
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New Ag8PtO6: synthesis, crystal structure, physical properties and theoretical analyses†
Gohil Singh
Thakur
 a,
Hans
Reuter
b,
Helge
Rosner
a,
Gerhard H.
Fecher
a,
Claudia
Felser
a and
Martin
Jansen
*ac
a,
Hans
Reuter
b,
Helge
Rosner
a,
Gerhard H.
Fecher
a,
Claudia
Felser
a and
Martin
Jansen
*ac
aMax-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Straße 40, 01187 Dresden, Germany
bInstitut für Chemie neuer Materialien, Universität Osnabrück, Barbarastraße 7, 49069 Osnabrück, Germany
cMax-Planck-Institut für Festkörperforschung, Heisenbergstr. 1, 70569 Stuttgart, Germany. E-mail: M.Jansen@fkf.mpg.de
First published on 18th March 2019
Abstract
We report the synthesis, crystal structure, and basic physical properties of Ag8PtO6, which represents the first silver platinum ternary oxide. The crystalline compound was obtained from appropriate mixtures of the binary constituents under alkaline conditions at high oxygen pressure, while applying relatively mild thermal conditions (573 K). Ag8PtO6 crystallizes in a new crystal structure in the triclinic system (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). The structure consists of slightly distorted, discrete PtO6 octahedra, which are linked via O–Ag–O dumbbells to form a three dimensional framework. It is a diamagnetic semiconductor with a band gap of 0.9 eV. DFT based calculations confirm an electronic ground state that corresponds to a 5d6 6s0 configuration of the Pt atoms, in accordance with the observed diamagnetism.
). The structure consists of slightly distorted, discrete PtO6 octahedra, which are linked via O–Ag–O dumbbells to form a three dimensional framework. It is a diamagnetic semiconductor with a band gap of 0.9 eV. DFT based calculations confirm an electronic ground state that corresponds to a 5d6 6s0 configuration of the Pt atoms, in accordance with the observed diamagnetism.
Introduction
The terrestrial abundance of the platinum group elements (PGEs) is quite low. However, this class of transition metals continues to play a prominent role in science and technology.1 Alloys based on such constituents are among the thermally and chemically most durable metallic structural materials.2 Catalysts, with PGEs as constituents, employed in large scale chemical production,3 for exhaust gas purification4 or in fuel cells5 are legendary, not to mention cis-platinum complexes in use as cancer drugs.6,7 In science, e.g., complexes of PGE helped to bring coordination chemistry as a new sub-discipline of inorganic chemistry to birth,8 or the so-called Krogmann's salts9 attracted considerable attention during at least two decades in the second half of the last century. In current condensed matter research, discovery of exotic electronic phases prevailing in certain PGE oxides has stimulated vibrant activity. Here, as a common thread, Hund's coupling, spin orbit coupling (SOC), crystal field (CF) splitting, hopping integrals, and electron correlations (UH) are energetically on a comparable scale, thus giving rise to diverse uncommon electronic and/or magnetic ground states.10,11Most of the phases so far studied represent ternary oxides of PGEs and group I and II elements. Monovalent cations of group XI, Cu1+ and Ag1+, which are known to interrupt or at least to weaken magnetic exchange paths, have attracted much less interest, yet. Aiming at keeping the electronic effects mentioned above as localized as possible by suppressing long range hopping, we started to explore the ternary systems spanned by silver(I) oxides and PGEs.
Here we report on Ag8PtO6, the first silver oxo-platinate, its crystal structure and basic physical properties. In our computational analyses, we place particular emphasis on the nature of the electronic ground state, which one would conventionally associate with a low spin 5d6, t2g6 configuration. However, due to relativistic contraction of the 6s orbitals, states derived from the 6s2, 5d4 (t2g4) configuration might be lower in energy and give rise to a Jeff = 0 scenario, as encountered e.g. with Ir5+ oxides.12 Applying density functional band structure calculations, we can clearly assign a 5d6 6s0 configuration to the Pt atoms, consistent with a non-magnetic state.
