
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRestricted rotation of an Fe(CO)2(PL3)-subunit in [FeFe]-hydrogenase active site mimics by intramolecular ligation†
Sonja
Pullen
 ab,
Somnath
Maji
ad,
Matthias
Stein
c and
Sascha
Ott
*a
ab,
Somnath
Maji
ad,
Matthias
Stein
c and
Sascha
Ott
*a
aDepartment of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se
bFaculty of Chemistry and Chemical Biology, TU Dortmund University, Otto Hahn Str. 6, 44227 Dortmund, Germany
cMax Planck Institute for Dynamics of Complex Technical Systems, Sandtorstraße 1, 39106 Magdeburg, Germany
dDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, 502285, Telangana, India
First published on 21st February 2019
Abstract
A new series of homodinuclear iron complexes as models of the [FeFe]-hydrogenase active site was prepared and characterized. The complexes of the general formula [Fe2(mcbdt)(CO)5PPh2R] (mcbdt = benzene-1,2-dithiol-3-carboxylic acid) feature covalent tethers that link the mcbdt ligand with the phosphine ligands which are terminally coordinated to one of the Fe centres. The synthetic feasability of the concept is demonstrated with the preparation of three novel complexes. A detailed theoretical investigation showes that by introducing a rigid covalent link between the phosphine and the bridging dithiolate ligands, the rotation of the Fe(CO)2P unit is hindered and higher rotation barriers were calculated compared to non-linked reference complexes. The concept of restricting Fe(L)3 rotation is an approach to kinetically stabilize terminal hydrides which are reactive intermediates in catalytic proton reduction cycles of the enzymes.
Introduction
[FeFe]-Hydrogenase enzymes are nature's most efficient catalysts for the reduction of protons, with turnover frequencies as high as 9000 s−1.1 The fact that the active site is held in place by the surrounding protein is crucial for this high activity, which has been revealed by analysis of the enzymes’ crystal structures.2 The binuclear active site holds a μ-bridging carbonyl ligand, and an open coordination site on one of the two iron centres. Already for a long time, it was anticipated that the reaction pathway for producing hydrogen operates via the formation of a terminal hydride at the iron centre that is distal to the Fe4S4 cluster (Fed). The presence of such a hydride species has first been spectroscopically implicated by H/D exchange experiments followed by FT-IR spectroscopy.3 Final proof for the terminal hydride has only recently been established by nuclear resonance vibrational spectroscopy (NRVS), FT-IR and DFT calculations in two native proteins.4 Biomimetic model compounds of the [FeFe]-hydrogenase active site could potentially also form terminal hydrides. DFT calculations by Hall et al. suggested that the first step of the catalytic cycle in such systems produces a terminal hydride as the kinetically favoured product. The terminal hydride, however, then rapidly transforms into a thermodynamically more stable bridging hydride.5 The resulting product is rather unreactive, which is one of the major reasons why synthetic model complexes till today cannot compete with the enzyme's activity. As mentioned above, the so-called rotated structure of Fed with a CO bridging to the neighbouring Fe and an open coordination site is stabilized through interactions of the carbonyl and cyanide ligands at Fed with amino acid residues that are provided by the surrounding protein. These interactions prevent rotation of the distal Fe(CO)2CN unit after protonation, and therefore stabilize the highly reactive terminal hydride.6 In order to design a more efficient synthetic proton reduction catalyst mimicking the active site, a similar strategy can be envisaged. This strategy to tether ligands at Fed models to the dithiolate linker is underexplored,7 and different to present literature examples where a terminal hydride could be detected. With respect to the latter, three approaches have been used so far: (1) introduction of sterically demanding phosphine ligands, (2) chelating phosphine