 (49380), 254 (13567), 286 (8882), 311 (6749). This compound was additionally characterized by X-ray crystallography. Crystals suitable for X-ray crystallography were obtained from a solution of H4L in MeOH.
:v/v, 30 mL) was added a solution of Sm(NO3)3·6H2O (128 mg, 0.289 mmol) at room temperature. The reaction mixture was stirred for 12 h and evaporated to ca. 10 mL to give a pale-yellow solid which was isolated by filtration, washed with MeOH (5 mL), and dried at 60 °C. The crude product was further purified by recrystallization from a mixed CH2Cl2/EtOH (1:1) mixture. Yield: 166 mg (82% based on H4L). mp > 245 °C (decomposes without melting). m/z (ESI−, CH2Cl2): C80H75N3O10Sm2 (1541.385) [2M − HNEt3+]− calcd: 1439.256; found 1439.2; [M]− calcd 719.124; found 719.1. Found: C 59.57, H 5.12, N 2.70; C80H75N3O10Sm2·4H2O (1539.21 + 72.06) requires: C 59.63, H 5.19, N 2.61. AT-IR (ZnSe): ν/cm−1 = 3580 (w), 3052 (w), 3025 (w), 2911 (w), 1635 (m, ν CN), 1593 (w, ν CC), 1548 (w), 1450 (s, ν CC), 1427 (m), 1399 (w), 1379 (w), 1316 (w), 1300 (m), 1288 (m), 1266 (w), 1246 (w), 1224 (w), 1186 (m), 1152 (m), 1124 (w), 1087 (m), 1072 (w), 1032 (m), 932 (w), 889 (m), 867 (m), 833 (w), 812 (w), 802 (w), 784 (w), 756 (s), 745 (s), 714 (m), 694 (w). Magnetic moment: μeff,dim = 2.40μB (per binuclear unit, 300 K), μeff = 1.70μB (per Sm3+). Single crystals of [HNEt3][Sm2(HL)(L)(MeCN)2]·MeCN were grown by diethyl ether vapor diffusion into a acetonitrile solution and analyzed by X-ray diffraction.
N), 1594 (w, ν CC), 1548 (w), 1465 (s, ν CC), 1450 (s, ν CC), 1427 (m), 1399 (w), 1317 (w), 1300 (m), 1288 (m), 1266 (w), 1246 (w), 1223 (w), 1188 (w), 1154 (w), 1123 (w), 1086 (w), 1071 (w), 1033 (w), 930 (w), 911 (w), 889 (m), 867 (m), 834 (w), 813 (w), 801 (w), 754 (s), 745 (s), 731 (w), 713 (w), 695 (w), 677 (w). Magnetic moment: μeff,dim = 4.73μB (per binuclear unit, 300 K), μeff = 3.34μB (per Eu3+).
N), 1595 (w, ν CC) 1582 (m), 1547 (w), 1496 (s, ν CC), 1454 (s, ν CC), 1425 (m), 1402 (w), 1325 (m), 1290 (m), 1245 (w), 1222 (w), 1189 (w), 1152 (w), 1085 (w), 1073 (w), 1035 (w), 934 (w), 900 (w), 868 (w), 849 (w), 834 (w), 814 (w), 803 (w), 757 (m), 732 (m), 715 (w). Magnetic moment: μeff,dim = 11.37μB (per binuclear unit, 300 K), μeff = 8.04μB (per Gd3+). Single crystals of [HNEt3][Gd2(HL)(L)(MeCN)2]·MeCN were grown by diethyl ether vapor diffusion into a acetonitrile solution and analyzed by X-ray diffraction.
N), 1596 (w, ν CC), 1589 (w, ν CC), 1548 (w), 1466 (s, ν CC), 1456 (s, ν CC), 1427 (m), 1440 (w), 1400 (w), 1385 (w), 1326 (w), 1303 (m), 1292 (m), 1265 (w), 1246 (w), 1224 (w), 1190 (w), 1152 (w), 1125 (w), 1085 (w), 1074 (w), 1049 (w), 1036 (w), 935 (w), 911 (w), 902 (w), 869 (m), 848 (w), 835 (w), 815 (w), 802 (w), 786 (w), 758 (m), 715 (w), 693 (w), 677 (w), 626 (w), 592 (w), 572 (w), 555 (w), 515 (w), 507 (w). Magnetic moment: μeff,dim = 13.82μB (per binuclear unit, 300 K), μeff = 9.77μB (per Tb3+).