Experimental section
Single crystal synthesis
Single crystals of Ag8PtO6 were prepared by reacting Ag2O and PtO2, 0.257 g and 0.0631 g, respectively, as a typical batch composition, in an alkaline medium at 573 K and under high oxygen pressure in a steel autoclave. Ag2O was pre-synthesized by precipitating from an aqueous solution of AgNO3 with an excess of NaOH. PtO2 was used as purchased (Sigma Aldrich, 99.9%) without any further purification. Well ground mixtures of the oxides were placed in a gold crucible of ∼4 cm height, welded from one end and mechanically crimped from the other. About 1.5 ml of 3 M KOH was used as a mineralizer to promote crystal growth. The crucible was placed in a 20 ml stainless steel autoclave13 and about 11.2 ml of liquid oxygen was condensed in it. The gas-tight sealed autoclave was heated at 573 K in a vertical furnace for 96 hours and cooled slowly to room temperature in about 24 hours. The O2 pressure generated in the autoclave at this temperature is estimated to be around 250 MPa. Large corrugated plate-like shiny black single crystals of maximum length ∼1.2 mm (see Fig. 1) were obtained along with small amounts of tiny polygonal crystals of Ag2O. The crystals were easily handpicked and sonicated in ethanol to remove any surface impurities. The crystalline product is insensitive to air and moisture. | ||
| Fig. 1 Photographs of few single crystals of Ag8PtO6, left: on a millimeter grid. | ||
Instruments for physical property measurements
Thermogravimetric (TGA) analysis of Ag8PtO6 was carried out on a Netzsch STA 449 C analyzer. About 30 mg of the sample was placed in a corundum crucible, which was heated and subsequently cooled at a rate of 5 K min−1 in the range of 300–1273 K under dynamic argon flow.Susceptibility of crushed single crystals was measured in applied magnetic fields up to μ0H = 7 T and in the temperature range between 2 and 400 K in a MPMS-XL7 magnetometer (Quantum Design).
Temperature dependent resistivity was measured on a plate-like single crystal of dimension 0.6 × 0.15 × 0.025 mm3 by a two-probe method in a PPMS instrument (Quantum Design).
Structure determination and refinement
A black plate-like crystal fragment of appropriate size was obtained by cutting a larger crystal under a microscope. It was fixed on a 50 micron Micromesh MiTeGen Micromount using FROMBLIN Y perflouropolyether (LVAC 16/6, Sigma-Aldrich). Diffraction data were collected at ambient temperature on a Bruker Kappa APEX II CCD-based 4-circle X-ray diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å) of a fine focus molybdenum target X-ray tube operating at 50 kV and 30 mA. Collection and reduction of data were carried out with the Bruker Suite software package.14 Intensities were corrected for absorption effects semi-empirically from equivalents via SADABS.15 The structure was solved by direct methods and refined by full-matrix least-squares fitting on F2 with the SHELXL software package.16,17 Figures were drawn using DIAMOND.18Crystallographic data, details of data collection and structure refinement are given in Table 1, atomic coordinates, and equivalent isotropic displacement parameters in Table 2, bond lengths in Table S1† and anisotropic displacement parameters Table S2 in the ESI.†
| Formula | Ag8O6Pt |
|---|---|
| Fw | 1154.05 (g mol−1) |
| T/K | 296(2) |
| λ/Å | 0.71073 |
| Space group |
P |
| a/Å | 6.1911(2) |
| b/Å | 6.9665(2) |
| c/Å | 10.3901(3) |
| α/° | 87.396(1) |
| β/° | 85.152(1) |
| γ/° | 71.751(1) |
| V/Å3 | 423.98(2) Å3 |
| Z | 2 |
| Calc. d/g cm−3 | 9.04 |
| μ(Mo-Kα)/mm−1 | 34.471 |
| F(000) | 1004 |
| Crystal size | 0.167 × 0.067 × 0.035 mm |
| Theta range for data collection | 3.079° to 29.991°. |
| Reflections collected/unique | 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 196/2467 [R(int) = 0.0495] 196/2467 [R(int) = 0.0495] |
| Completeness to theta = 25.242 | 99.9% |
| Absorption correction | Semi-empirical from equivalents |
| Refinement method | Full-matrix least squares on F2 |
| Data/restraints/parameters | 2467/0/140 |
| R 1/wR2 [all data] | 0.0135/0.0282 |
| R 1/wR2 [I > 2σ(I)] | 0.0137/0.0283 |
| Extinction coefficient | 0.00371(6) |
| Largest diff. peak/hole | 1.096/−1.067 e/Å3 |
| Atom | x | y | z | U(eq) |
|---|---|---|---|---|
| Ag(1) | 5000 | 0 | 5000 | 4(1) |
| Ag(2) | 1528(1) | 3618(1) | 4037(1) | 5(1) |
| Ag(3) | 4589(1) | 7214(1) | 388(1) | 6(1) |
| Ag(4) | 3812(1) | 6666(1) | 4016(1) | 6(1) |
| Ag(5) | 1585(1) | 4657(1) | 1343(1) | 5(1) |
| Ag(6) | −1482(1) | 7639(1) | 3046(1) | 6(1) |
| Ag(7) | −1378(1) | 8746(1) | 311(1) | 5(1) |
| Ag(8) | 0 | 0 | 5000 | 5(1) |
| Ag(9) | 3052(1) | 10319(1) |
2278(1) | 5(1) |
| Pt(1) | 7132(1) | 2898(1) | 2458(1) | 3(1) |
| O(1) | 5185(5) | 2617(4) | 4074(3) | 5(1) |
| O(2) | 8931(5) | 3463(4) | 870(3) | 6(1) |
| O(3) | 4480(5) | 5361(4) | 2052(3) | 6(1) |
| O(4) | 8087(5) | 4724(4) | 3578(3) | 5(1) |
| O(5) | 6038(5) | 1025(4) | 1418(3) | 6(1) |
| O(6) | 9615(5) | 356(4) | 2952(3) | 6(1) |
Further details of the crystal structure investigation may be obtained from FIZ Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: (+49)7247-808-666; email: crysdata(at)fiz-karlsruhe(dot)de, on quoting the deposition number CSD 434624. During all final calculations, the lattice parameters as obtained from X-ray powder pattern refinement were used.
