
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent functionalised naphthalimides and their Au(I)–NHC complexes for potential use in cellular bioimaging†‡
Lara M.
Groves
a,
Catrin F.
Williams
b,
Anthony J.
Hayes
c,
Benjamin D.
Ward
a,
Marc D.
Isaacs
c,
Nadine O.
Symonds
a,
David
Lloyd
c,
Peter N.
Horton
d,
Simon J.
Coles
 d and
Simon J. A.
Pope
*a
d and
Simon J. A.
Pope
*a
aSchool of Chemistry, Main Building, Cardiff University, Cardiff CF10 3AT, UK. E-mail: popesj@cardiff.ac.uk; Fax: +44 (0)29-20874030; Tel: +44 (0)29-20879316
bSchool of Engineering, Cardiff University, Cardiff, UK CF24 3AA
cSchool of Biosciences (and Bio-imaging Research Hub), Sir Martin Evans Building, Cardiff University, Cardiff, UK CF19 3AX
dUK National Crystallographic Service, Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, England, UK
First published on 19th December 2018
Abstract
A series of cationic, dihydroimidazolinium-functionalized 1,8-naphthalimide fluorophores have been isolated as their hexafluorophosphate salts, [HL]PF6. These pro-ligands react with [AuCl(tht)] in the presence of base to form N-heterocyclic carbene (NHC) complexes, [AuCl(L)]. Two X-ray structures represent a pro-ligand and complex pairing: the latter reveals the two-coordinate linear geometry of the NHC–Au(I) species, as well as intermolecular interactions supported by both ligand π–π stacking and a weak aurophilic interaction of 3.3205(6) Å. The luminescence properties of the pro-ligands and complexes are dominated by the ICT character of the substituted fluorophore at ca. 500 nm, which is further modulated via functionalization at the 4-position of the naphthalimide. Cytotoxicity assessments were performed for all [HL]PF6 and [AuCl(L)] species against LOVO, MCF-7, A549 and PC3 cell lines; added lipophilicity seems to correlate with increased cytotoxicity. Confocal fluorescence microscopy was undertaken on a selected [HL]PF6 and [AuCl(L)] species and showed that the intracellular distribution is dependent upon the specific ligand structure. More detailed co-localisation studies show that selected examples present a predominant lysosomal staining pattern. FLIM studies exemplified the applicability of these probes, and secondly suggest that fluorescence lifetime could be used to provide information on the integrity of the complex and thus liberation of gold in a biological environment.
Introduction
Although the use of gold in medicinal chemistry (known as chrysotherapy)1 has been recognised for millennia, recent developments have renewed interest in gold's use in metallodrugs. The variety of Au(I) complexes used in a clinical setting are those incorporating thiolate or phosphine ligands.2 The most widely utilised are auranofin, sodium aurothiomalate and sodium aurothioglucose, all of which have been applied to the treatment of inflammatory autoimmune conditions such as rheumatoid arthritis.3 Auranofin is also under investigation as a treatment to reduce the viral reservoir of HIV that lies latent in the body's T-cells.4 The mechanism of biological action of such clinical treatments remains extremely difficult to unravel.5 Over the last two decades, there have been several reports suggesting that a variety of Au(I) complexes possess antiproliferative properties in vitro against selected human tumour cell lines.6One must consider that the gold agents used clinically are not luminescent compounds and thus other, invasive and analytical techniques are required to understand the intracellular distribution of the gold.7 To support such studies, it is helpful therefore if the gold species in question is inherently luminescent, thus enabling non-invasive visualisation of live cells through fluorescence microscopy techniques.8
Whilst fluorescence wavelength and Stokes’ shift are important parameters for the application of probes to fluorescence microscopy, the use of time-resolved analyses via fluorescence lifetime imaging microscopy (FLIM) can provide even more detailed information. This current work describes the synthesis and characterisation of a series of dihydroimidazolinium functionalised 1,8-naphthalimide fluorophores which can be deprotonated to act as N-heterocyclic carbene (NHC) donors for coordination to Au(I). These new species are shown to be fluorescent in the visible region and show excellent utility as cell imaging agents including application to FLIM studies of MCF-7 cells. Importantly, the results suggest that fluorescence lifetime is significantly modulated by the presence of the coordinated Au(I), and thus provides further information on the integrity of intracellular metal–ligand complexes.
Results and discussion
Synthesis
The intermediates Cl-Nap and N-Nap, pro-ligands [HL]PF6 and complexes [AuCl(L)] are shown in Scheme 1. The di-substituted dihydroimidazolinium pro-ligand salts, [HL]PF6, were synthesized in three steps from commercially available 4-chloro-1,8-naphthalic anhydride. Reaction at the naphthalimide ring of Cl-Nap using either N-ethylethylenediamine or N-phenylethylenediamine (thus R2 = Et or Ph) gave the intermediate species, N-Nap. Subsequent ring closure of the N-Nap species using triethylorthoformate (both as reagent and solvent) in the presence of ammonium hexafluorophosphate yielded the cationic dihydroimidazolinium pro-ligands as their hexafluorophosphate salt, [HL]PF6. The corresponding gold(I) complexes, [AuCl(L)], were synthesised from [AuCl(tht)] (tht = tetrahydrothiophene) via deprotonation of the pro-ligand to yield the NHC Au(I) complex. Despite repeated attempts we were unable to isolate [AuCl(L5)] in sufficient quantities to allow unequivocal characterisation and further study. The yielded complexes (Scheme 2) are thus based upon an unsymmetrical substitution of a NHC donor at Au(I) that incorporates a conjugated 1,8-naphthalimide fluorophore. | ||
| Scheme 1 Synthetic route: (i) EtNH(CH2)2NH2, DMSO, heat; (ii) CH(OEt)3, NH4PF6, heat; (iii) [AuCl(tht)], KOtBu, MeOH. | ||
 | ||
| Scheme 2 Structures of the fluorescent Au(I) complexes targeted in this work. | ||
Spectroscopic characterisation of ligands and complexes
1H NMR spectra for N-Nap1–8 provided confirmation of substitution at the naphthalimide ring: in most cases, a characteristic NH resonance was noted ca. 6.0–6.5 ppm, which was assigned to the naphthalimide secondary amine substituent. Upon cyclisation to form the pro-ligands [HL1–8]PF6, the corresponding 1H NMR spectra showed subtle changes in the chemical shifts of the dihydroimidazolium ethyl backbone; in most cases these were observed as multiplets ca. 4.0–4.5 ppm. The NH resonance at 6.0–6.5 ppm also disappeared, and a new downfield peak at 9.0–10.0 ppm was assigned to the deshielded H2 position of the imidazolinium moiety: both observations were consistent with cyclisation. IR spectra of [HL1–8]PF6 showed ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) ca. 1650 cm−1 and ν(PF6) ca. 830 cm−1 consistent with the formation of the salts. HRMS were obtained for all intermediates and pro-ligands.