ligands or (3) increasing the basicity of the complex by using four monodentate phosphine ligands. Another family of synthetic models are mononuclear Fe-carbonyl complexes, mimicking only the Fed of the [FeFe]-hydrogenase active site.8Fig. 1 summarizes binuclear [FeFe]-hydrogenase model complexes that have been prepared in literature. The first evidence for a terminal hydride was given by Ezzaher et al.9 for the complex Fe2(pdt)(CO)4(κ-dppe) (κ-dppe = terminal diphenylphosphin-ethylene). Treating the complex with HBF4·Et2O at room temperature afforded a stable bridging hydride which could also/even be crystallized. Low temperature NMR studies gave evidence for two terminal hydride species, one with the hydride at the substituted Fe and the other one located at the Fe(CO)3 site. Following up, Adam et al.10 compared a series of complexes of the types Fe2(pdt)(CO)4(μ-Ph2P-X-PPh2) (x = 2–4 CH2) or Fe2(pdt)(CO)4(κ-Ph2P-X-PPh2) (x = 2–4 CH2), where the bidentate phosphine ligand was either bridging both iron atoms, or only attached to one iron centre. Bridging phosphine complexes did show that a high degree of ligand flexibility in the ligand (for n = 3 and 4) could stabilize a bridging hydride when acid was added, while the shorter dppe (x = 2) bridging ligand did not afford a detectable product. In the case where dppe or dppp were attached to only one iron atom, poorly stable terminal hydride species could be identified at low temperatures only, which rapidly transformed into the more stable bridging hydride.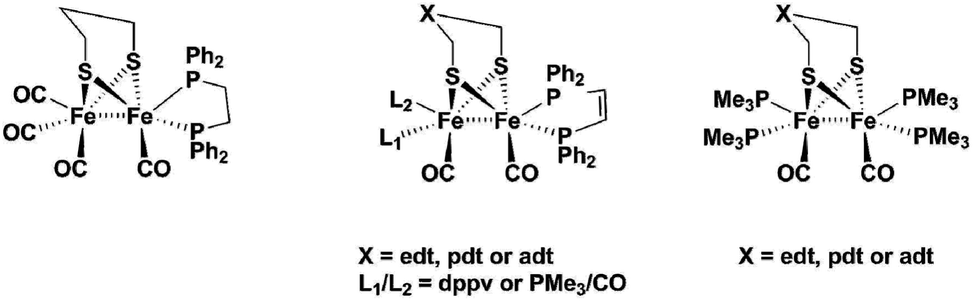 | ||
| Fig. 1 Literature complexes that can form terminal hydrides. | ||
Barton et al. worked on a series of complexes with either edt or pdt bridges and an increasing number of phosphine ligands.11 They used a chelating bisphosphine ligand dppv (= cis-1,2-bis(diphenylphosphino)ethene) and PMe3 to produce di-, tri- and tetra-substituted complexes. They found that pdt analogues showed terminal hydrides at low temperatures for the complexes with only one dppv ligand and another complex with dppv plus one PMe3 ligand, while edt bridged complexes only showed the bridging H−. Complexes of edt and pdt with two dppv ligands did both show terminal hydrides at low temperature and, in contrast to all earlier described models, they even exist at room temperature. The edt-analogue isomerizes within minutes, while the more stable pdt analogue has a half-life of about 10 minutes at 20 °C. They could even study the pdt analogue by cyclic voltammetry at 0 °C and show that the terminal hydride can be reduced at 200 mV milder potential than its bridging hydride isomer.12 In summary, they concluded that isomerization of terminal hydrides can be hindered both by increasing the basicity of the diiron centre and by increasing the steric bulk of the dithiolate ligand. Barton et al. also suggested aza- and oxa-analogues of the pdt complexes as proton relays.13 Carroll et al. picked up the suggestion and made an analogue using the adt-bridge: Fe2(adt)(dppv)2(CO2). With this complex, they were the first (and so far only ones) to publish a crystal structure containing a terminal hydride for the doubly protonated complex [(term-H)Fe2(adtH)-(CO)2(dppv)2]2+. In their study, they concluded that hydrogen evolution is greatly accelerated by the adt-cofactor.14
Finally, Zaffaroni et al. produced a series of tetra-PMe3 substituted complexes with edt, pdt and adt-bridge.15 For the edt- and pdt analogues, they propose that formation of terminal and bridging hydride are not sequential steps, but rather competing reactions. Their experiments on the edt analogue showed that at −90 °C the complex only displays the bridging hydride, while at room temperature they could observe a mixture of both the terminal and bridging hydride. For the pdt analogue, they further suggested protonation at sulfur prior to formation of the terminal hydride. Both studies used the very strong acid H(EtO2)2BArF4. According to their study, only the adt-analogue did exclusively produce the terminal hydride even with use of weaker acids.