Laboratory powder X-ray diffraction (PXRD) data (Fig. 2) were collected at room temperature on a HUBER G670 imaging plate Guinier camera with Cu-Kα1 radiation (λ = 1.5406 Å), in a 2θ range of 10–80 degrees. Rietveld refinement and Le Bail fitting were carried out with the TOPAS-4.2.0.2 (AXS) program.19 The refined parameters were the scale factor, zero point of θ, sample displacement (mm), background as a Chebyshev polynomial of 20th degree, 1/x function, crystallite size, micro-strain and cell constants.
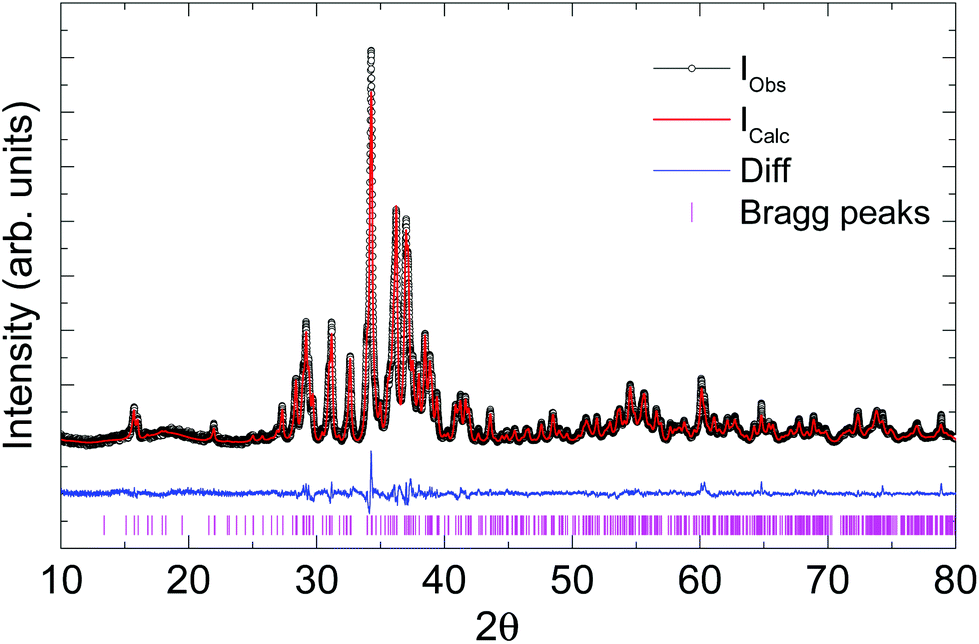 | ||
| Fig. 2 Le Bail fit of the powder X-ray diffraction data of Ag8PtO6 collected at 300 K. | ||
Theoretical calculations
Relativistic density functional (DFT) electronic structure calculations were performed using the full-potential FPLO code, version fplo18.00-52.20 For the exchange–correlation potential, within the general gradient approximation (GGA), the parametrization of Perdew–Burke–Ernzerhof was chosen.21The spin–orbit (SO) coupling was treated non-perturbatively solving the four component Kohn–Sham–Dirac equation.22 To obtain precise band structure information, the calculations were carried out on a well converged mesh of 600 k-points (10 × 10 × 6).
To test the reliability of the calculations, the internal positions of the atoms were optimized by minimizing forces with remaining forces smaller than 0.05 eV Å−1. For the shown electronic density of states, (DOS) the experimental crystal structure has been used.
Results and discussion
New Ag8PtO6 has been obtained from the binary constituent oxides under hydrothermal conditions, applying an elevated oxygen pressure of 250 MPa, however, rather mild thermal conditions of 573 K. It starts decomposing at about 673 K, loosing oxygen in a few steps and being completely reduced upon heating to 873 K, see Fig. S1 in the ESI.† The total weight loss is about 8.5% corresponding to complete loss of oxygen. The remaining product is a mixture of the constituent metals Ag and Pt.The title compound crystallizes in a triclinic system with space group P, cf.Tables 1 and 2. The unique crystal structure consists of discrete, virtually ideal PtO6 octahedra and linear, or slightly bent, respectively, AgO2 dumbbells (Fig. 3). The primary PtO6 structural units are linked by silver atoms in twofold coordination to form a three-dimensional framework structure. Oxygen is found in two different structural functions, μ3 and μ4, connecting either one platinum atom with two (O2 and O4) or three (O1, O3, O5, and O6) silver atoms, respectively. As a result, each PtO6 octahedron is connected to nine other surrounding octahedra through sixteen O–Ag–O linkages (Fig. 4).
