
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a miniaturized [FeFe] hydrogenase model system†
Charlène
Esmieu‡
a,
Meiyuan
Guo
b,
Holly J.
Redman
a,
Marcus
Lundberg
 b and
Gustav
Berggren
*a
b and
Gustav
Berggren
*a
aMolecular Biomimetics, Department of Chemistry – Ångström Laboratory, Uppsala University, 75120 Uppsala, Sweden. E-mail: gustav.berggren@kemi.uu.se
bTheoretical Chemistry, Department of Chemistry – Ångström Laboratory, Uppsala University, 75120 Uppsala, Sweden. E-mail: marcus.lundberg@kemi.uu.se
First published on 15th January 2019
Abstract
The reaction occurring during artificial maturation of [FeFe] hydrogenase has been recreated using molecular systems. The formation of a miniaturized [FeFe] hydrogenase model system, generated through the combination of a [4Fe4S] cluster binding oligopeptide and an organometallic Fe complex, has been monitored by a range of spectroscopic techniques. A structure of the final assembly is suggested based on EPR and FTIR spectroscopy in combination with DFT calculations. The capacity of this novel H-cluster model to catalyze H2 production in aqueous media at mild potentials is verified in chemical assays.
Renewable molecular hydrogen (H2) is often referred to as a future energy carrier. The interconversion between protons and molecular hydrogen is also central to the metabolism of a large number of microorganisms, and the enzymes that Nature has evolved for this purpose are the hydrogenases.1 In the case of [FeFe] hydrogenase (HydA), turnover takes place at the “H-cluster”. This cluster consists of a canonical [4Fe4S] cluster coordinated by four cysteine-derived thiols, coupled via a bridging thiolato ligand to a low valent dinuclear iron complex called the (catalytic) [2Fe] subsite. The two iron ions in the [2Fe] subsite are bridged by an azadithiolato ligand (adt = −SCH2NHCH2S−), and further decorated by CO and CN− ligands (Fig. 1A).2–4 A wide range of synthetic complexes inspired by the H-cluster have been prepared. These synthetic efforts have mainly focused on replicating the [2Fe] subsite and have delivered remarkably close mimics of this complex, but more elaborate models utilizing e.g. oligopeptides have also been reported.5–9
 | ||
| Fig. 1 Schematic representation of the inorganic cofactor constituting the active site of [FeFe]-hydrogenase and the model complexes employed in this study. (A): The complete H-cluster; (B): the [2Fe] subsite model [Fe2(adt)(CO)4(CN)2]2−, 1; (C): a truncated structure of P. aerogenes ferredoxin I, the -CIACGAC- core motif on which the FdM maquette is based is highlighted in yellow. Heteroatom colour coding: Fe = orange; S = yellow; N = blue and O = red. A and C generated from Protein Data Bank entries 3C8Y (selected amino acids removed to provide an unobstructed view of the cofactor) and 1DUR respectively. | ||
