
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A versatile mono-quaternary ammonium salt as a mesoporogen for the synthesis of hierarchical zeolites†
Aleksei
Bolshakov
 ,
Arno J. F.
van Hoof
,
Brahim
Mezari
,
Nikolay
Kosinov
and
Emiel
Hensen
*
,
Arno J. F.
van Hoof
,
Brahim
Mezari
,
Nikolay
Kosinov
and
Emiel
Hensen
*
Laboratory of Inorganic Materials and Catalysis, Schuit Institute of Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
First published on 31st October 2019
Abstract
Here we report a versatile method to synthesize hierarchically porous zeolites with FER, CHA and MFI topologies by using inexpensive mono-quaternary ammonium N-cetyl-N-methylpyrrolidinium (C16NMP) as a mesoporogen. Extensive characterization revealed that the mesoporous zeolites are crystalline, possess a high mesopore volume and exhibit comparable Brønsted acidity to their bulk counterparts. Due to the improved accessibility of the microporous domains, these hierarchical zeolites display enhanced performance as catalysts in various reactions such as the dehydration–isomerization of 1-butanol to iso-butene (FER) and methanol-to-hydrocarbons reaction (CHA and MFI).
1. Introduction
Crystalline porous materials, in particular zeolites applied as molecular sieves, sorbents, supports and heterogeneous catalysts, are a substantial part of the modern chemical industry.1 Zeolites are crystalline microporous aluminosilicates with regular microporous structural features such as channels, cages and pockets, constructed from SiO4 and AlO4− tetrahedra.2,3 Some of the 245 zeolite topologies known today4 have become indispensable heterogeneous catalysts for many industrial processes due to their strong acidity, high surface area and high (hydro)thermal stability.5–7 At the same time, the shape selectivity of zeolites with pores typically smaller than 1 nm comes at the price of inefficient pore space utilization due to severe diffusion limitations and rapid coke formation resulting in deactivation.5,8 One of the most effective approaches to overcome this issue is the incorporation of a secondary system of mesopores in the zeolite structure.9 A larger external surface and a decrease in diffusion lengths in the microporous domains can effectively mitigate micropore mass transport limitations.10–13There exist two strategies to integrate mesoporosity into zeolite crystals, namely “top-down” and “bottom-up” approaches. While economically feasible and scalable “top-down” methods such as desilication14 and dealumination15 are broadly applied in industry, “bottom-up” methods13,16–18 afford better control over the mesoporous structure of the final catalyst by applying soft-templates, such as cationic surfactants,19,20 soluble polymers21,22 and amphiphilic organosilanes.23,24 These soft templates usually make the “bottom-up” approach more costly than the “top-down” one.
Nowadays, there is increased attention paid to developing economically attractive mesoporogens that can be employed to prepare hierarchical zeolites on an industrial scale. One of the first successful examples of the synthesis of hierarchically porous ZSM-5 zeolite (MFI topology), with truly interconnected micro- and mesopores, was reported by Ryoo and co-workers in 2006 and involved the use of 3-(trimethoxysilyl) propyl-hexadecyldimethylammonium chloride (TPHAC).25 The affinity of the template's silyl group to the growing crystalline porous silica network resulted in the inclusion of the mesoporogen in the hybrid organic–inorganic precursor that after calcination gave a microporous–mesoporous zeolite.11,18 Besides MFI, organosilanes have also been successfully applied in the synthesis of other zeolite topologies, such as MWW,26 LTA,27 FAU,28 and MOR.29 However, the resulting zeolites typically contain some amorphous silica phase and the high cost of organosilanes and problems associated with organosilane hydrolysis limit the implementation of this approach on a practical scale.9
In recognizing that the hydrophilic head group should sufficiently interact with the silicate species, organic templates were designed and synthesized with more than one quaternary ammonium center. The first successful zeolite synthesis with such a template, a diquaternary ammonium surfactant C22H45–N+(CH3)2–C6H12–N+(CH3)2–C6H13(2Br), was also reported by the Ryoo group, who obtained MFI nanosheets with a crystal size along the b-axis of just a few unit cells.30 The same group used similar surfactants31 for the synthesis of MOR, FAU(X), CHA, MFI32 and Beta, MTW, MRE, and MFI33 zeolites. Despite the possibility to obtain different highly-crystalline zeolites via a one-pot synthesis approach, these multi-quaternary ammonium templates are expensive to synthesize, which leads to an unreasonably high cost of the final material.11,19 In some cases, hierarchical zeolites can also be obtained by combining multi-quaternary ammonium mesoporogens with conventional structure directing agents (SDAs), e.g. trimethyladamantylammonium hydroxide (TMAdaOH) and tetrapropylammonium bromide (TPABr), providing, respectively, access to CHA and MFI.32 Che and co-workers showed another approach for the formation of hierarchical MFI nanosheets in a single step in the presence of a mono-quaternary ammonium salt through the π–π interactions between aromatic (biphenyl and naphthyl) groups of the organic molecule and aluminosilicate species in the initial synthesis gel.34–36 Yet, also in this case, the high cost of template synthesis precludes commercialization.
