
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Twisting the arm: structural constraints in bicyclic expanded-ring N-heterocyclic carbenes†
Katharine R.
Sampford
a,
Jamie L.
Carden
a,
Edward B.
Kidner
a,
Abigail
Berry
a,
Kingsley J.
Cavell
a,
Damien M.
Murphy
a,
Benson M.
Kariuki
 a and
Paul D.
Newman
*ab
a and
Paul D.
Newman
*ab
aSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: newmanp1@cardiff.ac.uk; Fax: +44 (0) 2920 874030; Tel: +44 (0) 2920 870464
First published on 17th January 2019
Abstract
A series of diaryl, mono-aryl/alkyl and dialkyl mono- and bicyclic expanded-ring N-heterocyclic carbenes (ER-NHCs) have been prepared and their complexation to Au(I) investigated through the structural analysis of fifteen Au(NHC)X and/or [Au(NHC)2]X complexes. The substituted diaryl 7-NHCs are the most sterically encumbered with large buried volume (%VB) values of 40–50% with the less flexible six-membered analogues having %VB values at least 5% smaller. Although the bicyclic systems containing fused 6- and 7-membered rings (6,7-NHCs) are constrained with relatively acute NCN bond angles, they have the largest %VB values of the dialkyl derivatives reported here, a feature related to the fixed conformation of the heterocyclic rings and the compressional effect of a pre-set methyl substituent.
Introduction
The extensive literature on N-heterocyclic carbene ligands (NHCs) is dominated by the 5-membered derivatives.1 This preponderance is largely historical but has been propagated by their ease of synthesis and catalytic versatility. The larger ring 6-, 7-, and 8-membered analogues (so-called expanded-ring N-heterocyclic carbenes, ER-NHCs) have only come to prominence in the last 10 years or so and their coordination chemistry remains relatively under-explored.2 This apparent neglect would be understandable if the ER-NHCs behaved similarly to their 5-membered relatives but this is not the case. The larger rings have a quite distinct electronic and steric profile that makes them better donors and more bulky in comparison to their 5-NHC relatives.2,3 These effects are associated with a wider N–C–N angle that leads to a reduced s orbital contribution on the carbon ‘lone pair’ (electronic effect). The exo-substituents in ER-NHCs are thrust towards the metal centre (steric effect) which has repercussions on their coordination chemistry/catalytic application and can lead to improved performance over analogous complexes of 5-NHCs.2e,3e,4 Electronic product control in gold(I) catalysed cyclisations has been observed recently by Bielawski and coworkers5 whereas Aldridge et al.2b have noted the unusual occurrence of backbone C–H activation in ER-NHC complexes of iridium. This latter activity has been attributed to the large size of the ligands which preclude coordination of more than one ER-NHC at a single metal centre. These and related ER-NHCs have %VB values in excess of 50%.2b,h,3a,4The examples noted above serve to emphasise a need to understand and appreciate the stereoelectronic features associated with ER-NHCs to provide a holistic appreciation of stereoelectronic character and potential catalytic facility. Although the extent to which the wingtips impose upon a metal depends significantly on the NCN bond angle there are other, more subtle, factors that play a part. Six-membered monocyclic derivatives have little conformational freedom precluding structural promiscuity, whereas the larger 7-membered analogues show greater flexibility in the ring leading to less predictable features. Bicyclic NHCs derived from camphor contain fused 6,7-membered rings and are relatively rigid.6 They contain spatially-set methyl groups that can dictate the adoption of specific orientations of the exo-substituents on the N-atoms. However, to date, there is insufficient structural data to illuminate the distinctions between these bicyclic derivatives and their monocyclic counterparts.
A comprehensive database of crystallographically characterised NHC complexes is invaluable to understanding these differences. Although metal complexes of ER-NHCs do appear in the Cambridge Crystallographic Database (CCD), their number is still small compared to 5-NHCs. Of the ER-NHCs, bicyclic examples where the ligand is comprised of fused 6- and 7-membered rings (6,7-NHCs) are particularly rare. These ligands are anticipated to be structurally rigid as a consequence of the dimethylmethylene bridge that connects the 4 and 7 positions of the 7-membered ring (and hence creates the 6-membered ring) and previous studies have shown that they share some properties more akin to 5-NHCs than ER-NHCs.5 However these preliminary conclusions were based on a relatively small number of observations and further study is necessary in order to provide a satisfactory comparison. This current paper aims to go some way toward doing this through a structural examination of Au(I) complexes of dialkyl and alkyl/aryl substituted bicyclic 6,7-NHC ligands and appropriate comparison with related monocyclic 6- and 7-membered derivatives.
Results and discussion
Syntheses and solution studies
The synthesis of the monocyclic and bicyclic amidinium precursor salts followed published procedures.6b,e,7 As noted by ourselves and others, 1R,3S-1,2,2-trimethyl-1,3-diaminocyclopentane is an excellent scaffold for the construction of unsymmetrical amidinium salts as precursors to ER-NHCs. This is facilitated by the differential reactivity of the two amino groups with the 3-amino position being more readily functionalised than the 1-amino. While this is advantageous for the construction of unsymmetrical frameworks, it is a significant synthetic limitation as, to date, it has not been possible to introduce aryl groups at the 1-position and diarylated derivatives are necessarily absent. This contrasts with the known monocyclic ER-NHCs which are predominantly symmetrical, diarylated species as controlled construction of unsymmetrical derivatives is difficult for these compounds.The [Au(NHC)Cl] complexes were prepared by procedures similar to those reported by Dunsford et al.7g The NHC precursor is deprotonated using KHMDS in THF forming the free carbene in solution, which is then added dropwise to a stirred solution of either [Au(SMe2)Cl] or [Au(THT)Cl] (THT = tetrahydrothiophene) in THF at ambient temperature. The crude solids were purified by vapour diffusion techniques to give crystalline samples in isolated yields of 20 to 70%. Intriguingly, although several attempts were made to isolate mono-ligated (L)AuCl derivatives of the ligand shown in complex 13, this compound was all that could be isolated from reactions of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. It is unclear why this proved to be the case as no issues were encountered with the related pyridine derivative shown in 12. 14 and 15 which were deliberately targeted as attempts to isolate the mono-ligated (L)AuX compounds were frustrated by the formation of inseparable mixtures (14) and/or competing dimer formation (15).
:1 stoichiometry. It is unclear why this proved to be the case as no issues were encountered with the related pyridine derivative shown in 12. 14 and 15 which were deliberately targeted as attempts to isolate the mono-ligated (L)AuX compounds were frustrated by the formation of inseparable mixtures (14) and/or competing dimer formation (15).