In nature the synthesis and insertion of the [2Fe] subsite is dependent on a complex enzymatic machinery.10 However, it was recently shown how this can be circumvented and the hydrogenase enzyme artificially matured using synthetic mimics of the cofactor.3,11 When the [2Fe] subsite mimic [Fe2(adt)(CO)4(CN)2]2− (1) (Fig. 1B) is introduced into a form of the enzyme lacking the [2Fe] subsite ([4Fe4S]–HydA), the complex enters the active site and spontaneously reacts with the [4Fe4S] cluster to generate the H-cluster, thereby transforming 1 into a highly efficient and robust catalyst. Indeed, this reactivity appears quite promiscuous and complexes of the general structure [Fe2(xdt)(CO)(5−y)(CN)(1+y)](1+y)− (y = 1 or 0; xdt denotes different bridging dithiolate/-selenate ligands) all appear capable of reacting with the pre-assembled [4Fe4S] cluster to generate a wide range of semi-synthetic H-clusters.3,11–16
However, this reactivity has not been observed outside of HydA and the importance of the surrounding active-site pocket is not firmly established. Herein we show that the reaction observed between 1 and [4Fe4S]–HydA can be reproduced by a small synthetic peptide, providing a new synthetic route for the generation of complete H-cluster model systems.17
A model of [4Fe4S]–HydA was generated through the assembly of a [4Fe4S] cluster in a cysteine containing oligopeptide. More specifically, we employed the 16 amino acid synthetic peptide, or maquette, H2N-KLCEGGCIACGACGGW-CONH2 (FdM). This sequence is derived from the FeS cluster-binding motif of Peptococcus aerogenes ferredoxin I and has been reported by Dutton and coworkers as a prototype ferredoxin mimic (Fig. 1C).18,19
The FdM maquette features four cysteine residues capable of coordinating a [4Fe4S] cluster (highlighted in bold in the sequence), as observed for the H-cluster. Treating FdM with stoichiometric amounts of iron and sulfide in the presence of excess of β-mercaptoethanol under anaerobic conditions resulted in the formation of a [4Fe4S] cluster, in agreement with earlier reports.18,19 The as-prepared cluster resides in its EPR silent, oxidized, [4Fe4S]2+ state, observable by UV/Vis spectroscopy (Fig. 2, top and bottom, black spectra, ε385 ≈ 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L mol−1 cm−1). Chemical reduction of [4Fe4S]2+–FdM with sodium dithionite resulted in reduction of the [4Fe4S]2+ cluster (Fig. 2, top, red spectrum) with concomitant formation of the corresponding [4Fe4S]+ cluster, readily observable by its rhombic EPR signal (g = 2.06, 1.93 and 1.89; Fig. 2, bottom, red spectrum). Spin quantification using a copper standard indicated that approximately 55 ± 10% of the [4Fe4S] incorporated into the peptide generated an EPR visible [4Fe4S]+–FdM species. The reduction of a solution containing iron, sulfide and β-mercaptoethanol but lacking the peptide does not generate any EPR active species, highlighting the importance of the peptide environment for formation of a stable reduced [4Fe4S] cluster (Fig. 2, bottom, dashed spectrum).