A synthesis strategy using two templates can offer a better control of the different porosity levels and reduce the final cost of the material by decreasing the amount of the expensive mesoporogen. As such, it has been reported that mixing of zeolite-providing SDAs with a simple mono-quaternary ammonium surfactant containing N-methylpiperidine as a directing head group and a cetyl (C16) carbon tail allows introduction of a second level of porosity in MFI,37,38 CHA,37 and FER zeolites.39 Recently, we have reported that using a single mesoporogen with N-methylpyrrolidine as a hydrophilic group directs MOR growth into nanorods.40 An approach that allows several zeolite topologies to be obtained using relatively simple organic molecules would be attractive from the practical and economic point of view. Notably, applying the commercially available surfactant cetrimonium hydroxide (CTAOH) as an SDA that not only initiates zeolite crystallization but also introduces mesopores is limited to mesoporous ZSM-5 zeolite.41
In this work, we successfully report the synthesis of hierarchically porous zeolites with MFI, FER, and CHA topologies in a single step using N-cetyl-N-methylpyrrolidinium bromide (further denoted as C16NMP). The developed approach allows hierarchically porous variants of several commercially relevant zeolite topologies to be synthesized by using the mesoporogen directly in bromide form at a relatively low concentration.38 The simple preparation of C16NMP from a commercially available pyrrolidine derivative (estimated price 300 US$ per kg) together with its versatility for the synthesis of hierarchical materials is a promising starting point for further scale-up studies.
2. Experimental
2.1 Synthesis of materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : xC16NMP:0.22Na2O:0.1Al2O3:2SiO2:100H2O, where x is 0.1, 0.15, and 0.2. The resulting gel was loaded into a Teflon lined stainless-steel autoclave and tumbled at 50 rpm in an oven heated at 140 °C for 312–528 h. These zeolites were denoted as FER-x (x = 0.10, 0.15, 0.20) in accordance with the amount of C16NMP that partially substitutes NMP. A conventional ferrierite zeolite was synthesized without addition of C16NMP (x = 0, FER-C).
: xC16NMP:0.22Na2O:0.1Al2O3:2SiO2:100H2O, where x is 0.1, 0.15, and 0.2. The resulting gel was loaded into a Teflon lined stainless-steel autoclave and tumbled at 50 rpm in an oven heated at 140 °C for 312–528 h. These zeolites were denoted as FER-x (x = 0.10, 0.15, 0.20) in accordance with the amount of C16NMP that partially substitutes NMP. A conventional ferrierite zeolite was synthesized without addition of C16NMP (x = 0, FER-C).
The solid products were collected by filtration, followed by washing with distilled water until pH < 8, and drying at 110 °C overnight. The organic compounds were removed by calcination in air at 550 °C for 7 h.
:xC16NMP:0.375Na2O:0.125Al2O3:5SiO2:220H2O (x = 0.1, 0.2, 0.3, 0.4, 0.5). After vigorous stirring at room temperature for 2 h, the resulting gel was transferred into a 45 mL Teflon-lined stainless steel autoclave and crystallized at 160 °C for 168 h under rotation at 50 rpm. The obtained solids are represented as CHA-x (x = 0.1, 0.2, 0.3, 0.4, 0.5), where x indicates the percentage of C16NMP that replaces TMAdaOH. The CHA bulk (CHA-C) sample was synthesized without addition of the mesoporogen surfactant. After the crystallization process occurred, the products were filtered, washed with a copious amount of water and dried overnight at 110 °C. The zeolites were calcined at 550 °C for 7 h under flowing air.
:xC16NMP:1K2O:0.066Al2O3:6.67SiO2:280H2O (x = 0.1, 0.2, 0.3, 0.4). The resulting gel was stirred at room temperature for 4 h and then transferred into a Teflon lined stainless steel autoclave and crystallized at 140 °C for 144 h under rotation at 50 rpm. The obtained solids are represented as MFI-x (x = 0.1, 0.2, 0.3, 0.4), where x indicates the percentage of C16NMP that replaces DAH. The MFI bulk (MFI-C) sample was synthesized without addition of the mesoporogen surfactant. After crystallization, the zeolite products were filtered, thoroughly washed with deionized water and dried in air overnight at 110 °C. Finally, the zeolites were calcined at 550 °C for 7 h under flowing air.