Crystallography
Suitable single crystals of H2L, (HNEt3)[Sm2(HL)(L)(MeCN)2]·MeCN (4·3MeCN), and (HNEt3)[Gd2(HL)(L)(MeCN)2]·MeCN (6·3MeCN) were selected and mounted on the tip of a glass fibre using perfluoropolyether oil. The data sets were collected at 180(2) K using a STOE IPDS-2 diffractometer equipped with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The data were processed with the programs XAREA.109 The structure was solved by direct methods110 and refined by full-matrix least-squares techniques on the basis of all data against F2 using SHELXL-97.111 PLATON was used to search for higher symmetry.112 All non-hydrogen atoms were refined anisotropically. Graphics were produced with Ortep3 for Windows and PovRAY.399 reflections measured, 5261 unique, 3947 with I > 2σ(I). Final R1 = 0.0317, wR2 = 0.0757 (I > 2σ(I), 393 parameters/0 restraints, min./max. residual electron density = −0.093/0.115 e Å−3. The Flack x parameter (absolute structure parameter) was calculated to be −0.36(16) for the present structure and 1.36 for the inverted structure. The solvate molecule is disordered over two positions with site occupancy factors of 0.25 each (fixed). Hydrogen atoms were not calculated for this solvate molecule.
189.2(5) Å3, Z = 8, ρcalcd = 1.466 g cm−3, T = 180(2) K, μ(Mo Kα) = 1.595 mm−1 (λ = 0.71073 Å), 48009 reflections measured, 14906 unique, 11481 with I > 2σ(I). Final R1 = 0.0416, wR2 = 0.1137 (I > 2σ(I), 945 parameters/0 restraints, min./max. residual electron density = −0.611/1.995 e Å−3. The N–H hydrogen atom of the HNEt3+ cation and the OH hydrogen atom of the [Sm2(HL)(L)(MeCN)2]− anion were located unambiguously from final Fourier maps but were refined using a riding model.
175.7(10) Å3, Z = 8, ρcalcd = 1.466 g cm−3, T = 180(2) K, μ(Mo Kα) = 1.797 mm−1 (λ = 0.71073 Å), 51122 reflections measured, 16712 unique, 10439 with I > 2σ(I). Final R1 = 0.0485, wR2 = 0.1160 (I > 2σ(I), 942 parameters/6 restraints, min./max. residual electron density = −0.838/1.115 e Å−3.
CCDC 1880057 (H4L), 1880058 (4) and 1880059 (6)† contain the supplementary crystallographic data for this paper.
Spectrophotometric titrations/determination of stability constants
A series of UV-vis spectroscopic studies were performed in order to determine the composition and stability constants of the lanthanide complexes. The stoichiometry of the lanthanide complexes was determined by the mole ratio method. All titrations were performed at 298 K in Hellma 110-QS quartz cells of 1 cm optical path length containing solutions at constant ionic strength (N(nBu)4PF6 0.01 M) and constant ligand concentrations (5 × 10−5 M) in MeCN. For each experiment, 21 solutions were prepared by combining stock solutions of the ligand and the corresponding Ln(NO3)3·6H2O salts with an Eppendorf micropipette (volume range of 10–100 μL and 100–1000 μL; 0.71–0.10% error) and allowed to stir for 12 h. UV–vis absorption spectra were collected in the 190–650 nm range at uniform data point intervals of 1 nm with a doublebeam V-670 (Jasco) spectrophotometer. The multiwavelength data sets were analyzed by a nonlinear least-squares procedure implemented in the Hyperquad2008 v1.1.33 software.Synthesis of the polycarbonate films
Polycarbonate Z200 (0.30 g) was dissolved in CH2Cl2 (1.5 mL) and stirred for 10 min. A solution of the lanthanide complex (V = 1 mL, 8 × 10−3 M) in CH2Cl2 was added to the PC solution. The resulting mixture was spread on a Petri dish (d = 5 cm) and the solvent was allowed to evaporate in open air over night.Luminescence lifetime measurements
The luminescence lifetime of the Tb complex 7 was measured applying a Fluoromax4 (HoribaScientific) equipped with a Fluorohub (Horiba Scientific) and the DataStation (Version 2.7) software package for TCSPC applications. The sample, imbedded in a thin polycarbonate matrix, was installed at an 35° angle towards the incident excitation light beam. Excitation and emission wavelengths of 310 and 545 nm were chosen. The time resolution was 1.33 μs per channel. In order to determine the instrument response function we used the identical setup with a highly reflective spectralon sample. The photon count rate was well below 1 percent of the excitation count rate ruling out pile up effects.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are thankful to Prof. Dr H. Krautscheid for providing facilities for X-ray crystallographic measurements. Financial support from the German Federal Ministry of Education and Research (BMBF – project 02NUK046C) and the Deutsche Forschungsgemeinschaft (DFG – Priority Program SPP2102, “Light Controlled Reactivity of Metal Complexes”) is gratefully acknowledged.References
-
D. Gutsche, Calixarenes: An Introduction, RSC Publishing, Cambridge, 2nd edn, 2008 Search PubMed
.