The successful complexation of the NHC ligands is confirmed on inspection of the 1H and 13C{1H} NMR spectra of the isolated solids. Diagnostic features include the loss of the NCHN signal in the 1H NMR spectra and, when observable, a shift of the signal for the carbene carbon in the 13C{1H} NMR spectra. These are accompanied by chemical shift changes for the ring hydrogens in the monocyclic NHCs which move upfield by approximately 0.8 ppm upon coordination and similar, although generally smaller shifts, for the heterocyclic methine hydrogen in the bicyclic systems. These latter shifts are in line with those anticipated for the change from a cationic heterocycle to a neutral form. Although the NCN carbon was not seen in all cases in the 13C{1H} NMR spectra, where observed, the chemical shifts for this carbon in the monocyclic 7-membered systems were at least 10 ppm downfield (∼200 ppm) of the 6-membered analogues (∼190 ppm). These shifts mimic those observed previously with 6- and 7-membered NHCs.7g,8 Those complexes of the type Au(L)Cl where L is a bicyclic ER-NHC for which the NCN carbon signal could be assigned showed this resonance at 190.0 (11) and 188.8 (12) ppm respectively. Thus it would appear, based on this evidence alone, that these constrained NHCs resemble the 6-membered monocyclics more than the larger 7-NHCs. The NCN shifts in the 13C{1H} NMR spectra compare to like complexes of saturated 5-membered analogues such as 1,3-diethyl-4,5-dihydroimidazol-2-ylidene, 1,3-dibenzyl-4,5-dihydroimidazol-2-ylidene and 1,3-dipicolyl-4,5-dihydroimidazol-2-ylidene.9 The larger downfield shifts for complexes 13 and 15 are commensurate with those expected for cationic complexes compared to neutral species (Scheme 1).10
 | ||
| Scheme 1 Au(I) complexes of monocyclic ER-NHCs. | ||
Complex 11 was not isolated in a pure form as it always crystallised as a mixture of the complex and the amidinium salt, (1R,5S)-2,4-dibenzyl-1,8,8-trimethyl-4-aza-2-azoniabicyclo[3.2.1]oct-2-ene tetrafluoroborate, [Bz2-6,7-NHCH][BF4]. It was not certain whether this was the result of carry though of unreacted precursor salt or the result of partial in situ hydrolysis of the gold(I) complex. The NMR spectra clearly showed both species, one of which was readily identified as the amidinium salt.
The 31P{1H} NMR spectrum of 15 contained a very broad peak at a chemical shift little changed from the free ligand (−17.8 ppm). The fact that the resonance is broad does suggest some fluxional process in solution which may indicate a transient interaction of the phosphine groups with the Au(I) centre; this is not borne out in the solid state (see below). Exclusion of O2 was critical for the isolation of 15 as failure to do so resulted in rapid oxidation of the phosphine groups. This observation reinforces the suggestion that the P-donors remain unbound in the complex (Scheme 2).
 | ||
| Scheme 2 Au(I) complexes of bicyclic ER-NHCs. | ||
Solid-state studies
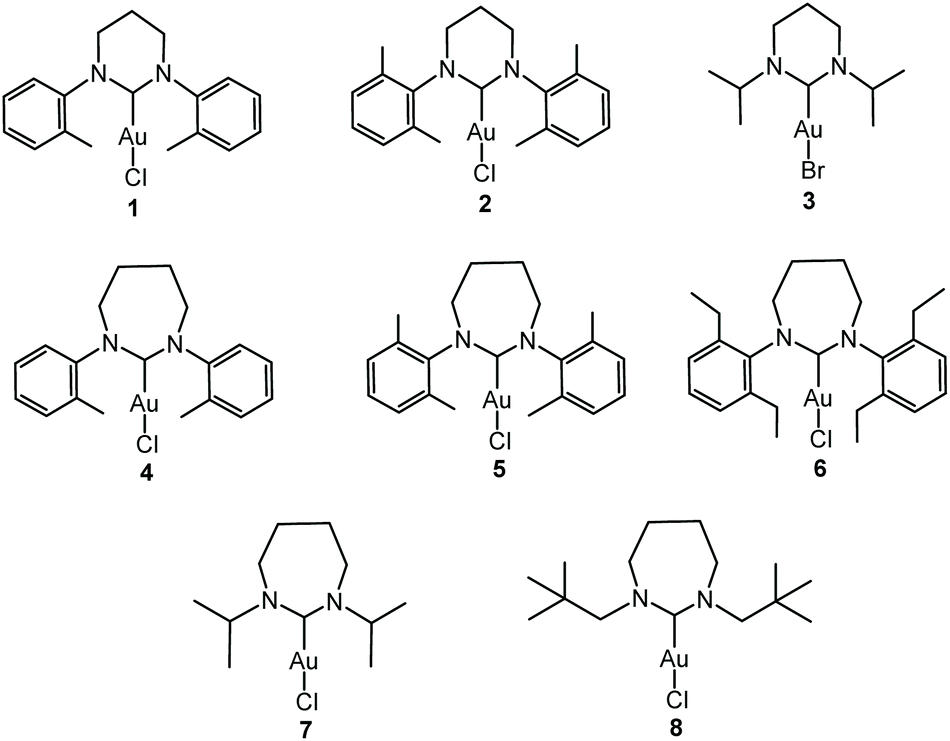
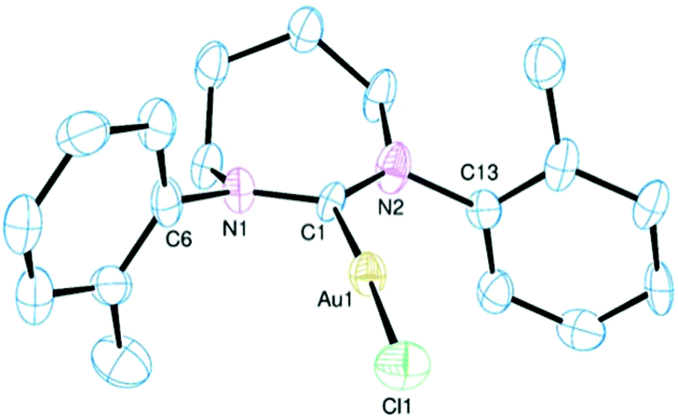
The principal aim of the current research was to establish to what extent fused bicyclic ER-NHCs derived from camphordiamine resembled monocyclic ER-NHCs as ligands. The bicyclic framework is restricting in that there is very little conformational freedom and the nitrogen atoms are relatively fixed with respect to each other. This latter limitation extends to the monocyclic derivatives as there is not much flexibility offered in the necessarily planar RNCNR′ unit. However, the remainder of the heterocycle has some conformational freedom in the monocyclic forms, particularly for the larger ring, which can impinge upon coordination behaviour. Furthermore, the bicyclic framework is embellished by the bridge and the three methyl groups in close proximity to the carbon donor which may lead to geometric differences with similar monocyclic ligands. In order to answer some of these questions we have structurally characterised all fifteen of the complexes reported here by single-crystal XRD. The structures of select complexes are shown in Fig. 1–4 (the remainder are available in the ESI†) with pertinent metrical data presented in Table 1. The synthetic nuances mentioned above limits direct comparison to complexes 3, 7 and 10 but structural relationships within each particular sub-group are important to establish (particularly for highlighting the impact of conformational variations). We will focus on the structural relationships within the three sub-groups of monocyclic 6-NHCs, monocyclic 7-NHCs and the bicyclic NHCs before considering comparisons between 3, 7 and 10.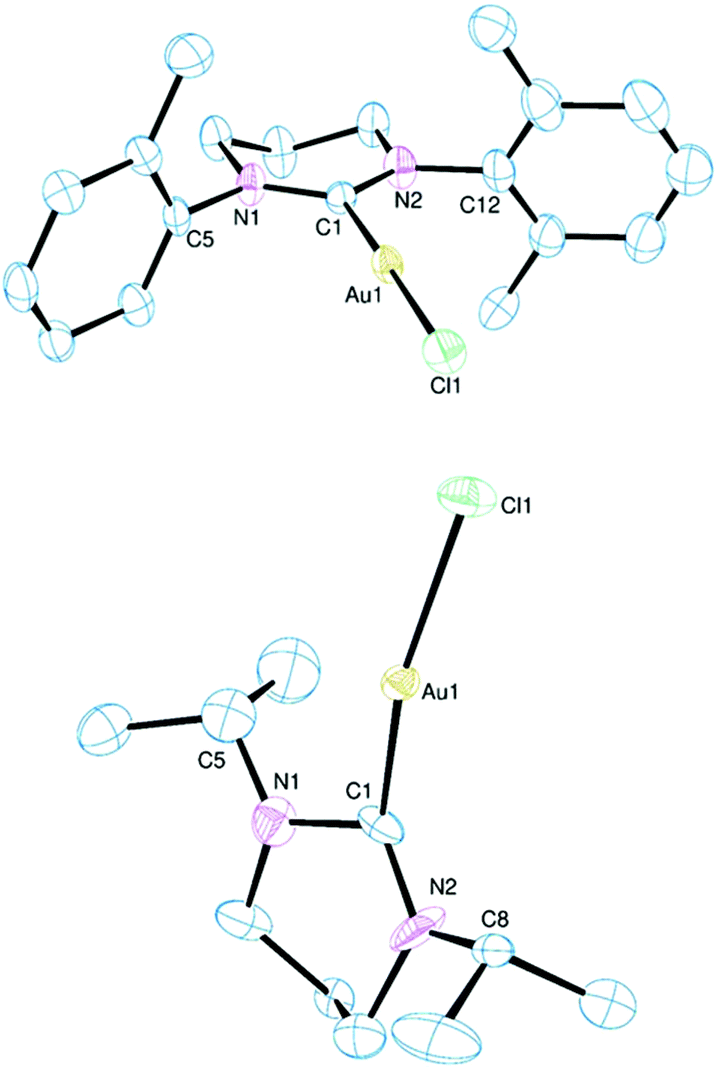 | ||
