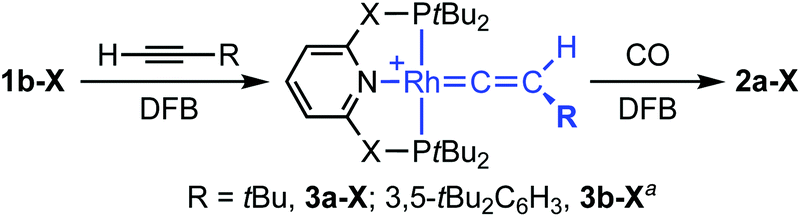DOI:
10.1039/C8DT05049J
(Communication)
Dalton Trans., 2019,
48, 2877-2880
A convenient method for the generation of {Rh(PNP)}+ and {Rh(PONOP)}+ fragments: reversible formation of vinylidene derivatives†
Received
21st December 2018
, Accepted 28th January 2019
First published on 7th February 2019
Abstract
The substitution reactions of [Rh(COD)2][BArF4] with PNP and PONOP pincer ligands 2,6-bis(di-tert-butylphosphinomethyl)pyridine and 2,6-bis(di-tert-butylphosphinito)pyridine in the weakly coordinating solvent 1,2-F2C6H4 are shown to be an operationally simple method for the generation of reactive formally 14 VE rhodium(I) adducts in solution. Application of this methodology enables synthesis of known adducts of CO, N2, H2, previously unknown water complexes, and novel vinylidene derivatives [Rh(pincer)(CCHR)][BArF4] (R = tBu, 3,5-tBu2C6H3), through reversible reactions with terminal alkynes.
Introduction
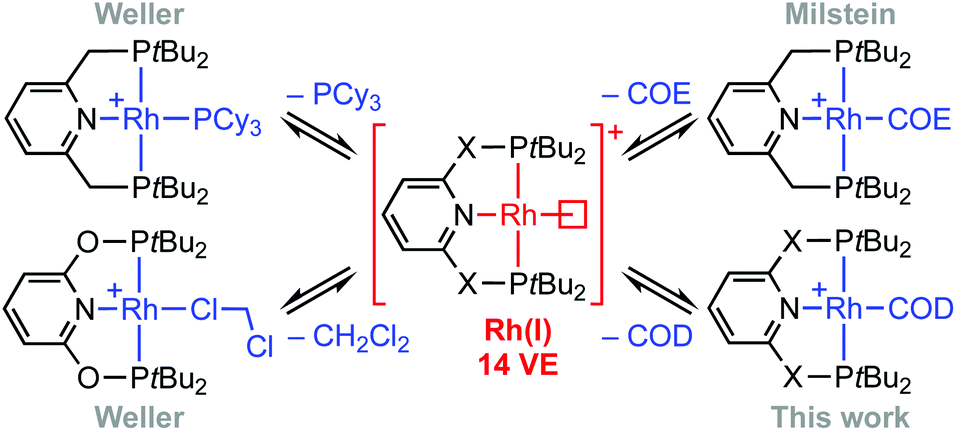
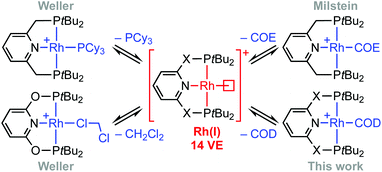
Phosphine-based pincers are a prominent ligand class in contemporary organometallic chemistry and catalysis, conferring thermal stability whilst permitting a wide range of metal-based reactivity.1,2 From a fundamental perspective, their capacity to support the generation of reactive low-coordinate metal fragments provides an attractive framework for gaining insights into the mechanism of small molecule activation reactions. The synthesis and characterisation of a rhodium(I) σ-methane complex in solution is a particularly outstanding example, with direct mechanistic relevance to C(sp3)–H bond activation and alkane dehydrogenation processes mediated at iridium homologues.2,3 Other group 9 highlights include the C(sp2)–H bond oxidative addition of aryl halides4,5 and the selective C(sp3)–F bond activation of fluorocarbons.6 In the context of supporting such endeavours and building on preceding work (Scheme 1),4,7–9 we herein report a convenient method for accessing reactive formally 14 VE rhodium(I) adducts of the widely studied PNP (2,6-bis(di-tert-butylphosphinomethyl)pyridine) and PONOP (2,6-bis(di-tert-butylphosphinito)pyridine) ligands, exploiting metastable coordination of 1,5-cyclooctadiene (COD) in the weakly coordinating 1,2-difluorobenzene solvent (DFB).10 We showcase this methodology for the preparation of previously unknown water complexes and the reversible formation of vinylidene derivatives.