O) ca. 1650 cm−1 and ν(PF6) ca. 830 cm−1 consistent with the formation of the salts. HRMS were obtained for all intermediates and pro-ligands.
The Au(I) complexes were again characterised by a range of spectroscopic and analytical techniques. 1H NMR spectra provided clear evidence of coordination to Au(I) through the absence of the downfield H2 imidazolinium resonance, and changes in the chemical shifts associated with the ethyl protons in the backbone of the NHC unit. 13C{1H} NMR spectra revealed resonances that correlated with the presence of the different R1 and R2 groups. In addition, two unique carbonyl resonances around 162–165 ppm were observed, consistent with the unsymmetrical nature of the 1,8-naphthalimide unit. A downfield signal ca. 192–195 ppm (Fig. S1, ESI‡) was assigned to the Au(I)-coordinated NHC carbon and is comparable with previous studies on Au(I)–NHC complexes which incorporate a saturated NHC backbone.9
DFT calculations were employed to further support this 13C{1H} NMR assignment, in which the NMR shielding constants were computed using the gauge including atomic orbital (GIAO) method.10 Sarotti and Pellegrinet11 have suggested that a judicious choice of reference compound gives superior chemical shift predictions, and to this end, since the carbene carbon can be considered sp2-hybridized, benzene was used as the reference standard and its shielding constants computed at the same level of theory as the carbene complexes. Using these calculations, the δC values of the coordinated carbene are predicted to be 192.1 ppm and 192.7 ppm for [AuCl(L3)] and [AuCl(L4)] respectively, both of which compare very favorably to the experimental data thus providing further evidence that the assignments are reliable.
HRMS data were also obtained for the complexes, often correlating with the formation of a cationic complex fragment (through loss of a chloride ligand, or addition of a cation such as Na+ or NH4+). Each spectrum was consistent with retention of the Au–NHC unit. IR spectra confirmed the presence of the ligand, primarily via the naphthalimide ν(CO) bands ca. 1600–1650 cm−1.
X-ray crystallography
During the synthesis of the ligands and complexes, diffraction quality crystals of [HL3]PF6 and [AuCl(L3)] were isolated. The crystals were obtained by the vapour diffusion of diethyl ether into either a concentrated solution of [HL3]PF6 (acetone) or [AuCl(L3)] (acetonitrile). Data collection parameters are shown in Table S1 (ESI‡), together with supporting bond length and bond angle data (Table 1). The resultant structures of [HL3]PF6 and [AuCl(L3)] are shown in Fig. 1 and 2. The structure of the complex (Fig. 1) shows an approximately linear (the C1–Au1–Cl1 angle is 173.76(6)°) two-coordinate geometry at Au(I). As expected, the C–N bond lengths within the dihydroimidazolinium moiety are extended upon coordination to Au(I). The Au–C bond length is 1.984(2) Å and thus comparable with structurally related NHC complexes of Au(I).9 The structure of [AuCl(L)] also reveals intermolecular π–π stacking interactions between neighbouring naphthalimide units (Fig. 2). Additional intermolecular interactions are also evident through an Au(I)⋯Au(I) distance of 3.31459(15) Å, which is at the weaker limit of an aurophilic interaction.12 | ||
| Fig. 1 Crystal structures of [HL3]PF6 (top) and [AuCl(L3)] (bottom). Ellipsoids drawn at 50% probability and counterion omitted for [HL3]PF6. | ||
 | ||
| Fig. 2 Representations of the crystal structure of [AuCl(L3)]. The π–π packing interactions where the angle between these two planes is 0.00(19)°, the centroid–centroid distance is 3.8001(17) Å and the shift distance is 1.644(3) Å. | ||
| [HL3]PF6 | Bond | [AuCl(L3)] | Bond |
|---|---|---|---|
| N(2)–C(16) | 1.318(7) | N(1)–C(1) | 1.359(2) |
| N(3)–C(16) | 1.299(7) | N(2)–C(1) | 1.318(3) |
| Au(1)–C(1) | 1.984(2) | ||
| Au(1)–Cl(1) | 2.2933 (5) | ||
| Au(1)–Au(1′) | 3.31459 (15) |
Electronic properties and DFT calculations
Solution state UV-vis. absorption and luminescence spectra were obtained for all pro-ligands and complexes (Table 2). The absorption spectra (MeCN) of the pro-ligands were characterised by shorter wavelength peaks (<300 nm) which are assigned to π → π* transitions associated with the naphthalimide chromophore and phenyl substituents. Between 300–450 nm the spectra also revealed weaker features due to different n → π* transitions including an intramolecular charge transfer (ICT) localised on the naphthalimide unit. The wavelength of the ICT band was relatively insensitive, firstly, to the nature of the imide (R1) substituent, and secondly, the R2 group on the dihydroimidazolinium group (Fig. 3). However, the ethyl variants ([HL1]PF6, [HL3]PF6, [HL5]PF6, [HL7]PF6) all showed substantial hyperchromic shifts in the ICT band relative to their phenyl ([HL2]PF6, [HL4]PF6, [HL6]PF6, [HL8]PF6) analogues. This may be due to the relative electron donating ability of the R2 substituent. | ||
| Fig. 3 A comparison of the UV-vis. absorption spectra for selected pro-ligands (recorded in acetone). | ||
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a /nm a /nm |
λ em /nm | τ obs /ns | ϕ /% | |
|---|---|---|---|---|
| a Measurements obtained in aerated 10−5 M acetone solutions. b λ ex = 405 nm. c λ ex = 372 nm. d Using aerated MeCN solution of [Ru(bipy)3](PF6)2 as a reference. | ||||
| [HL1]PF6 | 344, 426 | 504 | 7.3 | 4 |
| [HL2]PF6 | 342, 429 | 507 | 8.7 | 28 |
| [HL3]PF6 | 344, 426 | 502 | 9.1 | 4 |
| [HL4]PF6 | 344, 351, 432 | 503 | 7.2 | 4 |
| [HL5]PF6 | 344, 428 | 502 | 8.4 | 31 |
| [HL6]PF6 | 343, 435 | 504 | 8.8 | 17 |
| [HL7]PF6 | 344, 427 | 501 | 4.7 | 5 |
| [HL8]PF6 | 345, 433 | 503 | 9.0 | 12 |
| [AuCl(L1)] | 344, 351, 436 | 507 | 7.8 (45%), <1.0 (55%) | 2 |
| [AuCl(L2)] | 349, 433 | 487 | 7.2 (78%), 2.8 (22%) | 9 |
| [AuCl(L3)] | 345, 351, 439 | 500 | 1.1 (76%), <1.0 (24%) | 1 |
| [AuCl(L4)] | 354, 352 | 504 | 6.9 (33%), <1.0 (67%) | 1 |
| [AuCl(L6)] | 343, 436 | 503 | 8.3 (82%), 2.6 (18%) | 2 |
| [AuCl(L7)] | 345, 351, 439 | 499 | 4.3 (9%), 1.0 (91%) | 1 |
| [AuCl(L8)] | 344, 351, 441 | 504 | <1.0 | 8 |
Overall, when compared to other 4-amino-substituted 1,8-naphthalimides,13 the absorptivity of the ICT bands in these pro-ligands is relatively diminished and hypsochromically shifted. This is attributed to the cationic nature of the dihydroimidazolium moiety, which may well modulate the overall donor ability of the nitrogen atom at the 4-position of the naphthalimide chromophore. The absorption spectra of the related Au(I) complexes were thus dominated by the ligand-centred transitions. In general, a hypochromic shift of the ICT band relative to the pro-ligand was noted, again probably relating to the net donating ability of the ligated NHC substituent.