Schollhammer and coworkers also worked on alternative ligands for stabilizing terminal hydrides, using N-heterocyclic carbenes16 and a mixed phosphine-carbene complex.17 The former did not show any stable terminal hydrides even at low temperatures, while the phosphine carbene complex could hold a terminal hydride at the distal iron atom at low temperatures, which isomerized rapidly upon warming up.
All these studies show that it is indeed possible to form a terminal hydride if the complex is basic enough to be protonated rapidly. Bulky, chelating phosphine ligands further promote stabilization of the terminal hydrides, but all complexes eventually transform at room temperature into the thermodynamically more stable bridging hydride.
In the present study, we follow a new approach to hinder the rotation of the terminal ligands at one of the iron atoms. A new series of Fe2 complexes was designed that contain terminal mono-dentate phosphine ligands covalently linked to the dithiolate bridge. This fixation of the phosphine should be able to obstruct the ligand rotation and avoid the formation of the thermodynamically more stable bridging hydride. All complexes are prepared from the hexacarbonyl precursor [FeFe](mcbdt)(CO)61 (mcbdt = benzene-1,2-dithiol-3-carboxylic acid). 1 contains a carboxylic acid group at the bridging benzenedithiolate ligand that can synthetically be addressed by amino-phosphine ligands to form amide bonds. Complexes 2–5 (Fig. 2) contain a tethering alkyl chain of increasing length (ethyl to pentyl) that is linked via an amide bond to the bridge and holds a phosphine at the end which replaces one CO ligand. Thereby, the phosphine ligands are not just coordinating to the iron centre, but they are also linked to the bridging dithiolate to obtain a more rigid structure. The hypothesis is that through this tethering effect, the rotation of the Fe(CO)2P-unit will be hindered. As a result, structural rearrangements of the complex after formation of the kinetically favoured terminal hydride could potentially be prevented, or at least drastically slowed down, as compared to the situation in previous model complexes. Fig. 2(a) shows a schematic view of the [FeFe]-hydrogenase active site and its interactions with surrounding amino acid residues.18 In Fig. 2(b), the series of model complexes with tethering ligands and a reference compound 6 with a similar set of ligands lacking the linking unit are shown. In the present study, we discuss synthesis and characterization of complexes 1, 3, 4 and 6, along with a detailed theoretical investigation of all proposed compounds. In particular, we were interested in the effect of the tethering ligands on rotational energy barriers of the Fe(CO)2P unit.
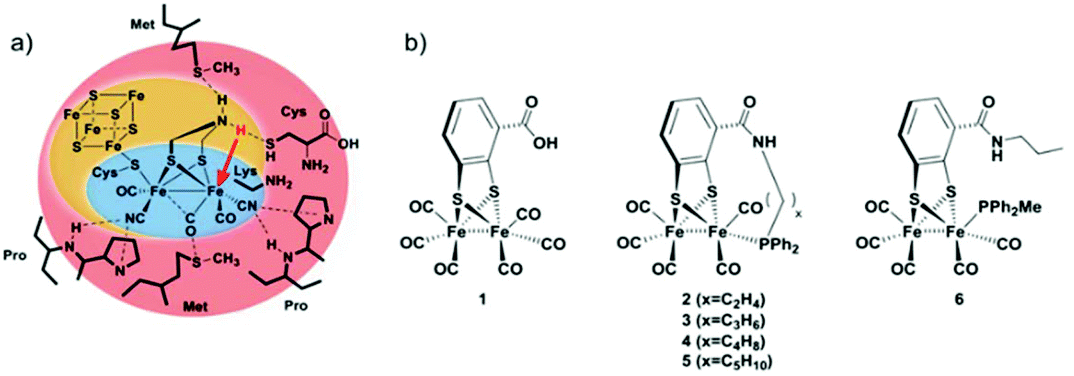 | ||
| Fig. 2 (a) Coordination spheres in the [FeFe]-hydrogenase active site. Amino acid residues in the outer shell (red) stabilize the rotated geometry to form a terminal hydride. (b) Series of model complexes presented in this study. Complexes 2–5 contain an alkyl-chain tethering the bridging dithiolate ligand and a phosphine ligand. | ||