 |
| | Scheme 1 Latent sources of the {Rh(PNP)}+ and {Rh(PONOP)}+ fragments (X = O, CH2). COE = cyclooctene; COD = 1,5-cyclooctadiene. | |
Results and discussion
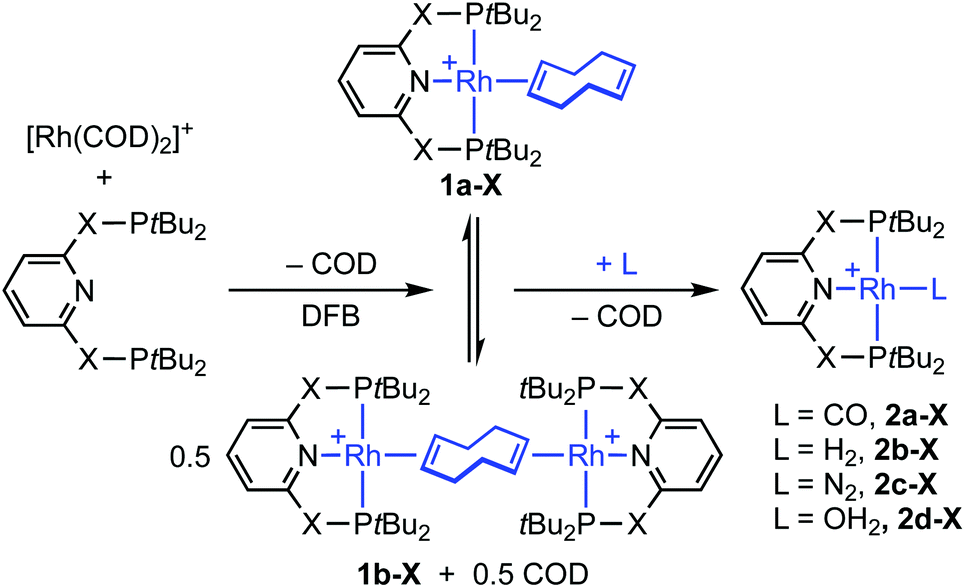
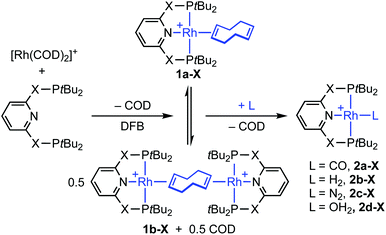
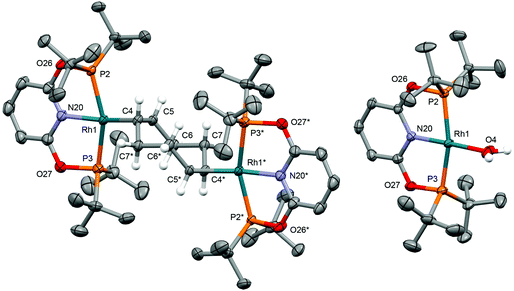
As an operationally simple entry point into the organometallic chemistry of cationic rhodium(I) pincer complexes, we targeted substitution reactions of the readily accessible (and commercially available) rhodium precursor [Rh(COD)2][BArF4] (ArF = 3,5-(CF3)2C6H3); postulating that alongside chelation of the pincer, steric buttressing from the phosphine substituents would promote efficient liberation of the comparatively bulky COD ligand and access to reactive {Rh(pincer)}+ fragments in solution.11 To this end, stoichiometric reactions between PNP/PONOP and [Rh(COD)2][BArF4] in DFB under an argon atmosphere were first studied in situ, resulting in quantitative coordination of the pincer and establishment of dynamic equilibrium mixtures containing monomeric 1a (PNP, δ31P 63.4, 52.6, 2JPP ∼320 Hz; PONOP, δ31P 202.5, 1JRhP = 134 Hz) as the major components and dimeric 1b as the minor components, alongside liberated COD (Scheme 2). The former were formed exclusively when the reactions were repeated in the presence of 10 equivalents of COD, while the latter dications were isolated in high yield by crystallisation from DFB/hexane (PNP, 90%; PONOP, 96%), structurally characterised in the solid state using X-ray diffraction and the bulk purity established by microanalysis (see Fig. 1 for 1b-O).† The synthetic utility of this methodology was then assessed by reacting mixtures of 1a/b, generated as described above in DFB, with CO, H2, and N2 (1 atm) and analysis in situ using 1H and 31P NMR spectroscopy. Introduction of strongly coordinating CO resulted in rapid (t < 5 min) and quantitative formation of the known carbonyl complexes 2a, with only free COD as the by-product.7,8 Reactions of H2 and N2 with 1a/b likewise afforded the corresponding (known) adducts 2b (with concomitant hydrogenation of COD) and 2c,7,8,12 respectively, but under disparately longer timeframes for the PONOP (t ∼ 2 days) compared to PNP complexes (t < 3 h). These reactions all proceeded to completion with the exception of 2c-O (ca. 95%), which instead required use of isolated 1b-O to presumably counter recoordination of COD (t ∼ 2 days).