2.2 Preparation of catalysts
The protonated form of the zeolites was obtained by three subsequent steps of ion-exchange of the as-synthesized samples with 1.0 M solutions of NH4NO3 (1 g of the solid per 100 mL) for 3 hours at 70 °C. The NH4-form of the zeolites was dried overnight at 110 °C and then calcined at 550 °C for 7 h to obtain the acidic form.2.3 Characterization
:1:1 (by weight) mixture of HF (40%):HNO3 (60%):H2O. The morphology of the zeolite crystals was observed by scanning electron microscopy (SEM, FEI Quanta 200F scanning electron microscope at an accelerating voltage of 3 kV and a spot size of 4.5) and transmission electron microscopy (TEM, FEI Tecnai 20 at 200 kV). Thermogravimetric analysis (TGA) of the zeolite samples was performed on a Mettler Toledo TGA/DSC instrument. About 10 mg of a catalyst sample was placed in an alumina crucible and heated up to 750 °C at a rate of 5 °C min−1 in 40 mL min−1 He and 20 mL min−1 O2 flow. To determine the differences in the adsorption behavior, the uptake of 1-butanol and benzene was followed by TGA for FER and FER/CHA/MFI zeolites, respectively. The samples were dehydrated at 550 °C before exposure to the adsorbates. Adsorption was performed at 30 °C for 1 h in a He flow (20 mL min−1) which was fed through a saturator kept at 20 °C (p1-butanol = 6.4 mbar; pbenzene = 100.2 mbar).
2.4 Catalytic activity measurements
3. Results and discussion
Several industrially important zeolites with different topologies and channel systems were chosen as the objects of our investigation of the versatility of C16NMP as a mesoporogen for zeolite synthesis. We started with the hydrothermal synthesis of conventional FER, MFI and CHA zeolites in the presence of N-methylpyrrolidine (NMP), 1,6-diaminohexane (DAH) and trimethyladamantylammonium hydroxide (TMAdaOH), respectively, as SDAs. Then, a dual-templating approach was used where the SDA was partially substituted by C16NMP in the initial gel. Using the same C16NMP as a single SDA in the hydrothermal synthesis resulted in the formation of MOR nanorods. After loading the Pd metal, the Pd/H-MOR catalyst displayed a more ideal hydrocracking selectivity than bulk MOR prepared solely with Na+ as the template.403.1 Ferrierite zeolite
We prepared bulk ferrierite zeolite using N-methylpyrrolidine as the SDA and explored the use of C16NMP to introduce mesopores. Ferrierite (framework type FER) is a medium-pore zeolite characterized by a two-dimensional (2D) pore system consisting of 10-ring (4.2 × 5.4 Å) channels intersected by 8-ring (3.5 × 4.8 Å) channels (Fig. S2†).4 Highly crystalline materials were obtained after 17 days of hydrothermal synthesis by substituting 10 or 15 mol% of NMP with C16NMP (Fig. 1). A further increase in the C16NMP concentration resulted in the formation of an amorphous material, even during a prolonged synthesis time of 22 days (Fig. 1). When compared to the reference bulk FER-C, the mesoporous samples displayed weaker XRD peaks, particularly Bragg reflections at 2θ = 9.45° belonging to [200] planes. The decreasing intensity of the [200] reflections can be attributed to the reduction of crystalline domain size in the a-direction.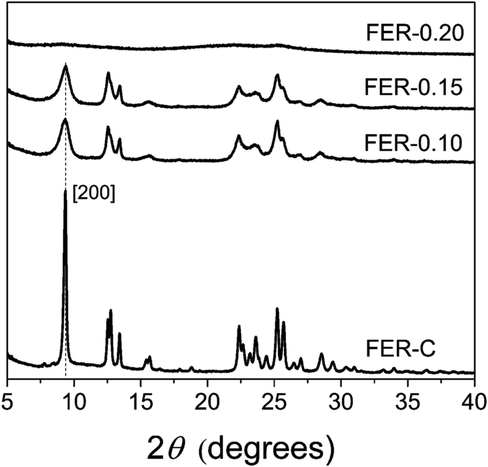 | ||
| Fig. 1 XRD patterns of the calcined ferrierite samples at different concentrations of C16NMP. | ||
Electron microscopy was used to detect the changes in the zeolite morphology after partial substitution of the NMP template with the C16NMP mesoporogen. The SEM and TEM images of FER-C (Fig. 2a and d) show a plate-like crystal morphology typical for ferrierite with a size of 1.5–2 μm,42 while mesoporous FER samples are made up of sheet-like structures of 0.6–0.8 μm in length (Fig. 2e and f). They are arranged in 3 μm and 5 μm agglomerates for FER-0.10 and FER-0.15, respectively (Fig. 2b and c). A careful TEM analysis of the FER-0.15 sample showed that the sheets are stacks with a width of about 15 nm (Fig. 3). The measured lattice distance of 0.91 nm can be attributed to the [200] planes, which establishes the relationship between the final structure and the direction of crystal growth. Formation of b-oriented nanosheets proceeds with the long hydrophobic tail sticking out the 10-ring channels, limiting the crystal growth in the c-direction. This result correlates well with the XRD data.
