
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microstructured p-Si photocathode outcompetes Pt as a counter electrode to hematite in photoelectrochemical water splitting†
Anurag
Kawde
 ab,
Alagappan
Annamalai
c,
Anita
Sellstedt
d,
Pieter
Glatzel
b,
Thomas
Wågberg
c and
Johannes
Messinger
*ae
ab,
Alagappan
Annamalai
c,
Anita
Sellstedt
d,
Pieter
Glatzel
b,
Thomas
Wågberg
c and
Johannes
Messinger
*ae
aUmeå University, Department of Chemistry, Sweden. E-mail: johannes.messinger@kemi.uu.se
bEuropean Synchrotron Radiation Facility (ESRF), Grenoble, France
cUmea University, Department of Physics, Sweden
dUmeå University, Department of Plant Physiology, Umeå Plant Science Centre (UPSC), Umeå, Sweden
eMolecular Biomimetics, Department of Chemistry, Ångström Laboratory, Uppsala University, Sweden
First published on 3rd December 2018
Abstract
Herein, we communicate about an Earth-abundant semiconductor photocathode (p-Si/TiO2/NiOx) as an alternative for the rare and expensive Pt as a counter electrode for overall photoelectrochemical water splitting. The proposed photoelectrochemical (PEC) water-splitting device mimics the “Z”-scheme observed in natural photosynthesis by combining two photoelectrodes in a parallel-illumination mode. A nearly 60% increase in the photocurrent density (Jph) for pristine α-Fe2O3 and a 77% increase in the applied bias photocurrent efficiency (ABPE) were achieved by replacing the conventionally used Pt cathode with an efficient, cost effective p-Si/TiO2/NiOx photocathode under parallel illumination. The resulting photocurrent density of 1.26 mA cm−2 at 1.23VRHE represents a new record performance for hydrothermally grown pristine α-Fe2O3 nanorod photoanodes in combination with a photocathode, which opens the prospect for further improvement by doping α-Fe2O3 or by its decoration with co-catalysts. Electrochemical impedance spectroscopy measurements suggest that this significant performance increase is due to the enhancement of the space-charge field in α-Fe2O3.
Harvesting solar energy to split water into solar fuels such as hydrogen and oxygen by employing catalysts made of Earth-abundant elements is the crux of “artificial photosynthesis”.1–3 Hitherto, extensive research has been performed on designing photoelectrochemical (PEC) water-splitting devices and the respective photocathodes4 and photoanodes.5 Arranging the photoelectrodes in the parallel-illumination mode replicating the “Z”-scheme of natural photosynthesis6 is one option for employing the abundant solar energy in an effective way.7 The major challenges for designing Z-scheme devices include the positioning of the respective band edges so that the conduction band minimum of the photoanode lies at a lower electrochemical potential than the valence band maximum of the photocathode.1,8 In addition, finding a favourable aqueous environment for both the photoelectrodes to achieve an efficient PEC performance for long time periods is difficult as most photoanodes are stable at alkaline pH, while the majority of the photocathodes work best at acidic pH.9