| Fig. 1 Ortep views of the molecular structures of 1 (top) and 3 (bottom). Hydrogen atoms and lattice solvent have been omitted for clarity. The upper structure shows two methyls on one of the phenyl rings as there is an equal distribution of cis and trans isomers in the crystal. Ellipsoids drawn at 50% probability. | ||
 | ||
| Fig. 2 Ortep view of the molecular structure of 4. Hydrogen atoms, lattice solvent and disordered atoms have been omitted for clarity. Ellipsoids drawn at 50% probability. | ||
 | ||
| Fig. 3 Ortep view of the molecular structure of 8. Hydrogen atoms, lattice solvent and disordered atoms have been omitted for clarity. Ellipsoids drawn at 50% probability. | ||
 | ||
| Fig. 4 Ortep views of the molecular structures of 10–12 (top to bottom). Hydrogen atoms, lattice solvent and disordered atoms have been omitted for clarity. Ellipsoids drawn at 50% probability. | ||
| Complex | Au–C, Å | N–C–N, ° | C–N–Cexo, ° | %Vbur |
|---|---|---|---|---|
| 1 | 1.984(9) | 117.8(8) | 121.2(7) | 41.1 |
| 119.3(8) | ||||
| 2 | 2.001(4) | 117.9(3) | 120.1(3) | 41.5 |
| 120.0(3) | ||||
| 3 | 2.072(11) | 116.8(13) | 119.3(12) | 32.7 |
| 123.3(9) | ||||
| 4 | 1.993(11) | 117.8(10) | 117.4(9) | 40.8 |
| 115.4(10) | ||||
| 5 | 2.001(7) | 119.9(7) | 119.9(6) | 42.9 |
| 117.9(6) | ||||
| 6 | 2.006(3) | 120.3(3) | 119.5(2) | 43.3 |
| 118.9(2) | ||||
| 7 | 2.026(4) | 120.7(4) | 118.2(4) | 34.4 |
| 120.8(4) | ||||
| 8 | 2.016(3) | 117.3(3) | 120.0(3) | 33.6 |
| 118.4(3) | ||||
| 9 | 2.024(5) | 117.7(5) | 120.4(5) | 29.4 |
| 122.5(5) | ||||
| 10 | 2.012(8) | 116.6(8) | 120.0(8) | 36.6 |
| 121.4(8) | ||||
| 11 | 2.102(9) | 121.6(8) | 119.2(7) | 36.9 |
| 123.4(7) | ||||
| 12 | 1.997(6) | 117.1(6) | 118.9(5) | 38.2 |
| 121.9(5) | ||||
| 13 | 2.037(10) | 118.1(7) | 118.1(7) | 37.9 |
| 2.045(9) | 117.4(8) | 118.8(7) | ||
| 118.2(7) | ||||
| 120.1(7) | ||||
| 14 | 2.051(8) | 118.8(7) | 120.0(6) | 35.3 |
| 2.063(7) | 117.8(7) | 122.1(6) | ||
| 119.1(7) | ||||
| 120.9(6) | ||||
| 15 | 2.064(10) | 117.2(9) | 117.0(8) | 43.1 |
| 2.065(11) | 117.7(9) | 122.7(9) | ||
| 119.1(9) | ||||
| 121.7(10) |
The molecular structures of two monocyclic 6-NHC compounds are shown in Fig. 1. All 6-NHC complexes share a common envelope conformation for the diazacycle with a close to co-planar CNCNC unit (most notably for 1 and 2) and the remaining CH2 group out of plane. The aromatic exo-substituents are orthogonal to the NCN plane in 1 and 2 with dihedral angles averaging 91.7° and 92.3° respectively. These structural features compare closely with those for complexes of similar ligands.11 The NCN angle is 117.3 ± 0.6° in all three complexes with the Au–C bond length showing some variation being at its longest in 3 (Table 1). This is also true for the Au–Cl bond length which is substantially longer in 3, an observation that might be attributable to the relatively acute C–Au–Cl bond angle of 166.9° (this angle is 179.1° in 1 and 176.3° in 2). Inspection of the unit cell contents shows no obvious reason for this distortion. The 6-membered ring in 3 is more distorted than in complexes 1 and 2, with intra-cycle CNCN dihedral angles of 32° and 13° respectively (cf. averages of 1° and 5° for 1 and 2). The 2-propyl groups on the nitrogens are arranged with their methyl groups directed away from the metal centre which leads to a reduced steric profile for this ligand (see later). The primary features observed here accord with other Au(I) complexes of 6-NHCs.2b,3a,b,12 Related complexes with 5-NHCs tend to show shorter Au–C bond lengths as a consequence of a reduced steric profile associated with more acute NCN bond angles (<110°).3a,13
A representative molecular structure of the complexes bearing diarylated 7-NHC ligands is shown in Fig. 2. Although there is very little variation in the ligand metrics in 4–6 the conformation of the 7-membered ring is not consistent across the three. This ring has an exaggerated envelope conformation in 6 with an essentially coplanar CNCN unit and the three other CH2 groups orientated on one side of this plane to form the flap of the envelope. A similar conformation is observed in 4, but the CH2CH2CH2 chain is skewed in 5 with one terminal carbon above the CNCN plane and the other below with the central carbon almost coplanar with the CNCN unit. In a like manner to the diarylated 6-NHC complexes the arene rings tend to orthogonal with regard to the central diazacycle although this is less pronounced than in 1 and 2 with relevant dihedral angles ranging from 73° to 105° in 4–6. Both the Au–C bond length and NCN bond angle increase along the series 4 < 5 < 6 as expected based on increasing steric profiles.
