
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring transition metal fluoride chelates – synthesis, properties and prospects towards potential PET probes†
Philip J.
Blower
 a,
William
Levason
b,
Sajinder K.
Luthra
c,
Graeme
McRobbie
c,
Francesco M.
Monzittu
*b,
Thomas O.
Mules
b,
Gillian
Reid
*b and
M. Nadeem
Subhan
b
a,
William
Levason
b,
Sajinder K.
Luthra
c,
Graeme
McRobbie
c,
Francesco M.
Monzittu
*b,
Thomas O.
Mules
b,
Gillian
Reid
*b and
M. Nadeem
Subhan
b
aSchool of Biomedical Engineering and Imaging Sciences, King's College London, St Thomas’ Hospital, London SE1 7EH, UK
bChemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk; F.Monzittu@soton.ac.uk
cPollards Wood, Nightingales Lane, Chalfont St Giles, Bucks, HP8 4SP, UK
First published on 24th April 2019
Abstract
The coordination chemistry of the first row transition metal trifluorides with terpy (2,2′:6′,2′′-terpyridine) and Me3-tacn (1,4,7-trimethyl-1,4,7-triazacyclononane) was explored to identify potential systems for 18F radiolabelling. The complexes [MF3(L)] (M = Cr, Mn, Fe, Co; L = Me3-tacn, terpy) were synthesised and fully characterised by UV-vis and IR spectroscopy, microanalysis, and, for the diamagnetic [CoF3(L)], using 1H, 19F{1H} and 59Co NMR spectroscopy. Single crystal X-ray analyses are reported for [MF3(Me3-tacn)] (M = Mn, Co), [FeF3(terpy)] and [FeF3(BnMe2-tacn)]. Stability tests on [MF3(Me3-tacn)] (M = Cr, Mn, Fe) and [M′F3(terpy)] (M′ = Cr, Fe) were performed and Cl/19F halide exchange reactions on [CrCl3(Me3-tacn)] using [Me4N]F in anhydrous MeCN solution, and [FeCl3(Me3-tacn)] using [Me4N]F in anhydrous MeCN or KF in aqueous MeCN solution were also carried out. Halide exchange reactions proved to be successful in forming [FeF3(Me3-tacn)] in aqueous MeCN solution within 30 minutes. Based upon the clean Cl/F exchange and the good stability observed for [FeF3(Me3-tacn)] in a range of competitive media, this was identified as a possible candidate for radiolabelling. 18F/19F isotopic exchange was achieved by addition of [18F]F− in the cyclotron target water to a MeCN solution of the benzyl-substituted analogue, [FeF3(BnMe2-tacn)], at a range of concentrations down to 24 nM with heating to 80 °C for 10 min.; the resulting [Fe18F19F2(BnMe2-tacn)] shows radiochemical purity (RCP) ≥90% after 2 h in a range of formulations, including 10% EtOH/phosphate buffered saline (PBS) and 10% EtOH/human serum albumin (HSA). This is the first reported complex with a transition metal directly bonded to [18F]F−.
Introduction
C–18F based radiotracers are extensively used in positron emission tomography (PET) for imaging purposes in oncology, cardiology and neurology.1 Thousands of PET scans are performed daily on cancer patients worldwide, providing important diagnostic and clinical information. Peptides that target receptors overexpressed on the surface of the diseased cells are becoming increasingly important in diagnostic PET imaging agents.2 However, the reaction conditions for the formation of the C–18F bond are often incompatible with biomolecules (e.g. peptides).3 Organic solvents that degrade the peptide, high temperatures and competing sites for the 18F-labelling reaction present in the biomolecule are the major problems encountered. As a result, often the [18F]F− is first incorporated into an organic molecule (prosthetic group) and then conjugated to a peptide, resulting in an increase of the total reaction time and the number of steps of the process.3 The need to improve these aspects has led to the investigation of alternative non-C–18F moieties. To date, several main group elements attached to [18F]F− have been studied, including boron-,4 aluminium-,5,6 gallium-,7,8 sulfur-,9 and silicon-fluoride10 systems. These are discussed in recent reviews.11 The two Group 13 metals are often coordinated to macrocyclic ligands (triazacyclononane derivatives) which confer thermodynamic and kinetic stability to the chelates and, in the case of the systems with 1,4-dimethyl-7-benzyl-1,4,7-triazacyclononane (BnMe2-tacn), the benzyl group provides a site for further functionalisation and bioconjugation.6,7 In this approach to PET radiotracers based on metal coordination complexes, the stability of the complex will be strongly dependant on the properties of the metal centre. The M–F bond dissociation energy has an important role since it should be stronger compared to M–Cl, hence allowing fluorination through halide exchange reactions and, ultimately, it should be resistant in physiological conditions. Other aspects related to the metal to be considered are its size (dictating the coordination number), its redox chemistry and oxophilicity (water or anions such as phosphate should not disrupt the coordination around the metal), its Lewis acidity and its lability, allowing sufficiently rapid substitution of fluoride into the coordination sphere (when the 18F half-life of 110 min is considered).We previously developed the chemistry of the Group 13 metal fluorides (Al, Ga) towards neutral nitrogen ligands and reported that radioactive 18F can be introduced into [MCl3(BnMe2-tacn)] (M = Al, Ga) through Cl/18F halide exchange reactions and that [GaF3(BnMe2-tacn)] can also be 18F-radiolabelled through 18F/19F isotopic exchange using a very small amount of material (27 nmol); both methods were successful in mild conditions, i.e. aqueous solution, room temperature or 80 °C.6,7,12 We also investigated the transition metal fluorides using Sc(III), Y(III), La(III) and Lu(III).13 Among these d- and f-block metal systems, only the [ScF3(RMe2-tacn)] (R = Me, Bn) were successfully synthesised through Cl/F halide exchange reactions. [ScF3(BnMe2-tacn)] was identified as a promising system for future 18F-radiolabelling, although, in contrast to the Group 13 systems, it could only be obtained from the trichloride analogue under anhydrous conditions using [NMe4]F or Me3SnF as the fluoride source. However, the trifluoride complex is very stable in water.13
In this work, we present an evaluation of the 3d transition metal (Cr3+, Mn3+, Fe3+ and Co3+) trifluoride complexes for possible PET applications. The distorted octahedral MF3 complexes bearing tridentate Me3-tacn and terpy ligands, are described and their stabilities in water probed by UV-vis spectroscopy. Their properties are discussed in order to identify the best prospects for fluorination for future possible applications in PET. Finally, we demonstrate successful 18F/19F isotopic exchange using [FeF3(BnMe2-tacn)], and confirm its stability in ethanolic phosphate buffered saline (PBS) or human serum albumin (HSA) over at least 2 h.
In setting out the scope for this study, Ti3+ and V3+ were not considered as the former is very readily oxidised, while the latter is also likely to form V(IV) in aqueous solution. Previous work has shown that [VOF2(Me3-tacn)] is readily obtained by adding a few drops of water to a methanol solution of [VF3(Me3-tacn)] in air.14 Nickel was also excluded as no trifluoride complexes are known and Ni(III) is reduced in water.15 The ligand substitution kinetics for the majority of second and third row transition metal complexes are expected to be too slow to allow sufficiently fast halide substitution, given the short half-life of the 18F radionuclide.
The electronic configurations of the 3d metal ions influence their reactivity and the kinetic robustness of the complexes formed. Neutral trifluoride complexes of these metals are scarce or unknown, in contrast to the heavier halides. Octahedral d3 Cr(III) mono-, di- and tri-fluoride complexes with N-donor ligands have been reported.16 In particular, complexes of ammonia and amines have been studied extensively for their absorption and emission properties, contributing to the development of inorganic electronic spectroscopy.17 The neutral species [CrF3(bipy)(OH2)],18fac-[CrF3(Me3-tacn)]19,20 and mer-[CrF3(terpy)]20 have also been reported and structurally characterised. Complexes with tetradentate N-donor ligands can also be found in the literature.21 Neutral complexes of Mn(III) trifluoride with N-donor ligands are known, with the structures of [MnF3(bipy)(OH2)],22 [MnF3(phen)(OH2)],23 [MnF3(terpy)]24 and [{MnF2(Me3-tacn)}2(μ-F)][PF6]25 reported. The d4 configuration associated with Mn(III) causes a Jahn–Teller distortion, a feature exploited in the synthesis of molecular magnets.26 Iron(III) trifluoride complexes reported in the literature include mer-[FeF3(NH3)3]27 and fac-[FeF3(Me3-tacn)].19 While octahedral Co(III) complexes with amine ligands have long been known,28 no crystal structures of neutral Co(III) trifluoride complexes with N-donor or O-donor ligands have been reported. mer-[CoF3(NH3)3] has been characterised spectroscopically.29 The coordination chemistry of transition metal fluoride complexes has been reviewed recently,30 including their complexes with neutral ligands.15
Experimental
Synthetic procedures and characterisation details are presented in the ESI†18F/19F isotopic exchange radiolabelling procedure
In a typical experiment, [FeF3(BnMe2-tacn)]·3.5H2O (1 mg, 2.36 μmol, 0.1 mg, 236 nmol or 0.01 mg, 24 nmol) was dissolved in MeCN (n = 8) (0.75 mL). To this solution was added 0.25 mL of an aqueous solution containing [18F]F− (40–180 MBq) and the vial was heated to 80 °C for 10 min. The crude reaction solution was diluted with water (20 mL) so that approximately 10% of the solvent composition was organic. A small sample (∼100 μL) of the diluted crude reaction solution was removed for analysis by analytical HPLC, which confirmed the percentage incorporation of [18F]F− into the metal complex (based upon integration of the radio peaks). Approximately 6% [18F]F− incorporation was observed when the radiolabelling experiment was performed using 1 mg of the iron complex in MeCN/H2O (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25) at room temperature. The product was purified by either a SPE process or by HPLC.