Recently, Jang et al.10 have reported a tandem PEC device that comprised ZnO/a-Si/TiO2/Pt as a photocathode and modified hematite (FTO/α-Fe2O3-NiFeOx) as a photoanode. This Pt-containing device had a solar-to-hydrogen efficiency of 0.91% and thus demonstrated that combining Si and hematite in a tandem device is a promising approach. Tandem PEC devices10–14 require a semi/transparent front photoelectrode (photoanode) with a wide bandgap energy and a rear photocathode with a narrow band gap energy. Thus, tandem PEC configurations are associated with strict fabrication constraints.15 On the other hand, the parallel-illumination mode selected in the present study gives the freedom to use opaque photoelectrodes for simultaneous illumination, adding considerable extra flexibility in the device design. We report here a Pt-free device that combines a well-studied pristine α-Fe2O3 photoanode16,17 in aqueous 1 M NaOH (pH 13.8; best conditions for α-Fe2O3) with a previously optimized micro-structured p-Si/TiO2/NiOx photocathode.18 We demonstrate that this Z-scheme device outcompetes a similar device featuring a Pt cathode. This approach has excellent potential for future optimization since the Earth-abundant Si and Fe2O3 photoelectrodes have thermodynamic solar-to-hydrogen conversion capabilities of ∼43% and ∼15%, respectively.19
The PEC performance of the pristine α-Fe2O3 photoanode was measured (see the ESI† for experimental details) against either a Pt or a microstructured p-Si cathode that was protected by a spin-coated TiO2 layer and functionalized with a Ni-oxide catalyst (p-Si/TiO2/NiOx).18 These single (α-Fe2O3) illumination configurations are compared to a Z-scheme configuration, in which the p-Si/TiO2/NiOx electrode was also illuminated at 1 sun and acted as a photocathode (Fig. 1a and b). The detailed and systematic half-cell characterization studies of both photoelectrodes have been presented recently.16–18 In agreement with these earlier studies, Fig. 1a shows that pristine α-Fe2O3 oxidizes, in conjunction with a Pt counter electrode, water with a photocurrent density (Jph) of 0.80 mA cm−2 in 1 M NaOH (pH 13.8) and 1.23VRHE. Consistent with the expectation that the hole diffusion length in the pristine α-Fe2O3 photoanode limits the overall PEC performance, we found that Jph barely changed (0.77 mA cm−2) when p-Si/TiO2/NiOx was used instead of Pt and only α-Fe2O3 was illuminated. Remarkably, an unprecedented increase in Jph to 1.26 mA cm−2 was recorded when both α-Fe2O3 and p-Si/TiO2/NiOx were illuminated simultaneously in the parallel-illumination mode (see the photograph of the PEC cell; Fig. S1†). The elemental and morphological characterization results of the photoelectrodes are shown in Fig. S2 and S3.