The molecular structure of complex 8 reveals a 7-membered ring conformation that is quite distinct to those observed in the other 7-NHC complexes reported here (Fig. 4). This ring adopts a chair conformation in 8 with the NCN atoms and the two carbon atoms β to the nitrogens being coplanar and the two α methylene groups being arranged on opposite sides of this plane. The driving force for the adoption of this conformation would appear to be, at least in part, the desire for the two out-of-plane CH2 groups to lie away from the neo-pentyl substituents on their neighbouring nitrogens. This necessarily leads to a mutually trans arrangement of the (CH3)3CCH2– groups. Although it is clear from the structures of the five 7-NHC complexes reported herein that the conformation of the diazacycle is conformationally flexible and sensitive to the nature of the exo-substituent, the effect of these differing geometries on the other metrics, notably the NCN angle and the Au–C bond length, would appear to be small (see below).
The structures in Fig. 4 exemplify the geometry observed in the bicyclic NHC ligand. The rigid conformation is defined by a five-atom (CNCNC) plane with the terminal carbon atoms supporting the dimethylmethine and dimethylene bridges which lie on opposing sides of the plane. All previously reported molecular structures of complexes containing this central NHC core with various wingtip functions conform to this general structure with NCN bond angles ranging from 115.4° to 120.2°.6e,f However, the majority of these contain secondary donors in one or more of the exo-groups which will have influenced these values. Part of the current remit was to extend the number of structurally characterised complexes containing simple alkyl or alkyl/aryl groups to try and understand inherent limitations on geometry, NCN bond angle etc. Expansion of the 6,7-NHC series was also critical for comparison with appropriate 6- and 7-NHCs.
The structure of the dimethyl-6,7-NHC Au(I) complex (see ESI†) shows very little distortion of the coordination sphere with an C–Au–Cl bond angle of 177.4° and Au–C and Au–Cl bond lengths of 2.024(5) and 2.2937(15) Å respectively. Although this ligand is the least sterically encumbered of those reported here, this does not lead to a particularly short Au–C bond which is presumably largely dictated by electronic factors. The molecular structure of the gold complex of the di-2-propyl derivative is shown in the upper part of Fig. 4. There is little distinction between the metrics seen with this complex and 9. However, one point of note is the relative orientation of the two 2-propyl groups with the methyls being presented towards the metal for the 1-amino substituent and away from the metal for those at the 3-amino position. This reflects the impact of the 6-methyl group of the bicyclic framework which forces the neighbouring 2-propyl group to orientate as shown to limit steric impedance. This has a significant effect on the steric profile of the ligand (see below).
The structural data recorded for 11 are distorted by the presence of the amidinium precursor salt [Bz2-6,7-NHCH][BF4] which co-crystallises with the complex. The metrics reported for 11 (Table 1) are thus an average of those for the two species and gives an inflated value for the NCN angle of 121.6(8)°; this is not a true representation of this metric in 11 and is more typical of amidinium salts of this molecular framework.
A noteworthy structural feature in all the complexes is the presence of disparate C–N–Cexo angles with one appreciably larger than the other. As might be expected, the larger angle is for the substituent at the 3-amino position as those off the 1-amino nitrogen experience a steric compression from the neighbouring methyl leading to more acute C–N–Cexo. This creates an asymmetric steric space by emboldening the substituent at the 1-position relative to that at the 3-amino. The greatest disparity in these bond angles are seen in 12 and 13.
The structures of the bis(ligand) complexes are not discussed explicitly here but details are available in the ESI.† Even though they are constitutionally distinct from the Au(L)Cl complexes and carry a formal positive charge, there appears to be little difference in the pertinent metrics (Table 1). The Au–C bond lengths are similar to but longer than those reported for [(5-NHC)2Au]+ (ref. 14) and [(6-NHC)2Au]+ complexes.2b,15 Compound 13 has a trans configuration in the solid-state which is largely dictated by the need for the bulky mesityl groups to reside away from one another. There is clear pi-stacking between pyridyl groups from two separate ligands in the structure of 14 with a distance of 3.682 Å between the centroids of each ring. The large wingtips evident in 15 lead to a distortion of the C–Au–C bond angle to 166.33° at the extreme and contribute to the disparate CNC angles mentioned above and shown in Table 1.
As much of the current study is to inform future ligand design, we are keen to identify structural characteristics that can be exploited to enhance the stereoelectronic profile of (especially) the bicyclic NHCs. While the electronic character is difficult to assess in the current series of complexes, the steric nature is easier to qualify through analysis of the buried volumes (%Vbur) of the ligands.16 The %Vbur values reported in Table 1 reveal, as noted previously, a controlling influence of the exo-substituents on the nitrogen atoms with conformational variations in the diazacyclic ring of the 7-NHCs having little obvious effect. Those ligands with common or closely related wingtips, e.g.1, 2, 4, 5 and 6, have very similar %Vbur values irrespective of the size and nature of the monocyclic ring. This analogy extends to the di-alkylated derivatives with 10 being an exception. As noted above, the steric impact of the methyl group at the position α- to one of the nitrogens forces the methyls on the 2-propyl group towards the metal. This has the effect of increasing the steric profile of the ligand relative to derivatives without this feature. This framework-induced enhancement of apparent bulk is useful as 3-(2-propyl)-3-mesityl-6,7-NHC should prove as space-hungry as monocyclic bis(mesityl)-6-NHC and bis(mesityl)-7-NHC derivatives; we are currently investigating the preparation of such a ligand and its metal complexes.