DFT calculations
To further probe the electronic properties of the substituted naphthalimide species, the structures of two pro-ligands [HL3]+ and [HL4]+ were calculated using density functional methods, and were subsequently used to probe the underlying electronic basis for the electronic absorption spectra. The structure of the ethyl congener [HL3]+ was compared to that obtained using X-ray data, and was found to provide good agreement between experiment and theory.Using these optimized structures for TD-DFT calculations gave a particularly noteworthy result: the low energy ICT band, experimentally observed at 425–430 nm, was not obtained in the calculations. We initially rationalized that the band ca. 350 nm might correspond to this transition, but with a large computational-experimental discrepancy. TD-DFT often underestimates transitions with a significant CT character,14 however, this hypothesis would involve an overestimation of the excited state energy, which is apparently contrary to known limitations in this area. Moreover, whilst the phenyl derivative [HL4]+ contains some CT character (vide infra), the ethyl derivative does not. In our earlier work, we have noticed that the accuracy of excited state calculations is only slightly dependent on basis set, but varies considerably with functional. Therefore, the calculations were performed using a number of functionals, namely CAM-B3LYP, B3LYP, PBE0, LC-wPBE, B3PW91, M06, and M06-2X. The lowest energy excited state varied between 291 nm (LC-wPBE) and 353 (B3LYP) for the ethyl derivative [HL3]+.
Upon comparison with the experimental spectra, it became apparent that the lowest energy transition corresponded to the experimental band at ca. 340–350 nm, and the low-energy band at 425 nm is not replicated in the TD-DFT calculations. One of the significant limitations of DFT calculations can be the lack of counterion or discrete solvent interactions with the substrate; attempts were made to model these, by introducing the anion, and solvent molecules (water and acetone) hydrogen bonding to the appropriate polar groups in the naphthalimide cation, but in every case only a slight (up to 10 nm) variation in the low energy excitation energy was observed.
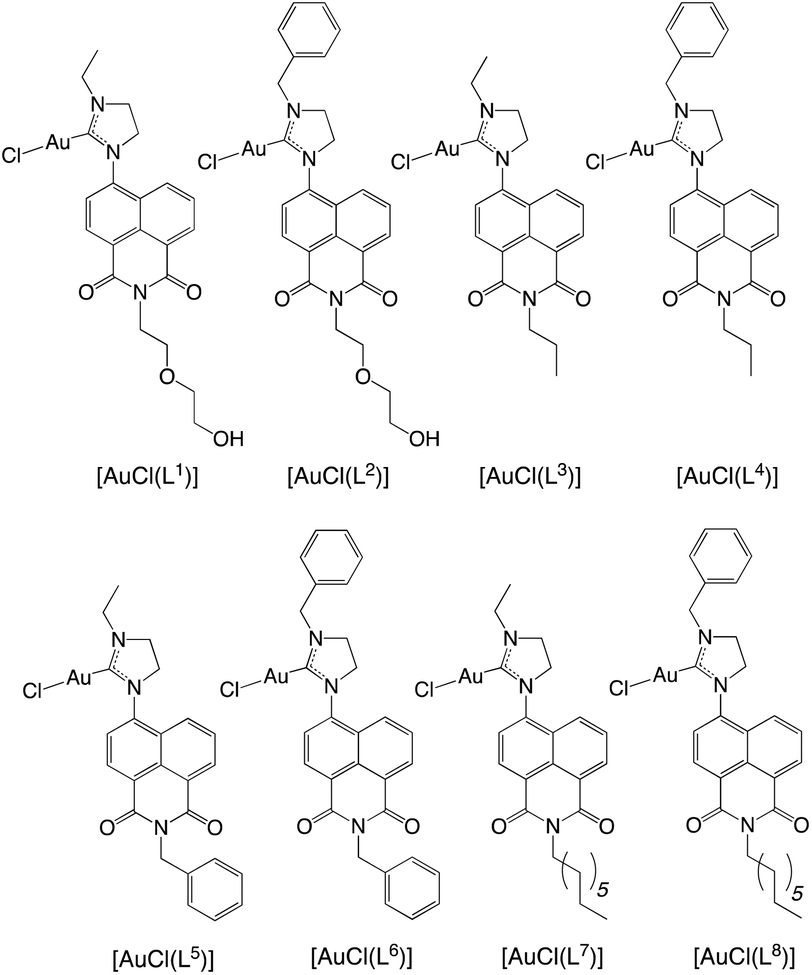
TD-DFT calculations were performed using the PBE0 hybrid functional, since this allows a closer comparison to related theoretical studies on 1,8-naphthalimides.15 In this regard, the calculations were in good agreement with the experimental data, since the computed energies were 342 nm ([HL3]+) and 348 nm ([HL4]+) versus 344 nm (for [HL3]PF6) and 351 nm (for [HL4]PF6). These maxima are in excellent agreement with the experimental data, and moreover the slight red-shift, experimentally observed for [HL4]+, is replicated (simulated spectra are provided in Fig. 4). The molar absorption coefficients have been estimated from the oscillator strengths using established methods,16 and their magnitudes agree with the experimentally observed values and are consistent with the expected π–π* transitions. Experimentally, the phenyl derivative has a higher absorptivity coefficient than the ethyl, which is replicated in the calculations. A further analysis of the excited states indicates that they correspond to π–π* transitions between the HOMO and LUMO; the HOMO and LUMO of [HL4]+ are displayed in Fig. 5. The principal difference between the HOMO of [HL3]+ and [HL4]+ is that there is a significant orbital coefficient corresponding to the phenyl group in [HL4]+; the LUMOs of [HL3]+ and [HL4]+ are essentially identical, with no significant orbital coefficient over the phenyl ring in [HL4]+. This difference in the HOMOs explains the subtle difference in intensity and excitation energy, with the longer excitation wavelength in [HL4]+ corresponding to the greater HOMO delocalization in [HL4]+, compared to [HL3]+.