Results and discussion
Synthesis
Synthesis of complexes 1, 3, 4 and 6 is described in detail in the Experimental section. A representative synthetic procedure for tethering phosphine containing complexes is outlined for the example of 3 in Fig. 3. Briefly, starting with [FeFe](mcbdt)(CO)61, an amide bond is formed by the reaction between the amine functionalized phosphine ligand and the carboxylic acid. A suitable coupling reagent for this transformation was found to be PyBop (benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluoro-phosphate). Continuous stirring for about five hours at room temperature leads to substitution of one carbonyl ligand at the iron centre. Reference compound 6 was synthesized in two steps, starting with the amide coupling of propylamine to the carboxylic acid and subsequent ligand exchange with PPh2Me. In order to selectively introduce only one P(Ph)2Me ligand, one equivalent Me3NO as decarbonylation reagent was added to the reaction. All complexes where characterized by IR, 31P-NMR and HR-MS in case of 3 and 4 (see ESI† and Experimental details).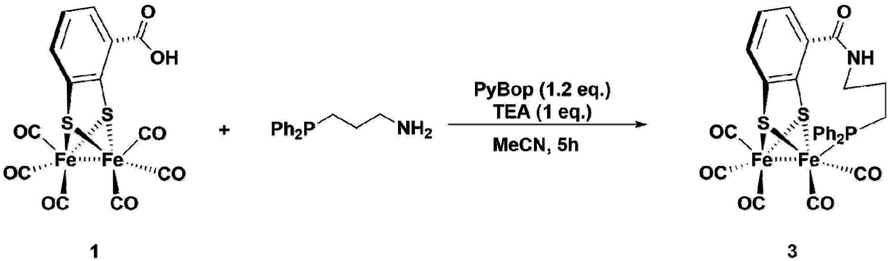 | ||
| Fig. 3 One-pot synthesis of the propyl-tethered complex 3. Complexes 4 was prepared in a similar fashion. | ||
IR spectroscopy
Coordination of the tethered phosphine ligands is clearly indicated in the IR spectra by a shift of the carbonyl stretching frequencies by about 30 cm−1 towards lower wavenumbers compared to that of the hexacarbonyl starting material 1 (Fig. 4 and Table 1). Spectra of 3, 4 and 6 are largely identical, suggesting that neither the presence of the tether, nor the presence of different tethers has a profound impact on the electronic properties of the Fe sites.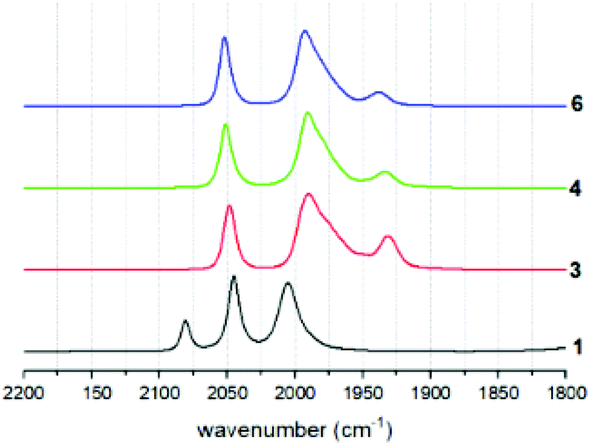 | ||
| Fig. 4 IR spectra of tethered complexes 3 and 4, reference complex 6 and hexacarbonyl precursor 1. | ||
| Compound | ν CO exp. cm−1 | ν CO calc. cm−1 |
|---|---|---|
| 1 | 2081, 2045, 2005 | 2061, 2030, 1998 |
| 3 | 2048, 1989 (b), 1931 | 2040, 1988, 1929 |
| 4 | 2051, 1992 (b), 1934 | 2037, 1989, 1935 |
| 6 | 2052, 1993, 1938 | 2041, 1984, 1943 |
As expected, also potentially different rotational isomers that arise from preferred apical or basal coordination modes in the different compounds cannot be clearly identified by the indistinguishable IR spectra. This behaviour is identical to all other mono-phosphine complexes of this type in the literature.19 Presumably, the rotational isomers rapidly interconvert, as supported by the fact that only one resonance is observed in the 31P solution NMR spectra of the compounds. Calculations of carbonyl stretching frequencies clearly show that the experimental spectra are a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 superposition of terminal and apical ligand species (see below).