Conclusions
A series of Au(I) complexes of monocyclic 6-, 7- and bicyclic 6,7-NHC ligands have been prepared and structurally characterised. Conformational flexibility was noted in the 7-membered ring with NCN angles ranging from 117.3° to 120.7° as opposed to the 6-NHCs which adopt a single envelope conformation and invariant NCN angles of 117.3 ± 0.6°. The bicyclic 6,7-NHC core is less rigid than expected with the NCN angle ranging from 117.1 to 121.6°, although the largest value is restricted to one complex. The unsymmetrical nature of the 6,7-NHC leads to differences in the CNCexo angles with, as predicted, the largest angle being observed at the 3-amino position. The constrained molecular core of the 6,7-NHCs also prevents certain substituents adopting preferred orientations, most notably in the di-(2-propyl) derivative where the presence of a neighbouring methyl forces the CH3 groups on the 2-propyl group at the 3-positon towards the metal. This arm-twisting is a direct consequence of the ligand framework as the monocyclic NHCs with 2-propyl wingtips have the methyl groups of the CH(CH3)2 arm projecting away from the metal. This inflates the steric impact of the 2-propyl group in 10 and engenders a larger steric profile to the 1-(2-propyl)-6,7-NHC compared to monocyclic analogues.Experimental
General information
Unless stated otherwise, all reactions were performed under a nitrogen atmosphere using standard Schlenk techniques and, where appropriate, an inert atmosphere glovebox. Solvents were dried and degassed by refluxing over standard drying agents under dinitrogen and distilled immediately prior to use or obtained from an MBraun SPS system. Infrared spectra were recorded as solid samples on a Shimadzu ATR spectrophotometer. Mass spectra were carried out on a VG Platform II Fisons mass spectrometer. The NMR spectra were recorded on Bruker Avance 400, 500 or 600 MHz instruments at the frequencies indicated. The amidinium precursor salts for the NHC precursors to 1,6b2,6c3,6d4,6b5,6c6,6b7,6a8,6a9,6e10,6e11,6f12,6b13,6b146f and 156f were prepared by the established routes. Percentage buried volumes were calculated using the SambVca 2 program.13b
Crystallography
Single-crystal XRD data were collected on single crystals mounted in paratone. Data were collected using either a Nonius Kappa CCD diffractometer using graphite monochromated Mo Kα radiation or on an Agilent SuperNova Dual Atlas three-circle diffractometer with a mirror monochromator using Mo (λ = 0.7107 Å) radiation. The samples were cooled to 150 K on using an Oxford Cryosystems apparatus. The structures were solved by direct methods using SHELXS17 and refined using refined against F2 using SHELXL.18 A summary of crystallographic data are available as ESI† and the structures deposited with the Cambridge Structural Database (CCDC deposition numbers 1877805–1877819†).Syntheses
1: Yield = 33%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 7.20–7.10 (8H, ArH, m) 3.50 (4H, CH2, m), 2.30 (2H, CH2, m), 2.25 (6H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 191.1 (NCH), 145.9 (C), 134.6 (C), 131.5 (CH), 128.8 (CH), 128.1 (CH), 127.3 (CH), 47.0 (CH2), 21.0 (CH2), 17.8 (CH3). MS(ES) m/z: [M+ − Cl + MeCN] 502.1567 (C20H23N3Au requires 502.1558). Analysis calculated for C18H20N2AuCl: C, 43.52; H, 4.06; N, 5.64. Found: C, 43.43; H, 3.98; N, 5.69.
2: Yield = 36%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 7.15 (2H, ArH, m) 7.05 (4H, ArH, d, 7.5 Hz), 3.40 (4H, CH2, t, 5.9 Hz), 2.30 (2H, CH2, m), 2.25 (12H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 144.3 (C), 134.8 (C), 129.2 (CH), 128.7 (CH), 45.3 (CH2), 20.7 (CH2), 18.0 (CH3). MS(ES) m/z: [M+ − Cl + MeCN] 530.1877 (C22H27N3Au requires 530.1871). Analysis calculated for C20H24N2AuCl: C, 45.77; H, 4.61; N, 5.34. Found: C, 45.86; H, 4.59; N, 5.46.
3: Yield = 42%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 5.25 (2H, CH2, m), 3.05 (4H, CH2, m), 1.85 (2H, CH2, m), 1.10 (12H, CH3, d, 6.8 Hz) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 187.0 (NCN), 60.1 (CH), 38.3 (CH2), 20.5 (CH2), 20.0 (CH3). MS(ES) m/z: [M+ − Cl + 2MeCN] 447.1926 (C14H26N4Au requires 447.1823). Analysis calculated for C10H20N2AuCl: C, 29.98; H, 5.03; N, 6.99. Found: C, 29.89; H, 4.97; N, 6.98.
4: Yield = 24%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 7.20–7.10 (8H, ArH, m), 4.10–3.65 (4H, CH2, m), 2.25 (6H, CH3, s), 2.15 (4H, CH2, m) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 146.9 (C), 131.7 (C), 128.6 (CH), 128.2 (CH), 127.6 (CH), 127.3 (CH), 54.3 (CH2), 24.6 (CH2), 18.1 (CH3). MS(ES) m/z: [M+ − Cl + MeCN] 516.1732 (C21H25N3Au requires 516.1714). Satisfactory elemental analysis could not be obtained for this compound.
5: Yield = 42%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 7.15 (2H, ArH, m) 7.05 (4H, ArH, d, 7.3 Hz), 3.85 (4H, CH2, m), 2.30 (12H, CH3, s), 2.25 (4H, CH2, m) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 201.1 (NCN), 146.6 (C), 134.5 (C), 129.3 (CH), 128.4 (CH), 53.1 (CH2), 25.1 (CH2), 18.6 (CH3). MS(ES) m/z: [M+ − Cl + MeCN] 544.2005 (C23H29N3Au requires 544.2027). Analysis calculated for C21H26N2AuCl: C, 46.81; H, 4.86; N, 5.20. Found: C, 46.78; H, 4.76; N, 5.17.
6: Yield = 38%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 7.20 (2H, ArH, t, 7.9 Hz) 7.10 (4H, ArH, d, 7.9 Hz), 3.85 (4H, CH2, m), 2.85 (4H, CH2, m), 2.60 (4H, CH2, m), 2.20 (4H, CH2, m), 1.30 (12H, CH3, t, 7.5 Hz) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 201.7 (NCN), 145.7 (C), 139.9 (C), 128.6 (CH), 126.7 (CH), 54.3 (CH2), 24.9 (CH2), 24.0 (CH2), 14.4 (CH3). MS(ES) m/z: [M + MeCN] 635.1718 (C23H29N3Au requires 635.1823). Analysis calculated for C25H34N2AuCl: C, 50.47; H, 5.76; N, 4.71. Found: C, 50.53; H, 5.79; N, 4.88.