 |
| | Scheme 2 Synthesis and reactivity of 1 (X = O, CH2). [BArF4]− counter anions omitted for clarity. | |
 |
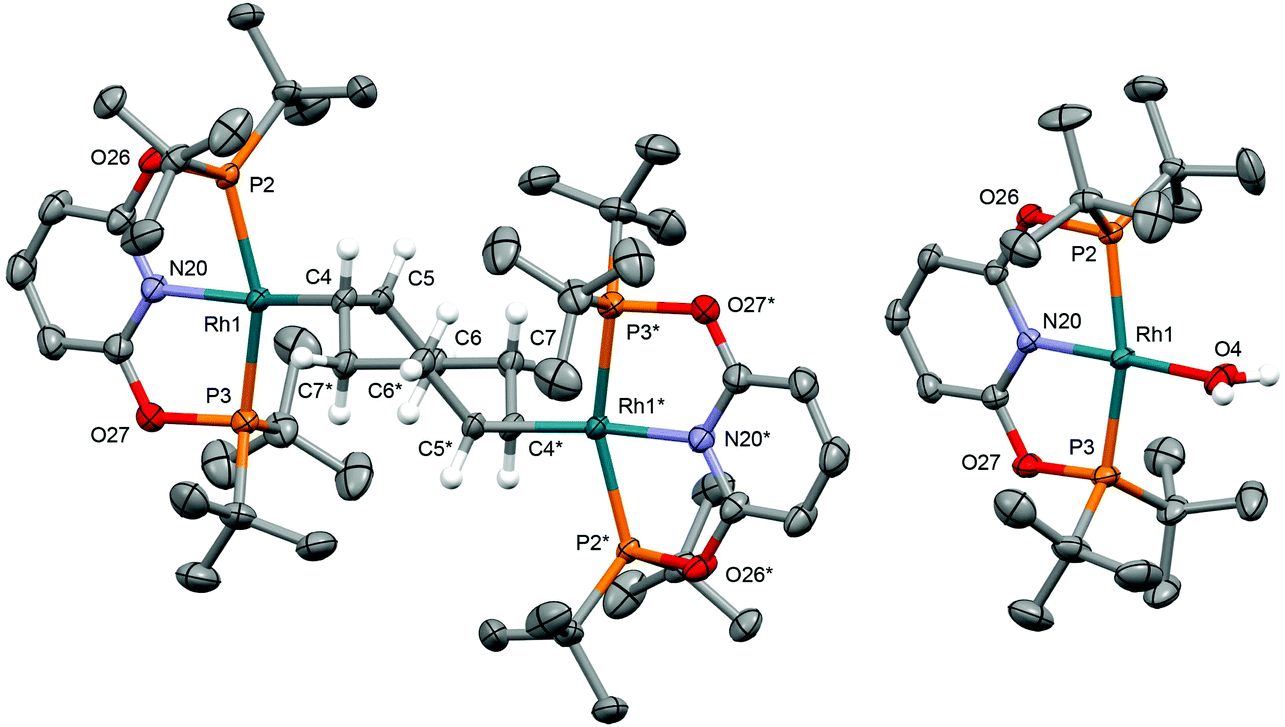
| | Fig. 1 Solid-state structures of 1b-O and 2d-O. Thermal ellipsoids drawn at 50% probability; anions and most hydrogen atoms omitted for clarity. The starred atoms are generated using the symmetry operation −x, 1 − y, 1 − z. Selected bond lengths (Å): 1b-O: Rh1–Cnt(C4,C5), 2.083(2); cf.1b-CH2, Rh1–Cnt(C4,C5), 2.058(2); 2d-O: Rh1–O4, 2.154(2); cf.2d-CH2, 2.152(7). | |
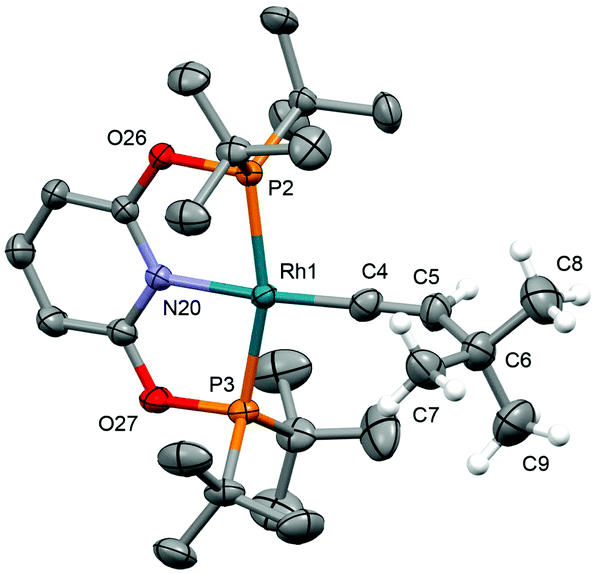
Application of our methodology allows access to novel rhodium(I) pincer adducts of water 2d. PNP variant 2d-CH2 (δ31P 63.1, 1JRhP = 144 Hz) was formed quantitatively on addition of excess degassed water (40 equiv.) to an equilibrium mixture of 1a/b-CH2 generated from stoichiometric reaction between [Rh(COD)2][BArF4] and PNP in DFB (t ∼ 3 h). Consistent with the aforementioned reactivity trends PONOP variant 2d-O (δ31P 199.1, 1JRhP = 150 Hz) was, however, only formed with 88% conversion after 3 days under these conditions. Analytically pure samples of the 2d-CH2 (71% yield) and 2d-O (49% yield) were obtained using 1a/b-CH2 or 1b-O, respectively, and excess water (500 equiv./Rh). The structural formulation of both new aqua complexes was confirmed using a combination of NMR spectroscopy, X-ray diffraction (see Fig. 1 for 2d-O) and satisfactory microanalysis. The solid-state structures of 2d are notable for and ![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) –
–![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) H2 bond lengths of ca. 2.15 Å and the presence of an additional hydrogen bonded water molecule in the lattice (RhH2⋯H2 = 2.7–2.8 Å).13
H2 bond lengths of ca. 2.15 Å and the presence of an additional hydrogen bonded water molecule in the lattice (RhH2⋯H2 = 2.7–2.8 Å).13
As a means of showcasing our new methodology and as part of our work exploring C(sp)–H activation reactions mediated by cationic rhodium(I) pincers,14 we set about studying the reactivity of the associated {Rh(pincer)}+ fragments with two bulky terminal alkynes (Scheme 3). Using isolated 1b-CH2 as the precursor, reaction with both HCCtBu and HCCAr′ (Ar′ = 3,5-tBu2C6H3) in DFB under an argon atmosphere resulted in rapid (t < 5 min) and quantitative formation of vinylidene derivatives 3a/b-CH2, the formation of which was readily apparent due to their striking deep green colours. Both complexes were subsequently isolated from solution and fully characterised in solution and the solid state.