 | ||
| Fig. 2 SEM (a–c) and TEM (d–f) images showing the morphology of the as-synthesized FER zeolite samples: (a and d) FER-C, (b and e) FER-0.10 and (c and f) FER-0.15. | ||
 | ||
| Fig. 3 TEM image of the mesoporous FER-0.15 sample, consisting of b-oriented FER nanosheets. The indicated lattice distance of 0.91 nm corresponds to [200] planes. | ||
Thermogravimetric analysis (Fig. 4) of the as-synthesized FER samples showed several combustion stages: (i) 150–380 °C due to decomposition and C–C bond breaking of the surfactant in mesopores43 and (ii) diffusion-limited combustion of organic molecules confined in micropores at 380–700 °C.44 The FER-C zeolite exhibited a weight loss of 12.4% which corresponds to the SDA located in the FER channels. An increase in the concentration of C16NMP in the initial gel led to the removal of more template in the “low temperature” regime (∼16% for FER-0.10 and ∼22% for FER-0.15) (Fig. 4b).
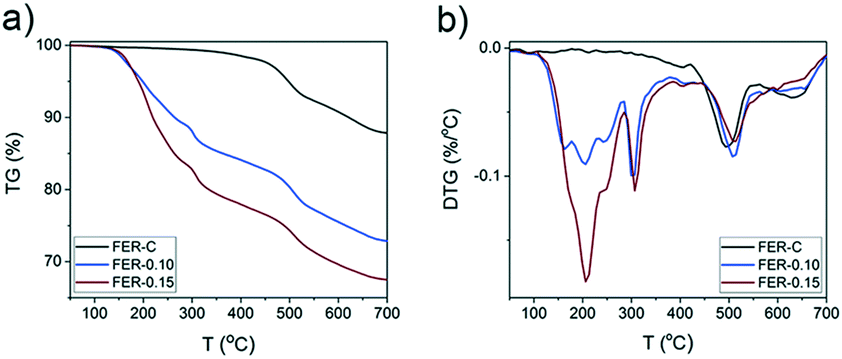 | ||
| Fig. 4 a) Thermogravimetric analysis (TGA) and b) derivative thermogravimetry (DTG) of the as-synthesized FER samples obtained at different concentrations of C16NMP (the weight losses for FER-C, FER-0.10 and FER-0.15 are 12.4%, 27.3% and 32.6%, respectively). | ||
Ar physisorption results demonstrated the formation of a second level of porosity in the samples synthesized in the presence of C16NMP (Fig. 5a). The isotherms can be characterized by type IV isotherms indicative of mesopores. Conventional FER-C has a type I isotherm, which is typical for microporous materials.45 The micropore volume of the obtained samples is in the 0.08–0.11 cm3 g−1 range, confirming their high crystallinity. An increase in the concentration of C16NMP resulted in a higher mesopore volume (0.21 cm3 g−1 and 0.25 cm3 g−1 for FER-0.10 and FER-0.15, respectively) and a more than three times higher external surface area (Table S1†) of the hierarchical zeolites. The pore size distributions determined using the NLDFT method show a broad distribution of mesopores (5–47 nm) for both mesoporous materials (Fig. 5b).
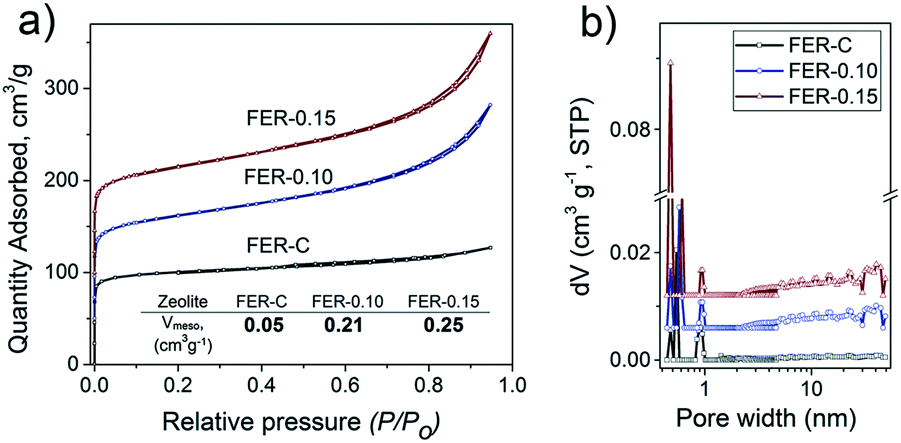 | ||
| Fig. 5 a) Ar physisorption isotherms and b) pore size distributions of calcined FER samples at different concentrations of C16NMP. The isotherms are vertically offset by equal intervals of 50 cm3 g−1. The pore size distributions were determined by the NLDFT method and vertically offset by equal intervals of 0.006 cm3 g−1 nm−1. | ||
27Al MAS NMR spectroscopy was used to examine the local environment of Al in the calcined zeolites (Fig. 6). The spectra can be characterized by two resonances: the first one at 56 ppm, typical for framework Al atoms in the tetrahedral position (AlIV), and the second at 0 ppm for extraframework Al (AlVI).46 Deconvolution of the spectra revealed that Al is mainly incorporated into the ferrierite framework during the hydrothermal synthesis (>72% framework Al) (Table S2†). The increased amount of EFAl in the mesoporous samples (∼28 vs. 21% for FER-C) can be explained by the easier extraction of framework Al atoms from the structure of the thin sheets during ion-exchange and calcination at 550 °C.47 The ICP-OES elemental analysis of all the samples showed similar Si/Al ratios of ∼10 (Table S2†).