7: Yield = 42%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 5.10 (2H, CH, m), 3.25 (4H, CH2, m), 1.75 (2H, CH2, m), 1.20 (12H, CH3, d, 6.8 Hz) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 199.0 (NCN), 60.2 (CH), 44.1 (CH2), 25.0 (CH2), 20.2 (CH3). MS(ES) m/z: [M + K+] 453.0777 (C11H22N2AuClK requires 453.0774). Analysis calculated for C11H22N2AuCl: C, 31.86; H, 5.35; N, 6.75. Found: C, 31.97; H, 5.46; N, 6.72.
8: Yield = 33%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 3.90 (4H, CH2, s), 3.45 (4H, CH2, t, 5.0 Hz), 1.75 (4H, CH2, m), 0.85 (18H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 204.5 (NCN), 74.7 (CH2), 56.0 (CH2), 33.3 (C), 28.8 (CH3), 24.7 (CH2). MS(ES) m/z: [M+ − Cl + 2MeCN] 517.37 (C19H36N4Au requires 517.26). Analysis calculated for C15H30N2AuCl: C, 38.27; H, 6.42; N, 5.95. Found: C, 38.41; H, 6.42; N, 6.05.
9: Yield = 38%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 3.40 (3H, CH3, s), 2.95 (1H, CH, d, 4.0 Hz), 2.20 (1H, CH2, m) 1.95 (2H, CH2, m), 1.75 (1H, CH2, m), 1.15 (3H, CH3, s), 0.95 (3H, CH3, s), 0.90 (3H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 69.1 (CH), 68.6 (C), 46.2 (CH3), 41.6 (CH3), 37.9 (C), 34.1 (CH2), 30.3 (CH2), 22.1 (CH3), 17.6 (CH3), 16.2 (CH3). MS(ES) m/z: [M+ − Cl + MeCN] 418.20 (C13H23N3Au requires 418.16). Analysis calculated for C11H20N2AuCl: C, 32.01; H, 4.88; N, 6.79. Found: C, 31.88; H, 4.73; N, 6.83.
10: Yield = 58%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 4.63 (1H, CH, m), 3.47 (1H, CH, m), 2.90 (1H, CH, d, 5.2 Hz), 2.02 (2H, CH2, m), 1.66 (2H, CH2, m), 1.35 (3H, CH3, d, 6.3 Hz), 1.28 (3H, CH3, d, 6.8 Hz), 1.13 (3H, CH3, s), 0.99 (3H, CH3, d, 6.8 Hz), 0.93 (3H, CH3, d, 6.7 Hz), 0.91 (3H, CH3, s), 0.86 (3H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 207.1 (NCN), 67.7 (CH), 57.6 (C), 45.5 (CH), 42.3 (C), 36.8 (CH2), 31.4 (CH2), 22.6 (CH3), 22.3 (CH3), 20.7 (CH3), 20.1 (CH3), 19.7 (CH3), 17.7 (CH3), 15.9 (CH3). MS(ES) m/z: [M+ − Cl + MeCN] 474.2161 (C17H31N3Au requires 474.2184). Analysis calculated for C15H28N2AuCl: C, 38.43; H, 6.02; N, 5.98. Found: C, 38.66; H, 6.16; N, 5.82.
11: Yield = 61%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 7.45 (2H, ArH, d, 7.0 Hz), 7.40 (2H, ArH, d, 7.2 Hz), 7.25 (4H, ArH, m), 7.20 (2H, ArH, m), 5.10 (1H, CH2, d, 14.1 Hz), 5.05 (1H, CH2, d, 16.3 Hz), 4.42 (1H, CH2, d, 16.3 Hz), 4.24 (1H, CH2, d, 14.1 Hz), 2.80 (1H, CH, d, 4.7 Hz), 1.98 (1H, CH2, m), 1.72 (1H, CH2, m), 1.59 (1H, CH2, m), 1.40 (1H, CH2, m), 0.96 (3H, CH3, s), 0.87 (3H, CH3, s), 0.52 (3H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 190.0 (NCN), 138.4 (C), 135.4 (C), 128.9 (CH), 128.7 (CH), 127.8 (CH), 126.9 (CH), 70.6 (CH), 66.3 (C), 62.3 (CH2), 58.5 (CH2), 40.5 (CH), 39.1 (C), 30.9 (CH2), 21.9 (CH3), 17.8 (CH3), 16.9 (CH3). MS(ES) m/z: [M + Na+] 587.2426 (C23H28N2AuClNa requires 587.2429). Satisfactory elemental analysis could not be obtained for this complex.
12: Yield = 61%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 8.41 (1H, ArH, d, 5.0 Hz), 7.80 (1H, ArH, d, 7.5 Hz), 7.73 (1H, ArH, t, 7.5 Hz), 7.46 (2H, ArH, d, 7.2 Hz), 7.33–7.17 (4H, ArH, m), 5.49 (1H, CH2, d, 16.0 Hz), 5.18 (1H, CH2, d, 16.0 Hz), 3.98 (1H, CH, d, 5.0 Hz), 2.25 (2H, CH2, m), 1.73 (2H, CH2, m), 1.22 (3H, CH3, s), 1.14 (3H, CH3, s), 0.99 (3H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 188.8 (NCN), 156.5 (C), 147.4 (CH), 136.6 (C), 127.4 (CH), 126.5 (CH), 125.5 (CH), 121.5 (CH), 120.0 (CH), 70.6 (CH), 67.8 (C), 57.9 (CH2), 39.5 (C), 38.4 (CH2), 30.1 (CH2), 20.6 (CH3), 16.8 (CH3), 15.6 (CH3). MS(ES) m/z: [M+ − Cl + MeCN] 557.1988 (C23H28N4Au requires 557.1980). Analysis calculated for C21H25N3AuCl: C, 45.70; H, 4.57; N, 7.61. Found: C, 45.41; H, 4.40; N, 7.61.