- V. Böhmer, Angew. Chem., 1995, 107, 785–818 (
Angew. Chem., Int. Ed. Engl.
, 1995
, 34
, 713–745
) CrossRef
-
R. Kumar, Y. Jung and J. S. Kim, in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer, 2016 Search PubMed
- C. Wieser, C. B. Dieleman and D. Matt, Coord. Chem. Rev., 1997, 165, 93–161 CrossRef CAS
- P. D. Beer and E. J. Hayes, Coord. Chem. Rev., 2003, 240, 167–189 CrossRef CAS
- C. Redshaw, Coord. Chem. Rev., 2003, 244, 45–70 CrossRef CAS
- C. J. Elsevier, J. Reedijk, P. H. Walton and M. D. Ward, Dalton Trans., 2003, 1869–1880 RSC
- W. Sliwa, J. Inclusion Phenom. Macrocyclic Chem., 2005, 52, 13–37 CrossRef CAS
- A. Casnati, N. Della Ca’, M. Fontanella, F. Sansone, F. Ugozzoli, R. Ungaro, K. Liger and J.-F. Dozol, Eur. J. Org. Chem., 2005, 2338–2348 CrossRef CAS
-
J. Vicens and J. Harrowfield, Calixarenes in the Nanoworld, Springer, Netherlands, 2007 Search PubMed
- B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893–962 CrossRef CAS
-
L. Baldini, F. Sansone, A. Casnati and R. Ungaro, Calixarenes in Molecular Recognition, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley & Sons, 2012, vol. 3 Search PubMed
- R. Gramage-Doria, D. Armspach and D. Matt, Coord. Chem. Rev., 2013, 257, 776–816 CrossRef CAS
- D. Semeril and D. Matt, Coord. Chem. Rev., 2014, 279, 58–95 CrossRef CAS
- D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed
- A. B. Chetry, T. Matsufuji, B. B. Adhikari, S. Morisada, H. Kawakita, K. Ohto, T. Oshima and Jumina, J. Inclusion Phenom. Macrocyclic Chem., 2015, 81, 301–310 CrossRef CAS
- J.-N. Rebilly, B. Colasson, O. Bistri, D. Over and O. Reinaud, Chem. Soc. Rev., 2015, 44, 467–489 RSC
-
M. A. McKervey, F. Arnaud-Neu and M.-J. Schwing-Weill, in Comprehensive Supramolecular Chemistry, ed. G. Gokel, Pergamon, Oxford, 1996 Search PubMed
- J. M. Harrowfield, M. I. Ogden and A. H. White, J. Chem. Soc., Dalton Trans., 1991, 2625–2632 RSC
- P. D. Beer, M. G. B. Drew, M. Kan, P. B. Leeson, M. I. Ogden and G. Williams, Inorg. Chem., 1996, 35, 2202–2211 CrossRef CAS PubMed
- A. Casnati, L. Baldini, F. Sansone, R. Ungaro, N. Armaroli, D. Pompei and F. Barigelletti, Supramol. Chem., 2002, 14, 281–289 CrossRef CAS
- G. Guillemot, B. Castellano, T. Prangé, E. Solari and C. Floriani, Inorg. Chem., 2007, 46, 5152–5154 CrossRef CAS PubMed
- G. B. Deacon, M. G. Gardiner, P. C. Junk, J. P. Townley and J. Wang, Organometallics, 2012, 31, 3857–3864 CrossRef CAS
- B. M. Furfy, J. M. Harrowfield, D. L. Kepert, B. W. Skelton, A. H. White and F. R. Wilner, Inorg. Chem., 1987, 26, 4231–4236 CrossRef
- S. Barboso, A. G. Carrera, S. E. Matthews, F. Arnaud-Neu, V. Böhmer, J.-F. Dozol, H. Rouquette and M.-J. Schwing-Weill, J. Chem. Soc., Perkin Trans. 2, 1999, 719–724 RSC