† The NMR spectra of 3a/b-CH2 are notable for the presence of vinylidene 13C resonances at δ 317.5/326.9 (located by HMBC experiments), time-averaged C2v symmetry – reconcilled by free rotation about the Rh![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC vector – and single phosphorus resonances with large coupling to 103Rh (δ31P 65.6/65.9, 1JRhP = 139/137 Hz). The solid-state structures affirm the structural assignments and, in addition to the RhC bond lengths of 1.81–1.84 Å that are fully inline with related rhodium precedents,15 notable for the adoption of both C2 (3a-CH2, Z′ = 2) and Cs (3a-CH2, Z′ = 2; 3b-CH2) symmetric pincer ligand conformations; demonstrating the flexibility of lutidene-based pincer backbones.16 In a similar manner, deep blue vinylidene complex 3a-O was obtained from HCCtBu and 1b-O, but over a more protracted time frame than the PNP analogue (ca. 18 h; δ31P 206.5, 1JRhP = 144 Hz, δ13C(RhC) 330.0, C2v solution symmetry; Fig. 2). Consistent with the aformetioned trends, the reaction between {Rh(PONOP)}+ and HCCAr′ only resulted in the formation of the green vinylidene derivative 3b-O (δ31P 208.4, 1JRhP = 143 Hz, δ13C(RhC) 339.5) as the minor component (30%) of a mixture with the balance made up by the corresponding π-complex [Rh(PONOP)(HCCAr′)][BArF4] (2e-O; δ31P 194.6. 1JRhP = 129 Hz).17 This ratio was found even in use of excess alkyne and the co-crystallisation of these two compounds (with the ratio reflected in the corresponding crystallographic occupancies) precluded separation in this manner.†
CC vector – and single phosphorus resonances with large coupling to 103Rh (δ31P 65.6/65.9, 1JRhP = 139/137 Hz). The solid-state structures affirm the structural assignments and, in addition to the RhC bond lengths of 1.81–1.84 Å that are fully inline with related rhodium precedents,15 notable for the adoption of both C2 (3a-CH2, Z′ = 2) and Cs (3a-CH2, Z′ = 2; 3b-CH2) symmetric pincer ligand conformations; demonstrating the flexibility of lutidene-based pincer