:1 superposition of terminal and apical ligand species (see below).
DFT calculations
Structural optimizations were conducted with the BP86 functional and a def2-TZVP basis set for all rotational isomers in order to identify the thermodynamically most favoured structures. This method has been shown to give accurate data for the precursor complex [FeFe](mcbdt)(CO)61 compared with the crystal structure (see Table S5 in ESI†). All calculated structures were true minima as indicated by vibrational frequency analysis (see Fig. 5 for comparison between experimental and calculated spectra). Additional single point energy calculations using the hybdrid B3LYP, the double-hybrid PWPB95 functional and local coupled-cluster DLPNO-CCSD(T) were performed in order to systematically increase the accuracy of calculated energy barriers B3LYP, PWPB95 and DLPNO-CCSD(T).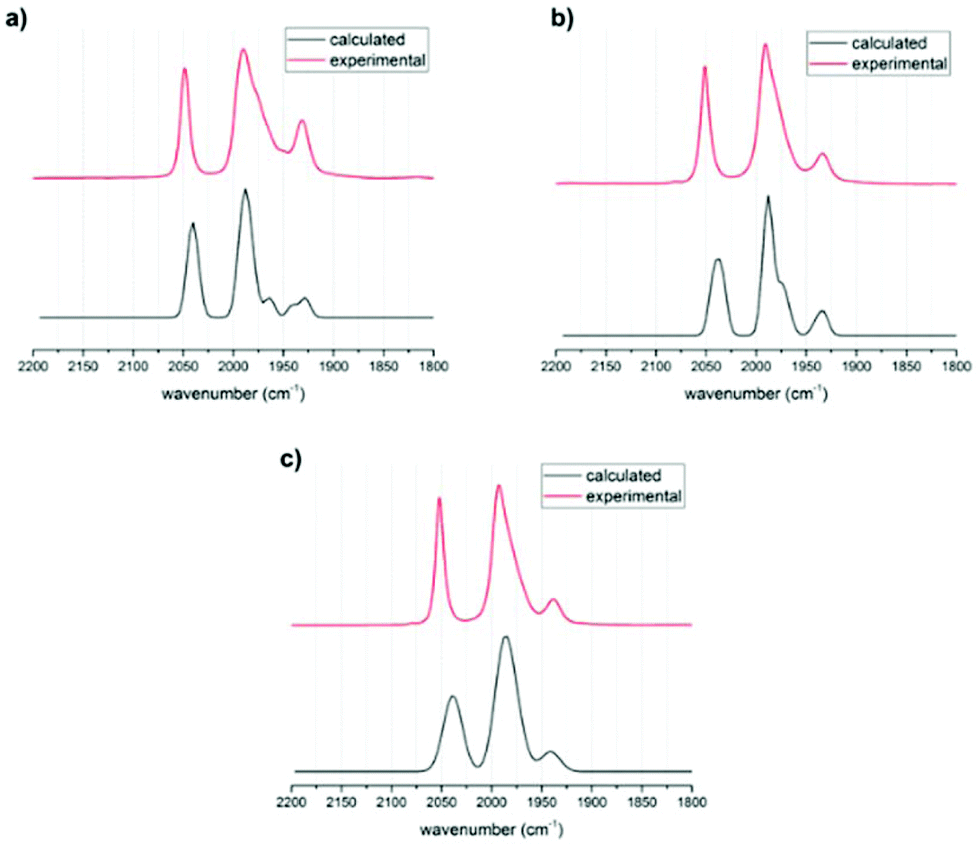 | ||
| Fig. 5 Calculated (black) and experimental (red) IR spectra of (a) 3, (b) 4 and (c) 6. The calculated spectra are 1:1 mixtures of the spectra obtained for the apical and basal isomers with BP-86 without use of scaling factor. | ||
First, the structures of complexes 3 and 4 as well as that of the reference complex 6 were optimized. Vibrational frequency analysis was performed for all three structures and the results are in good agreement with experimental data (Table 1). No scaling factor was used. The calculated IR spectra for the phosphine in apical versus basal position were very similar in shift, but there was an additional peak found for the basal structures (see Fig. 5). In the experimentally obtained spectrum, this peak in the centre of the spectrum around 1990 cm−1 is very broad and has a shoulder. Comparing the calculated spectra with experimental suggests that for all structures the apical and the basal isomers both are present in equilibrium. The visualized calculated spectra in Fig. 5 are a superposition of the calculated spectra of the apical and basal isomers in a ratio of 1:1. When the structures were calculated with PMe2 instead of PPh2, the shape and frequency of calculated IR absorption bands do not change significantly (ESI Fig. S6†). Since the PMe2-substituted complexes resemble the structure PPh2-substituted complexes very well, the following calculations concentrated on PMe2-substituted isomers. This computational modification is indicated by a “Me” as index to the compound number, e.g. (3Me).