† Z-scheme illumination is thus an efficient complementary approach for boosting the efficiency of the PEC performance of pristine α-Fe2O3. Previous studies have shown performance enhancements of up to ∼100% by introducing different dopants5,20–22 or suitable underlayers23–25 (see Table S1† with the PEC performance of functionalized α-Fe2O3 and different electrode assemblies under different illumination conditions). The proposed band energy schematic for overall Z-scheme water splitting is shown in Fig. 1b.1,8 As detailed below, the observed significant increase in the Jph (∼60%) in this configuration may be attributed to the enhanced generation of minority carriers at the p-Si/TiO2/NiOx photocathode, leading to an increased hole-utilization for water oxidation at the surface of the α-Fe2O3 photoanode.
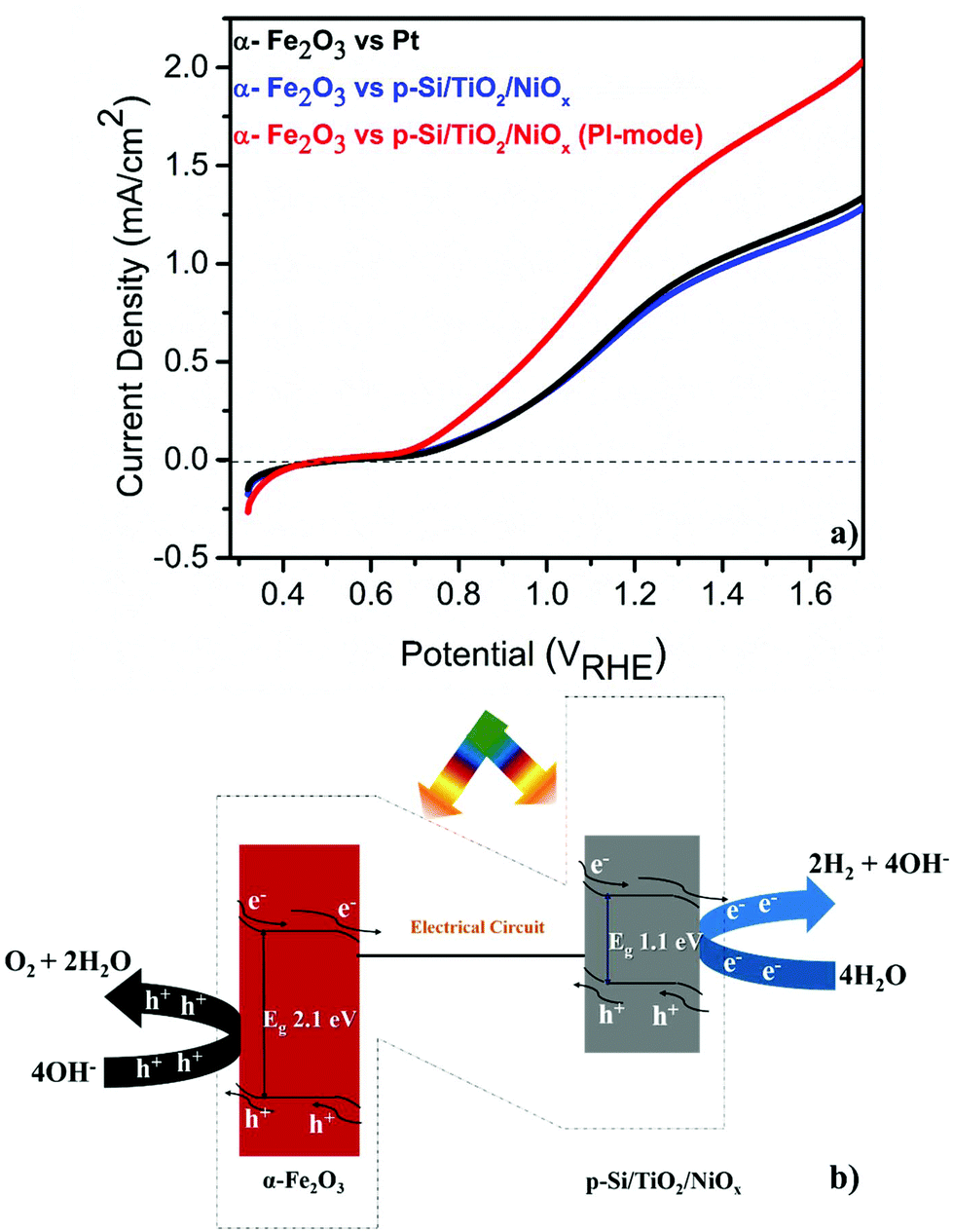 | ||
| Fig. 1 (a) Linear sweep voltammograms of a pristine α-Fe2O3 photoanode vs. Pt or p-Si/TiO2/NiOx in 1 M NaOH (pH 13.8) under standard (1 sun) single or parallel illumination (PI-mode). (b) Schematic band diagram illustrating Z-scheme overall water splitting. | ||
The stability test (Fig. 2a), performed at 1.4VRHE, demonstrated that the Z-scheme setup operated, after a slight decay in the first 30 min, at a stable photocurrent density of ∼1.5 mA cm−2 over the entire six-hour experiment. The sharp ‘spikes’ in Fig. 2a were produced by blocking the illumination of the p-Si/TiO2/NiOx electrode (Fig. 2b), or of both electrodes (Fig. 2c), demonstrating the photocurrent effects. The gases generated during this parallel-illumination PEC water-splitting experiment were collected and quantified using gas chromatography. The data in Fig. 2d show a stable and stoichiometric light-driven generation of O2 and H2, amounting to ∼158 μmol cm−2 H2 and ∼78 μmol cm−2 O2, which corresponded to a faradaic efficiency of ∼96% (the illuminated area of α-Fe2O3 was 0.09 cm2 and that of p-Si/TiO2/NiOx was 0.18 cm2). We additionally examined the photoelectrodes before and after the 6-hour PEC experiment employing X-ray spectroscopy. These high-energy resolution fluorescence detected X-ray absorption near edge structure (HERFD-XANES) and X-ray emission spectroscopy (XES) experiments did not reveal any indications for changes of the photoelectrodes (ESI Fig. S4†).
