
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly enantioselective epoxidation of olefins by H2O2 catalyzed by a non-heme Fe(II) catalyst of a chiral tetradentate ligand†
Mainak
Mitra‡
 a,
Olaf
Cusso
b,
Satish S.
Bhat
a,
Mingzhe
Sun
a,
Marco
Cianfanelli
b,
Miquel
Costas
*b and
Ebbe
Nordlander
*a
a,
Olaf
Cusso
b,
Satish S.
Bhat
a,
Mingzhe
Sun
a,
Marco
Cianfanelli
b,
Miquel
Costas
*b and
Ebbe
Nordlander
*a
aChemical Physics, Department of Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: Ebbe.Nordlander@chemphys.lu.se; Fax: +4646 22 24119; Tel: +46 46 22 28118
bQBIS, Universitat de Girona, Girona, E-17071, Spain. E-mail: Miquel.Costas@udg.edu; Fax: +34972 41 81 50; Tel: +34 972 41 98 42
First published on 5th April 2019
Abstract
The chiral tetradentate N4-donor ligand, 1-methyl-2-({(S)-2-[(S)-1-(1-methylbenzimidazol-2-yl methyl)pyrrolidin-2-yl]pyrrolidin-1-yl}methyl) benzimidazole (S,S-PDBzL), based on a chiral dipyrrolidine backbone, has been synthesized and its corresponding Fe(II) complex has been prepared and characterized. The X-ray structure of the complex reveals that the Fe(II) ion is in a distorted octahedral coordination environment with two cis-oriented coordination sites occupied by (labile) triflate anions. The ability of the iron complex to catalyze asymmetric epoxidation reactions of olefins with H2O2 was investigated, using 2-cyclohexen-1-one, 2-cyclopenten-1-one, cis-β-methylstyrene, isophorone, chalcones and tetralones as substrates. Different carboxylic acids were used as additives to enhance yields and enantioselectivities, and 2-ethylhexanoic acid was found to give the best results. The catalysis results indicate that the Fe(II) complex is capable of effecting comparatively high enantioselectivities (>80%) in the epoxidation reactions.
Introduction
Optically active (chiral) epoxides are widely used in organic synthesis and industry as intermediates and building blocks for the synthesis of drugs and agrochemicals.1 Amongst various methods developed over the years to synthesize chiral epoxides,1 catalytic asymmetric epoxidation of olefins has proven to be a very useful technique that is employed both in fine chemical and industrial syntheses. Since the pioneering work by Sharpless and coworkers in the 1980s,2 there have been numerous efforts to develop efficient catalytic systems,1a–c,3,4 including chiral metal complexes5,6 and organocatalysts,7,8 for such transformations. The development of environmentally benign and cheap catalysts and oxidants with wide ranges of applications remains a great challenge to synthetic chemists.9 With the growing demand for green and sustainable chemistry, the use of iron-based catalysts employing H2O2 as an oxidant has been an attractive research area because of the low cost, low toxicity and high abundance of iron in nature.10 Collman et al.11 reported in 1999 the first enantioselective epoxidation of styrene derivatives catalysed by iron-porphyrin complexes with iodosobenzene as an oxidant. Beller and co-workers12 have reported a methodology for the asymmetric epoxidation of stilbene derivatives using an in situ iron-based catalyst, giving up to 97% ee. Several non-heme iron-catalysed asymmetric epoxidation reactions are reported with various olefin substrates giving moderate to good yields and enantioselectivities.13–18The factors that indirectly control the yield and enantioselectivity in iron-catalysed asymmetric epoxidation need to be thoroughly investigated. The presence of catalytic amounts of a carboxylic acid has been found to enhance both the yield and enantioselectivity in epoxide formation.14b,16,18 A mechanistic scenario that has been proposed by Que and co-workers for the iron-catalysed epoxidation of alkenes with H2O2/acetic acid involves the formation of a carboxylate-Fe(V) species as the active oxidant via acetic acid-assisted heterolytic O–O bond cleavage of the hydro-peroxide ligand in an Fe(III)(OOH)(HOOCCH3) precursor (Scheme 1).19 Spectroscopic evidence for the formation of a formal Fe(V)-oxo-carboxylato species (A in Scheme 1) has been gained20,21a but its exact electronic structure is a matter of debate.21,22