13: Yield = 27%. 1H NMR (400 MHz, CDCl3, 298 K) δ = 6.91 (1H, ArH, s), 6.82 (1H, ArH, s), 3.05 (1H, CH, d, 3.6 Hz), 2.33–1.72 (4H, CH2, m), 2.22 (3H, CH3, s), 2.14 (3H, CH3, s), 2.08 (3H, CH3, s), 1.23 (3H, CH3, s), 1.06 (3H, CH3, s), 0.95 (3H, CH3, s) ppm. 13C{1H} NMR (CDCl3, 125 MHz, 298 K), δ 200.6 (NCN), 139.3 (C), 137.0 (C), 135.6 (C), 133.7 (C), 129.2 (CH), 129.0 (CH), 69.2 (CH), 67.8 (C), 40.6 (C), 38.6 (CH2), 38.0 (CH2), 29.4 (CH3), 21.3 (CH3), 19.9 (CH3), 18.6 (CH3), 18.5 (CH3), 17.9 (CH3), 15.5 (CH3). MS(ES) m/z: [M+] 765.4152 (C38H56N4Au requires 765.4170). Analysis calculated for C38H56N4Au2I2: C, 37.51; H, 4.64; N, 4.61. Found: C, 37.22; H, 4.44; N, 4.60.
14: Yield = 70%. 1H NMR (400 MHz, d6-DMSO, 298 K) δ = 8.34 (2H, ArH, d, 1.8 Hz), 8.27 (2H, ArH, d, 1.8 Hz), 7.88 (2H, ArH, t, 7.5 Hz), 7.69 (2H, ArH, t, 7.6 Hz), 7.52–7.25 (8H, ArH, m), 5.22 (4H, CH2, dd, 15.8, 14.8 Hz), 4.70 (4H, CH2, dd, 15.8, 14.8 Hz), 3.36 (2H, CH, d, 3.2 Hz), 1.90–1.70 (8H, CH2, m), 1.52 (6H, CH3, s), 1.40 (6H, CH3, s), 1.31 (6H, CH3, s) ppm. 13C{1H} NMR (d6-acetone, 125 MHz, 298 K), δ 203.5 (NCN), 158.9 (C), 156.7(C), 150.1 (CH), 137.5 (CH), 124.0 (CH), 123.8 (CH), 123.5 (CH), 122.2 (CH), 71.4 (CH), 68.7 (C), 62.8 (CH2), 59.0 (CH2), 41.2 (C), 39.7 (CH2), 31.0 (CH2), 22.0 (CH3), 18.4 (CH3), 16.6 (CH3). MS(ES) m/z: [M+] 865.3974 (C42H52N8Au requires 865.3980). Analysis calculated for C42H52N8AuBF4: C, 52.95; H, 5.50; N, 11.76. Found: C, 52.90; H, 5.39; N, 11.54.
15: Yield = 21%. 1H NMR (400 MHz, d6-DMSO, 298 K) δ = 7.53–6.85 (56H, ArH, m), 4.72 (2H, CH2, d, 14.5 Hz), 4.66 (2H, CH2, d, 14.5 Hz), 4.48 (2H, CH2, d, 14.9 Hz), 4.40 (2H, CH2, d, 14.9 Hz), 3.10 (2H, CH, br), 2.42 (2H, CH2, m), 1.82 (4H, CH2, m), 1.23 (6H, CH3, s), 1.15 (2H, m), 0.87 (6H, CH3, s), 0.83 (6H, CH3, s) ppm. 13C{1H} NMR (d6-acetone, 125 MHz, 298 K), δ 207.0 (NCN), 140–125 (16 × C, 36 × CH), 71.7 (CH), 65.3 (C), 53.5 (CH2), 41.2 (C), 37.9 (CH2), 29.4 (CH2), 21.0 (CH3), 17.9 (CH3), 14.7 (CH3). 31P{1H} NMR (162 MHz, CDCl3, 298 K) δ −17.8 (br) ppm. MS(ES) m/z: [M+] 1597.5940 (C94H92N4P4Au requires 1597.5938). Satisfactory elemental analysis could not be obtained for this complex.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EP/H023879/1) for financial support of some of this work (KRS).Notes and references
- For selected reviews see: (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed; (c) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef CAS PubMed; (d) W. Gil and A. M. Trzeciak, Coord. Chem. Rev., 2011, 255, 473 CrossRef CAS; (e) K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215 CrossRef CAS PubMed; (f) S. P. Nolan, Acc. Chem. Res., 2010, 44, 91 CrossRef PubMed; (g) D. Janssen-Müller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845 RSC.
- (a) N. Phillips, T. Dodson, R. Trifoin, J. I. Bates and S. Aldridge, Chem. – Eur. J., 2014, 20, 16721 CrossRef CAS PubMed; (b) N. Phillips, R. Trifoin and S. Aldridge, Chem. – Eur. J., 2014, 20, 3825 CrossRef CAS PubMed; (c) S. Pelties, E. Carter, A. Folli, M. F. Mahon, D. M. Murphy, M. K. Whittlesey and R. Wolf, Inorg. Chem., 2016, 55, 11006 CrossRef CAS PubMed; (d) L. R. Collins, T. M. Rookes, M. F. Mahon, I. M. Riddlestone and M. K. Whittlesey, Organometallics, 2014, 33, 5882 CrossRef CAS; (e) F. Sebest, J. J. Dunsford, M. Adams, J. Pivot, P. D. Newman and S. Diez-Gonzalez, ChemCatChem, 2018, 10, 2041 CrossRef CAS PubMed; (f) J. J. Dunsford, D. J. Evans, T. Pugh, S. N. Shah, N. F. Chiltern and M. J. Ingleson, Organometallics, 2016, 35, 1098 CrossRef CAS; (g) A. Sidiropoulos, B. Osborne, A. N. Simonov, D. Dange, A. M. Bond, A. Stasch and C. Jones, Dalton Trans., 2014, 43, 14858 RSC; (h) J. J. Dunsford and K. J. Cavell, Organometallics, 2014, 33, 2902 CrossRef CAS.
- (a) Q. Teng, W. Wu, H. A. Duong and H. V. Huynh, Chem. Commun., 2018, 54, 6044 RSC; (b) O. S. Morozov, A. V. Lunchev, A. A. Bush, A. A. Tukov, A. F. Asachenko, V. N. Khrustalev, S. S. Zalesskiy, V. P. Ananikov and M. S. Nechaev, Chem. – Eur. J., 2014, 20, 6162 CrossRef CAS PubMed; (c) C. Thie, C. Bruhn and U. Siemeling, Eur. J. Inorg. Chem., 2015, 5457 CrossRef CAS; (d) A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613 CrossRef CAS; (e) P. Hauwert, J. J. Dunsford, D. S. Tromp, J. J. Weigand, M. Lutz, K. J. Cavell and C. J. Elsevier, Organometallics, 2013, 32, 131 CrossRef CAS.
- J. W. Hall, D. M. L. Unson, P. Brunel, L. R. Collins, M. K. Cybulski, M. F. Mahon and M. K. Whittlesey, Organometallics, 2018, 37, 3102 CrossRef CAS.
- K. Arumugam, B. Varghese, J. N. Brantley, S. S. M. Konda, V. M. Lynch and C. W. Bielawski, Eur. J. Org. Chem., 2014, 493 CrossRef CAS.