000 L mol−1 cm−1). Chemical reduction of [4Fe4S]2+–FdM with sodium dithionite resulted in reduction of the [4Fe4S]2+ cluster (Fig. 2, top, red spectrum) with concomitant formation of the corresponding [4Fe4S]+ cluster, readily observable by its rhombic EPR signal (g = 2.06, 1.93 and 1.89; Fig. 2, bottom, red spectrum). Spin quantification using a copper standard indicated that approximately 55 ± 10% of the [4Fe4S] incorporated into the peptide generated an EPR visible [4Fe4S]+–FdM species. The reduction of a solution containing iron, sulfide and β-mercaptoethanol but lacking the peptide does not generate any EPR active species, highlighting the importance of the peptide environment for formation of a stable reduced [4Fe4S] cluster (Fig. 2, bottom, dashed spectrum).
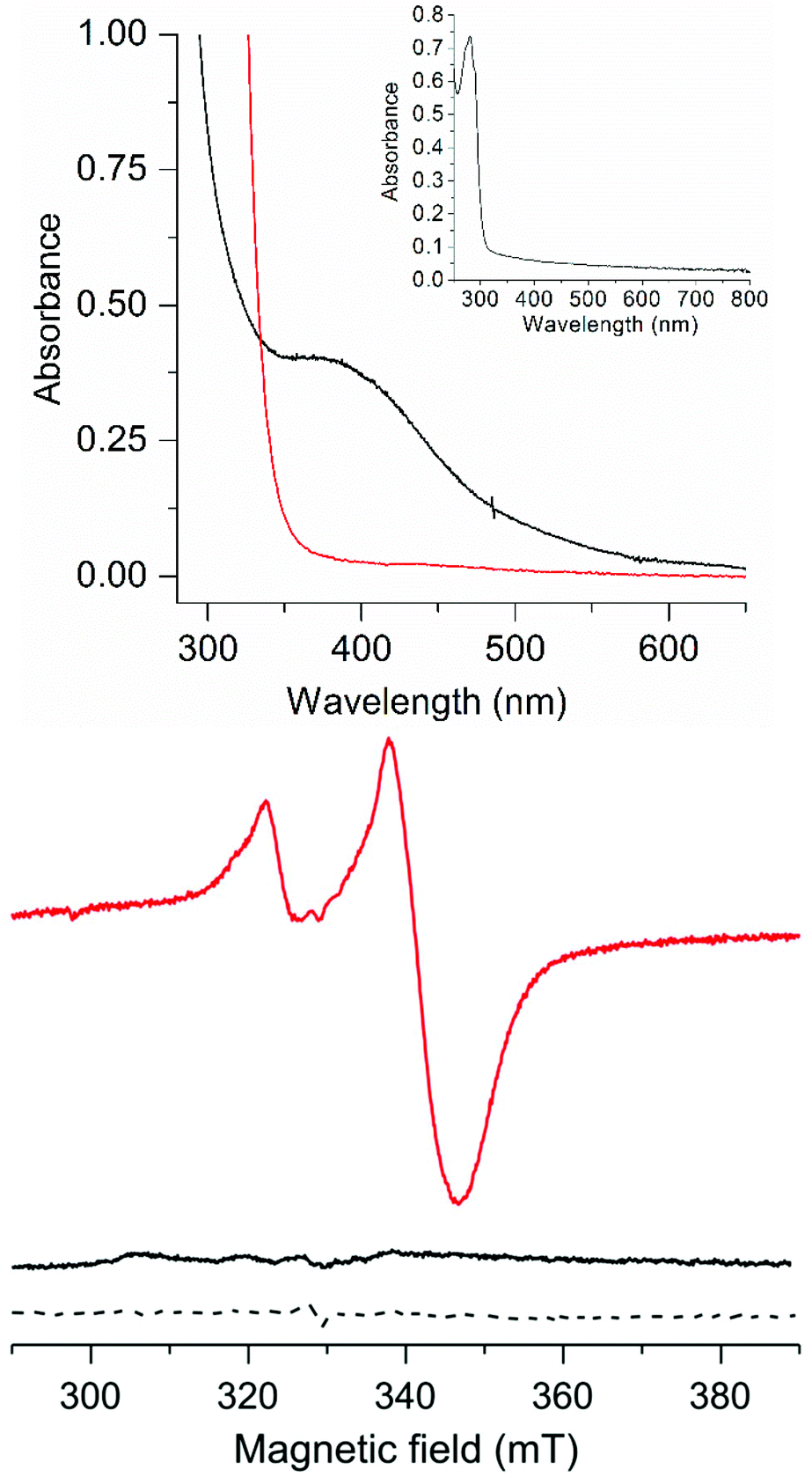 | ||
| Fig. 2 Spectroscopic properties of the as-prepared and reduced forms of [4Fe4S]–FdM; (Top): UV/Vis spectra, [4Fe4S]2+–FdM (20 μM black line) and [4Fe4S]+–FdM (20 μM, red line), (inset): apo-FdM (128 μM); (Bottom) X-band EPR spectra of [4Fe4S]2+–FdM (120 μM, black solid line), [4Fe4S]+–FdM (120 μM, red solid line) and a reduced solution of iron and sodium sulfide in the presence of β-mercaptoethanol (dashed line). All samples prepared in HEPES buffer (50 mM, pH 8.0). EPR spectra recorded at 10 K; microwave frequency 9.28 GHz; modulation amplitude: 10 G; microwave power: 1 mW. | ||
As described above, complex 1 reacts with the [4Fe4S] cluster present in HydA to spontaneously generate the H-cluster, via formation of a cysteine derived bridging thiolato ligand and concomitant release of a CO ligand.11,13 Analogously, the addition of [4Fe4S]–FdM to an aqueous solution of 1 in the presence of dithionite resulted in the release of a CO ligand, as determined via a deoxyhemoglobin (HHb) assay. A solution of 1 was treated with stoichiometric amounts of [4Fe4S]–FdM in a gas-tight reaction vessel at ambient temperature under reducing conditions. The addition of HHb to the reaction mixture after a 30 minutes incubation time resulted in an instantaneous shift of the HHb Soret band, as expected from the formation of COHb (Fig. S1†).20 The amount of CO released was found to vary linearly with the amount of 1 and [4Fe4S]–FdM, with approximately 0.67 mole of CO released per mole of [4Fe4S]–FdM added (Fig. S2 and S3†). The stoichiometry of the reaction agrees well with the fraction of [4Fe4S]+ observed by EPR upon reduction of [4Fe4S]–FdM, suggesting that the reaction only occurs with the reduced cluster. This is further supported by the observation that only trace amounts of CO could be detected from the reaction in the absence of [4Fe4S]–FdM or dithionite. Thus, [4Fe4S]2+–FdM does not appear nucleophilic enough to displace a CO ligand on 1, while the reaction occurs on a minute time-scale with [4Fe4S]+–FdM.