backbones.16 In a similar manner, deep blue vinylidene complex 3a-O was obtained from HCCtBu and 1b-O, but over a more protracted time frame than the PNP analogue (ca. 18 h; δ31P 206.5, 1JRhP = 144 Hz, δ13C(RhC) 330.0, C2v solution symmetry; Fig. 2). Consistent with the aformetioned trends, the reaction between {Rh(PONOP)}+ and HCCAr′ only resulted in the formation of the green vinylidene derivative 3b-O (δ31P 208.4, 1JRhP = 143 Hz, δ13C(RhC) 339.5) as the minor component (30%) of a mixture with the balance made up by the corresponding π-complex [Rh(PONOP)(HCCAr′)][BArF4] (2e-O; δ31P 194.6. 1JRhP = 129 Hz).17 This ratio was found even in use of excess alkyne and the co-crystallisation of these two compounds (with the ratio reflected in the corresponding crystallographic occupancies) precluded separation in this manner.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18
18
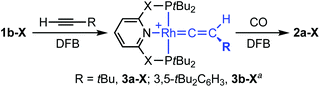 |
| | Scheme 3 Preparation of vinylidene complexes 3 (X = O, CH2). [BArF4]− counter anions omitted for clarity; a3b-O formed as mixture with the corresponding π-complex 2e-O. | |
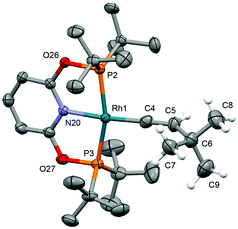 |
| | Fig. 2 Solid-state structure of 3a-O. Thermal ellipsoids drawn at 50% probability; anion, most hydrogen atoms and disordered component (vinylidene) omitted for clarity. Selected bond lengths (Å): 3a-O: Rh1–C4, 1.865(5); Rh1–C4A, 1.846(4); cf.3a-CH2, Rh–C, 1.826(5)–1.840(3); 3b-CH2, 1.812(2).18 | |
Curiously, we note reaction of isolated 3 (in the case of 3b-O, as a mixture with 2e-O) with CO (1 atm) resulted in complete conversion to 2a in all cases with concomitant liberation of the corresponding terminal alkynes (t < 90 min). Such reversible vinylidene formation is well documented in ruthenium complexes,19 and in line with related observations we have made when studying C(sp)–H bond activation reactions of cationic rhodium complexes of NHC-based pincer ligands.14
Conclusions
We have demonstrated that reaction between the readily accessible (and commercially available) rhodium precursor [Rh(COD)2][BArF4] and two phosphine-based pincer ligands (2,6-bis(di-tert-butylphosphinomethyl)pyridine and 2,6-bis(di-tert-butylphosphinito)pyridine) in 1,2-F2C6H4 provides a operationally simple method for generating the corresponding {Rh(pincer)}+ fragments, and subsequent