In general, complexes 3, 5, 6 prefer the apical ligand position due to steric reasons. In case of complex 4Me, however, the strain caused by the butyl tether is pronounced for the apical form and the basal isomer is energetically favoured. With the exception of complex 4Me, an increase in tethering chain length leads to an energetic preference of the apical over the basal rotamer (Table 2). Computational design of the tethering chain length thus allows to suggest suitable target complexes for the coordination preference of the phosphine ligand, and an estimate of the energy difference between the two isomers. Generally, BP86 and B3LYP underestimate the energy difference between the rotamers by 5–15 kJ mol−1.
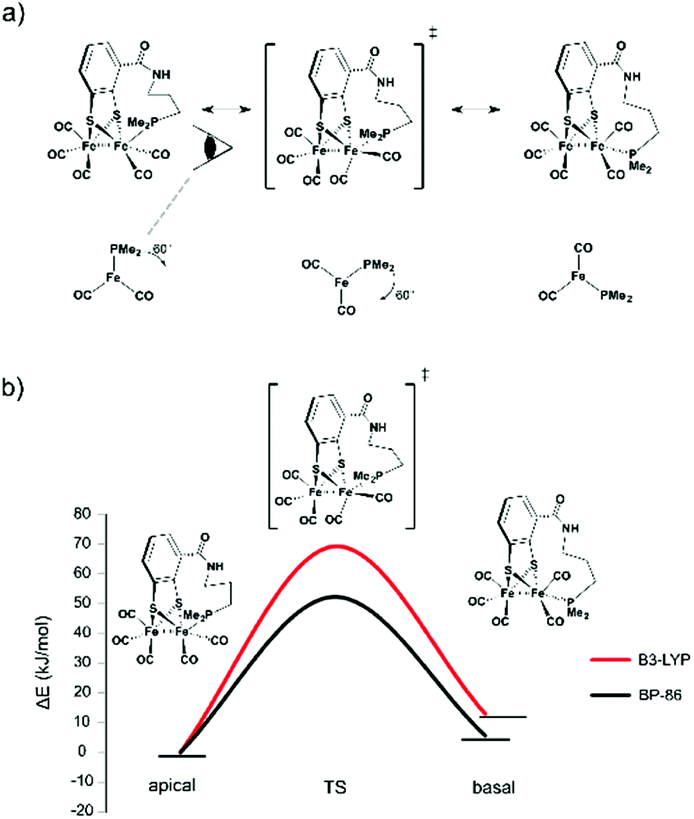 | ||
| Fig. 6 (a) Anticipated rotation from apical via transition state to basal form. (b) Calculated rotational barrier for the propyl-tethered complex. | ||
| BP86 | B3LYP | PWPB95 | DLPNO-CCSD(T) | |
|---|---|---|---|---|
| 1 | 41.4 | 54.5 | 32.2 | 57.1 |
| 2Me | n.a. | |||
| 3Me | 52.2 | 69.1 | 44.9 | 72.8 |
| 4Me | 23.5 | 37.4 | 21.9 | 45.4 |
| 6Me | 40.4 | 55.7 | 35.0 | 60.4 |
The starting compound 1 and the functionalized reference compound 6 display almost identical barriers to rotation. This shows that the terminal phosphine does not affect the rotational energies of the barrier. Since the ethyl-tethered complex 2 only affords an apical structure, no rotation barrier can be reported.