 | ||
| Fig. 4 Simulated UV-vis. absorption spectra of [HL3]+ (black), [HL4]+ (red), [AuCl(L3)] (green), and [AuCl(L4)] (blue) using TD-DFT data [PBE0: 6-311+G(d,p)/SDD]. | ||
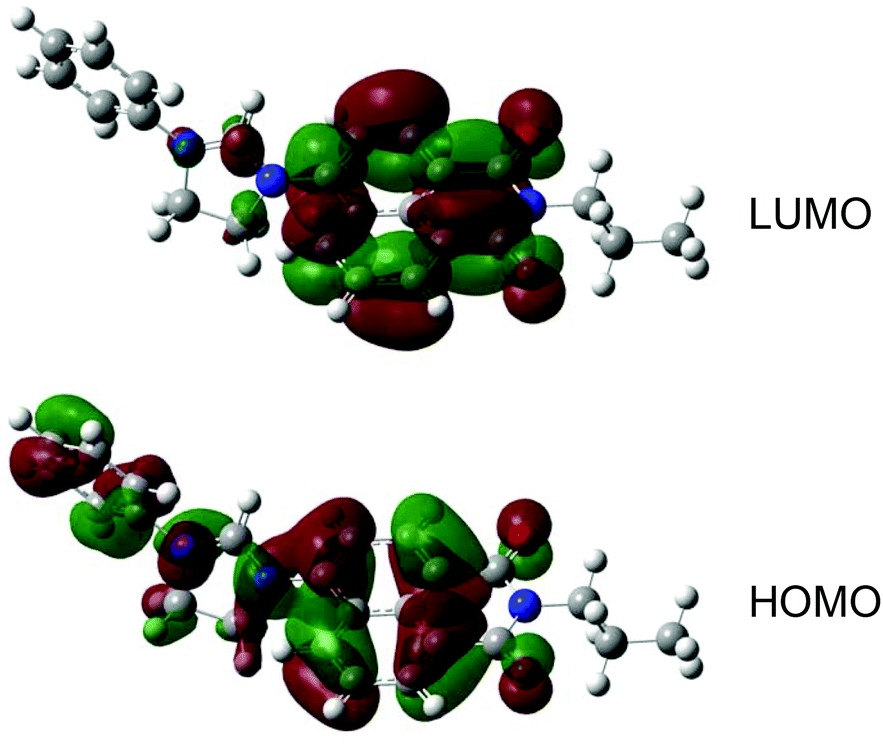 | ||
| Fig. 5 Calculated HOMO and LUMO of [HL4]+ [PBE0/6-311+G(d,p)]. | ||
The nature of the low energy bands do not change significantly upon coordination of the pro-ligands to Au(I), in that they are still dominated by HOMO–LUMO π–π* transitions. However, the maxima are predicted to undergo a small red-shift, which replicates the experimentally observed spectra. A comparison of the HOMO and LUMO orbital energies in the pro-ligands and complexes reveals that both HOMO and LUMO increase in energy as a result of coordination to the Au centre, but that the increase in the HOMO energies is marginally more than that found for the LUMO; for example in [HL3]+ the HOMO and LUMO lie at −7.32 eV and −3.06 eV respectively, and at −6.91 eV and −2.73 eV in [AuCl(L3)]. This has the effect of reducing the HOMO–LUMO difference in the complex to 4.18 eV from 4.26 eV in the imidazolium precursor. A similar observation is found for [HL4]+, for which the HOMO–LUMO energy difference decreases from 4.22 eV to 4.13 eV in [AuCl(L4)].
Fluorescence properties
The fluorescence properties of the ligands and complexes were assessed at room temperature in aerated acetone solution (Table 1). Irradiation with 405 nm (corresponding to the excitation line for confocal fluorescence microscopy) relates to excitation of the ICT absorption band. For the pro-ligands this yielded a broad structureless emission peak (Fig. 6) at around 490–510 nm with a corresponding lifetimes in the range 4–10 ns, consistent with previous reports on amino-functionalized 1,8-naphthalimides of this type.13 The variation in imide substituent had minimal influence upon emission wavelength (Fig. 6). Quantum yields were 4–31% implying some relatively bright fluorophores within the series. It was also noted that these species showed excitation wavelength dependent emission wavelengths: excitation at 345 nm, which corresponds to one of the π–π* absorptions gave a higher energy emission at ca. 440 nm, which is assumed to correlate with emission from a π–π* excited state. | ||
| Fig. 6 Normalised steady state emission spectra of selected ligands excited at 405 nm (recorded in aerated acetone). | ||
For the complexes, irradiation at 405 nm again generally revealed a ligand-based ICT peak ca. 500 nm, but in some cases an additional peak or shoulder at higher energies (440–450 nm) was also noted (Fig. 7). Again, this shorter wavelength peak was enhanced by using excitation at 345 nm. Comparison of the fluorescent lifetimes and quantum yield values for the complexes with the corresponding free ligands generally indicated a partial quenching of the ICT emission in the presence of a coordinated, heavy atom. For example, in the case of [AuCl(L3)] the fluorescence lifetime was dominated by a very short lifetime component ca. 1 ns, contrasting with, and differentiated from, the value obtained for [HL3]PF6 (Fig. 7).
 | ||
| Fig. 7 Top: Normalised emission spectra of selected complexes excited at 405 nm (recorded in aerated acetone). Bottom: Time-resolved measurements showing the comparative decay profiles for ligand [HL3]PF6 (red) and complex [AuCl(L3)] (blue) against the instrument response (green). | ||
Cytotoxicity and bioimaging studies
Au(I) NHC complexes have been the focus of several studies examining biological applications, including mitochondria-targeted, anti-tumour,17 and antimicrobial18 examples. Fine-tuning of the ligand architecture has allowed the development of lipophilic, cationic Au(I)–NHC complexes that selectively induce apoptosis in cancer cells and show selective targeting of mitochondrial selenoproteins, such as TrxR.19 Fluorophore-tethered Au(I)–NHC compounds have demonstrated apoptosis in cancer cells,20 as well as DNA binding capability.21 Au(I) NHC frameworks provide an excellent basis for the development of anticancer agents.22Prior to any imaging work, cytotoxicity assessments were obtained for complexes and pro-ligands using the MTT assay (Table 3). Data were recorded for four cancer cell lines: LOVO, A549, PC3 and MCF-7, the latter for correlation with the imaging studies. The data clearly revealed that the uncharged complexes were less toxic to LOVO, A549 and PC3 cells than the corresponding cationic pro-ligands. This is in contrast to our previous findings which have shown that gold complexes of the type [Au(PPh3)(L)] can often have enhanced cytotoxicity versus the free ligand, attributed to the presence of lipophilicity enhancing co-ligands (e.g. PPh3) at Au(I).23 The importance of charge has also been noted in cytotoxic iridium(III) complexes, with cationic species proving more toxic to cell lines24 and Gram positive bacteria25 than neutral analogues. For the pro-ligands, [HL6]PF6 was the least toxic with IC50 values >100 μM for each cell type. In contrast, closely related [HL5]PF6 was the most toxic, with strong activity against LOVO, PC3 and MCF-7s. Highly lipophilic [HL7]PF6 also showed strong activity against LOVO and MCF-7. Overall the results showed that relatively minor changes in the molecular structures had a profound influence upon the cytotoxicity.