In order to further monitor the formation of this new species, occurring by ligand exchange on 1, the reaction was probed by EPR spectroscopy. The addition of complex 1 to a solution of [4Fe4S]+–FdM resulted in a decrease in intensity of the S = ½ EPR spectrum. A complete disappearance of the signal was observed after addition of one equivalent of 1 relative to total peptide concentration, suggesting that a slight excess of 1 was required to drive the reaction to completion (Fig. S4†). The decrease of the rhombic signal due to oxidation of the cluster to [4Fe4S]2+ could be ruled out, as no increase of UV/Vis absorbance could be detected around 400 nm. Instead, the absence of an EPR signal suggests that complex 1 is oxidized into a mixed, FeIFeII, valence state upon formation of 1-[4Fe4S]+–FdM, resulting in an overall S = 0 species. Indeed, sampling of the headspace gas revealed the generation of 0.3 equivalents of H2 per [4Fe4S]–FdM added to 1. In combination with the amount of CO released, this observation strongly supports the notion that the reaction induces a one-electron oxidation of 1 with concomitant formation of 0.5 eq. of H2. The disappearance of the EPR signal is thus assigned to the formation of a new, EPR-silent, H-cluster like species [Fe2(I,II)(adt)(CO)3(CN)2]–[4Fe4S]+–FdM (2) (Scheme 1).
 | ||
| Scheme 1 Schematic representation of the formation of 2 proceeding via the binding of the reduced [4Fe4S] cluster to complex 1, resulting in the formation of a mixed valence complex with concomitant formation of H2 and release of a CO ligand. | ||
The reaction between [4Fe4S]–FdM and complex 1 is also clearly observable by FTIR spectroscopy. The FTIR spectrum of complex 1 in the presence of β-mercaptoethanol shows one CN− peak and three CO peaks at 2056, 1980, 1944 and 1910 cm−1 respectively (Fig. 3, spectrum C), in agreement with earlier reports for 1 in aqueous media. In the presence of [4Fe4S]2+–FdM the overall shape of the spectrum is retained with a shift of 5–7 cm−1 observed for the peaks in the CO region (Fig. 3, spectrum B). The absence of any significant changes to the FTIR spectrum underscores the inability of [4Fe4S]2+–FdM to displace a ligand on 1. The shifts in peak position is tentatively attributed to hydrogen bonding interactions between 1 and the oligopeptide, as a shift of similar magnitude has previously been reported for the protonation of the CN− ligands in the case of [Fe2(adt)(CO)5(CN)]− (3) and [Fe2(pdt)(CO)4(PMe3)(CN)]− (4, pdt = −SCH2CH2CH2S−).24,25