:25) at room temperature. The product was purified by either a SPE process or by HPLC.
SPE purification protocol
The diluted reaction mixture was trapped on a HLB cartridge, washed with water (5 mL × 3) to remove the residual [18F]F− and MeCN and then the product was eluted from the cartridge with ethanol (1 mL) into either (i) water to result in a formulated product in 80:20 water:EtOH; (ii) PBS to result in a formulated product in 90:10 PBS:EtOH or (iii) HSA to result in a formulated product in 90:10 HSA:EtOH. The formulated product was analysed by HPLC at t = 0 and various time intervals up to 120 min.
Experiments were analysed on an Agilent 1290 HPLC system with an Agilent 1260 DAD UV detector (G4212B) and a Bioscan FC3200 sodium iodide PMT with rate meter. Dionex Chromeleon 6.8 Chromatography data recording software was used to integrate the peak areas.
Analytical HPLC method
Column: Phenomenex Luna 5 μm C18(2) 250 × 4.6 mm. Mobile phase A = water, B = MeCN. Flow rate 1 mL min−1. Gradient 0–15 min (10–90% B), 15–20 min (90% B), 20–21 min (90–10% B), 21–26.5 min (10% B).HPLC purification
Column: Waters XBridge Prep Shield RP18, 5 μm, 10 × 100 mm (p/n 186003258); Dionex Ultimate 3000 pump; Knauer Smartline 2500 UV detector. Mobile phase A = water; B = MeCN. Flow rate 3 mL min−1. Gradient 0–10 min (0–10% B), 10–15 min (10–90% B), 15–20 min (90% B), 20–25 min (90–2% B).Results and discussion
The syntheses of the complexes [MF3(L)] (M = Cr, Mn, Fe, Co; L = terpy, Me3-tacn) were carried out in alcoholic (n-BuOH or MeOH) or dmf solutions at room temperature or under reflux (Scheme 1) and the products were characterised by IR spectroscopy, microanalysis and UV-vis spectroscopy (diffuse reflectance and solution).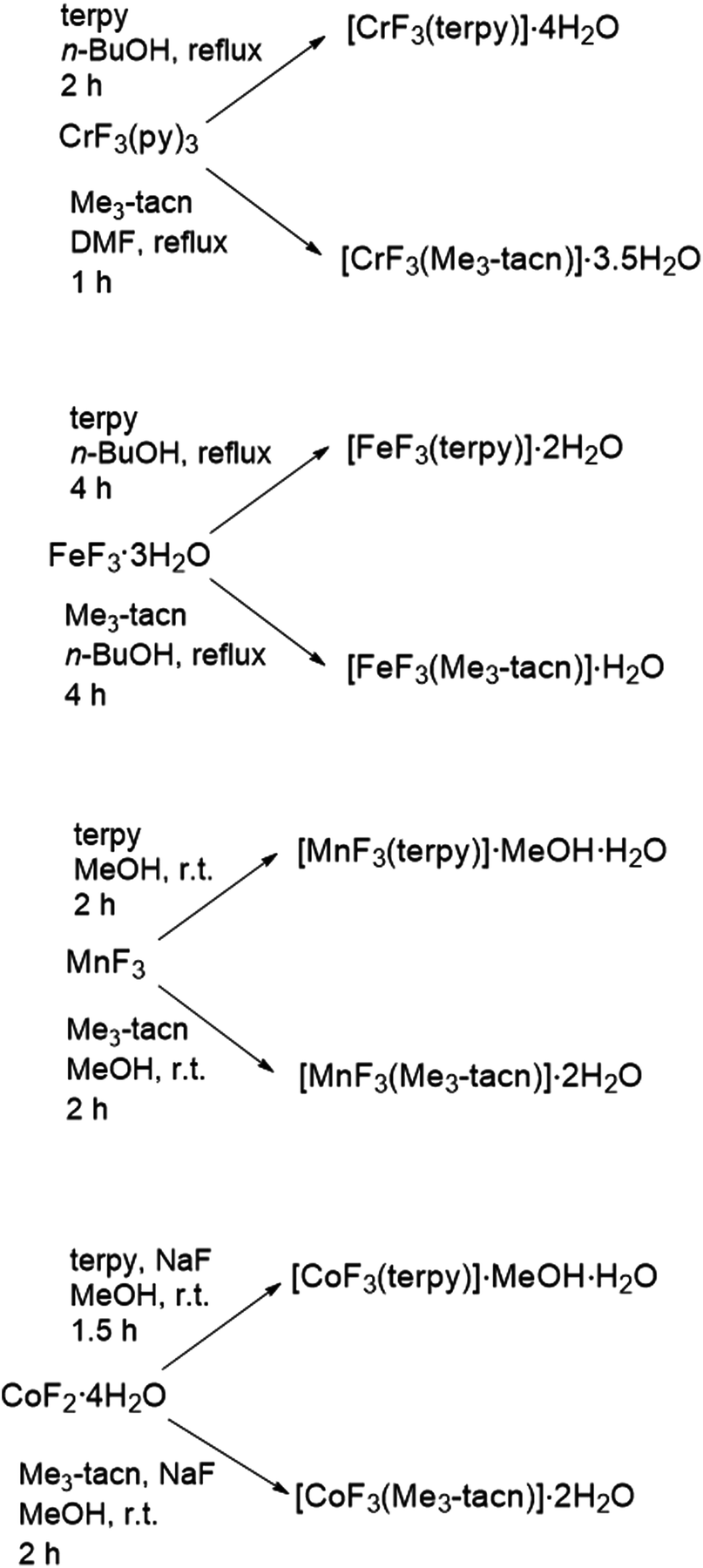 | ||
| Scheme 1 Reaction conditions for the preparation of the complexes. | ||
The trifluoro complexes have a strong tendency to form H-bonding interactions between the fluorides and water molecules in the lattice;8 this has often led to discrepancy in the number of water molecules co-crystallised in the lattice compared to the literature19,20,24 and in some cases in this work leading to differences in the number of associated water molecules between the bulk materials and the crystal structures (for example [MF3(Me3-tacn)], M = Mn, Co, have two water molecules in the bulk, but four in the crystal structure). This might be due to differences in the crystallisation methods employed and the length of time the bulk materials were dried in vacuo. The products are air stable and can be stored outside the glovebox for several weeks. The stability of the complexes was challenged in aqueous solution by the presence of up to 10-fold excess of competitive ions (NaCl, NaF, Na2CO3, Na3PO4, NaOAc), increased temperature and changes in pH at two different time points (t = 0 and t = 4 h). UV-vis spectroscopy was used to monitor the solutions, changes in the position of the relevant d–d transitions and or appearance/disappearance of bands were taken as an indication of the instability of the complex during the experiments.