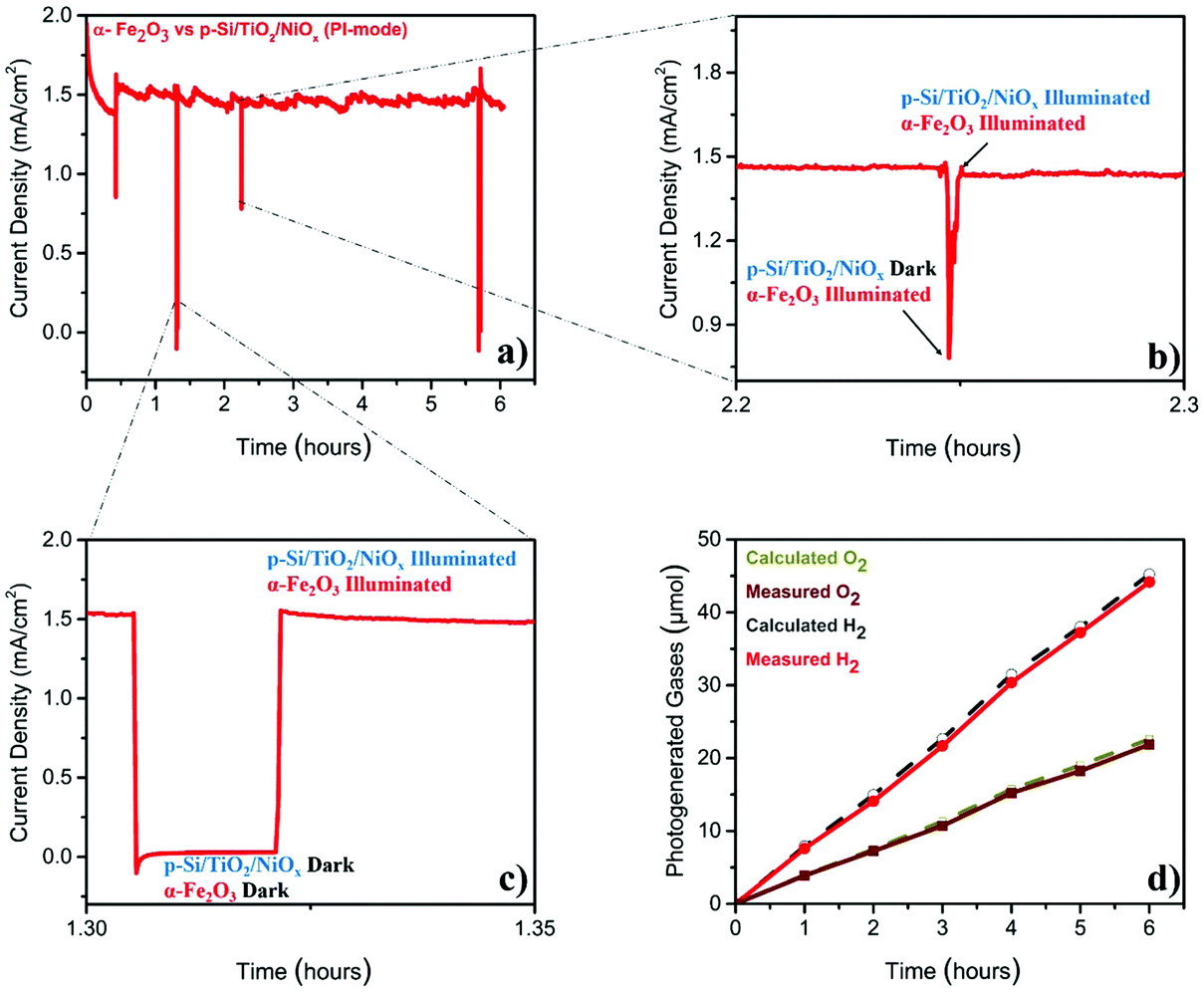 | ||
| Fig. 2 Photo-electrochemical characterization of α-Fe2O3vs. p-Si/TiO2/NiOx in 1 M NaOH (pH 13.8) under standard (1 sun) parallel illumination mode (PI-mode) (a) photocurrent density measured at 1.4VRHE; (b) magnified section showing the photocurrent when only α-Fe2O3 is illuminated; (c) magnified section showing the dark current; (d) comparison of calculated and measured photo-generated hydrogen and oxygen (illuminated area of α-Fe2O3 was 0.09 cm2 and that of p-Si/TiO2/NiOx was 0.18 cm2). | ||
Given this excellent stability, we determined the applied bias photocurrent efficiencies (ABPE) (Fig. 3(a)), the flat band potentials (Vfb) (Fig. 3(b)) and the onset potentials (Vonset) (Fig. 3(c)). In the parallel-illumination mode, a maximum ABPE of ∼0.14% was obtained, which is, taking the absorption of additional photons by the p-Si/TiO2/NiOx photocathode into account, ∼77% higher than that obtained with Pt as the counter electrode. Furthermore, Vfb and Vonset potentials deduced in the parallel-illumination mode were 100 mV and 40 mV lower as compared to using a Pt counter electrode. These remarkable results further substantiate the idea that the extra holes provided by the p-Si/TiO2/NiOx photocathode increased the hole flux towards water oxidation by enhancing the space-charge region in the α-Fe2O3 photoanode.13,26,27 Our observation is in agreement with earlier studies,28–31 wherein a large enough space-charge region (increased space-charge field) leads to the generation of relatively “long-lived” photogenerated holes that accumulate at the α-Fe2O3 electrode surface resulting in increased O2 evolution.