| Compound | LOVO | A549 | PC3 | MCF-7 |
|---|---|---|---|---|
| [HL1]PF6 | 30.07(3.19) | 33.62(5.09) | 56.40(3.08) | 68.35(3.95) |
| [HL2]PF6 | 73.36(2.36) | >100 | 98.1(3.91) | 66.83(2.85) |
| [HL3]PF6 | 42.75(8.66) | 56.83(4.06) | 71.27(2.89) | 61.21(3.02) |
| [HL4]PF6 | 51.81(5.34) | 75.68(1.10) | 72.82(3.03) | 56.38(2.84) |
| [HL5]PF6 | 7.27(1.19) | 47.10(0.55) | 8.58(0.03) | 7.12(0.36) |
| [HL6]PF6 | >100 | >100 | >100 | >100 |
| [HL7]PF6 | 5.22(2.49) | 53.95(1.41) | 42.00(5.69) | 9.98(4.77) |
| [HL8]PF6 | >100 | >100 | >100 | 33.23(5.84) |
| [AuCl(L1)] | 68.94(2.29) | >100 | >100 | 84.21(11.38) |
| [AuCl(L2)] | 91.64(3.20) | >100 | >100 | 62.88(5.58) |
| [AuCl(L3)] | >100 | >100 | >100 | 48.58(10.58) |
| [AuCl(L4)] | 41.54(2.88) | >100 | >100 | 27.79(11.89) |
| [AuCl(L6)] | 49.10(7.97) | >100 | >100 | 46.21(13.01) |
| [AuCl(L7)] | 50.92(6.20) | >100 | 46.63(0.71) | 41.75(9.34) |
| [AuCl(L8)] | 40.99(11.08) | >100 | >100 | 4.82(0.77) |
A limited number of Au(I) complexes incorporating fluorescent ligands have been reported26 for cellular imaging, although the fluorescent properties of such species are not always optimal for microscopy. Therefore, cellular microscopy studies including FLIM were undertaken with one of the pro-ligand/complex pairings, [HL3]PF6 and [AuCl(L3)], using MCF-7 cells. Fluorophore concentrations of 100 μg ml−1 (i.e. comparable to the IC50 values) and more dilute 10 ng ml−1 (well below the obtained IC50 values) in DMSO were used for [HL3]PF6 and [AuCl(L3)]. In all cases the cells were irradiated with 405 nm excitation which matches well with the ICT absorption band of these naphthalimide-based fluorophores; detection wavelengths were 500–550 nm.
Firstly, [HL3]PF6 demonstrated rapid uptake (<30 min) and internalisation by the MCF-7 cells (Fig. 8). Cytoplasmic fluorescence was concentrated in well-defined spheroidal volumes consistent with organelle localisation. There was no evidence of nuclear sequestration. The corresponding complex [AuCl(L3)] also showed reasonable uptake internalisation with bright foci of fluorescence observable within the cytoplasm, but not the nucleus (Fig. S2, ESI‡).
 | ||
| Fig. 8 Confocal fluorescence microscopy images of MCF-7 cells incubated with [HL3]PF6 showing localized signals (λex = 405 nm and λem = 525 nm). Cells incubated with 100 μg ml−1 of the fluorophore. | ||
More distinct localisation was observed for both [HL3]PF6 and [AuCl(L3)] at a diluted probe concentration of 10 ng ml−1 (1:10000 dilution when compared to the preliminary microscopy studies). Under these conditions, co-localisation studies were investigated using commercial fluorophores for staining of mitochondrial, endoplasmic reticulum (ER), Golgi and lysosomal compartments (Fig. 9 and Fig. S3 and S4, ESI‡). Fig. 9 shows the obtained superimposed images with [HL3]PF6 and [AuCl(L3)] respectively, and colocalisation is clearly evident (as indicated by the yellow-orange colouration) only with the lysosomal stain, LysoTracker Deep Red. A recently reported acridine-functionalized Au(I) compound has also shown lysosomal localisation.20
 | ||
| Fig. 9 Confocal fluorescence microscopy images of MCF-7 cells incubated with [HL3]PF6 (top) and [AuCl(L3)] (bottom). Representative superimposed images where yellow-orange signifies co-localisation (from left to right) with an ER stain, Golgi stain and lysosomal stain. Note strong lysosomal fluorescence co-localisation. | ||
Having fully elucidated the intracellular localisation of both probes, FLIM analyses was applied to MCF-7 cells which had been incubated with [HL3]PF6 and [AuCl(L3)]. For FLIM, the cells were imaged via a pulsed 440 nm diode laser excitation source, which is again well matched to the ICT absorption band of the naphthalimide fluorophores, and data collection was facilitated through time correlated single photon counting. Decay curves were fitted using an n-exponential tail fit, while applying an estimated instrument response function. In all cases the lowest χ2 square value was obtained by modelling three components to the exponential decay.
For [HL3]PF6, the FLIM images (Fig. 10) show the localised hot spots of fluorescence which were earlier attributed to lysosomal localisation. From the data analyses of the image, the dominant lifetime component was obtained as 9.2 ns. This value compares favourably (cf. 9.1 ns) with the solution state spectroscopic measurements (Table 1), validating the data fitting protocol. This value also implies that the fluorescence labelling of the lysosomal compartments of the cell can be clearly attributed to [HL3]PF6 and that the intrinsic structure of the fluorophore is intact following incubation with the cells.