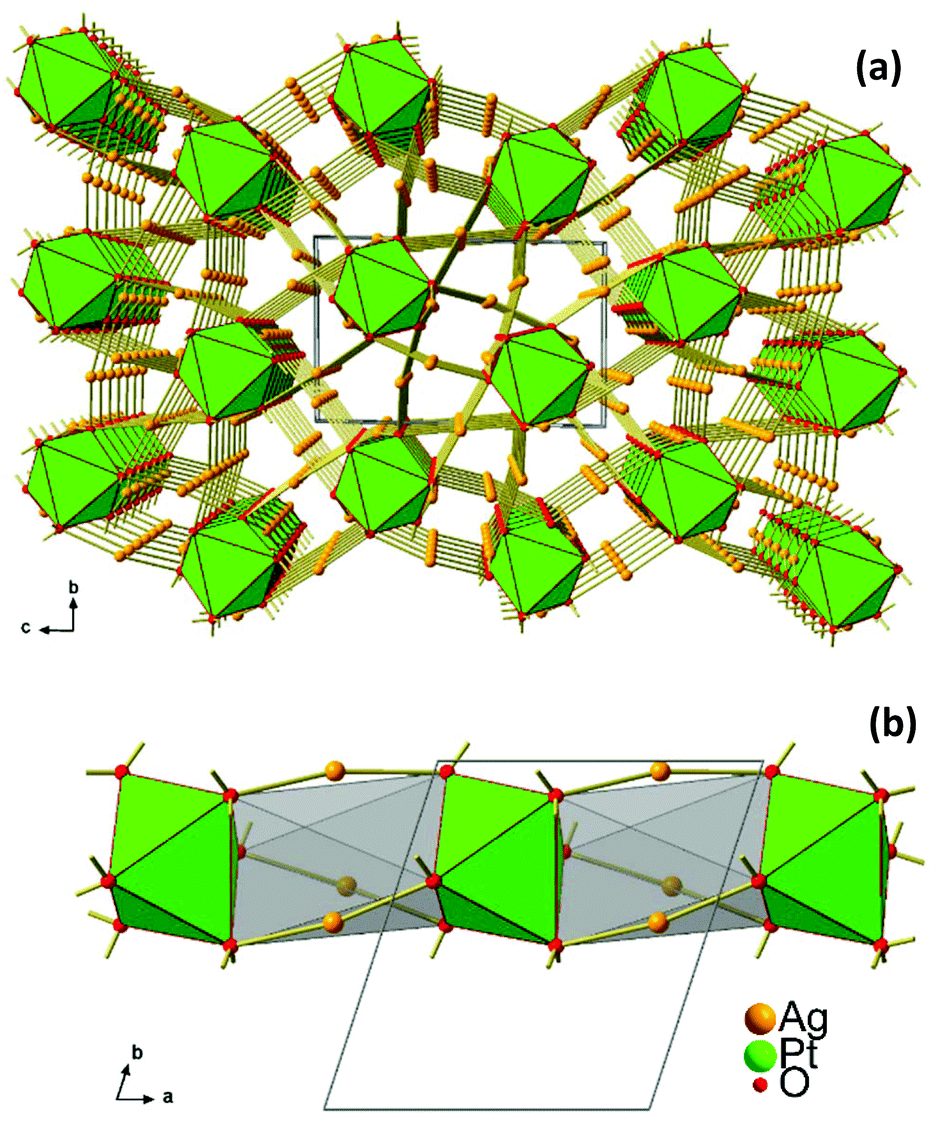 | ||
| Fig. 3 (a) Crystal structure of Ag8PtO6 viewed along the a-axis, (b) 1D stack of PtO6 octahedra extending along [100]. | ||
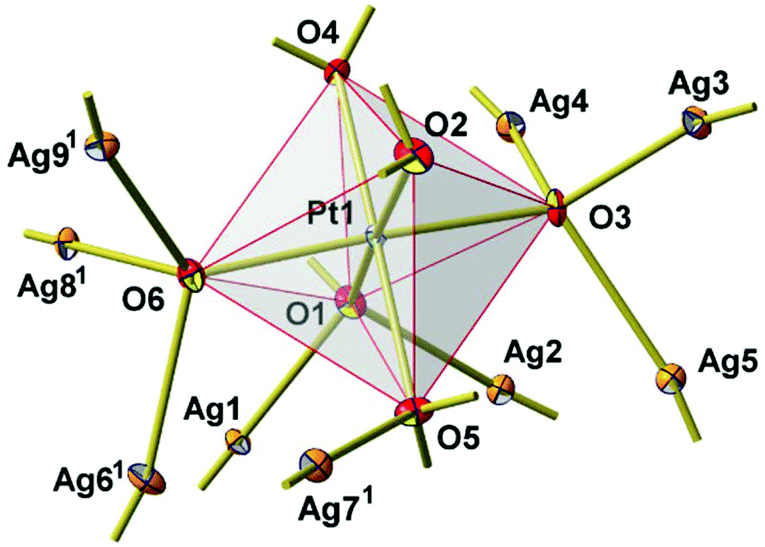 | ||
| Fig. 4 Primary PtO6 structural unit with connected AgO2 dumbbells. All atoms are drawn as thermal displacement ellipsoid at the 40% probability level. Symmetry code to generate equivalent atoms: (1) 1 + x, −1 + y, z. | ||
The Ag–O bond lengths range from 2.04 to 2.26 Å (Table S1†) and agree well with the values reported in the literature for Ag–O bonds.23–25 The Pt–O bond lengths vary in the narrow range of 1.99 to 2.03 Å (Table S1†) which are also in excellent agreement with other reported oxides with Pt in +4 oxidation.26–28 The shortest Ag–Ag distance is 2.85 Å (Ag3–Ag5) which is slightly shorter than that of the nearest neighbour distances in Ag metal (2.89 Å); this separation, and in addition those extending up to 2.95 Å, are indicative of the presence of weakly bonding d10–d10 interactions.29 At a first glance, the structure appears to be rather open; however, the calculated free volume amounts to only 11.8 Å3 or 8%.30
Interestingly, in spite of the underlying overall three dimensional connectivity pattern some eye catching one dimensional structural anisotropy is becoming apparent when viewing the crystal structure in the [100] direction, see Fig. 3a. In this perspective the octahedra appear to be stacked in a staggered fashion, like a column excised from a hcp packing of oxygen atoms where every second octahedral void is centred by platinum, while the empty ones are bridged by silver atoms, see Fig. 3b. This particular microscopic feature seems to reflect the fibrous appearance of the crystals’ surface.