The highest rotational energy barrier of all complexes was observed for propyl-tethered complex 3. Compared to the reference complex 6, an increase of the rotation barrier by 12–14 kJ mol−1 was calculated, depending on the level of calculation. This narrow range indicates the consistency of the relative energies although the absolute energies vary with the level of calculation. In contrast, complex 4 with a butyl tether did not show an increase in rotation energy barrier. The butyl tether allows too much structural flexibility and therefore the energy barrier is lower. As a matter of fact, it displays the lowest barrier of all complexes. For 4Me, also the energy difference between basal and apical structures is lowest and only for this complex the basal rotamer is preferred. The butyl chain in the apical rotamer is highly strained, which disfavours this structure. Presumably, the rotation energy is decreased due to this fact.
Conclusions
A new series of [FeFe]-hydrogenase model complexes with phosphine ligands that are covalently tethered to the bridging bdt ligand and coordinated to one iron centre has been prepared. The complexes have been characterized by FT-IR, 31P-NMR spectroscopy and HR-MS. A detailed theoretical investigation was performed on the model complexes to study the effect of tethering ligands on the rotation of the terminal Fe(CO)2PL3-subunit. The highest energy barrier for the rotation has been found for the propyl-tethered complex 3 compared to the hexacarbonyl precursor 1 and the reference compound 6 that is lacking the linkage. The propyl-tethered complex 3 is thus the most promising candidate in the series presented for investigating protonation and the formation of a terminal hydride. The butyl-tethered complex 4 was the only complex with the phosphine in basal position being the lowest rotamer. All other complexes preferred the phosphine ligands in apical position although the energy differences are small. Restricting the rotation can potentially lead to the stabilization of the reactive terminal hydride, which is considered to be an important intermediate in the catalytic cycle of hydrogen evolution.Experimental
Synthetic procedures
:3:0.2). The product was obtained as the second red band (the first one was the bdt-bridged complex).
Yield: 300 mg (18%).
IRCO (CH2Cl2): 2084, 2035, 2009, 1996, 1980, 1691 cm−1.
1H NMR: 7.20 (dd, J = 7.4, 0.9, 1H, Hortho), 7.06 (dd, J = 7.7, 0.8, 1H, Hpara), 6.69 (dd, J = 7.6, 1H, Hmeta).
IRCO (MeCN): 2079, 2044, 2005.
IRCO (MeCN): 2052, 1993, 1938.
31P-NMR: 38.94 ppm.
HR-MS: calculated for C28H24Fe2NO6PS2Na found 699.94 (product + Na), and for C28H24Fe2NO6PS2 (product + H) calculated 677.96, found 677.96 (product + H).
IRCO (MeCN): 2048, 1989, 1931.
31P-NMR: 32.8 ppm.
HR-MS: calculated for C27H20Fe2NO6PS2 660.92, found 661.92.
IRCO (MeCN): 2051, 1992, 1934.
31P-NMR: 49.9 ppm.
Computational methods
Turbomole 6.2 and 7.0 were used for GGA and hybrid DFT calculations (Ahlrichs, R. TURBOMOLE 6.2 and 7.0; Universität Karlsruhe: 2010/2015. See http://www.turbomole.com).22 Additional calculations used Orca 4.0.30Structural optimizations were performed at the BP8623 and the hybrid B3LYP24 functionals and structures were verified to be minima and transition states, respectively. The resolution of the identity (RI-J) approximation25 was used for the GGA calculations. The Ahlrichs’ def2-TZVP and def2-TZVPP basis sets26 were used as indicated. Additional single point calculations with the larger basis set at the BP86/def2-TZVP optimized structures using the double-hybrid PWPB9527 functional and the local coupled-cluster DLPNO-CCSD(T) method28 method were performed in order to obtain more accurate relative energies. For the CC calculation, tight threshold values (TightPNO) settings were employed.29
All the structures presented are geometry optimized in gas-phase. IR frequencies were obtained by analytical evaluation of the second derivative of the DFT energy expression. The lack of negative eigenvalues for all structures shows they are true minima. Transition state structures were optimized until the analytical vibrational frequency calculation resulted in only one imaginary frequency which corresponded to the terminal ligand rotation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the Swedish Research Council, the Knut & Alice Wallenberg Foundation, and the Swedish Energy Agency. MS is grateful to the Max Planck Society for the Advancement of Science (MPG) and the COST Action CM1305 ‘ECOSTBio’ for financial support. Financial support received from SERB-DST, India, under project no. (ECR/2016/000382 to SM) is gratefully acknowledged.References
- M. Frey, ChemBioChem, 2002, 3, 153–160 CrossRef CAS.