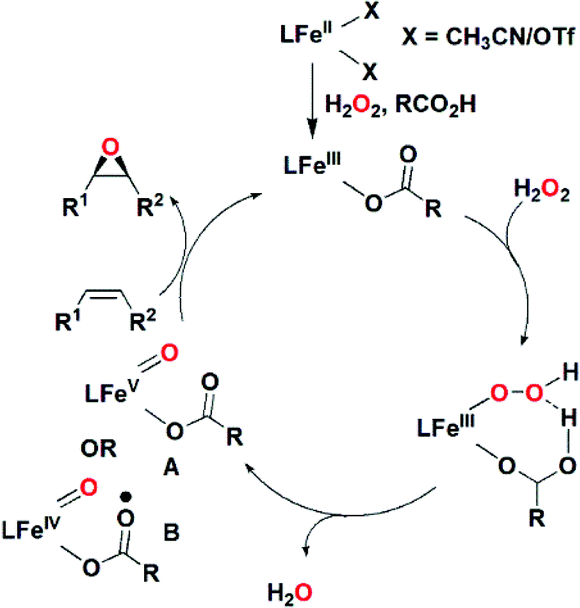 | ||
| Scheme 1 A proposed mechanism for the enhancement of olefin epoxidation catalyzed by a non-heme Fe(II) complex via the addition of a carboxylic acid, RCO2H.19 | ||
A recent computational study suggests that the carboxylic acid, owing to its non-innocent redox nature, can be oxidized by one electron and reduce the Fe ion to form a Fe(IV)-carboxyl radical intermediate23 similar to the cytochrome P450 compound I intermediate. In addition, the possible implication of a ferric peroxycarboxylate intermediate has been proposed24 and debated.25
Electronic factors imposed by the ligand on the iron ion play an important role in the activation of H2O2 and O-atom transfer, as observed in the epoxidation of different olefin substrates catalysed by a series of [FeII(PDP)(CF3SO3)2] complexes (PDP = 2-({(S)-2-[(S)-1-(pyridyl-2-ylmethyl)pyrrolidin-2-yl] pyrrolidin-1-yl}methyl)pyridine),26 where the pyridine rings are substituted with electron withdrawing or donating moieties (Me2N, MeO, Me, H, Cl, CO2Et) at the meta and para-positions, Fig. 1, giving up to 99% yield and 99% enantioselectivity.16 It was proposed that electron-rich ligands decrease the electrophilicity of the Fe-oxo entity, favouring the transition state to be shifted towards a more product-like complex, while electron-deficient ligands increase the electrophilicity of the Fe-oxo unit, making it more indiscriminate and less stereoselective.16
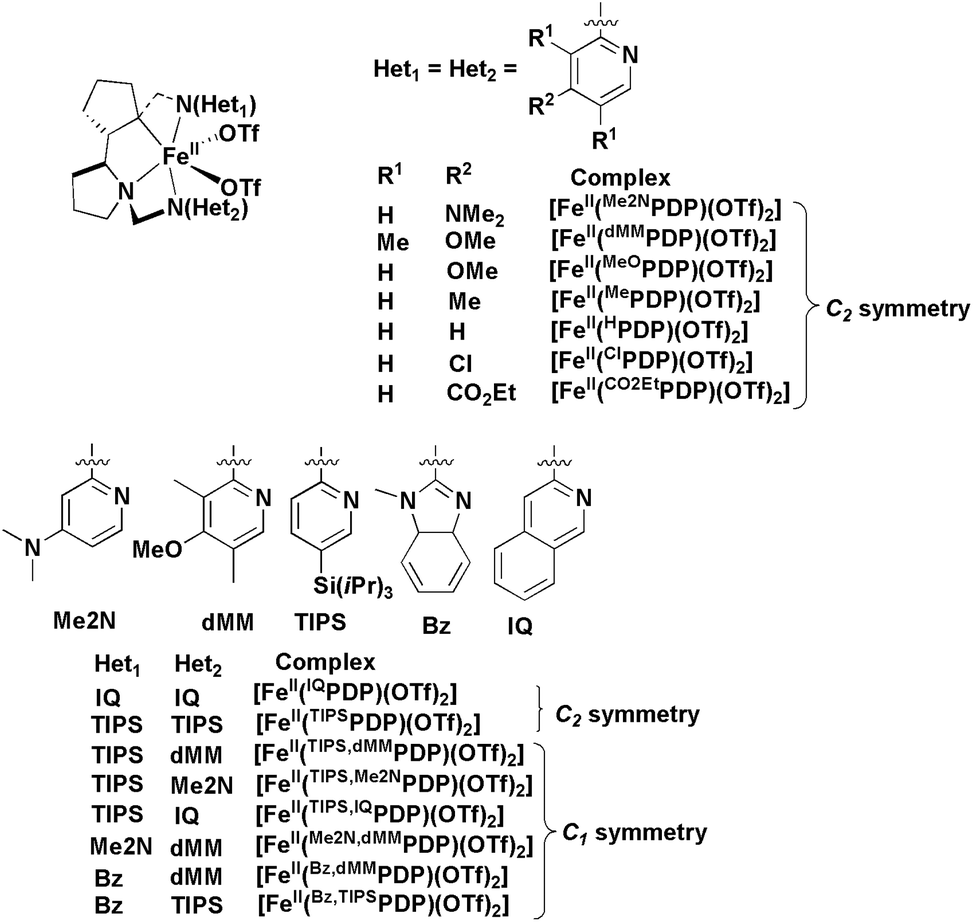 | ||
| Fig. 1 Structures of different Fe(II)-complexes based on the chiral (S,S)-2,2′-bispyrrolidine backbone employed for asymmetric epoxidation.16 | ||
In a recent study, the effects of different side arms attached to the chiral (S,S)-2,2′-bispyrrolidine backbone on the Fe(II)-catalyzed asymmetric epoxidation of olefins were investigated, and the study concluded that complexes containing (N-methyl)benzimidazole and electron rich pyridine or bulky pyridine resulted in excellent yields and enantioselectivities (Fig. 1).18 We have previously observed that an (N-methyl)benzimidazolyl-containing ligand can enhance the catalytic reactivity of an Fe(II) complex in stereospecific C–H hydroxylation reactions,27 and were therefore interested in investigating the influence of a chiral ligand containing (N-methyl)benzimidazolyl donor moieties on Fe(II)-catalyzed asymmetric epoxidation. For this purpose we have employed the tetradentate chiral ligand, S,S-PDBzL (1-methyl-2-({(S)-2-[(S)-1-(1-methylbenzimidazol-2-ylmethyl)pyrrolidin-2-yl]pyrrolidin-1-yl}methyl)benzimidazole) (Fig. 1), where the two (N-methyl)benzimidazolylmethyl arms are attached to the two nitrogen atoms of the chiral (S,S)-2,2′-bispyrrolidine backbone. Notably, Sun and co-workers reported high enantioselectivity (up to 96% ee) by the use of a Mn(II)-catalyst incorporating (R,R)-2,2′-bispyrrolidine and (N-methyl)benzimidazole into the ligand framework.28 Herein we report the synthesis and characterization of a non-heme Fe(II)-complex based on the ligand S,S-PDBzL and the results obtained on alkene epoxidation catalysed by this complex.