- R. Ludwig, Fresenius’ J. Anal. Chem., 2000, 376, 103–128 CrossRef
- J. F. Dozol and R. Ludwig, Ion Exch. Solvent Extr., 2010, 19, 195–318 CAS
- I. Vatsouro, A. Serebryannikova, L. Wang, V. Hubscher-Bruder, E. Shokova, M. Bolte, F. Arnaud-Neu, V. Böhmer and V. Kovalev, Tetrahedron, 2011, 67, 8092–8101 CrossRef CAS
- F. de María Ramírez, S. Varbanov, J.-C. G. Bünzli and R. Scopelliti, Inorg. Chim. Acta, 2011, 378, 163–168 CrossRef
- D. R. Raut, P. K. Mohapatra, S. A. Ansari, S. V. Godbole, M. Iqbal, D. Manna, T. K. Ghanty, J. Huskens and W. Verboom, RSC Adv., 2013, 3, 9296–9303 RSC
- M. Atanassova and V. Kurteva, RSC Adv., 2016, 6, 11303–11324 RSC
- S. A. Ansari, P. K. Mohapatra, S. M. Ali, A. Sengupta, A. Bhattacharyya and W. Verboom, Dalton Trans., 2016, 45, 5425–5429 RSC
- S. A. Ansari, P. K. Mohapatra, W. Verboom, Z. Zhang, P. D. Dau, J. K. Gibson and L. Rao, Dalton Trans., 2015, 44, 6416–6422 RSC
- A. Leydier, D. Lecerclé, S. Pellet-Rostaing, A. Favre-Réguillon, F. Taran and M. Lemaire, Tetrahedron, 2008, 64, 11319–11324 CrossRef CAS
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed
- J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS PubMed
- D. D'Alessio, S. Muzzioli, B. W. Skelton, S. Stagni, M. Massi and M. I. Ogden, Dalton Trans., 2012, 41, 4736–4739 RSC
- C. R. Driscoll, B. W. Skelton, M. Massi and M. I. Ogden, Supramol. Chem., 2016, 28, 567–574 CrossRef CAS
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652–1666 RSC
- M. L. Aulsebrook, B. Graham, M. R. Grace and K. L. Tuck, Coord. Chem. Rev., 2018, 375, 191–220 CrossRef CAS
- N. Sabbatini, M. Guardigli, A. Mecati, V. Balzani, R. Ungaro, E. Ghidini, A. Casnati and A. Pochini, J. Chem. Soc., Chem. Commun., 1990, 878–879 RSC
- N. Sato, I. Yoshida and S. Shinkai, Chem. Lett., 1993, 621–624 CAS
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408–9414 CrossRef CAS
- B. W. Ennis, S. Muzzioli, B. L. Reid, D. D'Alessio, S. Stagni, D. H. Brown, M. I. Ogden and M. Massi, Dalton Trans., 2013, 42, 6894–6901 RSC
- M. Massi and M. I. Ogden, Materials, 2017, 10, 1369–1381 CrossRef PubMed
- C. Lincheneau, E. Quinlan, J. A. Kitchen, T. McCabe, S. E. Matthews and T. Gunnlaugsson, Supramol. Chem., 2013, 25, 869–880 CrossRef CAS
- M. J. McIldowie, B. W. Skelton, M. Mocerino and M. I. Ogden, J. Inclusion Phenom. Macrocyclic Chem., 2015, 82, 43–46 CrossRef CAS
- P. D. Beer, M. G. B. Drew and M. I. Ogden, J. Chem. Soc., Dalton Trans., 1997, 1489–1492 RSC
- C. R. Driscoll, B. L. Reid, M. J. McIldowie, S. Muzzioli, G. L. Nealon, B. W. Skelton, S. Stagni, D. H. Brown, M. Massi and M. I. Ogden, Chem. Commun., 2011, 47, 3876–3878 RSC
- F. Glasneck, K. Kobalz and B. Kersting, Eur. J. Inorg. Chem., 2016, 3111–3122 CrossRef CAS