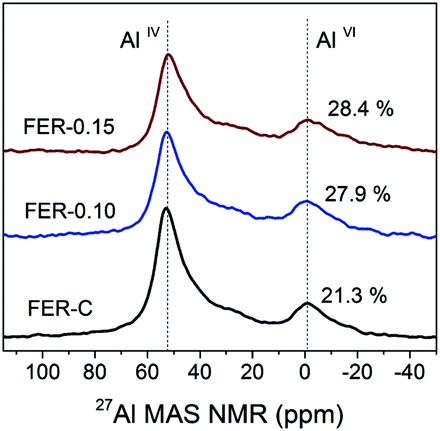 | ||
| Fig. 6 27Al MAS NMR spectra of hydrated H-FER zeolites. | ||
The nature of hydroxyl groups in the calcined FER samples was analyzed by IR spectroscopy of adsorbed pyridine and CO (Fig. 7). The IR spectra indicate the presence of several types of OH groups: bands at 3748 cm−1, 3720 cm−1 and 3606 cm−1 relate to external and internal (only in FER-C) silanols (Si–OH) and bridging hydroxyl groups (Si–OH–Al), resceptively.48,49 The shoulder at 3648 cm−1 can be attributed to the OH groups of extraframework aluminum (EFAl) and –OH species, which are grafted to the lattice.49–51 After adsorption of pyridine at 150 °C, characteristic signals corresponding to N–H vibrations appeared in the 1600–1400 cm−1 range.
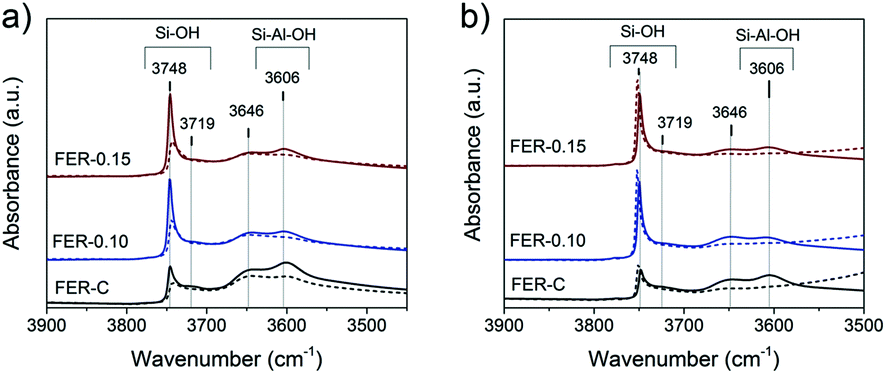 | ||
| Fig. 7 IR spectra of FER zeolites in the proton forms. IR spectra of the OH stretching region after a) pyridine and b) CO adsorption on the FER samples at 150 °C and −196 °C, respectively. The solid lines represent the spectra recorded at 150 °C under vacuum, while the dashed lines show the changes after adsorption of the probe molecules (spectra are normalized to the weight of the samples). | ||
The bands at 1545 cm−1 and 1455 cm−1 with a shoulder at 1445 cm−1 are related to pyridine interaction with strong Brønsted acid sites (BAS), Lewis acid sites (LAS) and terminal silanols, respectively (Fig. S3†). The band observed at 1490 cm−1 can be related to both types of acid sites.52,53 As bulky pyridine molecules cannot penetrate the small 8-ring FER channels52,54 and adsorb only on BAS located in 10-membered channels and the external surface of the zeolites, we did not observe complete disappearance of the bridging OH groups in the micro- and mesoporous FER samples (Fig. 7a). The calculated concentration of BAS after pyridine desorption showed a reduction (20–40%) after the introduction of the mesoporogen in the zeolites (Table S3†). This can be explained by a lower amount of framework Al in the hierarchical FER materials.