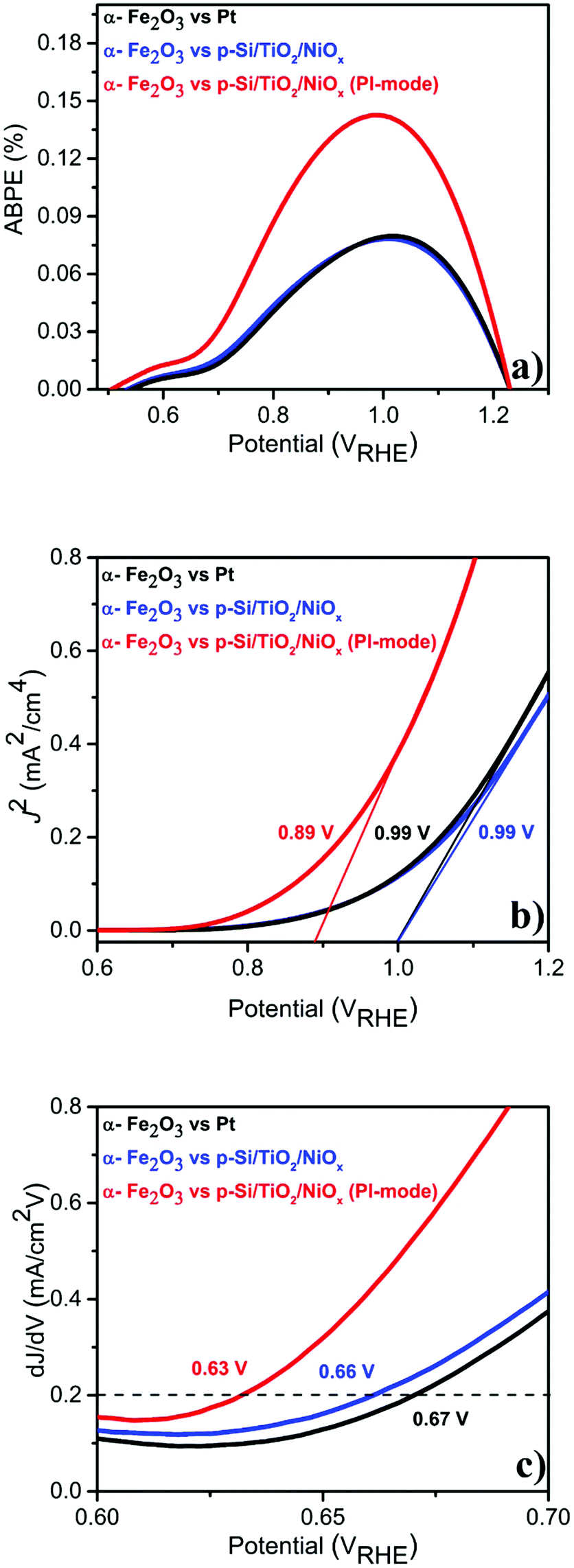 | ||
| Fig. 3 Photo-electrochemical performance of α-Fe2O3vs. Pt or p-Si/TiO2/NiOx in 1 M NaOH (pH 13.8) under standard (1 sun) single or parallel illumination (PI-mode) derived from LSVs shown in Fig. 1 (a) ABPE (%) (b) flat-band potential (Vfb) and (c) Vonset. | ||
To test this idea, electrochemical impedance spectroscopy (EIS) was performed. As shown in Fig. 4, replacing the Pt counter electrode by the p-Si/TiO2/NiOx photocathode improved the interfacial charge-transfer kinetics significantly. The charge-transfer resistance (RCT2 – larger semicircular region) across the photoanode/electrolyte interface reduced from 439.8 Ω to 298.2 Ω. The 33% decrease in RCT2 is attributed to the favorable band edge shifts in both α-Fe2O3 and p-Si/TiO2/NiOx under parallel illumination leading to a more effective transfer of photoexcited charge carriers to the surface of the respective photoelectrodes. These effective band edge shifts also increased the surface charges of the α-Fe2O3 photoanode, thereby allowing for a better surface adsorption of OH− onto α-Fe2O3, which improved the charge-transfer kinetics at the photoanode/electrolyte interface. However, the Z-scheme configuration also led to an undesired increase in hole recombination losses in the bulk of the α-Fe2O3 photoanode, as witnessed by the increased intrinsic charge-transfer resistance (RCT1 – smaller semicircular region) from 42.09 to 64.26 Ω. This suggests that the sluggish hole mobility in the hematite photoanodes and the resulting recombination losses remained a bottleneck for the overall performance, in agreement with several previous studies.5,27,32 However, such losses may be minimized in the future by using suitable dopants for α-Fe2O3.20,33,34
 | ||
| Fig. 4 Nyquist plots of α-Fe2O3vs. Pt or p-Si/TiO2/NiOx in 1 M NaOH (pH 13.8) under standard (1 sun) single or parallel illumination (PI-mode). The inset shows the equivalent circuit30 employed for analyzing the Nyquist plots. The fit data are provided in ESI Table S2.† | ||