Ag8PtO6 is semiconducting with an activated type behavior in the temperature range 400 K to 280 K with room temperature resistivity of the order of 50 kΩ cm (Fig. 5a). The Arrhenius fit in the high temperature region yields a band gap of 0.89 eV, which falls in line with the observed semiconducting nature of the sample.
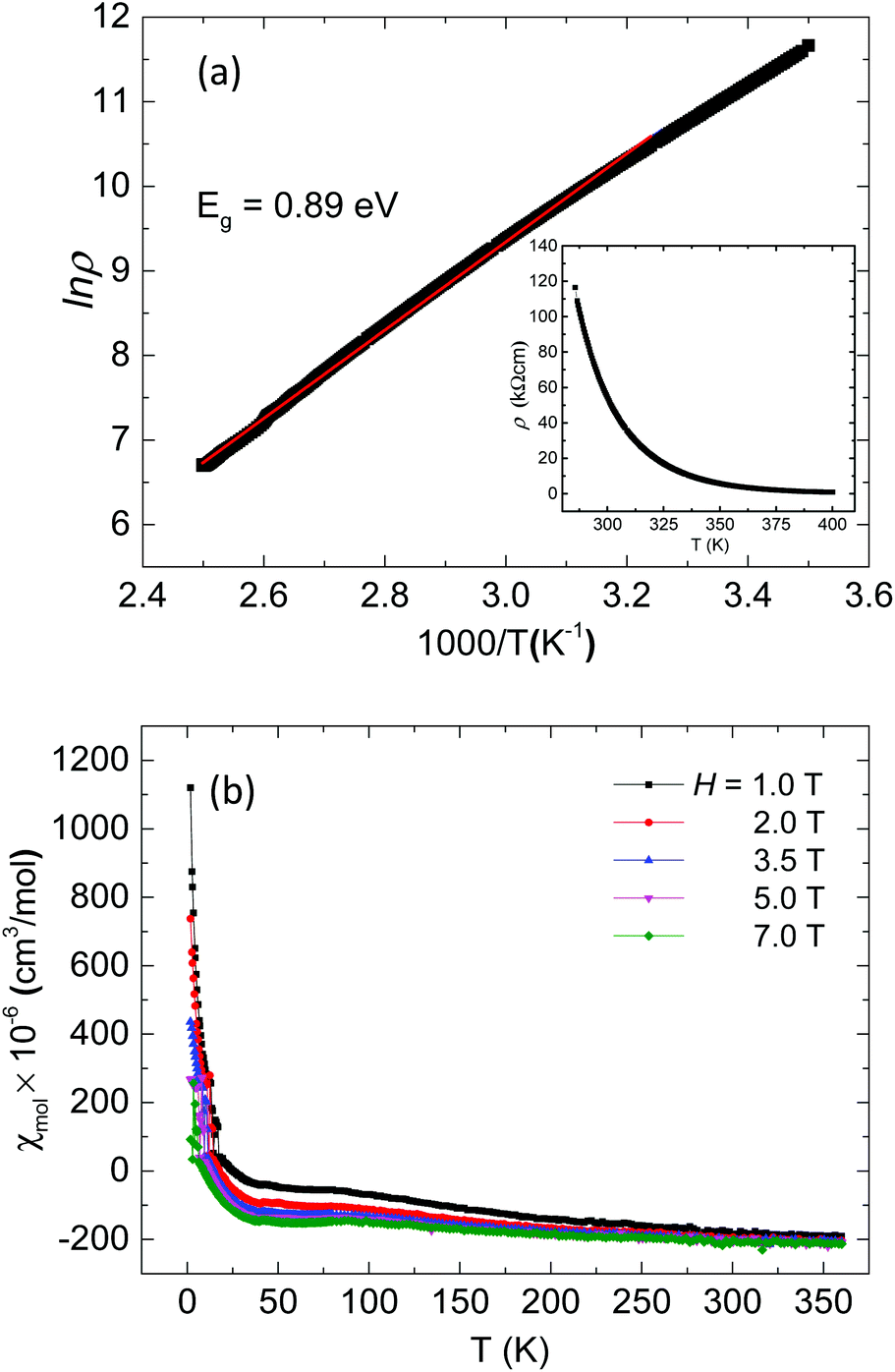 | ||
| Fig. 5 (a) Arrhenius plot of resistivity of Ag8PtO6 with variable temperature resistivity in the inset (b) variable temperature susceptibility of Ag8PtO6 at various applied magnetic fields. | ||
The compound is found to be diamagnetic in the entire temperature region studied from 360 K to 2 K, (Fig. 5b). The susceptibility is negative and temperature independent in the high temperature region from 360 to 50 K, below which a weak field dependent Curie–Weiss like behavior is observed, which is due to the paramagnetic contribution of trace molecular oxygen impurity, which corresponds to a total of 0.7% of a spin ½ species. The value of χ∞ estimated from the extrapolation of Honda–Owen plots to infinite field comes out to be −224 × 10−6 emu mol−1. The diamagnetic behavior is consistent with +4 oxidation state of Pt with all d-electrons paired as implied from the empirical formula.
In order to discriminate between the two possible diamagnetic ground states, as can be derived from the configurations 5d6 (t2g6), S = 0 and 6s2, 5d4 (t2g4), Jeff = 0, density functional band structure calculations have been performed. In a first step, we optimized the internal coordinates of the atomic positions with respect to the total energy, assuming the experimentally determined lattice parameters. As a result, we find a surprisingly good agreement of the experimentally refined and the calculated atomic positions, and changes of the Ag–O and Pt–O bond lengths are about 0.01 Å, only. The corresponding difference in total energy between both parameter sets yields 2.5 meV per atom, corresponding to a temperature of about 30 K. These findings provide independent strong support to the structural parameters from the XRD refinement.