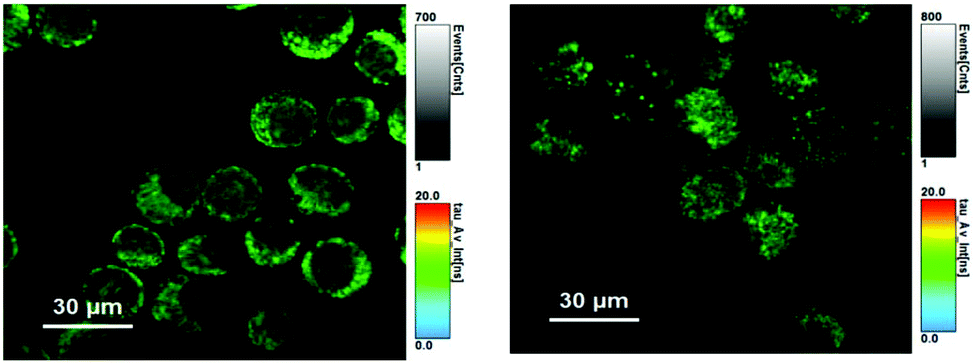 | ||
| Fig. 10 FLIM images of MCF-7 cells incubated with [HL3]PF6 (left) and [AuCl(L3)] (right) using pulsed excitation at 440 nm. Fitted parameters for left image: τ1 = 9.2 ns (77%), τ2 = 3.4 ns (20%), τ1 = 0.4 ns (3%); for right image, τ1 = 11.0 ns (68%), τ2 = 3.9 ns (25%), τ1 = 0.5 ns (7%). | ||
In the case of MCF-7 cells incubated with [AuCl(L3)], good quality FLIM images were again obtained. In this case, the fitted parameters suggest a dominant lifetime of ca. 11 ns (Fig. 10). This value is significantly longer than that spectroscopically obtained (1.1 ns), but more comparable with the fluorescence lifetime for [HL3]PF6. Therefore, the FLIM data suggests that the gold complex may dissociate under biological conditions to regenerate the dihydroimidazolinium-like fluorophore. In these cases, the fluorescence lifetime obtained from FLIM might be indicative of such an intracellular process.
The data from these microscopy measurements suggests that further work is required to understand the speciation of these Au(I) carbene complexes within a biological environment. In particular, it is important to consider the acidic nature of lysosomal compartments and the effect that this may have on such complexes. Since the solubility of [AuCl(L3)] is limited to organic solvents it has not been possible to elucidate the pH dependent stability of this species, but previous studies have noted how the gold–carbene bond can tolerate acidic conditions at elevated temperatures.27 Further, our studies have shown that such complexes are stable >6 h in DMSO (incubated at 37 celcius) solutions in the absence of light, as indicated by 13C NMR data (retention of coordinated carbene). However, it is well known that Au(I) carbene complexes can display photoinduced decomposition.28 Therefore one must exercise caution when seeking to correlate cytotoxicity data with confocal fluorescence microscopy imaging. Further work is required to unravel the influence that light irradiation may have upon the speciation and toxicity of gold(I) complexes within a cell.
Conclusions
The stepwise synthesis of a range of 4-substituted 1,8-naphthalimide derivatives that incorporate a dihydroimidazolium cationic unit give fluorescent species with emission dominated by an ICT emission band around 500 nm. Once deprotonated, these dihydroimidazolium salts can be utilised as NHC ligands for Au(I) to form complexes. In all cases, the fluorescence of these complexes is again dominated by the ligand, but often partially quenched by the presence of Au(I) leading to the possibility of discriminating ligand/complex pairings on the basis of fluorescence lifetime. The varying functionality of the ligands led to different cytotoxicity profiles across cancer cell lines, with the cationic pro-ligands proving generally more toxic than the corresponding complexes. Selected ligands and complexes were incubated with MCF-7 cells and fluorescence microscopy was undertaken, showing that these fluorophores are biocompatible. More detailed colocalisation studies were conducted on a ligand/complex pairing and identified that the species show definitive localisation in lysosomes. Finally, we have shown that the ICT fluorescence lifetime of these species can be utilised in FLIM. Lifetime imaging studies on [AuCl(L3)] indicate that this species probably dissociates from gold within the cell. Further studies are now required to fully elucidate the possible mechanism for this process, but FLIM appears to be a potential imaging tool for supporting such investigations.Experimental
General considerations
All reagents and solvents were commercially available and were used without further purification if not stated otherwise. For the measurement of 1H, 31P, and 13C NMR spectra a Bruker Fourier300 (300 MHz), Bruker AVANCE HD III equipped with a BFFO SmartProbe™ (400 MHz) or Bruker AVANCE III HD with BBO Prodigy CryoProbe (500 MHz) was used. The obtained chemical shifts δ are reported in ppm and are referenced to the residual solvent signal. Spin–spin coupling constants J are given in Hz.Low-resolution mass spectra were obtained by the staff at Cardiff University. High-resolution mass spectra were carried out at the EPSRC National Mass Spectrometry Facility at Swansea University. High resolution mass spectral (HRMS) data were obtained on a Waters MALDI-TOF mx at Cardiff University or on a Thermo Scientific LTQ Orbitrap XL by the EPSRC UK National Mass Spectrometry Facility at Swansea University. IR spectra were obtained from a Shimadzu IR-Affinity-1S FTIR. Reference to spectroscopic data are given for known compounds. UV-Vis studies were performed on a Shimadzu UV-1800 spectrophotometer as MeCN solutions (2.5 or 5 × 10−5 M). Photophysical data were obtained on a JobinYvon–Horiba Fluorolog spectrometer fitted with a JY TBX picosecond photodetection module as MeCN solutions. Quantum yield measurements were obtained on aerated MeCN solutions of the complexes using [Ru(bpy)3](PF6)2 in aerated MeCN as a standard (Φ = 0.016).29 Emission spectra were uncorrected and excitation spectra were instrument corrected. The pulsed source was a Nano-LED configured for 295 nm or 372 nm output operating at 1 MHz. Luminescence lifetime profiles were obtained using the JobinYvon–Horiba FluoroHub single photon counting module and the data fits yielded the lifetime values using the provided DAS6 deconvolution software.