Our results demonstrate that aqueous NaOH (pH 13.8) is a favorable environment for both the α-Fe2O3 photoanode and the p-Si/TiO2/NiOx photocathode, allowing stable Z-scheme device operation for overall light-driven water splitting with significantly better performance as compared to Pt counter electrodes.
Conclusions
A noteworthy photocurrent density (Jph) of 1.26 mA cm−2 at 1.23VRHE was achieved using a pristine α-Fe2O3 photoanode in combination with a micro-structured p-Si/TiO2/NiOx photocathode in parallel illumination, clearly outcompeting conventional device designs employing Pt counter electrodes. The micro-structured p-Si/TiO2/NiOx photocathode in this Z-scheme configuration improved the charge-transfer kinetics at the α-Fe2O3 photoanode/electrolyte interface, but also led to increased bulk recombination. This suggests that this Z-scheme device can be readily improved in future by suitable doping of the α-Fe2O3 photoanode and by functionalization with catalysts.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the European Synchrotron Radiation Facility for granting beamtime and the ID 26 beamline staff for the assistance during the experiments. The Knut and Alice Wallenbergs Foundation (Artificial Leaf Project; KAW 2011.0055) provided financial support. T. W. acknowledges support from Vetenskapsrådet (2017-04862), Energimyndigheten (45419-1), and Ångpanneföreningen (15-483).References
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511 CrossRef CAS.
- Y. Hou, B. L. Abrams, P. C. Vesborg, M. E. Björketun, K. Herbst, L. Bech, A. M. Setti, C. D. Damsgaard, T. Pedersen and O. Hansen, Nat. Mater., 2011, 10, 434 CrossRef CAS PubMed.
- A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS PubMed.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- S. Ida, K. Yamada, T. Matsunaga, H. Hagiwara, Y. Matsumoto and T. Ishihara, J. Am. Chem. Soc., 2010, 132, 17343–17345 CrossRef CAS PubMed.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90–105 CrossRef CAS.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- J.-W. Jang, C. Du, Y. Ye, Y. Lin, X. Yao, J. Thorne, E. Liu, G. McMahon, J. Zhu and A. Javey, Nat. Commun., 2015, 6, 7447 CrossRef.