- R. K. Pathak, A. G. Dikundwar, T. N. Guru Row and C. P. Rao, Chem. Commun., 2010, 46, 4345–4347 RSC
- R. K. Pathak, V. K. Hinge, M. Mondal and C. P. Rao, J. Org. Chem., 2011, 76, 10039–10049 CrossRef CAS PubMed
- R. Joseph, J. P. Chinta and C. P. Rao, Inorg. Chem., 2011, 50, 7050–7058 CrossRef CAS PubMed
- V. V. S. Mummidivarapu, V. K. Hinge and C. P. Rao, Dalton Trans., 2015, 44, 1130–1141 RSC
- R. K. Pathak, V. K. Hinge, A. Rai, D. Panda and C. P. Rao, Inorg. Chem., 2012, 51, 4994–5005 CrossRef CAS PubMed
- S. Ullmann, R. Schnorr, M. Handke, C. Laube, B. Abel, J. Matysik, M. Findeisen, R. Rüger, T. Heine and B. Kersting, Chem. – Eur. J., 2017, 23, 3824–3827 CrossRef CAS PubMed
- S. Ullmann, R. Schnorr, C. Laube, B. Abel and B. Kersting, Dalton Trans., 2018, 47, 5801–5811 RSC
- W.-C. Zhang and Z.-T. Huang, Synthesis, 1997, 1073–1076 CrossRef CAS
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, Hoboken, New Jersey, 6th edn, 2009, part B Search PubMed
- J. U. Ahmad, M. Nieger, M. R. Sundberg, M. Leskelä and T. Repo, J. Mol. Struct., 2011, 995, 9–15 CrossRef CAS
- C. Jaime, J. de Mendoza, P. Prados, P. M. Nieto and C. Sanchez, J. Org. Chem., 1991, 56, 3372–3376 CrossRef CAS
- Y. Liu, B.-T. Zhao, H.-Y. Zhang, T. Wada and Y. Inoue, J. Chem. Soc., Perkin Trans. 2, 2001, 1219–1223 RSC
- S. R. Menon and J. A. R. Schmidt, Tetrahedron, 2016, 72, 767–774 CrossRef CAS
- L. J. Charbonnière, C. Balsiger, K. J. Schenk and J.-C. G. Bünzli, J. Chem. Soc., Dalton Trans., 1998, 505–510 RSC
- M. La Deda, M. Ghedini, I. Aiello and A. Grisolia, Chem. Lett., 2004, 33, 1060–1061 CrossRef CAS
- R. D. Archer, H. Chen and L. C. Thompson, Inorg. Chem., 1998, 37, 2089–2095 CrossRef CAS
- A. Filarowski, J. Phys. Org. Chem., 2005, 18, 686–698 CrossRef CAS
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, Inc., Hoboken, New Jersey, 6th edn, 2009, part B Search PubMed
- W. Xu, R. J. Puddephatt, L. Manojlovic-Muir, K. W. Muir and C. S. Frampton, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 277–290 CrossRef CAS
- P. Thuéry, Z. Asfari, M. Nierlich and J. Vicens, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o223–o225 CrossRef
- J. M. Harrowfield, M. I. Ogden and A. H. White, Aust. J. Chem., 1991, 44, 1237–1247 CrossRef CAS
- S. Fleming, C. D. Gutsche, J. M. Harrowfield, M. I. Ogden, B. W. Skelton, D. F. Stewart and A. H. White, Dalton Trans., 2003, 3319–3327 RSC
- B. M. Furthy, J. M. Harrowfield, M. I. Ogden, B. W. Skelton, A. H. White and F. R. Wilner, J. Chem. Soc., Dalton Trans., 1989, 2217–2221 RSC
- I. D. Brown, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 24–31 CrossRef
-
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, UK, 1997 Search PubMed
- T. Steiner, Angew. Chem., 2002, 114, 50–80 (
Angew. Chem., Int. Ed.