In contrast to pyridine, the smaller CO probe molecule can reach all acid sites, which was confirmed by the complete disappearance of bands at 3648 cm−1 and 3606 cm−1 upon CO exposure at low temperature (Fig. 7b). The strength of the Brønsted acid sites was estimated by determining the shift of the bridging hydroxyl groups upon interaction with CO, which, as usual, was shifted to around 3300 cm−1 for zeolites.55,56 All the zeolite samples exhibited a CO-perturbed OH band at ∼3311 cm−1 and the formation of the CO stretching band at ∼2170 cm−1 (Fig. S4†). The ΔνOH of 295 ± 2 cm−1 indicates a similar acid strength of the bridging hydroxyl groups for all the samples (Fig. S4† left). Another CO-perturbed band at ∼3460 cm−1 was observed (Fig. S4† left), which is likely due to bridging hydroxyls partially linked to the framework.49,50 Garrone and coworkers57,58 reported that such grafted SiOAl(OH)OSi groups possess much higher acidity than Al–OH species (ΔνOH ∼210 cm−1 for MCM-22 and ITQ-2 zeolites) and are able to protonate ammonia.57 Upon CO adsorption, the strength of these grafted acidic –OH groups was measured by their shift to a region at ∼3455 cm−1 with a ΔνOH of 190 ± 2 cm−1 for all the FER samples. Detailed analysis of the νCO region revealed the initial appearance of the main band at 2172 cm−1, corresponding to CO adsorption on bridging hydroxyl groups together with grafted SiOAl(OH)OSi species, supported by a slight shift to low frequency (2168 cm−1) at high CO coverage. The presence of Lewis Al3+ sites with a different strength (bands at 2190, 2205 and 2233 cm−1) was also detected, indicative of the presence of extra-framework Al (Fig. S4† right).56
Rapid diffusion and desorption of products from the zeolite pores can also contribute to lowering the rate of coke formation.59 Shorter residence times of reactants and products within the pores generally imply a lower conversion into consecutive reaction products including those that cannot leave the pores. To probe the influence of intra-crystalline mesoporosity on the accessibility of the FER pores, the uptake of the two model organic molecules was followed for 1 h at 30 °C (Fig. 8). Benzene was chosen as a bulky molecule (kinetic diameter of 5.85 Å (ref. 60)), which only fits in the 10MR channels (4.2 × 5.4 Å) and not in the 8MR ones. On the other hand, the linear 1-butanol probe molecule can fit both types of FER pores.61
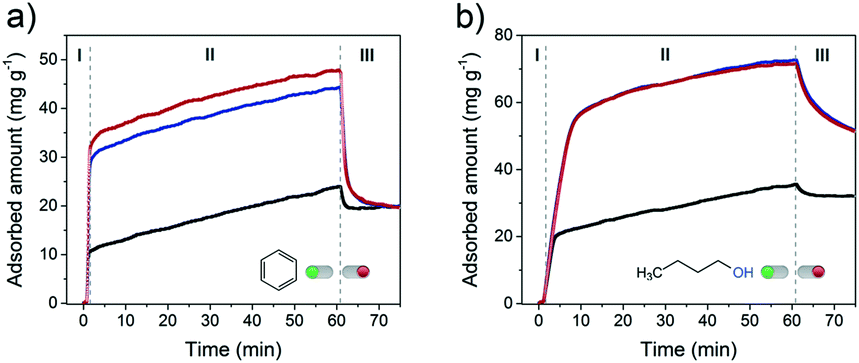 | ||
| Fig. 8 a) Benzene (pbenzene = 100 mbar) and b) 1-butanol (p1-butanol = 6.4 mbar) uptake experiments performed at 30 °C on FER zeolites: FER-C (black curve), FER-0.10 (blue curve) and FER-0.15 (red curve). The samples were dehydrated before the adsorption. | ||
Fig. 8a shows the uptake curves of benzene in the three different FER samples. A very steep uptake (stage I) is followed by a slower one (stage II) up to the point where the materials were flushed with pure He. Flushing in He led to rapid desorption of part of benzene. The three times higher uptake during stage I for the two mesoporous samples is due to the much higher external surface area of these zeolites (Fig. S5†), which increases the surface area relevant to adsorption in the micropores. During stage II, benzene uptake is limited by the slow diffusion of benzene through the 10MR channels. As a consequence, the uptake rate is independent of the texture of the material. Likely not all micropores are occupied after exposure to He/benzene flow for 1 h. The total benzene adsorption capacity amounted to 0.024 cm3 g−1, 0.051 cm3 g−1 and 0.055 cm3 g−1 for FER-C, FER-0.10 and FER-0.15, respectively. These values correspond to 20% (bulk FER-C) and 60–65% (hierarchical FER materials) of the micropore volumes (Table S1†). Desorption in He leads to a similar residual level of ∼20 mg g−1, suggesting that predominantly benzene remains in the micropores as can be expected from the stronger adsorption in micropores.
With 1-butanol, the uptake profiles were qualitatively similar (Fig. 8b). The total uptake of 1-butanol is higher in all cases, which is due to the possibility to also adsorb n-butyl alcohol in the 8MR channels. The maximum observed 1-butanol uptakes correspond to 41% (FER-C), 100% (FER-0.1) and 100% (FER-0.15) of the respective micropore volumes. Altogether, these findings point to a significantly improved accessibility of both 10MR and 8MR pores in hierarchical FER materials. This higher accessibility will improve the utilization degree of the microporous domains in catalytic reactions.