- Y. Qiu, W. Liu, W. Chen, G. Zhou, P.-C. Hsu, R. Zhang, Z. Liang, S. Fan, Y. Zhang and Y. Cui, Sci. Adv., 2016, 2, e1501764 CrossRef PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS.
- C. Ding, W. Qin, N. Wang, G. Liu, Z. Wang, P. Yan, J. Shi and C. Li, Phys. Chem. Chem. Phys., 2014, 16, 15608–15614 RSC.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412 CrossRef.
- Y. Kuang, T. Yamada and K. Domen, Joule, 2017, 12, 290–305 Search PubMed.
- A. Annamalai, A. Subramanian, U. Kang, H. Park, S. H. Choi and J. S. Jang, J. Phys. Chem. C, 2015, 119, 3810–3817 CrossRef CAS.
- A. Annamalai, P. S. Shinde, T. H. Jeon, H. H. Lee, H. G. Kim, W. Choi and J. S. Jang, Sol. Energy Mater. Sol. Cells, 2016, 144, 247–255 CrossRef CAS.
- A. Kawde, A. Annamalai, L. Amidani, M. Boniolo, W. L. Kwong, A. Sellstedt, P. Glatzel, T. Wagberg and J. Messinger, Sustainable Energy Fuels, 2018, 2, 2215–2223 RSC.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske and M. Sunkara, J. Mater. Res., 2010, 25, 3–16 CrossRef CAS.
- A. Annamalai, R. Sandström, E. Gracia-Espino, N. Boulanger, J.-F. Boily, I. Mühlbacher, A. Shchukarev and T. Wågberg, ACS Appl. Mater. Interfaces, 2018, 10, 16467–16473 CrossRef CAS.
- A. Y. Ahmed, M. G. Ahmed and T. A. Kandiel, J. Phys. Chem. C, 2016, 120, 23415–23420 CrossRef CAS.
- A. Kay, D. A. Grave, D. S. Ellis, H. Dotan and A. Rothschild, ACS Energy Lett., 2016, 1, 827–833 CrossRef CAS.
- K. Sivula, F. L. Formal and M. Gratzel, Chem. Mater., 2009, 21, 2862–2867 CrossRef CAS.
- A. Annamalai, P. S. Shinde, A. Subramanian, J. Y. Kim, J. H. Kim, S. H. Choi, J. S. Lee and J. S. Jang, J. Mater. Chem. A, 2015, 3, 5007–5013 RSC.
- I. S. Cho, H. S. Han, M. Logar, J. Park and X. Zheng, Adv. Energy Mater., 2016, 6, 1501840 CrossRef.
- J. Brillet, J.-H. Yum, M. Cornuz, T. Hisatomi, R. Solarska, J. Augustynski, M. Graetzel and K. Sivula, Nat. Photonics, 2012, 6, 824 CrossRef CAS.
- P. S. Shinde, S. H. Choi, Y. Kim, J. Ryu and J. S. Jang, Phys. Chem. Chem. Phys., 2016, 18, 2495–2509 RSC.
- D. A. Wheeler, G. Wang, Y. Ling, Y. Li and J. Z. Zhang, Energy Environ. Sci., 2012, 5, 6682–6702 RSC.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef CAS PubMed.
- T. Lopes, L. Andrade, F. Le Formal, M. Gratzel, K. Sivula and A. Mendes, Phys. Chem. Chem. Phys., 2014, 16, 16515–16523 RSC.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2015, 137, 6629–6637 CrossRef CAS.
- L. Jia, K. Harbauer, P. Bogdanoff, I. Herrmann-Geppert, A. Ramírez, R. van de Krol and S. Fiechter, J. Mater. Chem. A, 2014, 2, 20196–20202 RSC.
- W. L. Kwong, C. C. Lee, A. Shchukarev, E. Björn and J. Messinger, J. Catal., 2018, 365, 29–35 CrossRef CAS.
- R. Zhang, Y. Fang, T. Chen, F. Qu, Z. Liu, G. Du, A. M. Asiri, T. Gao and X. Sun, ACS Sustainable Chem. Eng., 2017, 5, 7502–7506 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt03653e |
| This journal is © The Royal Society of Chemistry 2019 |