Results and discussion
Synthesis and characterization of the ligand and metal complex
The chiral ligand S,S-PDBzL was synthesized by the reaction of one eq. of (S,S)-2,2′-bispyrrolidine tartrate with two eq. of 2-(chloromethyl)-1-methylbenzimidazole29 in the presence of a base (NaOH), and was characterized by 1H NMR and 13C NMR spectroscopy as well as high resolution mass spectrometry (cf. Experimental section).The reaction of equimolar amounts of S,S-PDBzL and [FeII(CH3CN)2(CF3SO3)2] in THF under an inert atmosphere gave the metal complex [FeII(S,S-PDBzL)(CF3SO3)2] (1OTf) as a light yellow solid (Scheme 2, cf. Experimental section). The analogous acetonitrile derivative [FeII(S,S-PDBzL)(CH3CN)2](SbF6)2 (1SbF6) was prepared in two steps: the initial reaction of one eq. S,S-PDBzL with one equivalent of FeCl2 in MeCN to afford [FeII(S,S-PDBzL)Cl2], followed by the reaction of two eq. AgSbF6 with one eq. of [FeII(S,S-PDBzL)Cl2] in CH3CN to form the desired complex, 1SbF6 as a red microcrystalline solid.
 | ||
| Scheme 2 Synthesis of the metal complex [FeII(S,S-PDBzL)(CF3SO3)2] (1OTf). | ||
Complexes 1OTf and 1SbF6 were characterized by high resolution mass spectrometry (HRMS). The HRMS of 1OTf in CH3CN showed a prominent mass peak at m/z 242.1042 corresponding to the formulation [FeII(S,S-PDBzL)]2+ (i.e. z = 2, calc. 242.1014) and at m/z 633.1517 corresponding to the formulation of [FeII(S,S-PDBzL)(CF3SO3)]+ (calc. 633.1558) (Fig. S1–S3, ESI†). The HRMS of complex 1SbF6 in MeCN also showed prominent mass peaks at m/z 242.1022 and 719.0956 corresponding to the formulations of [FeII(S,S-PDBzL)]2+ (calc. 242.1014) and [FeII(S,S-PDBzL)(SbF6)]+ (calc. 719.0980), respectively (Fig. S4 and S5, ESI†).
The 1H-NMR spectra of 1OTf and 1SbF6 were recorded in CD3CN. The spectral window exhibited by both complexes ranges from −20 to 140 ppm, which is indicative of a high spin Fe(II) complex (Fig. S6 and S7, ESI†). The number of resonances and relative integration are consistent with a C2-symmetric complex,18 as shown by its crystal structure (vide infra).
Crystal and molecular structure of complex 1OTf
The solid state structure of complex 1OTf was confirmed by X-ray crystallography. The details of the structure determination are found in the Experimental Section and selected bond distances and bond angles are listed in Table 1. The molecular structure (Fig. 2) shows that the iron ion adopts a distorted octahedral coordination geometry. Four coordination sites are occupied by the nitrogen atoms of the tetradentate S,S-PDBzL ligand while the remaining two cis-sites are occupied by the oxygen atoms of the triflate anions. The two (N-methyl)benzimidazole rings remain above and below the plane containing the iron, the two nitrogens of the S,S-bis-pyrrolidine backbone and the two oxygen atoms of the triflate anions, and are almost perpendicular with respect to each other. The Fe–N bond distances range from 2.15 to 2.25 Å and the Fe–O bond distances range from 2.12 to 2.17 Å, which are in agreement with a high spin Fe(II) ion.30 The bulky (N-methyl)benzimidazolyl moieties introduce steric strain making the O–Fe–O angle smaller (96.7(2)°) relative to the corresponding angle in [FeII(MeOPDP)(CF3SO3)2] (108.47(5)°).16| Fe(1)–N(1) | 2.148(4) |
| Fe(1)–O(1) | 2.115(6) |
| Fe(1)–N(3) | 2.249(6) |
| Fe(1)–N(4) | 2.237(6) |
| Fe(1)–O(4) | 2.168(6) |
| Fe(1)–N(5) | 2.149(5) |
| N(1)–Fe(1)–O(1) | 96.8(2) |
| N(1)–Fe(1)–N(3) | 76.5(2) |
| N(1)–Fe(1)–N(4) | 98.1(2) |
| N(1)–Fe(1)–O(4) | 90.0(2) |
| N(1)–Fe(1)–N(5) | 170.8(2) |
| O(1)–Fe(1)–N(3) | 91.1(2) |
| O(1)–Fe(1)–N(4) | 160.6(2) |
| O(1)–Fe(1)–O(4) | 96.7(2) |
| O(1)–Fe(1)–N(5) | 87.5(2) |
| N(3)–Fe(1)–N(4) | 80.3(2) |
| N(3)–Fe(1)–O(4) | 165.1(2) |
| N(3)–Fe(1)–N(5) | 95.4(2) |
| N(4)–Fe(1)–N(5) | 76.1(2) |
| O(4)–Fe(1)–N(5) | 97.6(2) |
 | ||
| Fig. 2 A Mercury plot of the molecular structure of 1OTf, showing the atom numbering scheme. The thermal ellipsoids are drawn with 30% probability and the hydrogen atoms are omitted for clarity. | ||
Catalytic asymmetric epoxidation studies
Relatively simple cyclic enones have been generally less explored as substrates for asymmetric epoxidation than α,β-unsaturated aromatic ketones. Therefore, this study focuses on using the challenging substrate 2-cyclohexene-1-one for asymmetric epoxidation. In a typical catalytic experiment, H2O2 was delivered using a syringe pump to a stirred solution of CH3CN containing the Fe catalyst, 1OTf, and the substrate (Scheme 3, cf. Experimental section for detailed catalytic conditions). The catalytic reactions were carried out in air and at a low temperature (−30 °C). As mentioned above, previous studies have shown that the presence of a carboxylic acid enhances the efficiency of the catalyst, and the choice of carboxylic acid plays an important role in tuning the efficiency and selectivity of the asymmetric epoxidation reactions. In recent studies, Bryliakov, Talsi and co-workers showed that racemic 2-ethylhexanoic acid (2-eha) resulted in high enantiomeric excess (ee) and relative good yields in iron-catalyzed asymmetric epoxidation reactions.13b Therefore, the study of epoxidation of 2-cyclohexe-1-one was performed using 2-eha. | ||