Chromium
mer-[CrF3(terpy)]·4H2O and fac-[CrF3(Me3-tacn)]·3.5H2O were obtained as purple solids in good yields by reaction of [CrF3(py)3] with the ligands in n-BuOH or dmf, respectively, following literature methods.20 The IR spectra of the solids confirm the presence of water and show two ν(Cr–F) bands for [CrF3(Me3-tacn)]·3.5H2O, as expected for a fac octahedral configuration in C3v symmetry, whereas one very broad band is seen for [CrF3(terpy)]·4H2O (three bands are expected in a mer C2v symmetry, but not resolved). The diffuse reflectance spectra of the complexes are shown in Fig. S8 and S12,† and resemble those reported previously.20Manganese
The reaction of MnF3 with Me3-tacn or terpy in anhydrous MeOH at room temperature produces the species [MnF3(Me3-tacn)]·2H2O and [MnF3(terpy)]·MeOH·3H2O, respectively. The presence of MeOH in [MnF3(terpy)]·MeOH·3H2O was confirmed by 1H NMR spectroscopy (CD3CN, δ = 3.28 (CH3OH), 2.15 (CH3OH)). The molecular composition of [MnF3(Me3-tacn)] was confirmed by a single crystal X-ray structure analysis (Fig. 1).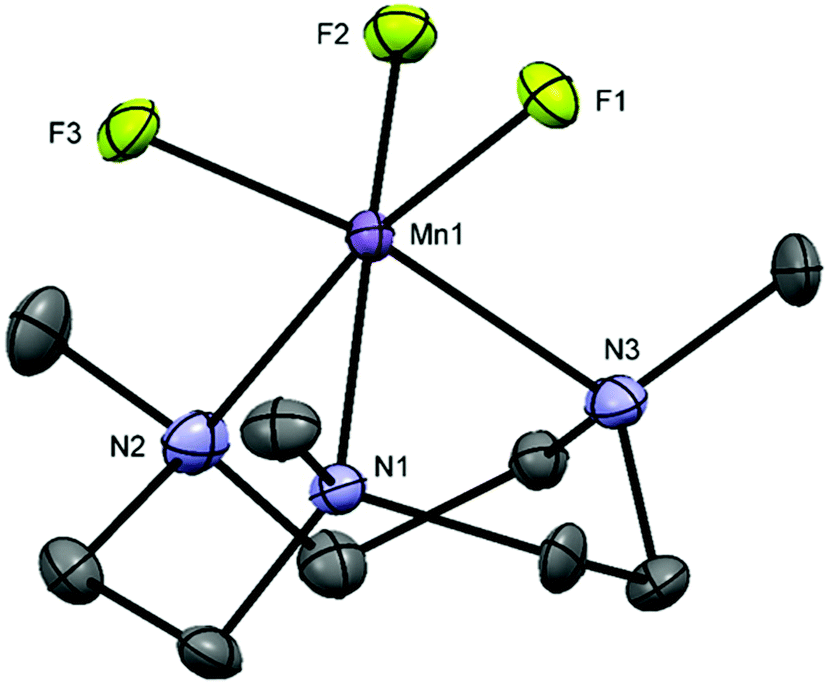 | ||
| Fig. 1 Crystal structure of fac-[MnF3(Me3-tacn)]·4H2O with ellipsoids drawn at the 50% probability level. H atoms and water molecules are omitted for clarity. Select bond lengths (Å) and angles (°): Mn1–F1 = 2.017(2), Mn1–F2 = 1.852(2), Mn1–F3 = 1.848(2), Mn1–N1 = 2.080(3), Mn1–N2 = 2.267(3), Mn1–N3 = 2.096(3), F1–Mn1–N1 = 91.29(10), F3–Mn1–N3 = 170.31(11), F2–Mn1–N1 = 172.65(10), F1–Mn1–F3 = 94.88(9), F1–Mn1–F2 = 94.43(9), N1–Mn1–N2 = 80.92(11), N1–Mn1–N3 = 83.43(11). | ||
The structure shows a distorted octahedral environment with the fluorides facially coordinated to the metal. The Mn–F3 and Mn–N3 bond lengths are elongated by ∼0.17 Å and ∼0.18 Å compared to the other Mn–F and Mn–N distances, respectively. This significant difference in the bond lengths is consistent with a Jahn–Teller distortion in the high spin d4 configuration.25 As confirmation of this, the complex has a magnetic moment of 4.94 BM.31 The tetragonal elongation in the related complex [{MnF2(Me3-tacn)}2(μ-F)][PF6]25 is observed along the axis on which Mn–Fbridging and Mn–N trans to it lie. These bond lengths are ∼0.18 and ∼0.23 Å longer than the other Mn–N and Mn–F bonds, respectively.25 The Mn–Fterminal distance in [{MnF2(Me3-tacn)}2(μ-F)][PF6] is ∼0.03 Å shorter than Mn1–F1 and Mn1–F2 in [MnF3(Me3-tacn)]·2H2O. Extensive H-bonding involving the water molecules and the fluorides is also present (Fig. 2). The same H-bonding pattern was observed in [GaF3(Me3-tacn)]·4H2O.7
 | ||
| Fig. 2 H-Bonding interaction present in [MnF3(Me3-tacn)]·4H2O. | ||
The diffuse reflectance UV-vis spectrum of [MnF3(Me3-tacn)]·4H2O (Fig. S19†) shows intense bands in the UV region due to ligand to metal charge transfer transitions (σN → Mn) and a single d–d transition at ∼520 nm, generically assigned to 5Eg → 5T2g. Although in an octahedral d4 high spin configuration only one spin-allowed transition is predicted (5Eg → 5T2g), Jahn–Teller distortions can often lower the symmetry, resulting in splitting of the single transition. However, in this case splitting is not resolved. These data resemble those of the dimer [{MnF2(Me3-tacn)}2(μ-F)][PF6].25
Splitting is more resolved in [MnF3(terpy)]·MeOH·3H2O (Fig. S15†) and the three bands shown in the spectra are tentatively assigned to 5B1g → 5A1g, 5B1g → 5B2g, 5B1g → 5Eg. The metal centre symmetry is C2v and it is possible that both this and the Jahn–Teller effect results in greater splitting of the bands compared to [MnF3(Me3-tacn)]·2H2O. Strong absorptions at high energy, due to the LMCT transitions, σN → Mn, and π–π* transitions within the aromatic rings, are also present.
Iron
[FeF3(Me3-tacn)]·H2O was obtained as a pale yellow solid (in contrast to the green colour reported previously in the literature19) after reaction of FeF3·3H2O with Me3-tacn in refluxing n-BuOH. Its IR spectrum shows the expected two ν(Fe–F) bands at 512 and 529 cm−1. [FeF3(terpy)]·2H2O, made using the same method, was obtained as a light purple powder and characterised similarly. The diffuse reflectance UV-vis spectra of the complexes are shown in Fig. S26 and S30.† The electronic transitions in a d5 high spin system with a ground state 6A1g are all spin-forbidden and weak bands in the visible region are therefore seen.Crystals suitable for single crystal X-ray analysis of [FeF3(terpy)]·3H2O were obtained by slow evaporation of a concentrated solution of the complex in water (Fig. 3).
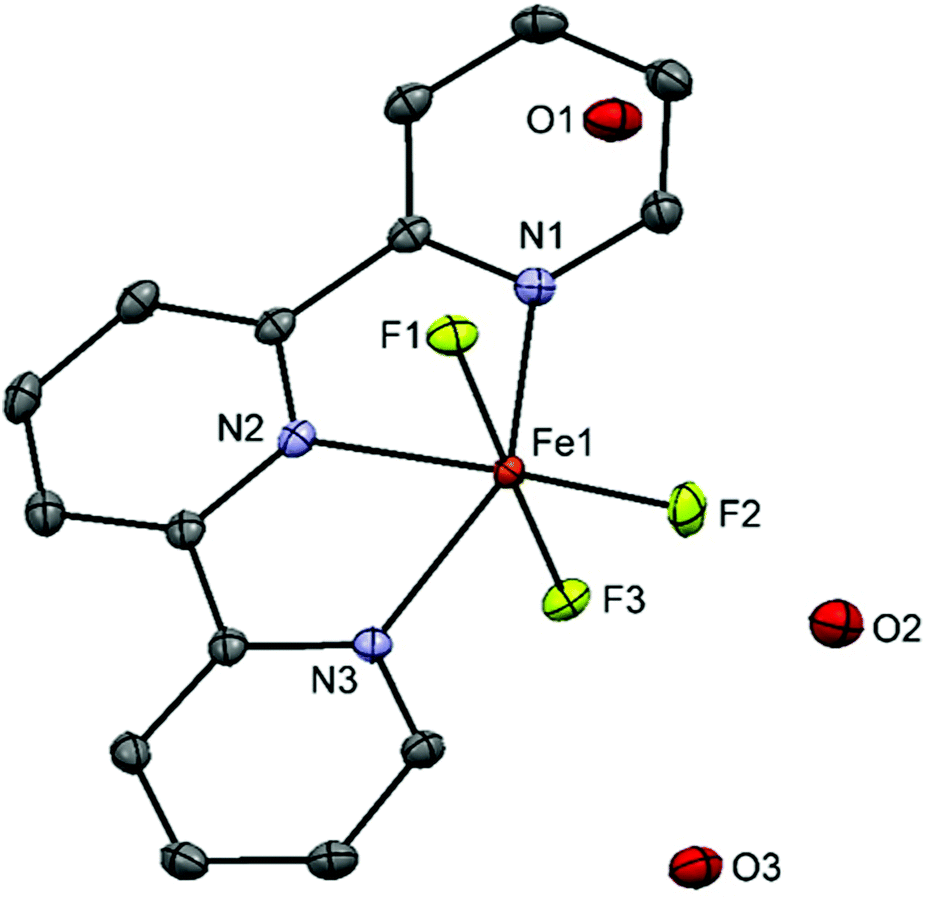 | ||
| Fig. 3 Crystal structure of mer-[FeF3(terpy)]·3H2O with ellipsoids drawn at the 50% probability level. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Fe1–F1 = 1.8774(8), Fe1–F2 = 1.8563(8), Fe1–F3 = 1.9062(8), Fe1–N1 = 2.1564(12), Fe1–N2 = 2.1144(12), Fe1–N3 = 2.1410(11), F1–Fe1–N1 = 88.03(4), F3–Fe1–N3 = 0.89.78(4), F2–Fe1–N1 = 110.00(4), F1–Fe1–F3 = 176.33(4), F1–Fe1–F2 = 91.20(4), N1–Fe1–N2 = 74.69(4), N1–Fe1–N3 = 149.17(4), F2–Fe1–N2 = 174.92(4). | ||
The structure shows a meridional configuration around the metal. The Fe–F bond lengths are slightly longer than those in [FeF3(Me3-tacn)]·H2O19 (1.878–1.907 Å vs. 1.850–1.866 Å) whereas the opposite trend is seen in the Fe–N bonds with the terpy complex having the shortest Fe–N distance (2.142–2.156 vs. 2.223–2.228). The rigidity of the terpy ligand forces some of the angles to deviate from the 180/90° expected for an octahedron. Extensive H-bonding and π-stacking interactions are also present in the lattice (Fig. S1†).