X-ray diffraction
For both samples, a suitable crystal30 was selected and mounted on a MITIGEN holder in oil on a Rigaku FRE+ (45.0 kV, 55.0 mA) equipped with HF Varimax confocal mirrors (70 μm focus) and an AFC12 goniometer and HG Saturn 724+ detector ([HL3]PF6) or a HyPix 6000 detector ([AuCl(L3)]). The crystals were kept at T = 100(2) K during data collection. Data were measured using profile data from ω-scans using MoKα radiation. Cell determination and data collection were carried out using CrystalClear31 ([HL3]PF6) or CrystalisPro32 ([AuCl(L3)]). With the data reduction, cell refinement and absorption correction using CrystalisPro. Using Olex2,33 the structures were solved with the ShelXT34 structure solution program and the models were refined with version 2014/7 of ShelXL35 using Least Squares minimisation. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model. For sample [HL3]PF6, the PF6 anion was disordered over two positions and for [AuCl(L3)] the propyl chain was disordered over two positions. As such various geometrical (SADI) and displacement (SIMU, RIGU) restraints were applied to the disordered atoms. Diffractometer: Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator with VHF Varimax optics (70 μm focus). Cell determination and data collection: CrystalClear-SM Expert 3.1 b27 (Rigaku, 2013). Data reduction, cell refinement and absorption correction: CrystAlisPro. Structure solution: SUPERFLIP.36 For sample L3, the PF6 anion was disordered over two positions. As such various geometrical (SAME) and displacement (SIMU) restraints were applied.Cytotoxicity assessment via MTT assay
The cytotoxicity of the complexes was assessed using the colourimetric and quantitative MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay, first reported by Mosmann.37 Quantification was achieved using a multi-well scanning spectrophotometer and reported as an IC50 value.Method for cytotoxicity analysis
Anti-tumor evaluation in MCF7, LOVO, A549 and PC3 cell lines was performed by MTT assay. Compounds were prepared as 0.1–100 mM stock solutions dissolved in DMSO and stored at −20 °C. Cells were seeded into 96-well microtitre plates at a density of 5 × 103 cells per well and allowed 24 h to adhere. Decimal compound dilutions were prepared in medium immediately prior to each assay (final concentration 0.1–100 μM). Experimental medium was DMEM +10% FCS (PC3 and Lovo) or RPMI +10% heat inactivated FCS (A549 and MCF7). Following 96 h compound exposure at 37 °C, 5% CO2, MTT reagent (Sigma Aldrich) was added to each well (final concentration 0.5 mg ml−1). Incubation at 37 °C for 4 h allowed reduction of MTT by viable cells to an insoluble formazan product. MTT was removed and formazan solubilized by addition of 10% Triton X-100 in PBS. Absorbance was read on a Tecan Sunrise spectrophotometer at 540 nm as a measure of cell viability; thus inhibition relative to control was determined (IC50) from four independent sets of data and the standard deviations calculated from these data sets.Confocal imaging of MCF-7 cells
:1000 dilution from the commercial kit; λex = 577 nm; λem = 609 nm); the endoplasmic reticulum with ER Tracker red (1 μM; λex = 587 nm; λem = 615 nm); the Golgi apparatus with BODIPY TR ceramide (5 μM; λex = 590 nm; λem = 620 nm) and lysosomes with LysoTracker Red (1 μM; λex = 577 nm; λem = 590 nm). All fluorescent compounds were incubated for 30 minutes at 37 °C then washed in warmed PBS before imaging.
Density functional calculations
Calculations were performed using the Gaussian 09 program.38 Calculated structures were optimized without symmetry constraints and the nature of the stationary point verified using a frequency calculation; in all cases no imaginary frequencies were observed. The PBE039 functional was used along with the 6-311+G(d,p) triple-ζ basis set40 on all centres except for Au, for which the Stuttgart-Dresden basis set was used with a relativistic effective core potential.41 Solvent was included in all calculations as the polarized continuum model, with the molecular cavity defined by a united atom model that incorporates hydrogen into the parent heavy atom.42 TD-DFT calculations were carried out with at the same level of theory as the geometry optimizations; the first 24 excited states were calculated. NMR shielding constants were computed using the gauge including atomic orbital method incorporated into Gaussian 09;1013C chemical shifts were computed relative to benzene (δC = 129.2 ppm).Reagents and precursors
Cl-Nap derivatives43 and [AuCl(tht)]44 were prepared according to the literature. All other reagents were used as received and general and standard precautions were taken during their handling and manipulation.Ligands and precursors
920). HRMS found m/z 382.1752, calcd m/z 382.1761 for [C21H24N3O4]+. IR (solid) ν/cm−1: 2978, 2947, 2875, 1697, 1651, 1581, 1503, 1462, 1450, 1346, 1157, 1123, 1090, 1057, 825, 783, 754, 739, 556, 422, 405, 392.
980). HRMS found m/z 336.1709, calcd m/z 336.1707 for [C20H22N3O2]+. IR (solid) ν/cm−1: 3381, 3089, 2968, 1697, 1651, 1591, 1514, 1443, 1429, 1408, 1389, 1350, 1269, 1234, 1201, 1155, 1086, 1069, 962, 891, 876, 825, 789, 760, 738, 556, 463, 434, 409.
320), 344 (0.599). HRMS found m/z 384.1700, calcd m/z 384.1707 for [C24H22N3O2]+. IR (solid) ν/cm−1: 2974, 1699, 1651, 1626, 1585, 1494, 1391, 1364, 1350, 1271, 1236, 1072, 8423, 829, 787, 758, 689, 557, 469, 434.
560). HRMS found m/z 384.1718, calcd m/z 384.1712 for [C24H22N3O2]+. IR (solid) ν/cm−1: 2372, 2341, 1699, 1651, 1582, 1549, 1497, 1454, 1423, 1381, 1346, 1290, 1234, 1182, 1155, 1072, 1028, 970, 876, 827, 783, 754, 734, 702, 665, 613, 583, 556, 469, 407.
620). HRMS found m/z 406.2482, calcd m/z 406.2489 for [C25H32N3O2]+. IR (solid) ν/cm−1: 2959, 2924, 2855, 1703, 1651, 1589, 1508, 1391, 1354, 1254, 1232, 1155, 1093, 827, 785, 754, 555, 416.
760), 345 (11860). HRMS found m/z 597.1551, calcd m/z 597.1559 for [C24H21N3O2AuCl–HCl + NH4]+. IR (solid) ν/cm−1: 2967, 2955, 2873, 1703, 1655, 1622, 1591, 1506, 1475, 1433, 1389, 1354, 1340, 1286, 1276, 1238, 1068, 1018, 958, 908, 858, 785, 764, 754, 692, 673, 588, 546.
160), 344 (14560). HRMS found m/z 650.2071, calcd m/z 650.2076 for [C29H31AuN3O2–Cl]+. IR (solid) ν/cm−1: 3059, 2949, 2918, 2851, 1703, 1655, 1620, 1589, 1502, 1477, 1438, 1404, 1392, 1357, 1340, 1321, 1288, 1234, 1176, 1097, 1074, 1045, 1024, 858, 785, 754, 692, 671, 651, 613, 584, 559, 543, 482, 410.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Cardiff University for an Endowment Studentship and EPSRC for DTP funding (L. M. G). Dr C. Williams holds a Sêr Cymru II Fellowship part funded by the European Regional Development Fund through the Welsh Government. The EPSRC Mass Spectrometry National Service (Swansea University) and EPSRC National Crystallographic Service (University of Southampton) are also thanked. Dr E. Lloyd-Evans and Dr P. Watson (Cardiff University) are thanked for commercial fluorophores and Mr H. Mottram for cytotoxicity data.Notes and references
- G. J. Higby, Gold Bull., 1982, 15, 130 CrossRef CAS PubMed; P. J. Sadler, Struct. Bonding, 1976, 29, 171 CrossRef.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550 CAS.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540 CrossRef CAS PubMed.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863 RSC.
- E. R. T. Tiekink, Crit. Rev. Oncol. Hematol., 2002, 42, 225 CrossRef CAS PubMed; S. S. Gunatilleke and A. M. Barrios, J. Med. Chem., 2006, 49, 3933 CrossRef PubMed.