| Scheme 3 Schematic depiction of the epoxidation reaction of 2-cyclohexe-1-one by 1OTf using H2O2, with 2-ethylhexanoic acid as an additive. | ||
The complex 1OTf (4 mol%) oxidized 2-cyclohexen-1-one to form the epoxide with a low yield (16%) in the presence of H2O2 (2 eq. w.r.t. substrate) and 2-eha (3 eq. w.r.t. substrate; Scheme 3), although the conversion of the substrate into oxidized products was 43% (based on the oxidant). The ee value obtained was 89%. An increase of the carboxylic acid loading did not lead to any significant change in the epoxide yield or the ee value (with 5 eq. of acid the yield of epoxides was 10% and ee was 86% and with 10 eq. of acid the yield of epoxides was 13% and ee was 87%; entries 1–3, Table 2). The effect of different amount of catalyst loading on the substrate conversion, yield of epoxides and ee value was examined. In all cases, the amounts of H2O2 (2 eq. w.r.t. substrate) and carboxylic acid (10 eq. w.r.t. substrate) were kept fixed. Changing the catalyst loading from 4 mol% to 2 mol% resulted in a decrease in the conversion of the substrate into oxidized products (30%) and the yield of epoxides (10%), while maintaining a high ee value (87%; entry 4, Table 2). Increasing the catalyst loading from 4 mol% to 8 mol% (or 10 mol%) increased the conversion of the substrate into products (66 and 67% for 8 and 10 mol% catalyst loading, respectively), but the epoxide yield was more or less similar (entry 5, Table 2). Finally, the amount of H2O2 was varied while the amounts of the catalyst (4 mol%) and carboxylic acid (10 eq. w.r.t. substrate) were kept fixed. The addition of 1.3 eq. of H2O2 resulted in a conversion of 55% with a yield of epoxides of 11% and an ee value of 84%, while the addition of 3 eq. of H2O2 resulted in a conversion of 68% but poor yield of epoxides (7%) and lowering of the ee value (74%; entries 6–8, Table 2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Entry | Catalyst (mol%) | CAb (eq.) | Eq. of H2O2 | Conversion of substrate (%) | Yield of epoxides (%) | eec (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: cf. Experimental section. b CA = carboxylic acid. c Enantiomeric excess. | ||||||
| 1 | 1OTf (4) | 2-eha (3) | 2 | 43 | 16 | 89 |
| 2 | 1OTf (4) | 2-eha (5) | 2 | 52 | 10 | 86 |
| 3 | 1OTf (4) | 2-eha (10) | 2 | 64 | 13 | 87 |
| 4 | 1OTf (2) | 2-eha (10) | 2 | 30 | 10 | 87 |
| 5 | 1OTf (8) | 2-eha (10) | 2 | 66 | 11 | 84 |
| 6 | 1OTf (4) | 2-eha (10) | 1.3 | 55 | 11 | 87 |
| 7 | 1OTf (4) | 2-eha (10) | 1.6 | 59 | 12 | 87 |
| 8 | 1OTf (4) | 2-eha (10) | 3 | 68 | 7 | 74 |
| 9 | 1SbF6 (4) | 2-eha (10) | 2 | 64 | 13 | 86 |
| 10 | 1SbF6 (4) | (CH3)3CO2H (10) | 2 | 72 | 2 | 60 |
| 11 | 1SbF6 (4) | S-Ibuprofen (10) | 2 | 2 | 1 | 58 |
Product analysis by GC and GC-MS spectrometry of the reaction of 1OTf with cyclohexenone in the presence of H2O2 (2 eq. w.r.t. substrate) and 2-eha or acetic acid (10 eq. w.r.t. substrate in each case) revealed that in addition to the desired (chiral) epoxide, small amounts of a number of oxidized products were formed, viz. 3-hydroxycyclohexanone, cyclohexene-1,4-dione, and 4-hydroxycyclohexenone when eha was used (in addition to small amounts of lactones from the oxidation of eha). When acetic acid was used, the minor side products were cyclohexene-1,4-dione, 4-hydroxycyclohexenone and 2,3-hydroxycyclohexanone (cf. ESI, Fig. S10–S13†). The relatively low yields of these side products cannot explain the overall low yield in the reactions (Table 2). No ring-opened products were detected, but it is possible that ring-opening occurs and that further substrate degradation leads to (oxidized) products of low molecular weight that could not be identified.
As expected, complex 1SbF6 exhibited the same behaviour as 1OTf (entries 3 and 9, Table 2). On changing the carboxylic acid, both the yields and enantioselectivities were diminished (2% yield and 60% ee for pivalic acid and 1% yield and 58% ee for S-ibuprofen; entries 9–11, Table 2).
Complex 1OTf was further investigated in the oxidation of 2-cyclopenten-1-one where it provided a low conversion of the substrate (52%) with a very low yield of the epoxide (4%) and moderate enantioselectivity (ee 76%).