synthesis of adducts of CO, H2, N2 and H2O (2). This methodology is underpinned by reversible binding of COD, that leads to dynamic equilibrium mixtures of monomeric [Rh(pincer)(η2-COD)][BArF4] 1a and dimeric [{Rh(pincer)}2(μ–η2:η2-COD)][BArF4]21b, the latter of which were readily isolated by recrystallisation. This methodology was used to study the reaction between {Rh(pincer)}+ and bulky terminal alkynes, which resulted in the (reversible) formation of novel vinylidene derivatives [Rh(pincer)(CCHR)][BArF4] (R = tBu, 3a; 3,5-tBu2C6H3, 3b).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313) and Royal Society (UF100592, UF150675, A. B. C.) for financial support. Crystallographic data for 3a-O were collected using an instrument purchased through support from Advantage West Midlands and the European Regional Development Fund. All other crystallographic data were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 637313).
References
-
(a)
Pincer Compounds: Chemistry and Applications, ed. D. Morales-Morales, Elsevier, 2018, vol. 1 Search PubMed
 ;
(b)
Organometallic Pincer Chemistry, in Topics in Organometallic Chemistry, ed. G. van Koten and D. Milstein, Springer, 2013, vol. 40 Search PubMed ;
(c) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed ;
(d) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS .
;
(b)
Organometallic Pincer Chemistry, in Topics in Organometallic Chemistry, ed. G. van Koten and D. Milstein, Springer, 2013, vol. 40 Search PubMed ;
(c) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed ;
(d) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS .
- A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed .
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed .
-
(a) E. Ben-Ari, M. Gandelman, H. Rozenberg, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2003, 125, 4714–4715 CrossRef CAS PubMed ;
(b) E. Ben-Ari, R. Cohen, M. Gandelman, L. J. W. Shimon, J. M. L. Martin and D. Milstein, Organometallics, 2006, 25, 3190–3210 CrossRef CAS .
-
(a) S. A. Hauser, J. Emerson-King, S. Habershon and A. B. Chaplin, Chem. Commun., 2017, 53, 3634–3636 RSC ;
(b) L. Fan, S. Parkin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 16772–16773 CrossRef CAS PubMed .
- J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Science, 2011, 332, 1545–1548 CrossRef CAS PubMed .