Our calculations reveal that Ag8PtO6 is a narrow band gap semiconductor. The calculated band gap has a size of about 150 meV. No instability towards magnetism was found. These findings are consistent with the experimental observations. The experimentally observed gap of 0.9 eV is, however, significantly larger than the calculated one. This is in line with the usual underestimation of the electronic gap size applying standard DFT functionals.
The calculated electronic density of states is shown in Fig. 6 (upper panel). The valence band has a width of about 8 eV, typical of this type of oxide system. The density of states is dominated by the silver 4d bands that are concentrated in the energy range from −6 to −2 eV. The oxygen states are distributed rather uniformly across the complete valence bands. A more detailed analysis shows that these states are of rather different character. The states below −6 eV are bonding states derived from the Pt–O octahedra. Their counterpart is separated by a large covalency split of about 5 eV between −4 eV and the Fermi level at zero energy. The Pt states of the valence band are almost exclusively of 5d character (see Fig. 6, lower panel). They are found in three groups: at the bottom of the valence band below −6 eV, in the middle of the valence band, and close to the Fermi energy above −1.5 eV. The low and high lying states are hybridized with the oxygen states, originating from the covalent PtO6 octahedra, whereas the states between −3.5 and −2 eV fall in the energy range where the majority of Ag 4d-states are located.