cis-β-Methylstyrene was also employed as a substrate and the conditions for catalysis were used as reported earlier,16 in order to enable direct comparison of 1OTf with related Fe(II) complexes. Under catalytic conditions (cf. Experimental section for details), complex 1OTf oxidized cis-β-methylstyrene to form the epoxide with 60% yield and an ee of 43% (Table 3). Under the same conditions, the catalyst [FeII(HPDP)(CF3SO3)2] (H1) (HPDP = 2-({(S)-2-[(S)-1-(pyridyl-2-ylmethyl)pyrrolidin-2-yl]pyrrolidin-1-yl}methyl)pyridine) and [FeII(Me2NPDP)(CF3SO3)2] (Me2N1) (Me2NPDP is the modified PDP ligand where the para-positions of the two pyridyl rings are occupied by Me2N groups, cf. Fig. 1) provided an epoxide yield of 26% with an ee of 19% and 87% with an ee of 62%, respectively.16
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | CAb (mol%) | Temp. (°C) | Eq. of H2O2 | Conversion of substrate (%) | Yield of epoxides (%) | eec (%) |
| a Reaction conditions: Experimental section. b Carboxylic acids. c Enantiomeric excess determined by GC. | |||||||
| 1 | 1OTf (1) | AcOH (3) | −30 | 1.6 | 41 | 27 | 43 |
| 2 | 1OTf (1) | AcOH(1.4) | −30 | 1.6 | 84 | 60 | 43 |
| 3 | 1OTf (2) | AcOH(1.4) | −30 | 1.6 | 99 | 52 | 46 |
| 4 | 1OTf (2) | AcOH(1.4) | 0 | 1.6 | 99 | 49 | 40 |
| 5 | 1OTf (2) | 2-eha (1.4) | −30 | 1.6 | 100 | 61 | 61 |
| 6 | 1OTf (2) | 2-eha (1.4) | 0 | 1.6 | 100 | 60 | 55 |
Very poor epoxidation yield and enantioselectivity were obtained for other substrates such as trans-β-methylstyrene and 4-chloro-α-methylstyrene (Table S1†). For isophorone a very good enantioselectivity of 91% was obtained but the epoxide yield was unfortunately poor (12%); attempts to optimise the epoxide yield proved futile (Table S2†).
In organic synthesis, the trisubstituted cyclic α,β-epoxyketones are resourceful intermediates because of the presence of a chiral quaternary carbon center.31 The development of an iron catalyst for the enantioselective construction of such quaternary carbon centers coordinated to an oxygen atom is still highly attractive.32 Using tetralone derivatives as substrates for this type of reaction, we obtained good yields of trisubstituted α,β-epoxyketones with quaternary carbon centers with excellent enantioselectivities (up to 97%, Table 4).

Furthermore, the epoxidation of α,β-unsaturated ketones was evaluated. For chalcone and its derivatives, a higher epoxide yield and ee (up to 97%) were obtained with 2-eha than AcOH as the auxiliary carboxylic acid (Table 5, entries 1–5). Having electron withdrawing or donating groups at the para position of the substrate phenyl ring did not affect the enantioselectivity. As expected, the complex with the corresponding R,R-PDBzL ligand (with an (R,R)-2,2′-bispyrrolidine backbone) gave 84% chalcone epoxide formation with an ee value of 96% under catalytic conditions similar to those of complex 1OTf (entry 1, Table 2) but with the opposite absolute configuration.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | CAb (mol%) | Yieldc (%) | eed (%) |
| a Reaction conditions: Experimental section. b Carboxylic acids. c Isolated product yield. d Enantiomeric excess determined by HPLC analysis. | |||||
| 1 | Ph | Ph | 2-eha (1.4) | 76 | 97 |
| 2 | Ph | Ph | AcOH (1.4) | 71 | 91 |
| 3 | p-Me-C6H4 | Ph | 2-eha (1.4) | 56 | 97 |
| 4 | p-Cl-C6H4 | Ph | 2-eha (1.4) | 84 | 97 |
| 5 | Ph | p-CF3-C6H4 | 2-eha (1.4) | 66 | 94 |

Conclusions
A tetradentate N4 ligand S,S-PDBzL, with an S,S-dipyrrolidine chiral backbone, has been synthesized. The corresponding Fe(II) complexes, [FeII(S,S-PDBzL)(CF3SO3)2] (1OTf) and [FeII(S,S-PDBzL)(CH3CN)2](SbF6)2 (1SbF6), were synthesized and characterized. The complex 1OTf exhibited high enantioselectivity (ee > 80%) in the epoxidation of the cyclic olefin 2-cyclohexen-1-one, albeit with low yields of the chiral epoxides (10–15%). It has previously been shown that while the complex [FeII(Me2NPDP)(CF3SO3)2] provided a poor yield (12%) and moderate enantioselectivity (ee 76%) in the epoxidation of 2-cyclohexen-1-one, the related complex [FeII(MeOPDP)(CF3SO3)2] (Fig. 1) showed excellent yield (99%) and high enantioselectivity (ee 84%) under similar conditions. This difference was attributed to the rapid deactivation of the former catalyst during the course of oxidation of the substrate, a phenomenon that takes place when the substrate oxidizes slowly under the reaction conditions. The similar reactivities of 1OTf and [FeII(Me2NPDP)(CF3SO3)2] towards the substrate 2-cyclohexen-1-one might therefore be due to similar rapid deactivation of the Fe-catalyst. With cis-β-methylstyrene, 1OTf provided moderate yield (60%) and lower enantioselectivity (ee 41%). In the case of tetralone and chalcones excellent epoxide yield and enantioselectivity (up to 97%) were obtained. The relative orientation of the bulky (N-methyl)benzimidazole groups may also provide excessive steric constraints against the approach of the incoming substrate to the reactive metal oxo species (cf.Scheme 1), causing a poor yield of the epoxides and unwanted side products. Further studies on the catalytic properties of 1OTf and 1SbF6 to explore a wider range of substrates are warranted.Experimental section
Reagents and materials
Reagents and solvents were of at least 99% purity and used as received without any further purification. All reagents and solvents were purchased from Sigma Aldrich or Fisher Scientific. Dichloromethane and acetonitrile were dried by distillation from CaH2; diethyl ether was dried by distillation from Na/benzophenone. The starting material 2-(chloromethyl)-1-methylbenzimidazole was synthesized according to a literature procedure.29Instrumentation
Infrared spectra were recorded on a Nicolet Avatar 360 FTIR spectrometer. UV-Visible spectroscopy was performed in a 1 cm quartz cell using an Agilent Technology 8453 UV-Vis spectrophotometer equipped with a diode-array detector. NMR spectra were recorded on a Bruker DPX 400 MHz spectrometer in CDCl3 or CD3CN solvent under standard conditions and were referenced to the residual proton signal of the solvent. Elemental analysis was performed on a 4.1 Vario EL 3 elemental analyzer from Elementar. The ESI-MS experiments were performed with a Bruker Esquire 6000 LC/MS chromatograph, using acetonitrile as a mobile phase. The product analyses after catalysis experiments were carried out on an Agilent Technology 7820A gas chromatograph equipped with a 16-sample automatic liquid sampler, flame ionization detector and EzChrom Elite Compact software.Syntheses
:2% 1 M NaOH:82% CH2Cl2:11% petroleum ether. The collected fractions were combined, washed with 1 M NaOH solution, dried over Na2SO4 and the solvent was evaporated to obtain the ligand as a pale orange solid. Yield: 0.366 g (63.2%). HRMS: 429.2760 [M + H]+, calc. 429.2761. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.72 (m, 2H), 7.33–7.21 (m, 6H), 4.23 (d, 2H), 3.80 (s, 6H), 3.66 (d, 2H), 2.80 (dt, 4H), 2.32 (m, 2H), 1.79 (m, 4H), 1.68 (m, 4H). 13C-NMR (100 MHz, CDCl3) δ (ppm): 152.4, 142.2, 136.1, 122.4, 121.8, 119.6, 109.0, 65.2, 55.6, 52.5, 29.9, 26.2, 24.0.