FER zeolites are promising catalysts for the skeletal isomerization of linear butenes. Iso-butene is a useful intermediate in the production of methyl tert-butyl ether (MTBE), amongst other applications.62 Recently, Chadwick and co-workers63 reported an alternative approach for C4-olefin production on acid sites of zeolites such as theta-1, ZSM-23 and FER via one-step dehydration and isomerization of 1-butanol (BuOH). As BuOH can be obtained by fermentation of biomass, a route to biobased iso-butene is available. The authors reported that FER exhibited the highest iso-butene selectivity. A problem is that dealumination takes place under reaction conditions (400 °C), induced by water generated from n-butyl alcohol dehydration, resulting in catalyst deactivation. Given this issue, we compared the performance of FER-0.15 with that of FER-C in the combined dehydration and isomerization of 1-butanol to iso-butene reaction at a lower temperature of 300 °C. FER-0.15 was chosen because it had a higher external surface area (Sext = 153.8 m2 g−1) than FER-0.10.
Fig. 9a shows the 1-butanol conversion as a function of time on stream. At the start of the reaction, FER-C converts the BuOH feed completely, whereas the initial conversion for FER-0.15 was 96%. The lower activity can be explained by the lower BAS concentration in the mesoporous sample, as followed from our characterization. Comparing the time-on-stream behavior of these two catalysts shows that the deactivation for FER-0.15 is much lower than that of FER-C. After 4.5 days, the conversion was still 86% for FER-0.15. We explain the lower rate of deactivation due to a shorter residence of the C4-products in the zeolite pores, thereby lowering the rate of oligomerization and coke deposition.53 The lower rate of coke deposition is confirmed by thermogravimetric analysis (TGA) (Fig. S6†), demonstrating that FER-C contains a higher amount of coke after 4.5 days than FER-0.15. The prepared FER catalysts were very selective towards the formation of C4 olefins with a total C4 product selectivity of ∼99% (Fig. 9b). The selectivity to iso-butene was 48 ± 2%, nearly similar for bulk and hierarchical ferrierite catalysts.
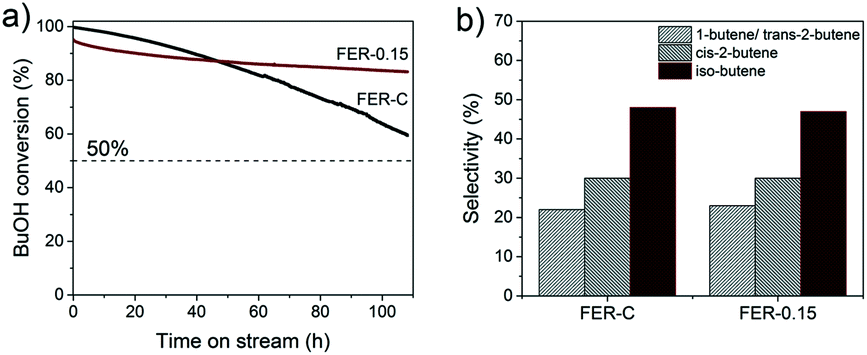 | ||
| Fig. 9 a) Conversion as a function of time-on-stream for 1-butanol to iso-butene for FER-C and FER-0.15 (reaction conditions: WHSV = 69.1 h−1, 300 °C). b) Butene product distribution at 90% 1-butanol conversion. | ||
3.2 CHA and MFI zeolites
SSZ-13 is a zeolite with the chabazite (CHA) topology, which has a three-dimensional pore system with cages of 6.7 Å × 10.9 Å size interconnected through 8-ring windows with pore apertures of 3.8 Å (Fig. S7†).4,64 ZSM-5 (MFI topology) is characterized by 10-ring pores and two types of channel systems with straight channels of 5.4 × 5.6 Å and sinusoidal channels of 5.1 × 5.5 Å (Fig. S8†).4,65We used TMAdaOH and 1,6-diaminohexane (DAH) as SDAs for obtaining bulk forms of CHA and MFI, respectively. 1,6-Diaminohexane was chosen for this dual-template approach, because the commonly used SDA tetrapropylammonium hydroxide (TPAOH) was reported not to be compatible in an earlier report.38 Highly crystalline mesoporous SSZ-13 and ZSM-5 zeolites were obtained after 6 days of synthesis by partial substitution of the original SDA with C16NMP in the range from 10–50 mol%. All the samples were phase-pure, as judged from the XRD patterns in Fig. 10.