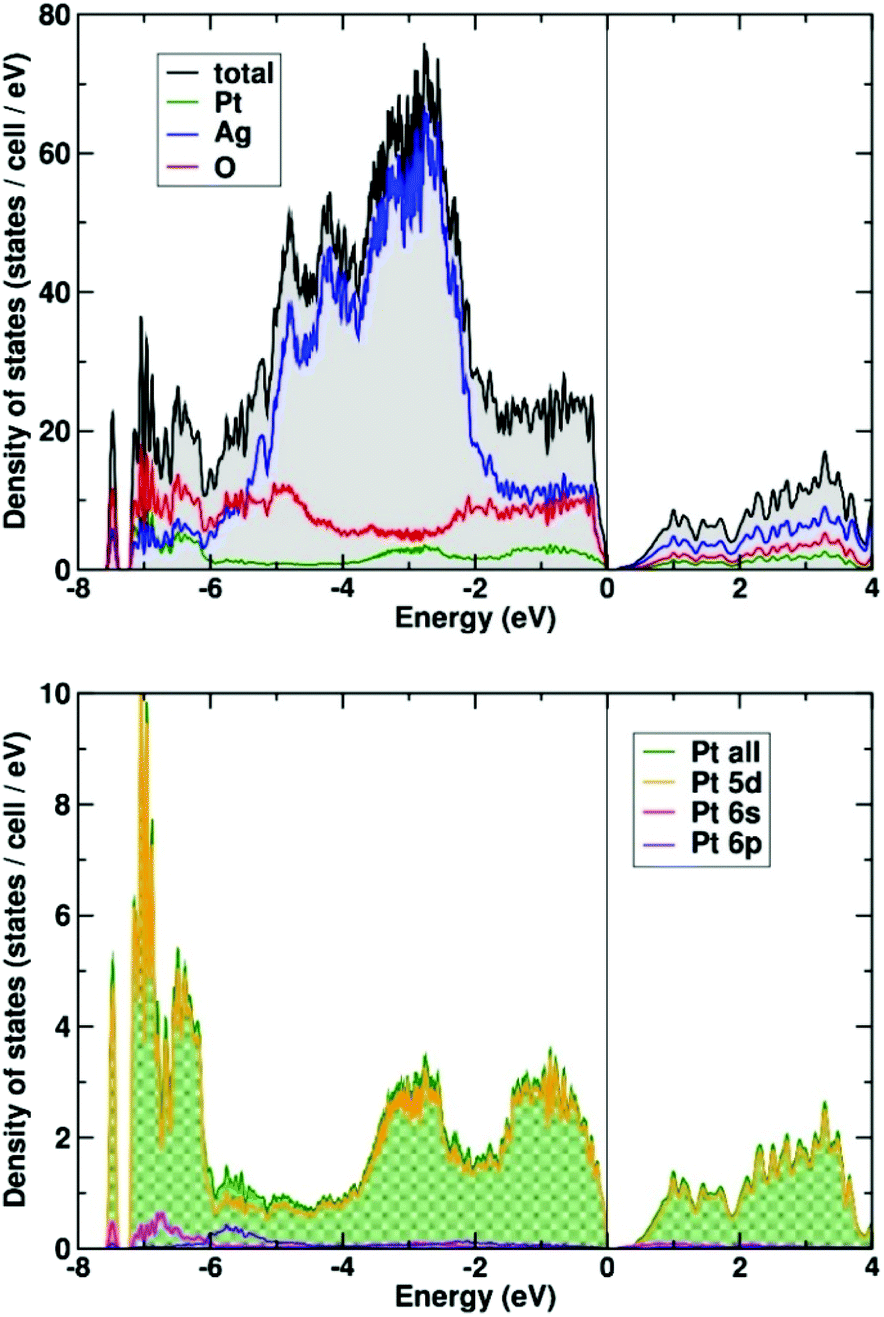 | ||
| Fig. 6 Total and partial electronic density (DOS) of states of Ag8PtO6 (upper panel). The lower panel shows the orbital decomposed DOS of the Pt atom. | ||
A small amount of Pt 6s (and 6p) states is found at the bottom of the valence band (see Fig. 6, lower panel). A population analysis shows that 6s states are occupied by about 0.15 electrons (on-site contribution). The majority of (unoccupied) Pt 6s states appear about 10 eV above the Fermi energy (not shown here).
The electronic ground state of Pt in Ag8PtO6 is 5d6, 6s0 in very good approximation. Thus, the findings are clearing the suspicion that the 6s might have submerged below the 5d states, as was found earlier for Cs2Pt31 and other platinides.32
Conclusions
A new silver-platinate, Ag8PtO6, has been synthesized as single crystals. This ternary oxide is inaccessible via solid state synthesis or solution chemistry, unless elevated oxygen pressure is applied, sufficiently high to suppress decomposition of the binary starting oxides. This finding is accentuating again the great potential of oxygen high pressure techniques for the synthesis of multinary oxide materials based on noble metals, be it in an explorative or purposeful approach. Such advanced preparation techniques have opened an entrance to a widely unexplored area of the energy landscape of chemical matter33 spanned e.g. by the oxides of PGEs and coinage metals. Compounds made of appropriate combinations of noble metals are offering interesting prospects for basic research as well as for applications. The title compound will for sure show activity as a heterogeneous catalyst, yet, its pertinent performance in comparison with catalysts actually in use needs to be checked.The title compound crystalizes in a triclinic unit cell, and represents a new structure type. The structure consists of isolated PtO6 octahedra connected by linear and/or slightly bent Ag–O linkages. It is found to be a diamagnetic semiconductor with an experimental band gap of ∼0.9 eV. Band structure calculations clearly assign a 5d6 6s0 state to the Pt atom, consistent with its non-magnetic state.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Walter Schnelle and Dr Reinhard Kremer for the magnetic measurements. Open Access funding provided by the Max Planck Society.References
- Gmelin Handbook of Inorganic and Organometallic Chemistry - 8th edition, Supplement Volume A 1 Technology of Platinum-Group Metals, ed. G. J. K. Acres and K. Swars, Springer, Berlin Heidelberg, 1985 Search PubMed.