- S. B. Aher, P. N. Muskawar, K. Thenmozhi and P. R. Bhagat, Eur. J. Med. Chem., 2014, 81, 408 CrossRef CAS PubMed; A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23 RSC; H. G. Raubenheimer and S. Cronje, Chem. Soc. Rev., 2008, 37, 1998 RSC; J. F. Arambula, R. McCall, K. J. Sidoran, D. Magda, N. A. Mitchell, C. W. Bielawski, V. M. Lynch, J. L. Sessler and K. Arumugam, Chem. Sci., 2016, 7, 1245 RSC.
- (a) L. E. Wedlock and S. J. Berners-Price, Aust. J. Chem., 2011, 64, 692 CrossRef CAS; (b) L. E. Wedlock, M. R. Kilburn, J. B. Cliff, L. Filgueira, M. Saunders and S. J. Berners-Price, Metallomics, 2011, 3, 917 RSC; (c) O. Karaca, V. Scalcon, S. M. Meier-Menches, R. Bonsignore, J. M. J. L. Brouwer, F. Tonolo, A. Folda, M. P. Rigobello, F. E. Kuhn and A. Casini, Inorg. Chem., 2017, 56, 14237 CrossRef CAS PubMed.
- E. E. Langdon-Jones and S. J. A. Pope, Chem. Commun., 2014, 50, 10343 RSC.
- J. J. Dunsford, K. J. Cavell and B. M. Kariuki, Organometallics, 2012, 31, 4118 CrossRef CAS; P.-H. Lanoe, B. Najjari, F. Hallez, G. Gontard and H. Amouri, Inorganics, 2017, 5, 58 CrossRef.
- (a) R. J. Ditchfield, J. Chem. Phys., 1972, 56, 5688 CrossRef CAS; (b) A. M. Lee, N. C. Handy and S. M. Colwell, J. Chem. Phys., 1995, 103, 10095 CrossRef CAS.
- A. M. Sarotti and S. C. Pellegrinet, J. Org. Chem., 2009, 74, 7254 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- (a) S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601 RSC.
- A. Vlček and S. Zalis, Coord. Chem. Rev., 2007, 251, 258 CrossRef.
- D. Jacquemin, E. A. Perpète, G. Scalmani, I. Ciofini, C. Peltier and C. Adamo, Chem. Phys., 2010, 372, 61 CrossRef CAS.
- P. J. Stephens and H. Nobuyuki, Chirality, 2010, 22, 229 CAS.
- P. J. Barnard, M. V. Baker, B. W. Skelton, A. H. White and S. J. Berners-Price, J. Inorg. Biochem., 2003, 96, 99 CrossRef CAS; P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642 CrossRef PubMed.
- I. Özdemir, A. Denizci, H. T. Öztürk and B. Çetinkaya, Appl. Organomet. Chem., 2004, 18, 318 CrossRef.
- (a) J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570 CrossRef CAS PubMed; (b) B. A. Stanley, V. Sivakumaran, S. Shi, I. McDonald, D. Lloyd, W. H. Watson, M. A. Aon and N. Paolocci, J. Biol. Chem., 2011, 286, 33669 CrossRef CAS PubMed; (c) C. F. Williams, N. Yarlett, M. A. Aon and D. Lloyd, Mol. Biochem. Parasitol., 2014, 196, 45 CrossRef CAS PubMed.
- R. Visbal, V. Fernández-Moreira, I. Marzo, A. Laguna and M. C. Gimeno, Dalton Trans., 2016, 45, 15026 RSC.
- A. Meyer, L. Oehninger, Y. Geldmacher, H. Alborzinia, S. Wölfl, W. S. Sheldrick and I. Ott, ChemMedChem, 2014, 9, 1794 CAS.
- M. Porchia, M. Pellei, M. Marinelli, F. Tisato, F. Del Bello and C. Santini, Eur. J. Med. Chem., 2018, 146, 709 CrossRef CAS PubMed.
- L. A. Mullice, H. J. Mottram, A. J. Hallett and S. J. A. Pope, Eur. J. Inorg. Chem., 2012, 3054 CrossRef CAS.
- C. Carporale, C. A. Bader, A. Sorvina, K. D. M. Magee, B. W. Skelton, T. A. Gillam, P. J. Wright, P. Raiteri, S. Stagni, J. L. Morrison, S. E. Plush, D. A. Brooks and M. Massi, Chem. – Eur. J., 2017, 23, 15666 CrossRef PubMed.
- V. Fiorini, I. Zannoni, A. L. Costa, A. Hochkoeppler, V. Zanotti, A. Ranieri, M. Masi, A. Stefan and S. Stagni, Dalton Trans., 2017, 46, 12328 RSC.
- For recent examples: L. H. Davies, R. W. Harrington, W. Clegg and L. J. Higham, Dalton Trans., 2014, 43, 13485 RSC; A. Meyer, C. P. Bagowski, M. Kokoschka, M. Stefanopoulou, H. Alborzinia, S. Can, D. H. Vlecken, W. S. Sheldrick, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2012, 51, 8895 CrossRef CAS PubMed.
- M. E. Garner, W. Niu, X. Chen, I. Ghiviriga, K. A. Abboud, W. Tan and A. S. Veige, Dalton Trans., 2015, 44, 1914 RSC.
- G. D. Frey, R. D. Dewhurst, S. Kousar, B. Donnadieu and G. Bertrand, J. Organomet. Chem., 2008, 693, 1674 CrossRef CAS PubMed.
- M. Frank, M. Nieger, F. Vogtle, P. Belser, A. von Zelewsky, L. De Cola, V. Balzani, F. Barrigelletti and L. Flamigni, Inorg. Chim. Acta, 1996, 242, 281 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- CrystalClear-SM Expert 3.1 b27, Rigaku, 2013 Search PubMed.
- CrystAlisPro 1.171.38.41, Rigaku Oxford Diffraction, 2015 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 27, 3 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- D. Andrae, U. Häußermann, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- (a) E. E. Langdon-Jones, D. Lloyd, A. J. Hayes, S. D. Wainwright, H. J. Mottram, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2015, 54, 6606 CrossRef CAS PubMed; (b) E. E. Langdon-Jones, N. O. Symonds, S. E. Yates, A. J. Hayes, D. Lloyd, R. Williams, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2014, 53, 3788 CrossRef CAS PubMed.
- R. Uson, A. Laguna and J. Vicente, J. Organomet. Chem., 1977, 131, 471 CrossRef CAS.
Footnotes |
| † Information on the data underpinning the results presented here, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2018.0064817469. |
| ‡ Electronic supplementary information (ESI) available: DFT Cartesian coordinates of all calculated structures in a format for convenient visualization; details of all excited states and examples of NMR spectra. CCDC 1831224 and 1831225 for [HL]PF6 and [AuCl(L)]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04069a |
| This journal is © The Royal Society of Chemistry 2019 |