Cobalt
[CoF3(Me3-tacn)]·2H2O and [CoF3(terpy)]·MeOH·H2O were prepared by oxidising CoF2·4H2O to Co(III) in air in the presence of NaF and the appropriate ligand. The reactions were carried out in MeOH at room temperature. The complexes were characterised by UV-vis and IR spectroscopy and by 1H, 19F{1H} and 59Co NMR spectroscopy, given the diamagnetic d6 low spin electronic configuration of the complexes. The 19F{1H} NMR spectra of the complexes in CD3OD show broad resonances, with no resolved 1JF–Co coupling, at −149 ([CoF3(Me3-tacn)]·2H2O) and −116 ppm ([CoF3(terpy)]·MeOH·H2O). The 59Co (I = 7/2, Q = 0.42 × 10−28 m2)32 NMR spectra show very broad peaks at ∼ 9000 and 8600 ppm respectively, which are in the range of other octahedral Co(III) complexes, such as [Co(NH3)6]3+ and [Co(en)3]3+.33 However, when the spectrum of [CoF3(Me3-tacn)]·2H2O is recorded in D2O solution, the 19F{1H} NMR resonance associated with the complex is lost and only F− is present (118.9 ppm).The UV-vis diffuse reflectance spectra of the complexes are shown in Fig. S34 and S40.† The two spin-allowed transitions predicted for a low spin d6 system are present in the spectrum of fac-[CoF3(Me3-tacn)] (Fig. S40†) at ∼570 and ∼375 nm and are assigned to 1A1g → 1T1g and 1A1g → 1T2g, respectively. The mer-[CoF3(terpy)]·MeOH·H2O has C2v symmetry and splitting is greater than in the Me3-tacn complex (C3v). The 1T1g level split into three components and the transitions observed are tentatively assigned to 1A1g → 1B1g, 1A1g → 1B2g and 1A1g → 1B3g (Fig. S34,† from low to high energy).17,34 In this case, the third (higher energy) spin-allowed transition 1A1g → 1T2g is masked by the ligand to metal charge transfer and/or π–π* bands involving the terpy ligand. Crystals of [CoF3(Me3-tacn)]·4H2O suitable for single crystal X-ray analysis were obtained by slow evaporation of a concentrated solution of the complex in water (Fig. 4). The complex is isostructural to [MnF3(Me3-tacn)]·4H2O and shows the same H-bonding pattern in the lattice (Fig. S2†).
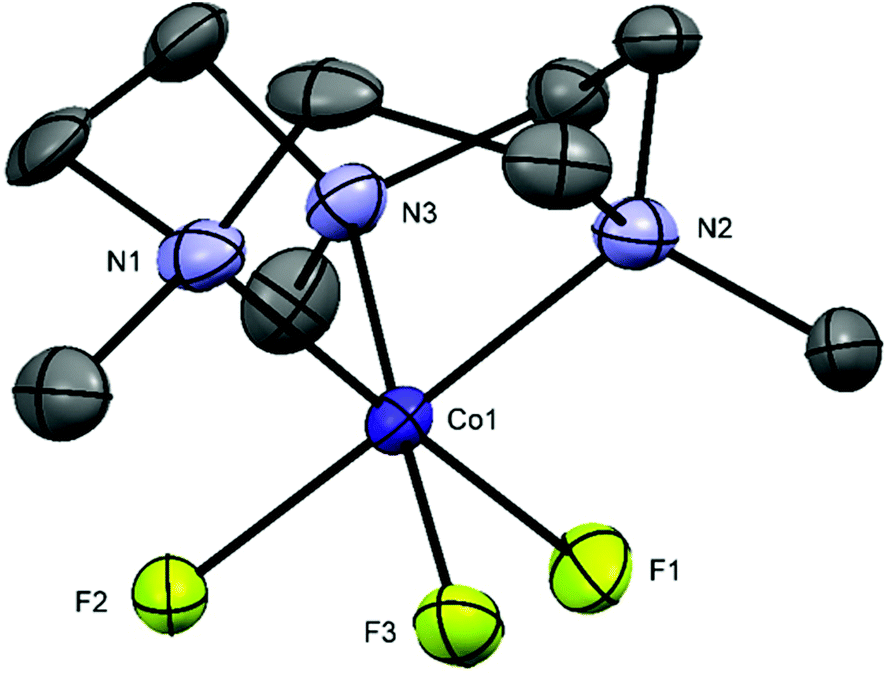 | ||
| Fig. 4 Crystal structure of fac-[CoF3(Me3-tacn)]·4H2O with ellipsoids drawn at the 50% probability level. H atoms and water molecules are omitted for clarity. Selected bond lengths (Å) and angles (°): Co1–F1 = 1.857(3), Co1–F2 = 1.871(2), Co1–F3 = 1.871(3), Co1–N1 = 1.960(4), Co1–N2 = 1.960(4), Co1–N3 = 1.958(4), F1–Co1–N1 = 178.30(14), F3–Co1–N3 = 178.13(14), F2–Co1–N1 = 91.39(14), F1–Co1–F3 = 88.29(13), F1–Co1–F2 = 89.92(12), N1–Co1–N2 = 87.62(16), N1–Co1–N3 = 87.95(15). | ||
Stability tests
The stability of a 10−3 M solution of the complexes [MF3(Me3-tacn)] (M = Cr, Mn, Fe) and [M′F3(terpy)] (M = Cr, Fe) was challenged in the presence of up to a 10-fold excess of competitive ions (F−, Cl− CO32−, PO43−, AcO−, added as sodium salts), pH variations (4–11), temperature (80 °C for 2 h) and time. The spectra acquired were compared with the spectra of the relevant reference complex. Given the limited stability of [CoF3(Me3-tacn)]·2H2O in water, stability tests were not carried out on this complex.[CrF3(Me3-tacn)]·3.5H2O appears to be stable under all conditions tested (Fig. 5), whereas [CrF3(terpy)]·4H2O is not stable in the presence of PO43− after 4 h (the 558 nm band shifts to lower energy by ∼10 nm). Both complexes are unaffected by heating at 80 °C for 2 h and after standing for an extended period (one week) in aqueous solution (Fig. S11† and Fig. 5).