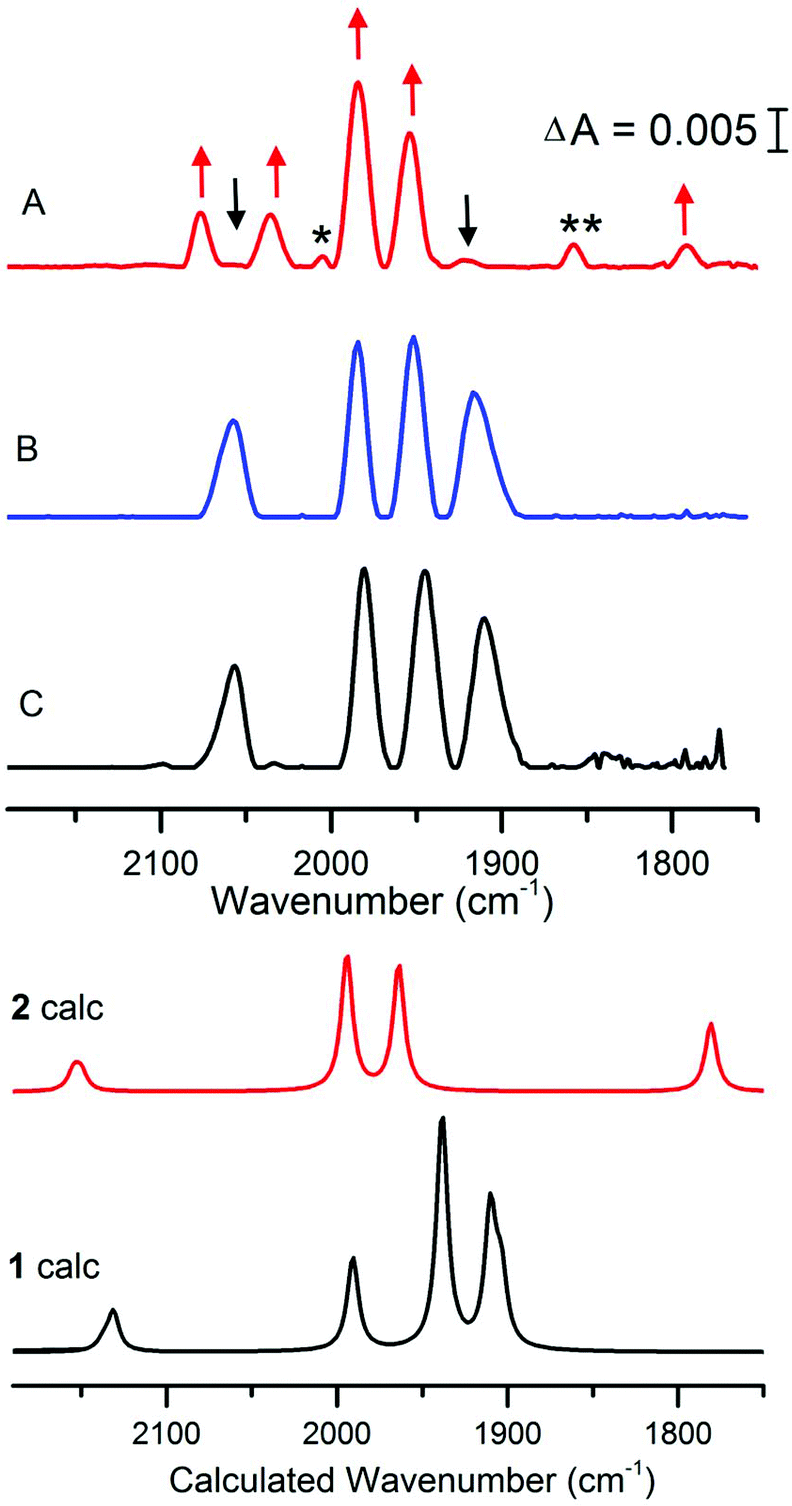 | ||
| Fig. 3 FTIR spectra of 1 treated with [4Fe4S]–FdM under reducing or non-reducing conditions. Complex 1 in aqueous buffer (spectrum C); 1 + 2 eq. of [4Fe4S]2+–FdM (300 μM) (spectrum B); 1 + 2 eq. of [4Fe4S]+–FdM (300 μM) (spectrum A). Arrows indicate the new peaks assigned to 2 (red) and the consumption of 1 (black); peaks not assigned to 1 or 2 indicated with asterisk (see main text). All samples prepared in HEPES buffer (50 mM, pH 8) and spectra recorded after 30 minutes incubation time with 1. Calculated spectra are obtained using an [Fe2(adt)(CO)4(CN)2]2− model of 1 and an [4Fe-4S]-S(Cys)-[2Fe] model of 2 (see Fig. S9 and S13† for details). Results for alternative isomers and protonation states are given in the ESI.† | ||
Conversely, under reducing conditions the reaction between 1 and [4Fe4S]+–FdM results in the formation of new species, readily observed by the disappearance of the features at 2056 and 1908 cm−1. The new spectrum features two bands of equal intensity in the region generally associated with CN− at 2078 and 2036 cm−1, four new signals in the terminal CO region at 2005 (w), 1984 (s), 1953 (s), 1856 (w) and a band at 1792 cm−1 indicative of a bridging CO (Fig. 3, spectrum A). The observed FTIR spectrum is consistent with the reaction proposed in Scheme 1 for the formation of 2. In contrast, no changes were observed in the FTIR spectrum of complex 1 if the peptide was omitted from the reaction (Fig. S5†), further underscoring the requirement for the reduced [4Fe4S]+–FdM cluster for the reaction to proceed. The main features of the spectrum (2078, 2036, 1984, 1953 and 1792 cm−1, indicated with red arrows in Fig. 3) agrees well with data reported for the HydA enzyme. More specifically, for states of the H-cluster where the [2Fe] subsite resides in an oxidized mixed valence Fe2(I,II) configuration, i.e. the so-called Hox (Fe2(I,II)–[4Fe4S]2+) or Hred′ (Fe2(I,II)–[4Fe4S]+) states (Table 1).21–23 The weak feature at 1856 cm−1 appears in varying intensity in the preparations of 2, whether it reflects a side-product during the synthesis or a different protonation/redox state of the complex is currently not firmly established. Finally, the weak feature at 2005 cm−1 is similar to what has been reported for the CO inhibited Hox-CO state. This latter feature disappears upon addition of HHb to the solution, showing that this extra CO ligand can be readily removed (Fig. S6†). Thus, we attribute it to a small fraction of 2 retaining a weakly bound fourth CO ligand.