- A. S. Darling, Platinum Metals Rev., 1960, 4, 18–26 CAS.
- J. E. Perea Buceta, I. Fernández, S. Heikkinen, K. Axenov, A. W. T. King, T. Niemi, M. Nieger, M. Leskelä and T. Repo, Angew. Chem., Int. Ed., 2015, 54, 14321–14325 CrossRef CAS PubMed.
- H. Hirata, Catal. Surv. Asia, 2014, 18, 128–133 CrossRef CAS.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- CISPLATIN – Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, WILEY-VCH, Weinheim, 1999 Search PubMed.
- L. H. Gade, Z. Anorg. Allg. Chem., 2012, 638, 247–248 CrossRef CAS.
- H. Wagner, M. P. Geserich, R. V. Baltz and K. Krogmann, Solid State Commun., 1973, 13, 659–663 CrossRef CAS.
- Y. Tokura and N. Nagaosa, Science, 2000, 288, 462–468 CrossRef CAS PubMed.
- S. V. Streltsov and D. I. Khomskii, Phys.-Usp., 2017, 60, 1121 CrossRef CAS.
- B. E. Prasad, Th. Doert, C. Felser and M. Jansen, Chem. – Eur. J., 2018, 24, 16762–16765 CrossRef CAS PubMed.
- C. Linke and M. Jansen, Z. Anorg. Allg. Chem., 1997, 623, 1441–1446 CrossRef CAS.
- Bruker Suite, version 2013/1, Bruker AXS Inc., Madison, WI, 2013 Search PubMed.
- G. M. Sheldrick, SADABS —Bruker AXS area detector scaling and absorption, version 2012/1, University of Göttingen, Germany, 2012 Search PubMed.
- G. M. Sheldrick, Short History of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Brandenburg, Diamond-Crystal and Molecular Visualization, Crystal Impact, Bonn, Germany, 2006 Search PubMed.
- TOPAS (version 4.2.0.2), General Profile and Structure Analysis Software for Powder Diffraction Data, Bruker AXS GmbH, Karlsruhe (Germany), 2012 Search PubMed.
- K. Koepernik and H. Eschrig, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1743–1757 CrossRef CAS; I. Opahle, K. Koepernik and H. Eschrig, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14035–14040 CrossRef http://www.fplo.de .
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. Eschrig, M. Richter and I. Opahle, Relativistic Solid State Calculations, in Relativistic Electronic Structure Theory, (Part II, Applications), ed. P. Schwerdtfeger, Theoretical and Computational Chemistry, Elsevier, 2004, vol. 13, p. 723 Search PubMed.
- S. Deibele, J. Curda, E.-M. Peters and M. Jansen, Chem. Commun., 2000, 679–680 RSC.
- J. Curda, W. Klein and M. Jansen, J. Solid State Chem., 2001, 162, 220–224 CrossRef CAS.
- S. Deibele and M. Jansen, J. Solid State Chem., 1999, 147, 117–121 CrossRef CAS.
- W. Urland and R. Hoppe, Z. Anorg. Allg. Chem., 1972, 392, 23–36 CrossRef CAS.
- J. J. Randall and L. Katz, Acta Crystallogr., 1959, 12, 519–521 CrossRef CAS.
- P. Kroeschell, R. Wolf and R. Hoppe, Z. Anorg. Allg. Chem., 1986, 536, 81–91 CrossRef CAS.
- M. Jansen, Angew. Chem., 1987, 99, 1136–1149 ( Angew. Chem., Int. Ed. Engl. , 1987 , 26 , 1098–1110 ) CrossRef CAS.
- Mercury CSD 2.0 - New Features for the Visualization and Investigation of Crystal Structures: C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. S. Karpov, J. Nuss, U. Wedig and M. Jansen, Angew. Chem., 2003, 115, 4966–4969 ( Angew. Chem., Int. Ed. Engl. , 2003 , 42 , 4818–4821 ) CrossRef.
- A. S. Karpov, J. Nuss, U. Wedig and M. Jansen, J. Am. Chem. Soc., 2004, 126, 14123–14128 CrossRef CAS PubMed.
- M. Jansen, Angew. Chem., 2002, 114, 3896–3917 ( Angew. Chem., Int. Ed. Engl. , 2002 , 41 , 3746–3766 ) CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt05081c |
| This journal is © The Royal Society of Chemistry 2019 |