Crystal structure determination
Orange crystals of 1OTf were grown by slow diffusion of diethyl ether into a CH2Cl2 solution of the compound. Collection of diffraction data was carried out at (100(2) K) on a BRUKER SMART APEX CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The measurements were made in the θ range 2.172 to 27.744°. A full-sphere data collection was carried out with ω and φ scans. A total of 82922 reflections were collected of which 8370 were unique (Table 6). Programs used: data collection, Smart;33 data reduction, Saint+;34 absorption correction, SADABS.35 Structure solution and refinement were done using SHELXTL.36
| Empirical formula | C28H32F6FeN6O6S2·CH2Cl2 (1OTf) |
| Formula weight | 867.49 |
| Temperature | 100(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Hexagonal |
| Space group | P65 |
| Unit cell dimensions | a = 10.8264(3) Å |
| b = 10.8264(3) Å | |
| c = 52.532(2) Å | |
| α = 90° | |
| β = 90° | |
| γ = 120° | |
| Volume | 5332.4(4) Å3 |
| Z | 6 |
| Density (calculated) | 1.621 Mg m−3 |
| Absorption coefficient | 0.775 mm−1 |
| F(000) | 2664 |
| Crystal size | 0.3 × 0.3 × 0.25 mm3 |
| Theta range for data collection | 2.172 to 27.744° |
| Index ranges | −14 ≤ h ≤ 14, −14 ≤ k ≤ 14, −68 ≤ l ≤ 68 |
| Reflections collected | 82922 |
| Independent reflections | 8370 |
| Completeness | 100% (to theta = 25.242) |
| Absorption correction | Empirical |
| Max. and min. transmission | 1.0 and 0.900775 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 8370/3/471 |
| Goodness-of-fit on F2 | 1.053 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0592, wR2 = 0.1622 |
| R indices (all data) | R 1 = 0.0632, wR2 = 0.1668 |
| Largest diff. peak and hole | 2.223 and −1.601 e Å−3 |
The structure was solved by direct methods and refined by full-matrix least-squares methods on F2. The non-hydrogen atoms were refined anisotropically. The H-atoms were placed at geometrically optimized positions and forced to ride on the atom to which they are attached. The structure crystallized in the chiral space group P65.
Reaction conditions for catalysis
:1 (v:v) H2O2 (30% wt/wt in H2O):CH3CN was delivered to the reaction solution over a period of 30 min, using a syringe pump. After the addition of H2O2, the reaction solution was further stirred at 30 °C for 30 min. At this point, a known amount of biphenyl solution was added as the internal standard. The solution was passed through a small alumina column and the column was rinsed with 2 × 1 ml ethyl acetate and the resultant elute was subjected to GC analysis. The racemic products were identified by their GC retention times and the yields were determined from the integration area of the GC spectrum.
:1 (v:v) H2O2 in water (30% wt/wt):CH3CN was delivered to the reaction solution over a period of 30 min using a syringe pump. After this addition, the reaction solution was further stirred at 30 °C for 30 min. At this point, a known amount of biphenyl solution was added as the internal standard. The solution was passed through a small alumina column and the column was rinsed with 2 × 1 ml ethyl acetate, and the resultant elute was subjected to GC analysis. The racemic products were identified by their GC retention times and the yields were determined from the integration area of the GC spectrum.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This paper is dedicated to the memory of Dr Dominque Mandon, a good friend and colleague, and a thoughtful chemist whose life ended too early. The research has been carried out within the frameworks of the International Research Training Group Metal sites in biomolecules: structures, regulation and mechanisms (http://www.biometals.eu), the Marie Sklodowska-Curie Innovative Training Network MSCA-ITN-2015-ETN 675020 and COST Action CM1003. M. M. thanks the European Commission for an Erasmus Mundus fellowship. We thank Dr Ahibur Rahaman and Mr Yong Li for assistance with the preparation of the (S,S)-PDBzL ligand, and Prof. Wei Sun for valuable discussions and help in performing the epoxidation experiment with the Fe(II)-complex of the (R,R)-PDBzL ligand.Notes and references
- (a) M. J. Porter and J. Skidmore, Chem. Commun., 2000, 1215–1225 RSC; (b) B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2473 CrossRef CAS PubMed; (c) Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed; (d) O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958–3987 CrossRef CAS PubMed; (e) D. Diez, M. G. Nunez, A. B. Anton, P. Garcia, R. F. Moro, N. M. Garrido, I. S. Marcos, P. Basabe and J. G. Urones, Curr. Org. Synth., 2008, 5, 186–216 CrossRef CAS; (f) K. Matsumoto and T. Katsuki, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 3rd edn, 2010, pp. 839–890 Search PubMed; (g) G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722–1760 RSC.
- K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2024–2032 CrossRef CAS PubMed.