, 2002
, 41
, 48–76
) CrossRef
- G. L. Nealon, M. Mocerino, M. I. Ogden and B. W. Skelton, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65, 25–30 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- M. Andruh, E. Bakalbassis, O. Kahn, J.-C. Trombe and P. Porcher, Inorg. Chem., 1993, 32, 1616–1622 CrossRef CAS
-
A. F. Orchard, Magnetochemistry, Oxford University Press, Oxford, 2007 Search PubMed
- J.-P. Costes, F. Dahan, A. Dupuis, S. Lagrave and J.-P. Laurent, Inorg. Chem., 1998, 37, 153–155 CrossRef CAS PubMed
- J. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed
- P.-H. Lin, M. Leclère, J. Long, T. J. Burchell, I. Korobkov, R. Clérac and M. Murugesu, Dalton Trans., 2010, 39, 5698–5704 RSC
- P. Comba, M. Großhauser, R. Klingeler, C. Koo, Y. Lan, D. Müller, J. Park, A. Powell, M. J. Riley and H. Wadepohl, Inorg. Chem., 2015, 54, 11247–11258 CrossRef CAS PubMed
- L. E. Roy and T. Hughbanks, J. Am. Chem. Soc., 2006, 128, 568–575 CrossRef CAS PubMed
- S. Liu, L. Gelmini, S. J. Rettig, R. C. Thompson and C. Orvig, J. Am. Chem. Soc., 1992, 114, 6081–6087 CrossRef CAS
- J. H. Van Vleck and A. Frank, Phys. Rev., 1929, 34, 1494–1499 CrossRef CAS
- A. Jäschke, M. Kischel, A. Mansel and B. Kersting, Eur. J. Inorg. Chem., 2017, 894–901 CrossRef
- J.-X. Xu, Y. Ma, D. Liao, G.-F. Xu, J. Tang, C. Wang, N. Zhou, S.-P. Yan, P. Cheng and L.-C. Li, Inorg. Chem., 2009, 48, 8890–8896 CrossRef CAS PubMed
- M. Reng, Z.-L. Xu, S.-S. Bao, T.-T. Wang, Z.-H. Zheng, R. A. Ferreira, L.-M. Zheng and L. D. Carlos, Dalton Trans., 2016, 2974–2982 Search PubMed
- J. H. Yoe and A. E. Harvey, J. Am. Chem. Soc., 1948, 70, 648–654 CrossRef CAS
- P. Di Bernardo, A. Melchior, M. Tolazzi and P. L. Zanonato, Coord. Chem. Rev., 2012, 256, 328–351 CrossRef CAS
- A. F. Danil de Namor and O. Jafou, J. Phys. Chem. B, 2001, 105, 8018–8027 CrossRef CAS
- P. M. Marcos, J. R. Ascenso, M. A. P. Segurado, P. J. Cragg, S. Michel, V. Hubscher-Bruder and F. Arnaud-Neu, Supramol. Chem., 2011, 23, 93–101 CrossRef CAS
- P. M. Marcos, J. D. Fonseca, C. S. Proença, J. R. Ascenso, R. J. Bernardino, J. Kulesza and M. Bochenska, Supramol. Chem., 2016, 28, 367–376 CrossRef CAS
- A. F. Danil de Namor, K. Baron, S. Chahine and O. Jafou, J. Phys. Chem. A, 2004, 108, 1082–1089 CrossRef CAS
- N. E. Borisova, A. A. Kostin, E. A. Eroshkina, M. D. Reshetova, K. A. Lyssenko, E. N. Spodine and L. N. Puntus, Eur. J. Inorg. Chem., 2014, 2014, 2219–2229 CrossRef CAS
- The Sm complex 4 is not emissive at 77 K neither in solution nor in polycarbonate.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS
- A. E. V. Gorden, J. D. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed
- G. Vicentini, L. B. Zinner, J. Zukerman-Schpector and K. Zinner, Coord. Chem. Rev., 2000, 196, 353–382 CrossRef CAS
- M. Hilder, P. C. Junk, U. H. Kynast and M. M. Lezhnina, J. Photochem. Photobiol., A, 2009, 202, 10–20 CrossRef CAS
-
J.-C. G. Bünzli and S. Eliseeva, Basics of Lanthanide Photophysics, in Lanthanide Luminescence, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, 2011, vol. 7, pp. 1–45 Search PubMed
- D. Parker, Coord. Chem. Rev., 2000, 205, 109–130 CrossRef CAS
- N. Felorzabihi, J. C. Haley, G. R. Bardajee and M. A. Winnik, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2333–2343 CrossRef CAS
- A. T. Bui, A. Grichine, A. Duperray, P. Lidon, F. Riobé, C. Andraud and O. Maury, J. Am. Chem. Soc., 2017, 139, 7693–7696 CrossRef CAS PubMed
- M. Strobel, K. Kita-Tokarczyk, A. Taubert, C. Vebert, P. A. Heiney, M. Chami and W. Meier, Adv. Funct. Mater., 2006, 16, 252–259 CrossRef CAS
-
Stoe & Cie, X-AREA and X-RED 32; V1.35, Stoe & Cie, Darmstadt, Germany, 2006 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef
-
G. M. Sheldrick, SHELXL-97, Computer program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed
-
A. L. Spek, PLATON - A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2000 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1880057–1880059. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt00292h |
| This journal is © The Royal Society of Chemistry 2019 |