- A. B. Chaplin and A. S. Weller, Organometallics, 2011, 30, 4466–4469 CrossRef CAS .
- G. M. Adams, F. M. Chadwick, S. D. Pike and A. S. Weller, Dalton Trans., 2015, 44, 6340–6342 RSC .
- D. Hermann, M. Gandelman, H. Rozenberg, L. J. W. Shimon and D. Milstein, Organometallics, 2002, 21, 812–818 CrossRef CAS .
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC .
- This approach notably avoids the need for a late stage halogen abstraction step, which can limit the choice of regents or solvents. For instance, Na[BArF4] is not a very effective chloride abstractor in DFB due to appreciable solubility of NaCl (see ref. 10). Silver salts are also sufficiently oxidising to enact one electron oxidation of rhodium(I) complexes (see: M. Feller, E. Ben-Ari, T. Gupta, L. J. W. Shimon, G. Leitus, Y. Diskin-Posner, L. Weiner and D. Milstein, Inorg. Chem., 2007, 46, 10479–10490 CrossRef CAS PubMed .).
- M. Findlater, K. M. Schultz, W. H. Bernskoetter, A. Cartwright-Sykes, D. M. Heinekey and M. Brookhart, Inorg. Chem., 2012, 51, 4672–4678 CrossRef CAS PubMed .
-
(a) M. B. Hursthouse, K. M. A. Malik, E. W. Evans, M. B. H. Howlader and M. T. Atlay, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1782–1784 CrossRef ;
(b) P. J. Stang, L. Song, Y.-H. Huang and A. M. Arif, J. Organomet. Chem., 1991, 405, 403–406 CrossRef CAS .
- C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–12006 CrossRef CAS PubMed .
-
(a) O. Nürnberg and H. Werner, J. Organomet. Chem., 1993, 460, 163–175 CrossRef ;
(b) B. Windmüller, O. Nürnberg, J. Wolf and H. Werner, Eur. J. Inorg. Chem., 1999, 1999, 613–619 CrossRef .
- For comparison to a related NHC-based pincer system see: R. E. Andrew, D. W. Ferdani, C. A. Ohlin and A. B. Chaplin, Organometallics, 2015, 34, 913–917 CrossRef CAS .
- These complexes are presumably in equilibrium (and this suggestion is support by subsequent reaction with CO). See: Y. Wakatsuki, J. Organomet. Chem., 2004, 689, 4092–4109 CrossRef CAS .
- Due to presence as the minor disordered component in the solid state, the metrics associated with the vinylidene in 3b-O are unreliable.
-
(a) G. Albertin, S. Autoniutti, E. Bordignon, F. Cazzaro, S. Ianelli and G. Pelizzi, Organometallics, 1995, 14, 4114–4125 CrossRef CAS ;
(b) M. I. Bruce, B. C. Hall and E. R. T. Tiekink, Aust. J. Chem., 1997, 50, 1097–1100 CrossRef CAS ;
(c) C. Slugovc, V. N. Sapunov, P. Wiede, K. Mereiter, R. Schmid and K. Kirchner, J. Chem. Soc., Dalton Trans., 1997, 4209–4216 RSC ;
(d) V. Cadierno, M. P. Gamasa and J. Gimeno, Organometallics, 1999, 18, 2821–2832 CrossRef CAS ;
(e) N. J. Beach, J. M. Walker, H. A. Jenkins and G. J. Spivak, J. Organomet. Chem., 2006, 691, 4147–4152 CrossRef CAS ;
(f) M. Bassetti, V. Cadierno, J. Gimeno and C. Pasquini, Organometallics, 2008, 27, 5009–5016 CrossRef CAS ;
(g) Y. Mutoh, K. Imai, Y. Kimura, Y. Ikeda and Y. Ishii, Organometallics, 2011, 30, 204–207 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and NMR spectra of new compounds and selected reactions (PDF). Primary NMR data (MNOVA). CCDC 1886657–1886664. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt05049j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Thomas M.
Hood
,
Thomas M.
Hood