- J. W. Peters, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- (a) D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed; (b) M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 16894–16902 CrossRef CAS PubMed.
- C. Liu, J. N. T. Peck, J. A. Wright, C. J. Pickett and M. B. Hall, Eur. J. Inorg. Chem., 2011, 2011, 1080–1093 CrossRef.
- A. R. Finkelmann, M. T. Stiebritz and M. Reiher, Chem. Sci., 2014, 5, 215–221 RSC.
- (a) L.-C. Song, J. Cheng, J. Yan, H.-T. Wang, X.-F. Liu and Q.-M. Hu, Organometallics, 2006, 25, 1544–1547 CrossRef CAS; (b) M. Razavet, S. C. Davies, D. L. Hughes and C. J. Pickett, Chem. Commun., 2001, 847–848 RSC; (c) S. J. George, Z. Cui, M. Razavet and C. J. Pickett, Chem. – Eur. J., 2002, 8, 4037–4046 CrossRef CAS PubMed.
- (a) A. Orthaber, M. Karnahl, S. Tschierlei, D. Streich, M. Stein and S. Ott, Dalton Trans., 2014, 43, 4537–4549 RSC; (b) M. Beyler, S. Ezzaher, M. Karnahl, M.-P. Santoni, R. Lomoth and S. Ott, Chem. Commun., 2011, 47, 11662–11664 RSC.
- S. Ezzaher, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer, J. Talarmin, R. Pichon and N. Kervarec, Inorg. Chem., 2007, 46, 3426–3428 CrossRef CAS.
- F. I. Adam, G. Hogarth, S. E. Kabir and I. Richards, C. R. Chim., 2008, 11, 890–905 CrossRef CAS.
- B. E. Barton, G. Zampella, A. K. Justice, L. de Gioia, T. B. Rauchfuss and S. R. Wilson, Dalton Trans., 2010, 39, 3011–3019 RSC.
- B. E. Barton and T. B. Rauchfuss, Inorg. Chem., 2008, 47, 2261–2263 CrossRef CAS PubMed.
- B. E. Barton, M. T. Olsen and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 16834–16835 CrossRef CAS PubMed.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef CAS PubMed.
- R. Zaffaroni, T. B. Rauchfuss, D. L. Gray, L. de Gioia and G. Zampella, J. Am. Chem. Soc., 2012, 134, 19260–19269 CrossRef CAS PubMed.
- D. Morvan, J.-F. Capon, F. Gloaguen, A. Le Goff, M. Marchivie, F. Michaud, P. Schollhammer, J. Talarmin, J.-J. Yaouanc, R. Pichon and N. Kervarec, Organometallics, 2007, 26, 2042–2052 CrossRef CAS.
- D. Morvan, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer, J. Talarmin, J.-J. Yaouanc, F. Michaud and N. Kervarec, J. Organomet. Chem., 2009, 694, 2801–2807 CrossRef CAS.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
- (a) D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed; (b) C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- A. K. Vannucci, S. Wang, G. S. Nichol, D. L. Lichtenberger, D. H. Evans and R. S. Glass, Dalton Trans., 2010, 39, 3050–3056 RSC.
- (a) S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed; (b) W. W. Seidel, F. E. Hahn and T. Lügger, Inorg. Chem., 1998, 37, 6587–6596 CrossRef CAS PubMed; (c) D. Sellmann, T. Becker and F. Knoch, Chem. Ber., 1996, 129, 509–519 CrossRef CAS; (d) L. Schwartz, P. S. Singh, L. Eriksson, R. Lomoth and S. Ott, C. R. Chim., 2008, 11, 875–889 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- S. Grimme, J. Chem. Phys., 2006, 124, 34108 CrossRef PubMed.
- (a) C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed; (b) C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 24109 CrossRef PubMed.
- F. Pavošević, P. Pinski, C. Riplinger, F. Neese and E. F. Valeev, J. Chem. Phys., 2016, 144, 144109 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt05148h |
| This journal is © The Royal Society of Chemistry 2019 |