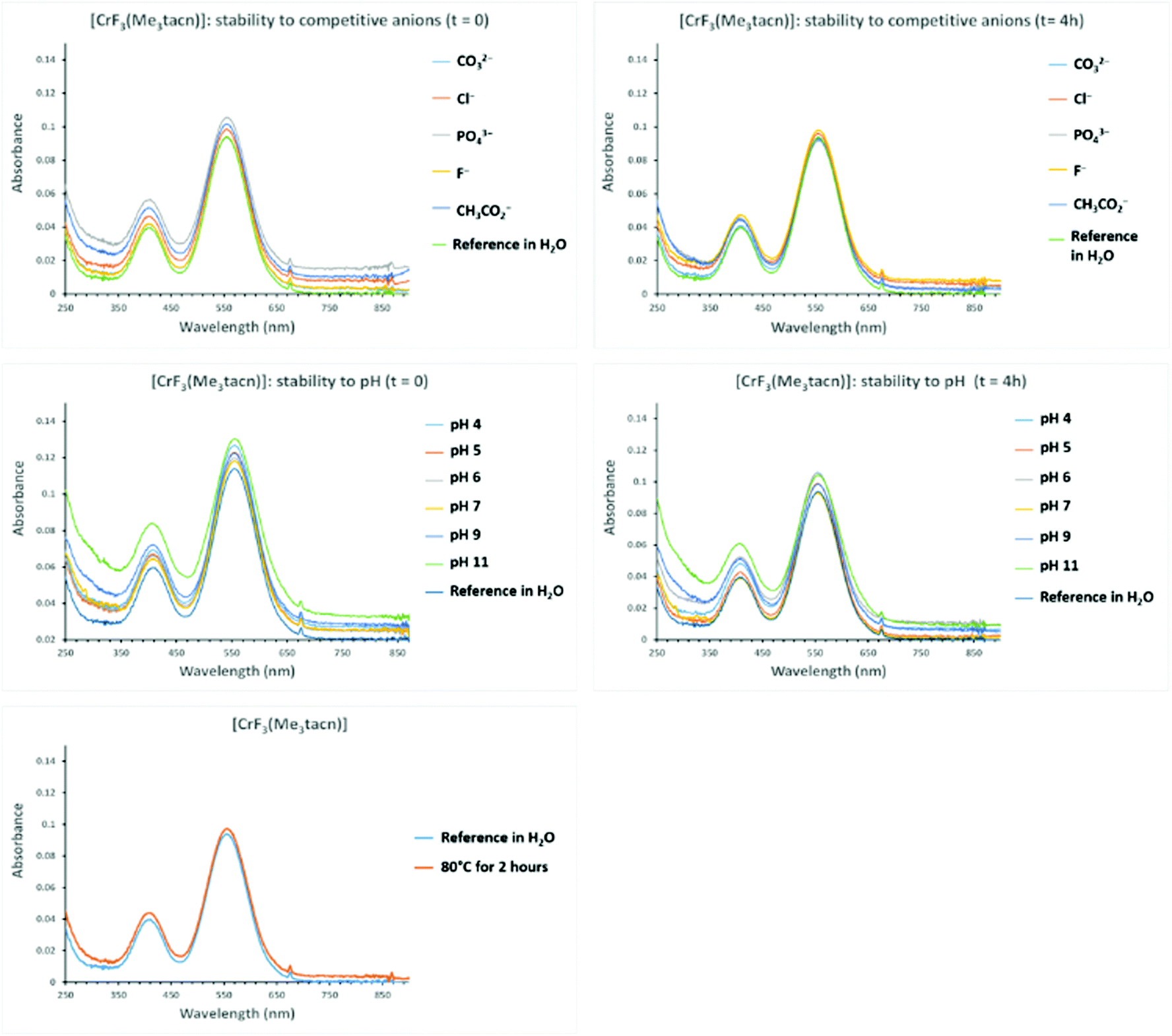 | ||
| Fig. 5 Stability test of [CrF3(Me3-tacn)]·3.5H2O. First row from the top: UV-vis spectra of the complex in the presence of a 10-fold excess anions at (left) t = 0 and (right) t = 4 hours; middle row: UV-vis spectra of the complex at different pH conditions at (left) t = 0 and (right) t = 4 hours; bottom row: UV-vis spectra of the complex after two hours at 80 °C. | ||
The stability of the Mn(III) complex is inferior to that of the Cr(III) analogue, as expected for a d4 system. [MnF3(Me3-tacn)]·2H2O (Fig. S23†) is stable to the presence of excess chloride and fluoride, but is unstable to phosphate, acetate and carbonate even at t = 0, with no significant further change observed after 4 h. Moreover, pH > 4 also results in the decomposition of the complex, as also observed upon heating and prolonged exposure in aqueous solution. Decomposition of the sample was clearly observable as a black solid formed, most likely MnO2 or MnO(OH). Considering the already problematic stability of [MnF3(Me3-tacn)]·2H2O, the stability tests on [MnF3(terpy)]·MeOH·3H2O were not pursued.
The results of the stability tests on [FeF3(Me3-tacn)]·H2O are shown in Fig. S33.† The complex is stable to the presence of all the anions studied at t = 0. However, the spectra acquired after 4 h show that the presence of carbonate anions causes decomposition of the complex. Similar behaviour was observed in the pH 7 experiment (stable at t = 0 and unstable at t = 4 h), whereas the complex is unstable at pH 11 from t = 0. The complex is stable after 2 h at 80 °C in water and is unchanged after one week in aqueous solution. The stability tests on [FeF3(terpy)]·2H2O in the same conditions (Fig. S29†) showed that the positions of the peaks were unchanged, however a change in their intensity and in the colour of the solution of the complex in water from red to pink was observed.
Halide exchange reactions on [MCl3(Me3-tacn)] (M = Cr, Fe)
Since radioactive [18F]F− can be incorporated into [AlCl3(BnMe2-tacn)], [GaCl3(BnMe2-tacn)] and [GaCl(Bn(CH2COO)2-tacn)] through Cl−/F− exchange reactions,6,8 this possibility was explored for [MCl3(Me3-tacn)] (M = Cr, Fe) through preliminary experiments using non-radioactive [NMe4]19F or K19F as test reactions on a preparative scale. Given the instability of [MnF3(Me3-tacn)] in the majority of the conditions tested and the limited stability of [CoF3(Me3-tacn)]·2H2O in water, fluorination reactions to form these complexes were not pursued.The Cl/F substitution on [CrCl3(Me3-tacn)] was incomplete after 24 h under reflux in MeCN in the presence of 4 mol. equiv. of [NMe4]F. The crude solid obtained was analysed by IR spectroscopy and compared with the IR spectra of [CrCl3(Me3-tacn)] and [CrF3(Me3-tacn)] (Fig. S3†), showing that the CrF3-complex is formed during the reaction (ν Cr–F 539, 507 cm−1) but it appears that some CrCl3-complex and/or mixed chloride/fluoride species are still present (ν Cr–Cl 343, 333 cm−1). This is not surprising given the slow substitution kinetics in the d3 systems. However, much more promisingly, [FeCl3(Me3-tacn)] was successfully fluorinated in the presence of either 4 mol. equiv. of [NMe4]F in anhydrous MeCN under reflux for 4 h or using 4 mol. equiv. of KF in aqueous MeCN at room temperature within 30 min. [FeF3(Me3-tacn)] was isolated after work-up and characterised by IR spectroscopy. Comparison of the IR spectra of the parent [FeCl3(Me3-tacn)] and [FeF3(Me3-tacn)] with the product obtained from the halide exchange reaction is shown in Fig. 6.
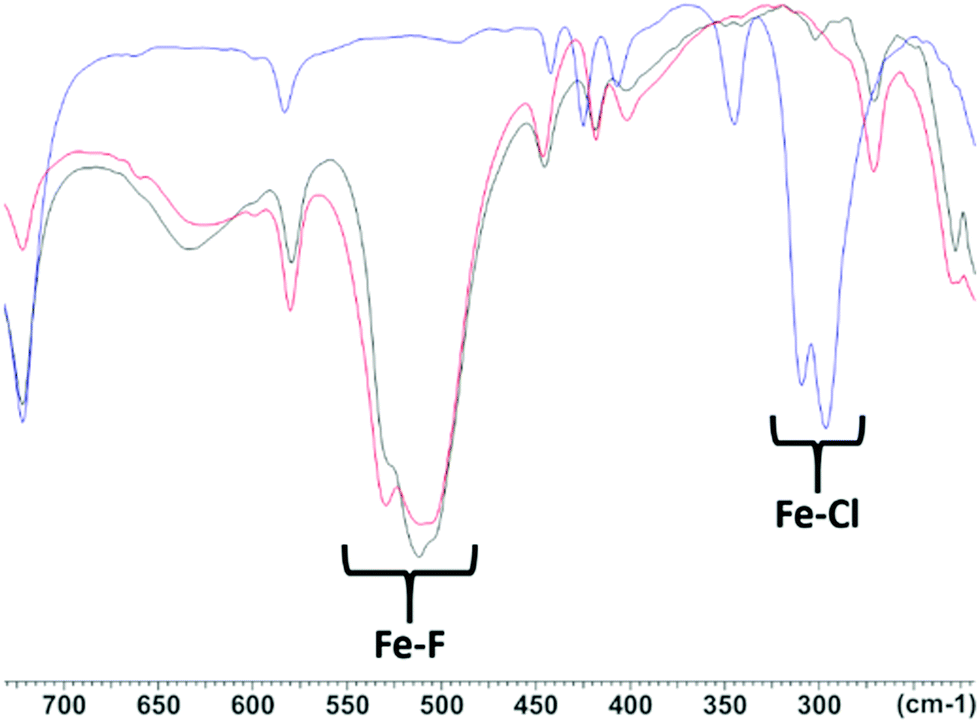 | ||
| Fig. 6 Comparison of the IR spectra of [FeCl3(Me3-tacn)] (blue), [FeF3(Me3-tacn)] (black) and (FeF3(Me3-tacn)) from the halide exchange reaction (red). | ||
It can be seen that the IR spectrum of [FeF3(Me3-tacn)] (Fig. 6, red line) obtained by the halide exchange reaction in aqueous MeCN shows the disappearance of the Fe–Cl stretches (∼300 and ∼310 cm−1, blue line) and the peaks at ∼510 and ∼525 cm−1 match the Fe–F stretches in the spectrum of the reference compound, [FeF3(Me3-tacn)] (black line), obtained directly from the reaction of FeF3·3H2O and Me3-tacn.