- (a) E. Rais, U. Floerke and R. Wilhelm, Synthesis, 2017, 49, 2852 CrossRef CAS; (b) M. Koppenwallner, E. Rais, M. Uzarewicz-Baig, S. Tabassum, M. A. Gilani and R. Wilhelm, Synthesis, 2015, 47, 789 CAS; (c) P. V. G. Reddy, S. Tabassum, A. Blanrue and R. Wilhelm, Chem. Commun., 2009, 39, 5910 RSC; (d) L. C. Wilkins, R. L. Melen, J. A. Platts and P. D. Newman, Dalton Trans., 2017, 46, 14234 RSC; (e) M. Bouche, M. Mordan, B. M. Kariuki, S. J. Coles, J. Christensen and P. D. Newman, Dalton Trans., 2016, 45, 13347 RSC; (f) P. D. Newman, K. J. Cavell and B. M. Kariuki, Organometallics, 2010, 29, 2724 CrossRef CAS.
- (a) J. J. Dunsford, D. S. Tromp, K. J. Cavell, C. J. Elsevier and B. M. Kariuki, Dalton Trans., 2013, 42, 7318 RSC; (b) M. Iglesias, D. J. Beetstra, B. Kariuki, K. J. Cavell, A. Dervisi and I. A. Fallis, Eur. J. Inorg. Chem., 2009, 1913 CrossRef CAS; (c) M. Iglesias, D. J. Beetstra, J. C. Knight, L. L. Ooi, A. Stasch, S. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279 CrossRef CAS; (d) B. Bantu, D. Wang, K. Wurst and M. R. Buchmeister, Tetrahedron, 2005, 61, 12145 CrossRef CAS; (e) J. Lam, B. A. R. Gunther, J. M. Farrell, P. Eisenberger, B. P. Bestvater, P. D. Newman, R. L. Melen, C. M. Crudden and D. W. Stephan, Dalton Trans., 2016, 45, 15303 RSC; (f) P. D. Newman, K. J. Cavell and B. M. Kariuki, Chem. Commun., 2012, 48, 6511 RSC; (g) J. J. Dunsford, K. J. Cavell and B. M. Kariuiki, Organometallics, 2012, 31, 4118 CrossRef CAS.
- (a) O. S. Morozov, A. V. Lunchev, A. A. Bush, A. A. Tukov, A. F. Asachenko, V. N. Khrustalev, S. S. Zalesskiy and V. P. Ananikov, Chem. – Eur. J., 2014, 20, 6162 CrossRef CAS PubMed.
- (a) S.-T. Liu, C.-I. Lee, C.-F. Fu, C.-H. Chen, Y.-H. Liu, C. J. Elsevier, S.-M. Peng and J.-T. Chen, Organometallics, 2009, 28, 6957 CrossRef CAS; (b) D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385 CrossRef CAS PubMed.
- (a) C. Hirtenlehner, C. Krims, J. Holbling, M. List, M. Zabel, M. Fleck, R. J. F. Berger, W. Schoefbergera and U. Monkowius, Dalton Trans., 2011, 40, 9899 RSC; (b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed.
- J. J. Dunsford, D. S. Tromp, K. J. Cavell, C. J. Elsevier and B. M. Kariuki, Dalton Trans., 2013, 42, 7318 RSC.
- (a) X. Hu, D. Martin, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 13594 CrossRef CAS PubMed; (b) T. Wurm, F. Mulks, C. R. N. Böhling, D. Riedel, P. Zargaran, M. Rudolph, F. Rominger and A. S. K. Hashmi, Organometallics, 2016, 35, 1070 CrossRef CAS; (c) M. Bouché, M. Mordan, B. M. Kariuki, S. J. Coles, J. Christensen and P. D. Newman, Dalton Trans., 2016, 45, 13347 RSC.
- (a) P. Zargaran, T. Wurm, D. Zahner, J. Schießl, M. Rudolph, F. Rominger and A. S. Hashmi, Adv. Synth. Catal., 2018, 360, 106 CrossRef CAS; (b) M. Pažický, A. Loos, M. João Ferreira, D. Serra, N. Vinokurov, F. Rominger, C. Jäkel, A. S. K. Hashmi and M. Limbach, Organometallics, 2010, 29, 4448 CrossRef; (c) R. M. P. Veenboer, D. Gasperini, F. Nahra, D. B. Cordes, A. M. Z. Slawin, C. S. J. Cazin and S. P. Nolan, Organometallics, 2017, 36, 3645 CrossRef CAS; (d) R. M. P. Veenboer, A. Collado, S. Dupuy, T. Lebl, L. Falivene, L. Cavallo, D. B. Cordes, A. M. Z. Slawin, C. S. J. Cazin and S. P. Nolan, Organometallics, 2017, 36, 2861 CrossRef CAS.
- (a) K. Nomiya, S. Morozumi, Y. Yanagawa, M. Hasegawa, K. Kurose, K. Taguchi, R. Sakamoto, K. Mihara and N. C. Kasuga, Inorg. Chem., 2018, 57, 11322 CrossRef CAS PubMed; (b) V. J. Catalano, M. A. Malwitz and A. O. Etogo, Inorg. Chem., 2004, 43, 5714 CrossRef CAS PubMed; (c) P. J. Barnard, M. V. Baker, S. J. Berners-Price, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1038 RSC; (d) A. H. Mageed, B. W. Skelton and M. V. Baker, Dalton Trans., 2017, 46, 7844 RSC; (e) S. Kobialka, C. Müller-Tautges, M. T. S. Schmidt, G. Schnakenburg, O. Hollóczki, B. Kirchner and M. Engeser, Inorg. Chem., 2015, 54, 6100 CrossRef CAS PubMed; (f) V. J. Catalano, A. L. Moore, J. Shearer and J. Kim, Inorg. Chem., 2009, 48, 11362 CrossRef CAS PubMed; (g) P. Ai, M. Mauro, L. De Cola, A. A. Danopoulos and P. Braunstein, Angew. Chem., Int. Ed., 2016, 55, 3338 CrossRef CAS PubMed; (h) A. Collado, J. Bohnenberger, M.-J. Oliva-Madrid, P. Nun, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Eur. J. Inorg. Chem., 2016, 4111 CrossRef CAS; (i) C. Kaub, S. Lebedkin, S. Bestgen, R. Köppe, M. M. Kappes and P. W. Roesky, Chem. Commun., 2017, 53, 9578 RSC.
- M. J. López_Gómez, D. Martin and G. Bertrand, Chem. Commun., 2013, 49, 4483 RSC.
- (a) H. Claver and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC; (b) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all the complexes together with crystallographic details for those compounds characterized by single crystal X-ray techniques. CCDC 1877805–1877819. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04462g. Information on the data underpinning the results presented here, including how to access them, can be found in the Cardiff University data catalogue at DOI: 10.17035/d.2018.0065259122 |
| This journal is © The Royal Society of Chemistry 2019 |