| Species | CN− | Terminal CO | Bridging CO | Ref. |
|---|---|---|---|---|
| a Observed for HydA1 from Chlamydomonas reinhardtii. b Native H-cluster. c Semi-synthetic H-cluster incorporating the [2Fe]pdt (= [Fe2(pdt)(CO)3(CN2)]2−) cofactor. | ||||
| 1 | 2056 | 1980, 1944, 1910 | — | This work and ref. 3 |
| 2 | 2078, 2036 | (2005), 1984, 1953 (1856) | 1792 | This work |
| Hoxa,b | 2088, 2072 | 1964, 1940 | 1800 | 21 |
| Hox-pdta,c | 2090, 2072 | 1966, 1941 | 1810 | 22 |
| Hox-COa,b | 2092, 2084 | 2013, 1970, 1964 | 1810 | 21 |
| Hred′a,b | 2083, 2067 | 1962, 1933 | 1791 | 23 |
| Hred′-pdta,c | 2084, 2065 | 1963, 1933 | 1798 | 22 |
In combination, the aforementioned data strongly support the hypothesis that the reaction observed during maturation of the native hydrogenase enzyme can be reproduced using small model systems. The binding of thioether ligands to cyanide containing [2Fe] subsite mimics has previously been reported, albeit as transient species.26,27 Considering the requirement for reducing conditions, the disappearance of the S = ½ EPR signal with concomitant formation of H2, the appearance of a bridging CO band and the observed release of a CO ligand we hypothesize that the reaction occurs via coordination of a cluster coordinating cysteinyl ligand to an iron ion of complex 1, analogously to what has been observed in the case of artificial maturation of HydA (Scheme 1).11
To test the feasibility of these assignments, we simulated IR spectra of cluster models of 1 and 2 using the density functional TPSSh and the TZVP basis set.28 The sensitivity of the results with respect to method and structure are shown in the ESI† computational methods. Comparing 1 to 2 confirms the appearance of the bridging CO as well as the loss of the low-frequency CO band (Fig. 3), and a model of 2 with a weakly bound water at the proposed catalytic site gives good agreement with experiment. The major difference is an underestimation of the split of the CN− bands in 2 in the cluster models. This discrepancy could either be due to excessive delocalization of the mixed-valence metal dimer in the DFT calculation, or the lack of a good model of the heterogenous environment in the real system.29 A reduced Fe0FeI version of 2 would show red-shifted bands compared to 1, see Fig. S16,† which is not consistent with experiments. These results thus further support the FeIFeII assignment for 2.
Cyanide ligated [2Fe] subsite derivatives, e.g.1, are generally inefficient proton reduction catalysts in solution. Still, they transform into efficient catalysts upon incorporation into HydA.3,14,25,30,31 In order to determine whether the coupling of 1 to the [4Fe4S]–FdM cluster improved its catalytic properties we assayed hydrogen evolution capacity under aqueous conditions, in the presence of reduced methyl viologen (MV+) (E0, MV+/2+ ≈ −0.45 V). As expected no H2 was detected with complex 1 in isolation, and [4Fe4S]–FdM produced only trace amounts. Conversely, catalytic H2 evolution was observed in the case of 2, with a total of ≈10 turnovers after 1 h (Fig. S17†) at which point the reaction slows down. FTIR spectra recorded after 1 h show a loss of 2, indicating degradation of the catalyst. This instability shows that further stabilization of the cluster is required for efficient catalysis, either via manipulation of the peptide or by embedding the catalyst in a more rigid framework. Still, not only does this, to the best of our knowledge, represent the first report of catalytic H2 evolution from a dicyanide mimic of the [2Fe] subsite, the catalyst is also active at very mild potentials.