These studies suggest that the [FeF3(R3-tacn)] system may be worth further investigation as a possible platform for PET applications. In order to test this, the Bn-substituted analogue, [FeF3(BnMe2-tacn)] was prepared; the presence of the Bn group aids identification of the final radio-product via UV-vis spectroscopy. The crystal structure (Fig. 7) confirms the formulation [FeF3(BnMe2-tacn)]·2H2O, with two co-crystallised water molecules in the lattice. Two crystallographically independent molecules are present in the asymmetric unit, although the bond distances are not significantly different.
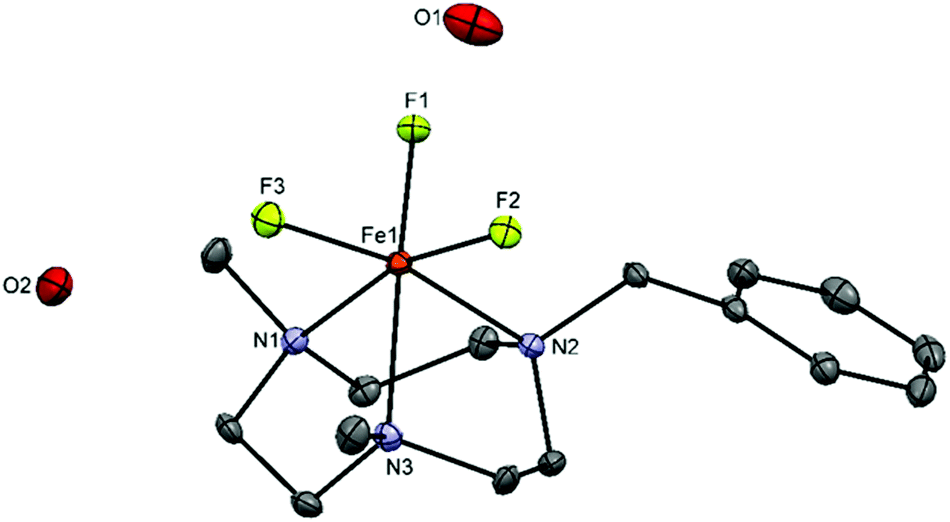 | ||
| Fig. 7 Crystal structure of [FeF3(BnMe2-tacn)·2H2O showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Fe1–F1 = 1.8878(19), Fe1–F2 = 1.8673(17), Fe1–F3 = 1.8778(19), Fe1–N1 = 2.206(2), Fe1–N2 = 2.237(3), Fe1–N3 = 2.197(3), F1–Fe1–F2 = 99.00(8), F1–Fe1–F3 = 95.91(8), F2–Fe1–F3 = 99.13(8), F1–Fe1–N3 = 167.58(9), F1–Fe1–N2 = 90.30(9), F1–Fe1–N1 = 91.36(9), F3–Fe1–N2 = 167.08(9), F2–Fe1–N1 = 165.67(8), N1–Fe1–N2 = 78.96(9), N1–Fe1–N3 = 79.32(9), N2–Fe1–N3 = 79.92(10). | ||
Radiofluorination
18F-radiofluorination of [FeF3(BnMe2-tacn)] was achieved through 18F/19F isotopic exchange reactions in unbuffered 75:25 MeCN:H2O and heating the mixture to 80 °C for 10 min (Scheme 2). The water used in the reaction is the target water containing [18F]F− directly received from the cyclotron, without further purification. The reaction produces [Fe18F19F2(BnMe2-tacn)] in a ∼40% radiochemical yield (RCY) when starting with 1 and 0.1 mg (2360 and 236 nmol respectively) of [Fe19F3(BnMe2-tacn)], along with unreacted [18F]F−. The identity of the radio-product was confirmed by comparison with the UV trace of the inactive reference standard. [Fe19F3(BnMe2-tacn)]. [18F]F− incorporation also occurs within 10 min at room temperature, although (unsurprisingly) this leads to a considerably lower RCY (6 ± 1%) (Table 1 and Fig. S52 in ESI†). Radiofluorination was also achieved using 0.01 mg (24 nmol) of precursor (80 °C per 10 min), resulting in 13 ± 5% RCY.
 | ||
| Scheme 2 18F radiolabelling conditions. | ||
:25 MeCN/target water. All experiments were performed at least 2 times
| [FeF3(BnMe2-tacn)] (mass/mg) | Scale (nmol) | T/°C (time/min) | RCY (%) |
|---|---|---|---|
| 1 | 2360 | 25 (10) | 6 ± 1 |
| 1 | 2360 | 80 (10) | 44 ± 6 |
| 0.1 | 236 | 80 (10) | 40 ± 6 |
| 0.01 | 24 | 80 (10) | 13 ± 5 |
The [Fe18F19F2(BnMe2-tacn)] was purified using a solid phase extraction (SPE) protocol through an HLB cartridge to leave the 18F-fluorinated metal-chelate as the single product. The stability of [Fe18F19F2(BnMe2-tacn)] was investigated in different formulation solutions (20:80 EtOH:water, 10:90 EtOH:PBS and 10:90 EtOH:HSA, Fig. 8 and 9, Fig. S53 and S54†), showing good stability over at least 2 hours with RCP = 99% for the EtOH/H2O and EtOH/PBS, and RCP = 90% for EtOH/HSA. The target radioproduct could also be purified through a prep. HPLC system, giving the same RCP at t = 0 as in the SPE purification protocol.