- Representative examples: (a) H. Wynberg and B. J. Greijdanus, J. Chem. Soc., Chem. Commun., 1978, 427–428 RSC; (b) S. Julia, J. Guixer, J. Masana, J. Rocas, S. Colonna, R. Annuziata and H. J. Molinari, J. Chem. Soc., Perkin Trans. 1, 1982, 1317–1324 RSC; (c) D. Enders, J. Zhu and G. Raabe, Angew. Chem., Int. Ed. Engl., 1996, 35, 1725–1728 CrossRef CAS; (d) D. Yang, Y.-C. Yip, M.-W. Tang, M.-K. Wong, J.-H. Zheng and K.-K. Cheung, J. Am. Chem. Soc., 1996, 118, 491–492 CrossRef CAS; (e) M. Bougauchi, S. Watanabe, T. Arai, H. Sasai and M. Shibakasi, J. Org. Chem., 1998, 63, 8090–8091 CrossRef; (f) E. J. Corey and F.-Y. Zhang, Org. Lett., 1999, 1, 1287–1290 CrossRef CAS; (g) O. Jacques, S. J. Richards and R. F. W. Jackson, Chem. Commun., 2001, 1712–1713 Search PubMed; (h) C. Li, E. A. Pace, M. C. Liang, E. Lobkovsky, T. D. Galimore and J. A. Porco Jr., J. Am. Chem. Soc., 2001, 123, 11308–11309 CrossRef CAS PubMed; (i) A. Berkessel, M. Guixa, F. Schimdt, J. M. Neudorfl and J. Lex, Chem. – Eur. J., 2007, 13, 4482–4498 CrossRef PubMed; (j) A. Erkkila, P. M. Pihko and M.-R. Clarke, Adv. Synth. Catal., 2007, 349, 802–806 CrossRef CAS; (k) G. Peris, C. E. Jakobsche and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 8710–8711 CrossRef CAS PubMed; (l) W. Zhang and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 286–287 CrossRef CAS PubMed.
- (a) W. Adam, C. R. Saha-Moller and P. A. Ganeshpure, Chem. Rev., 2001, 101, 3499–3548 CrossRef CAS PubMed; (b) A. Lattanzi, Curr. Org. Synth., 2008, 5, 117–133 CrossRef CAS.
- (a) T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS; (b) W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS; (c) R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef CAS; (d) K. Matsumoto, Y. Sawada, B. Saito, K. Sakai and T. Katsuki, Angew. Chem., 2005, 117, 5015–5019 CrossRef; (e) Y. Sawada, K. Matsumoto, S. Kondo, H. Watanabe, T. Ozawa, K. Suzuki, B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2006, 45, 3478–3480 CrossRef CAS PubMed; (f) A. U. Barlan, A. Basak and H. Yamamato, Angew. Chem., Int. Ed., 2006, 45, 5849–5852 CrossRef CAS PubMed.
- E. M. McGarrigle and D. G. Gilheany, Chem. Soc. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed.
- (a) Y. Shi, Acc. Chem. Res., 2004, 37, 488–496 CrossRef CAS PubMed; (b) D. Yang, Acc. Chem. Res., 2004, 37, 497–505 CrossRef CAS PubMed; (c) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199–8256 CrossRef CAS.
- (a) T. Ooi, D. Ohara, M. Tamura and K. Maruoka, J. Am. Chem. Soc., 2004, 126, 6884–6845 CrossRef PubMed; (b) M. Marigo, J. Franzen, T. B. Poulsen and K. A. Jorgensen, J. Am. Chem. Soc., 2005, 127, 6964–6965 CrossRef CAS PubMed; (c) X. W. Wang, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2008, 130, 6070–6071 CrossRef CAS PubMed; (d) X. J. Lu, Y. Liu, B. F. Sun, B. Cindric and L. Deng, J. Am. Chem. Soc., 2008, 130, 8134–8135 CrossRef CAS PubMed; (e) C. M. Reisinger, X. W. Wang and B. List, Angew. Chem., Int. Ed., 2008, 47, 8112–8115 CrossRef CAS PubMed.
- F. G. Gelalcha, Adv. Synth. Catal., 2014, 356, 261–299 CrossRef CAS.
- (a) L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed; (b) E. B. Bauer, Curr. Org. Chem., 2008, 12, 1341–1369 CrossRef CAS; (c) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed; (d) A. Correa, O. G. Mancheno and C. Bolm, Chem. Soc. Rev., 2008, 8, 1108–1117 RSC; (e) L.-X. Liu, Curr. Org. Chem., 2010, 14, 1099–1126 CrossRef CAS; (f) C.-L. Sun Chang-Liang, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef PubMed; (g) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 2, 243–255 RSC; (h) K. Gopalaiah, Chem. Rev., 2013, 113, 3248–3296 CrossRef CAS.
- J. P. Collman, Z. Wang, A. Straumanis, M. Quelquejeu and E. Bose, J. Am. Chem. Soc., 1999, 121, 460–461 CrossRef CAS.
- F. G. Gelalcha, B. Bitterlich, G. Anilkumar, M. K. Tse and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 7293–7296 CrossRef CAS.
- (a) F. G. Gelalcha, G. Anilkumar, M. K. Tse, A. Bruckner and M. Beller, Chem. – Eur. J., 2008, 14, 7687–7698 CrossRef CAS PubMed; (b) O. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196–1202 CrossRef CAS; (c) T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538–13541 CrossRef CAS PubMed; (d) V. A. Yazerski, A. Orue, T. Evers, H. Kleijn and R. J. M. Klein Gebbink, Catal. Sci. Technol., 2013, 3, 2810–2818 RSC.
- (a) M. Wu, C.-X. Miao, S. Wang, X. Hu, C. Xia, F. E. Kühn and W. Sun, Adv. Synth. Catal., 2011, 353, 3014–3022 CrossRef CAS; (b) X. Wang, C. Miao, S. Wang, C. Xia and W. Sun, ChemCatChem, 2013, 5, 2489–2494 CrossRef CAS; (c) W. Wang, Q. Sun, D. Xu, C. Xia and W. Sun, ChemCatChem, 2017, 9, 420–424 CrossRef CAS.