Conclusions
In this communication we have shown how [4Fe4S] binding oligopeptides can be employed to prepare catalytically active complexes with structures approaching Nature's H-cluster, when combined with [2Fe] subsite mimics. Our results indicate that the reactivity observed during the maturation of HydA can be reproduced in small molecular systems, providing a new route to explore this chemistry. Moreover, although the rate of hydrogen production observed for 2 is moderate, it emphasizes the importance of the [4Fe4S] cluster for the catalytic properties of this family of organometallic [2Fe] complexes. Moving forward, the possibility to generate completely synthetic “miniaturized hydrogenases” opens up a wide design space, providing the opportunity to readily modify both the organometallic complex as well the ligation motif of the [4Fe4S] cluster and their surroundings.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research performed in the authors laboratories are supported by grants from the Swedish Research Council, VR (G. B. contract no. 621-2014-5670), the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning, Formas (G. B. contract no. 213-2014-880), The Wenner-Gren Foundations (C. E. and G. B.) and the ERC (G. B. contract no. 714102).Notes and references
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- S. Roy, T.-A. D. Nguyen, L. Gan and A. K. Jones, Dalton Trans., 2015, 44, 14865–14876 RSC.
- A. K. Jones, B. R. Lichtenstein, A. Dutta, G. Gordon and P. L. Dutton, J. Am. Chem. Soc., 2007, 129, 14844–14845 CrossRef CAS PubMed.
- A. Roy, C. Madden and G. Ghirlanda, Chem. Commun., 2012, 48, 9816–9818 RSC.
- V. Artero, G. Berggren, M. Atta, G. Caserta, S. Roy, L. Pecqueur and M. Fontecave, Acc. Chem. Res., 2015, 48, 2380–2387 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- L. Kertess, F. Wittkamp, C. Sommer, J. Esselborn, O. Rudiger, E. J. Reijerse, E. Hofmann, W. Lubitz, M. Winkler, T. Happe and U. P. Apfel, Dalton Trans., 2017, 46, 16947–16958 RSC.
- C. F. Megarity, J. Esselborn, S. V. Hexter, F. Wittkamp, U.-P. Apfel, T. Happe and F. A. Armstrong, J. Am. Chem. Soc., 2016, 138, 15227–15233 CrossRef CAS PubMed.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Biochemistry, 2015, 54, 1474–1483 CrossRef CAS PubMed.
- C. Esmieu, P. Raleiras and G. Berggren, Sustainable Energy Fuels, 2018, 2, 724–750 RSC.
- J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, Joule, 2017, 1, 61–76 CrossRef CAS.
- C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. D. Gioia, S. C. Davies, X. Yang, L.-S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610–613 CrossRef CAS PubMed.
- S. E. Mulholland, B. R. Gibney, F. Rabanal and P. L. Dutton, J. Am. Chem. Soc., 1998, 120, 10296–10302 CrossRef CAS.
- B. R. Gibney, S. E. Mulholland, F. Rabanal and P. L. Dutton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15041–15046 CrossRef CAS.
- E. M. Shepard, B. R. Duffus, S. J. George, S. E. McGlynn, M. R. Challand, K. D. Swanson, P. L. Roach, S. P. Cramer, J. W. Peters and J. B. Broderick, J. Am. Chem. Soc., 2010, 132, 9247–9249 CrossRef CAS PubMed.
- A. Silakov, C. Kamp, E. Reijerse, T. Happe and W. Lubitz, Biochemistry, 2009, 48, 7780–7786 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 9476–9477 CrossRef CAS PubMed.
- C. Esmieu and G. Berggren, Dalton Trans., 2016, 45, 19242–19248 RSC.
- M. Razavet, S. C. Davies, D. L. Hughes and C. J. Pickett, Chem. Commun., 2001, 847–848 RSC.
- M. Razavet, S. J. Borg, S. J. George, S. P. Best, S. A. Fairhurst and C. J. Pickett, Chem. Commun., 2002, 700–701 RSC.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- M. Lundberg and P. E. M. Siegbahn, J. Chem. Phys., 2005, 122, 224103 CrossRef PubMed.
- S. Roy, M. Bacchi, G. Berggren and V. Artero, ChemSusChem, 2015, 8, 3632–3638 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence, M. Schmidt, S. R. Wilson and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 12518–12527 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details on CO release and H2 production assays and computational and experimental methods. See DOI: 10.1039/c8dt05085f |
| ‡ Current address: CNRS, LCC (Laboratoire de Chimie de Coordination), 205 route de Narbonne, BP 44099 31077 Toulouse cedex 4, France. |
| This journal is © The Royal Society of Chemistry 2019 |