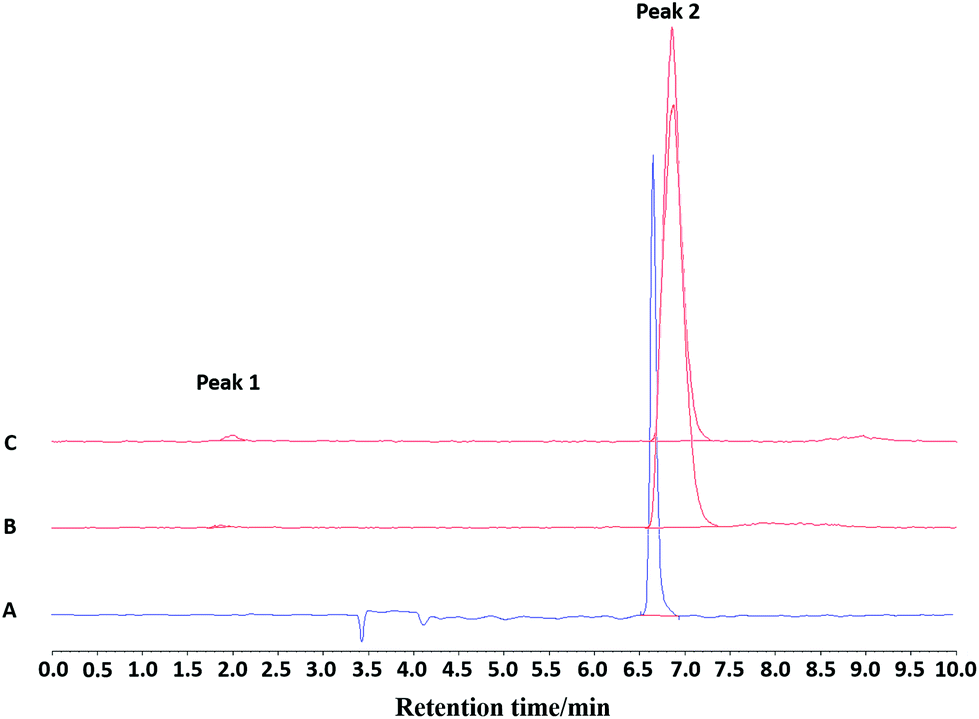 | ||
| Fig. 8 (A) Analytical UV-HPLC chromatogram of the reference standard compound [Fe19F3(BnMe2-tacn)] (Rt = 6.67 min); (B) analytical radio-HPLC chromatogram of the purified product eluted from a HLB cartridge and formulated in 20:80 EtOH:water at t = 0. Peak 2: Rt = 6.88 min >99% ([Fe18F19F2 (BnMe2-tacn)]); (C) analytical radio-HPLC chromatogram of the purified product eluted from a HLB cartridge and formulated in 20:80 EtOH:water at t = 120 min. Peak 1: Rt = 1.99 min 1% ([18F]F−). Peak 2: Rt = 6.85 min 99% ([Fe18F19F2 (BnMe2-tacn)]). | ||
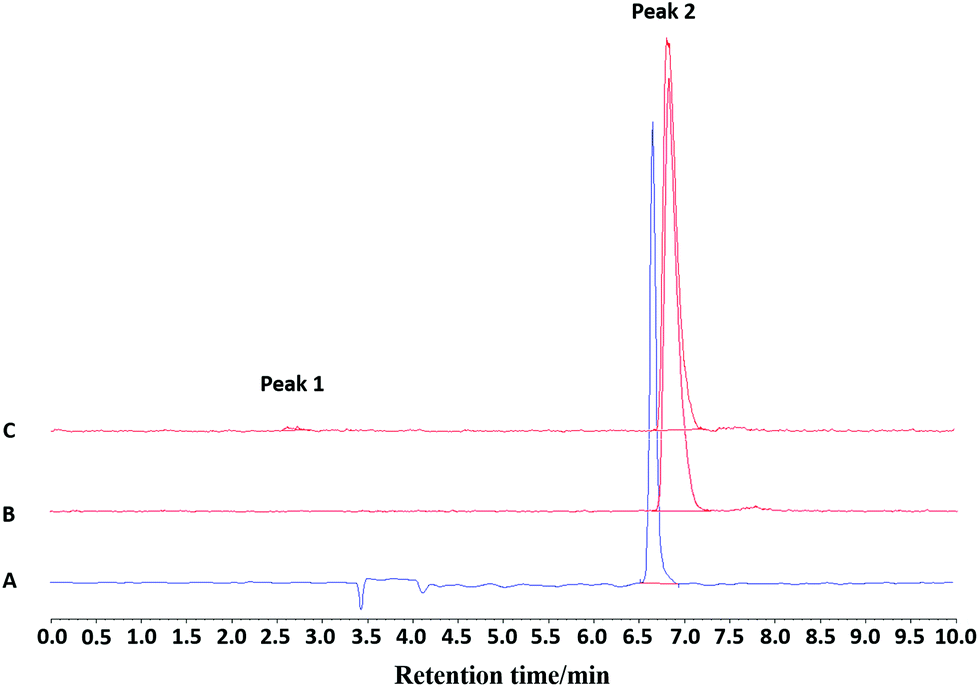 | ||
| Fig. 9 (A) Analytical UV-HPLC chromatogram of the reference standard compound [Fe19F3(BnMe2-tacn)] (Rt = 6.67 min); (B) analytical radio-HPLC chromatogram of the purified product eluted from a HLB cartridge and formulated in 10:90 EtOH:PBS at t = 0. Peak 2: Rt = 6.88 min >99% ([Fe18F19F2(BnMe2-tacn)]); (C) analytical radio-HPLC chromatogram of the purified product eluted from a HLB cartridge and formulated in 10:90 EtOH:PBS at t = 120 min. Peak 1: Rt = 2.73 min 1% ([18F]F−). Peak 2: Rt = 6.82 min 99% ([Fe18F19F2 (BnMe2-tacn)]). | ||
Conclusions
Evaluation of the stability of complexes of the type [MF3(L)], where M = Cr(III), Mn(III), Fe(III), Co(III) and L = terpy, Me3-tacn, has been carried out by means of UV-vis spectroscopy. The compounds, including the new [MnF3(Me3-tacn)], [FeF3(terpy)], [FeF3(BnMe2-tacn)], [CoF3(L)] and [CoCl3(Me3-tacn)], have been synthesised and fully characterised. [CoF3(Me3-tacn)] represents the first structurally characterised Co(III) fluoride complex with a neutral N-donor ligand.The possibility of obtaining the fluoride complexes from the chloride analogues by Cl/F exchange reactions on [MF3(Me3-tacn)] (M = Cr, Fe) was also explored. Although the bond dissociation energies of the M–F bonds are greater than the M–Cl bonds in all cases, the combination of thermodynamic and kinetic stability greatly affects the complexes and the outcome of the halide exchange reactions.
Stability tests performed on [MF3(Me3-tacn)] and [MF3(terpy)] (M = Cr, Fe) have shown that the terpy complexes do not have the stability required to be a contender for future PET applications. This was also observed in the Group 13 (Al, Ga) and ScF3 terpy complexes.8,13 Among the complexes with Me3-tacn as ligand, [FeF3(Me3-tacn)] and [CrF3(Me3-tacn)] showed good stability in most conditions; however, the reaction kinetics of the Cl/F exchange on [CrCl3(Me3-tacn)] using 4 mol. equiv. of [Me4N]F in MeCN under reflux proved to be slow, with a mixture of the chloride and fluoride complexes present after 24 h. The d5 system, [FeF3(Me3-tacn)], proved to be more successful. Fluorination was achieved within 30 min in aqueous MeCN at room temperature using KF as the fluoride source, causing complete conversion to [FeF3(Me3-tacn)].
Stability tests indicated that tacn derivatives bearing the FeF3 fragment may be suitable for radiofluorination, and this was borne out by 18F/19F isotopic exchange reactions using [FeF3(BnMe2-tacn)] at concentrations down to 24 nM. The resulting [Fe18F19F2(BnMe2-tacn)] is stable in ethanolic PBS or HAS over at least 2 h. This constitutes the first example of a transition metal complex where radiofluorination is achieved through formation of a direct M–18F bond.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC and GE Healthcare for funding through a CASE studentship to F. M. M. (EP/L505651/1) and EPSRC for a doctoral prize (EP/R513325/1).References
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS PubMed.
- J. D. G. Correia, A. Paulo, P. D. Raposinho and I. Santos, Dalton Trans., 2011, 40, 6144 RSC; R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670 RSC.
- G. E. Smith, H. L. Sladen, S. C. G. Biagini and P. J. Blower, Dalton Trans., 2011, 40, 6196 RSC; M. Glaser, P. Iveson, S. Hoppmann, B. Indrevoll, A. Wilson, J. Arukwe, A. Danikas, R. Bhalla and D. Hiscock, J. Nucl. Med., 2013, 54, 1981 CrossRef CAS PubMed.
- R. Ting, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2005, 127, 13094 CrossRef CAS PubMed; T. W. Hudnall and F. P. Gabbaï, J. Am. Chem. Soc., 2007, 129, 11978 CrossRef PubMed; A. Khoshnevisan, M. Jauregui-Osoro, K. Shaw, J. B. Torres, J. D. Young, N. K. Ramakrishnan, A. Jackson, G. E. Smith, A. Gee and P. J. Blower, EJNMMI Res., 2016, 6, 34 CrossRef PubMed; J. O'Doherty, M. Jauregui-Osoro, T. Brothwood, T. Szyszko, P. K. Marsden, M. J. O'Doherty, G. J. R. Cook, P. J. Blower and V. Lewington, J. Nucl. Med., 2017, 58, 1666 CrossRef PubMed; Z. Li, K. Chansaenpak, S. Liu, C. R. Wade, P. S. Conti and F. P. Gabbaï, MedChemComm, 2012, 3, 1305 RSC; K. Chansaenpak, M. Wang, Z. Wu, R. Zaman, Z. Li and F. P. Gabbaï, Chem. Commun., 2015, 51, 12439 RSC; Z. Liu, M. Pourghiasian, M. A. Radtke, J. Lau, J. Pan, G. M. Dias, D. Yapp, K. S. Lin, F. Bénard and D. M. Perrin, Angew. Chem., Int. Ed., 2014, 53, 11876 CrossRef PubMed.
- P. Leverman, W. McBride, R. Sharkey, A. Eek, L. Joosten, W. Oyen, D. Goldenberg and O. Boerman, J. Nucl. Med., 2010, 50, 454 CrossRef CAS PubMed; W. McBride, C. D'Souza, R. Sharkey, H. Karacay, E. Rossi, C. Chang and D. Goldenberg, Bioconjugate Chem., 2016, 21, 1331 CrossRef PubMed; W. McBride, R. Sharkey, H. Karacay, C. D'Souza, E. Rossi, P. Laverman, C. Chang, O. Boerman and D. Goldenberg, J. Nucl. Med., 2009, 50, 991 CrossRef PubMed; W. McBride, C. D'Souza, H. Karacay, R. Sherkey and D. Goldenberg, Bioconjugate Chem., 2012, 23, 538 CrossRef PubMed.