- Y. Nishikawa and H. Yamamoto, J. Am. Chem. Soc., 2011, 133, 8432–8435 CrossRef CAS PubMed.
- O. Cusso, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871–14878 CrossRef CAS.
- O. Cusso, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285–14298 RSC.
- (a) O. Cusso, M. Cianfanelli, X. Ribas, R. J. M. Klein Gebbink and M. Costas, J. Am. Chem. Soc., 2016, 138, 2732–2738 CrossRef CAS; (b) O. Cussó, X. Ribas, J. Lloret-Fillol and M. Costas, Angew. Chem., Int. Ed., 2015, 54, 2729–2733 CrossRef PubMed; (c) O. Cussó, M. W. Giuliano, X. Ribas, S. J. Miller and M. Costas, Chem. Sci., 2017, 8, 3660–3667 RSC.
- R. Mas-Balleste and L. Que Jr., J. Am. Chem. Soc., 2007, 129, 15964–15972 CrossRef CAS PubMed.
- (a) A. M. Zima, O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2016, 6, 5399–5404 CrossRef CAS; (b) O. Y. Lyakin, A. M. Zima, D. G. Samsonenko, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2015, 5, 2702–2707 CrossRef CAS; (c) W. N. Oloo, R. Banerjee, J. D. Lipscomb and L. Que Jr., J. Am. Chem. Soc., 2017, 139, 17313–17326 CrossRef CAS.
- (a) J. Serrano-Plana, W. N. Oloo, L. Acosta-Rueda, K. K. Meier, B. Verdejo, E. Garcia-Espana, M. G. Basallote, E. Munck, L. Que Jr., A. Company and M. Costas, J. Am. Chem. Soc., 2015, 137, 15833–15342 CrossRef CAS PubMed; (b) R. Fan, J. Serrano-Plana, W. N. Oloo, A. Draksharapu, E. Delgado-Pinar, A. Company, V. Martin-Diaconescu, M. Borrell, J. Lloret-Fillol, E. Garcia-Espana, Y. Guo, E. L. Bominaar, L. Que Jr., M. Costas and E. Munck, J. Am. Chem. Soc., 2018, 140, 3916–3928 CrossRef CAS PubMed.
- B. Mondal, F. Neese, E. Bill and S. Ye, J. Am. Chem. Soc., 2018, 140, 9531–9544 CrossRef CAS PubMed.
- Y. Wang, D. Janardanan, D. Usharani, K. Han, L. Que Jr. and S. Shaik, ACS Catal., 2013, 3, 1334–1341 CrossRef CAS.
- A. M. Zima, O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2017, 7, 60–69 CrossRef CAS.
- O. Cusso, J. Serrano-Plana and M. Costas, ACS Catal., 2017, 7, 5046–5053 CrossRef CAS.
- (a) E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063–7064 CrossRef CAS; (b) M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed.
- M. Mitra, J. Lloret-Fillol, M. Haukka, M. Costas and E. Nordlander, Chem. Commun., 2014, 50, 1408–1410 RSC.
- D. Shen, C. Miao, S. Wang, C. Xia and W. Sun, Eur. J. Inorg. Chem., 2014, 5777–5782 CrossRef CAS.
- S. V. Amrutkar, U. D. Bhagat, P. Pargharmol, S. S. Kotgire and M. S. Ranawat, Int. J. Pharm. Pharm. Sci., 2010, 2, 84–92 CAS.
- (a) A. Diebold and K. S. Hagen, Inorg. Chem., 1998, 37, 215–223 CrossRef CAS; (b) D. W. Blakesley, S. C. Payne and K. S. Hagen, Inorg. Chem., 2000, 39, 1979–1989 CrossRef CAS; (c) K. Chen and L. Que Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed; (d) G. J. P. Britovsek, J. England and A. P. J. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed; (e) A. J. Simaan, S. Dopner, F. Banse, S. Bourcier, G. Bouchoux, A. Boussac, P. Hildebrandt and J. J. Girerd, Eur. J. Inorg. Chem., 2000, 1627–1633 CrossRef CAS; (f) I. Prat, A. Company, T. Corona, T. Parella, X. Ribas and M. Costas, Inorg. Chem., 2013, 52, 9229–9244 CrossRef CAS PubMed.
- (a) K. Fuji, Chem. Rev., 1993, 93, 2037–2066 CrossRef CAS; (b) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef; (c) B. M. Wang and Y. Q. Tu, Acc. Chem. Res., 2011, 44, 1207–1222 CrossRef CAS PubMed.
- (a) B. Wang, S. Wang, C. Xia and W. Sun, Chem. – Eur. J., 2012, 18, 7332–7335 CrossRef CAS PubMed; (b) E. J. Corey and F.-Y. Zhang, Org. Lett., 1999, 1, 1287–1290 CrossRef CAS PubMed; (c) Y. Liu, B. A. Provencher, K. J. Bartelsonaand and L. Deng, Chem. Sci., 2011, 2, 1301–1304 RSC.
- Bruker Advanced X-ray Solutions. SMART: Version 5.631, 1997–2002 Search PubMed.
- Bruker Advanced X-ray Solutions. SAINT+, Version 6.36A, 2001 Search PubMed.
- G. M. Sheldrick, Empirical Absorption Correction Program, Universität Göttingen, 1996 Search PubMed; Bruker Advanced X-ray Solutions. SADABS Version 2.10, 2001 Search PubMed.
- G. M. Sheldrick, Program for Crystal Structure Refinement, Universität Göttingen, 1997 Search PubMed; Bruker Advanced X-ray Solutions, SHELXTL Version 6.14, 2000–2003. SHELXL-2013 (Sheldrick, 2013) Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR, ESI-MS and FTIR spectra of the ligand and complexes. CCDC 1453881. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04449j |
| ‡ Present Address: Department of Chemistry, Burdwan Raj College, Purba Bardhaman, West Bengal-713104, India. |
| This journal is © The Royal Society of Chemistry 2019 |