- W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu and G. Reid, Dalton Trans., 2017, 46, 14519 RSC.
- F. M. Monzittu, I. Khan, W. Levason, S. K. Luthra, G. McRobbie and G. Reid, Angew. Chem., Int. Ed., 2018, 57, 6658 CrossRef CAS PubMed; R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, G. Sanderson and G. Reid, Chem. – Eur. J., 2015, 21, 4688 CrossRef PubMed; R. Bhalla, C. Darby, W. Levason, S. K. Luthra, G. McRobbie, G. Reid and G. Sanderson, Chem. Sci., 2014, 5, 381 RSC.
- T. K. Venkatachalam, P. V. Bernhardt, D. H. R. Stimson, G. K. Pierens, R. Bhalla and D. C. Reutens, Aust. J. Chem., 2018, 71, 81 CrossRef CAS.
- G. Pascali, L. Matesic, B. Zhang, A. T. King, A. J. Robinson, A. T. Ung and B. H. Fraser, EJNMMI radiopharm. chem., 2017, 2, 9 CrossRef CAS PubMed; A. Khoshnevisan, K. Chuamsaamarkkee, M. Boudjemeline, A. Jackson, G. E. Smith, A. D. Gee, G. D. Fruhwirth and P. J. Blower, J. Nucl. Med., 2017, 58, 156 CrossRef PubMed.
- R. Schirrmacher, G. Bradtmoller, E. Schirrmacher, O. Thews, J. Tillmanns, T. Siessmeier, H. G. Buchholz, P. Bartenstein, B. Wängler, C. M. Niemeyer and K. Jurkschat, Angew. Chem., Int. Ed., 2006, 45, 6047 CrossRef CAS PubMed; C. Wängler, B. Waser, A. Alke, L. Iovkova, H. G. Buchholz, S. Niedermoser, K. Jurkschat, C. Fottner, P. Bertenstein, R. Schirrmacher, J. C. Reubi, H. J. Wester and B. Wängler, Bioconjugate Chem., 2010, 21, 2289 CrossRef PubMed; S. Niedermoser, J. Chin, C. Wängler, A. Kostikov, V. Bernard-Gauthier, N. Vogler, J. P. Soucy, A. J. McEwan, R. Schirrmacher and B. Wängler, J. Nucl. Med., 2015, 56, 1100 CrossRef PubMed.
- K. Chansaenpak, B. Vabre and F. P. Gabbaï, Chem. Soc. Rev., 2016, 45, 954 RSC; V. Bernard-Gauthier, J. J. Bailey, Z. Liu, B. Wängler, C. Wängler, K. Jurkschat, D. M. Perrin and R. Schirrmacher, Bioconjugate Chem., 2016, 27, 267 CrossRef CAS PubMed; V. Bernard-Gauthier, M. L. Lepage, B. Waengler, J. J. Bailey, S. H. Liang, D. M. Perrin, N. Vasdev and R. Schirrmacher, J. Nucl. Med., 2018, 59, 568 CrossRef PubMed.
- R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu, J. Palmer, G. Reid, G. Sanderson and W. Zhang, Dalton Trans., 2015, 44, 9569 RSC.
- E. Curnock, W. Levason, M. E. Light, S. K. Luthra, G. McRobbie, F. M. Monzittu, G. Reid and R. N. Williams, Dalton Trans., 2018, 47, 6059 RSC.
- P. Knopp and K. Wieghardt, Z. Naturforsch., 1991, 46b, 1077 Search PubMed.
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC.
- D. A. House and V. McKee, Polyhedron, 1993, 12, 2335 CrossRef CAS; J. V. Brencic, B. Ceh and I. Leban, Monatsh. Chem., 1981, 12, 1359 CrossRef; J. Glerup, J. Josephsen, K. Michelsen, E. Pedersen and C. E. Schaffer, Acta Chem. Scand., 1970, 24, 247 CrossRef; J. V. Brencic and I. Leban, Z. Anorg. Allg. Chem., 1981, 480, 213 CrossRef; J. W. Vaughn and J. Marzowski, Inorg. Chem., 1973, 12, 2346 CrossRef; J. W. Vaughn, Inorg. Chem., 1981, 20, 2397 CrossRef; H. L. Schlaefer, H. Gausmann and H. U. Zander, Inorg. Chem., 1967, 6, 1528 CrossRef; C. E. Schaffer, J. M. Lang and H. G. Drickamer, Inorg. Chem., 1996, 35, 5072 CrossRef PubMed.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1984 Search PubMed.
- H. X. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 65, m1093 CrossRef PubMed.
- K. S. Pedersen, G. Lorusso, J. J. Morales, T. Weyhermuller, S. Piligkos, S. K. Singh, D. Larsen, M. Schau-Magnussen, G. Rajaraman, M. Evangelisti and J. Bendix, Angew. Chem., Int. Ed., 2014, 53, 2394 CrossRef CAS PubMed.
- T. Birk, M. J. Magnussen, S. Piligkos, H. Weihe, A. Holten and J. Bendix, J. Fluorine Chem., 2010, 131, 898 CrossRef CAS.
- J. H. Choi and U. Lee, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1186 CrossRef CAS PubMed; J. H. Choi, I. G. Oh, K. S. Ryoo, W. T. Lim, Y. C. Park and M. H. Habibi, Spectrochim. Acta, Part A, 2006, 65, 1138 CrossRef PubMed.
- P. Nunez, C. Elias, J. Fuentes, X. Solans, A. Tressaud, M. C. M. DeLucas and F. Rodriguez, J. Chem. Soc., Dalton Trans., 1997, 4335 RSC.
- A. R. Biju and M. V. Rajasekharan, J. Mol. Struct., 2008, 875, 456 CrossRef CAS.
- C. Mantel, S. K. Hassan, J. Pecaut, A. Deronzier, M. N. Collomb and C. Duboc-Toia, J. Am. Chem. Soc., 2003, 125, 12337 CrossRef CAS PubMed.
- K. S. Pedersen, M. Sigrist, H. Weihe, A. D. Bond, C. A. Thuesen, K. P. Simonsen, T. Birk, H. Mutka, A. L. Barra and J. Bendix, Inorg. Chem., 2014, 53, 5013 CrossRef CAS PubMed.
- K. S. Pedersen, M. A. Sorensen and J. Bendix, Coord. Chem. Rev., 2015, 299, 1 CrossRef CAS.
- F. Kraus and S. A. Baer, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 865 CrossRef CAS.
- J. A. McCleverty and T. J. Meyer, Comprehensive Coordination Chemistry II, Elsevier, Oxford, 2004, vol. 6 Search PubMed.
- Y. Mitsutsuka, E. Tursun and M. Nakahara, Bull. Chem. Soc. Jpn., 1990, 63, 260 CrossRef CAS.
- E. F. Murphy, R. Murugavel and H. W. Roesky, Chem. Rev., 1997, 97, 3425 CrossRef CAS PubMed; N. M. Doherty and N. W. Hoffman, Chem. Rev., 1991, 91, 553 CrossRef; W. Massa and D. Babel, Chem. Rev., 1988, 88, 275 CrossRef; M. J. Molski and K. Seppelt, Dalton Trans., 2009, 3379 RSC; M. Leblanc, V. Maisonneuve and A. Tressaud, Chem. Rev., 2015, 115, 1191 CrossRef PubMed.
- B. N. Figgis and J. Lewis, Prog. Inorg. Chem., 1967, 6, 37 Search PubMed.
- J. Mason, Multinuclear NMR, Plenum, New York, 1987 Search PubMed.
- N. Juranic, J. Chem. Soc., Dalton Trans., 1984, 1537 RSC.
- S. Bagger and H. P. Jensen, Acta Chem. Scand., 1978, 32, 659 CrossRef; T. Laier, C. E. Schaffer and J. Springborg, Acta Chem. Scand., 1980, 34, 343 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic parameters are given in Table S1. The ESI also contains UV-vis, IR and NMR spectra of the complexes, UV-vis spectra of the stability tests for [MF3(Me3-tacn)] (M = Mn, Fe) and [MF3(terpy)] (M = Cr, Fe), figures showing the π-stacking and H-bonding in [FeF3(terpy)] and H-bonding in [CoF3(Me3-tacn)] and [FeF3(BnMe2-tacn)], and radio and UV-traces for the radiofluorination experiments. CCDC 1860121–1860123 and 1897030. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt03696a |
| This journal is © The Royal Society of Chemistry 2019 |