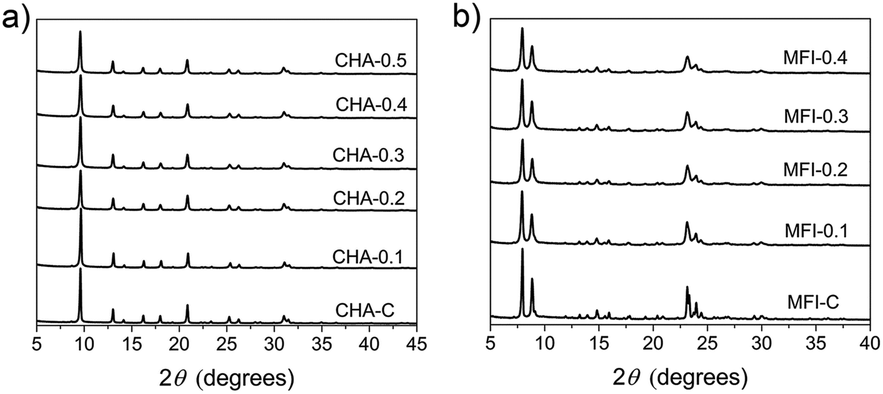 | ||
| Fig. 10 XRD patterns of calcined a) CHA and b) MFI samples at different concentrations of C16NMP. | ||
The morphology of the samples was investigated by electron microscopy (Fig. 11 and 12). The conventional CHA-C sample is composed of typical cube-shaped crystals66 with a size of ∼6 μm (Fig. 11a) with a rough surface attributed to primary intergrown microporous particles (Fig. 12a). Addition of C16NMP to the gel led to reduction of the crystal size to the 0.5–1.5 μm range with a higher amount of the mesoporogen giving smaller crystals (Fig. 11b–d). A further increase in the substitution level resulted in agglomeration of the primary crystals into 3–4 μm particles (Fig. 11e and f). The TEM images of the CHA samples show that the crystals were constructed from stacked crystallites with ∼50 nm size separated by mesoporous voids (Fig. 12b and c). The CHA structure allows the location of the hydrophilic head group of the mesoporogen in its supercages (6.7 Å × 10.9 Å), while the long hydrocarbon tail can protrude from the 8MR windows. The crystals of conventional ZSM-5 are characterized by round-edged hexahedra of 1–1.5 μm size (Fig. 13a and d). Addition of the mesoporogen resulted in agglomeration of uneven sized nanocrystals (Fig. 13b and e) or nano-needles of 15–30 nm width (Fig. 13c and f) by preserving the morphology of the conventional MFI sample.
 | ||
| Fig. 11 SEM images of calcined CHA zeolite samples: a) CHA-C, b) CHA-0.1, c) CHA-0.2, d) CHA-0.3, e) CHA-0.4 and f) CHA-0.5. | ||
 | ||
| Fig. 12 TEM images of calcined CHA zeolites: a) CHA-C, b) CHA-0.1 and c) CHA-0.3. | ||
 | ||
| Fig. 13 (a–c) SEM and (d–f) TEM images of calcined MFI zeolite samples: (a and d) MFI-C, (b and e) MFI-0.1, and (c and f) MFI-0.2. | ||
We speculate here that the pyrrolidine head group can fit in both the straight and zig-zag 10MR pores of the MFI structure, resulting in non-uniform nano-crystals/needles. An increase in C16NMP concentration in the initial gel leads to the formation of an amorphous phase (Fig. S9†).
Similar to results obtained for FER synthesis, the C16NMP molecule starts to decompose between ∼200 °C and 380 °C (Fig. S10b and d†), while the SDA/surfactant mixture in micropores combusts at a higher temperature (>380 °C). Substitution of more than 20% of TMAdaOH with the mesoporogen in the synthesis gel did not lead to significant incorporation of C16NMP molecules in the CHA structure (mass loss in mesopores: CHA-0.1 – 2.8%, CHA-0.2 – 15.4%, CHA-0.3 – 17.4%, and CHA-0.4 – 20.5%) (Fig. S10b†). We observed a gradual increase in the weight loss in the “low temperature” regime (∼230 °C) for hierarchical MFI samples with an increase of the amount of C16NMP in the initial gel up to 30% (Fig. S10d†).
Fig. 14 shows the Ar physisorption isotherms and NLDFT-derived pore size distributions of the CHA and MFI samples. The isotherms of the conventional CHA-C and MFI-C materials obtained without adding C16NMP are of type I45 (Fig. 14a and c). The isotherms of the mesoporous samples (Fig. 14a and c) evidenced a larger pore volume, a shift of uptake to higher pressure and the development of hysteresis loops due to the capillary condensation in the mesopores. These features correspond to the type IV isotherm.67 The micropore volumes of hierarchical CHA (0.18–0.19 cm3 g−1) and MFI (0.12–0.13 cm3 g−1) samples are similar to those of the crystalline bulk materials. The mesopore volume of CHA zeolites increased when more C16NMP was added and attained a maximum of 0.24 cm3 g−1 for CHA-0.5 (Table S4†). The highest external surface area (211 m2 g−1) was obtained for CHA-0.3. On the other hand, the external surface area of CHA-0.5 was lower compared to that of the reference (Table S4†), which is due to morphological changes. The EM images show the development of additional inter-crystal voids formed through an agglomeration of the particles (Fig. 11 and 12). These results are also supported by the presence of secondary mesopores with a size in the 30–50 nm range for CHA-0.4 and CHA-0.5, together with rather uniform pores of ∼5–7 nm calculated by the NLDFT method (Fig. 14b).
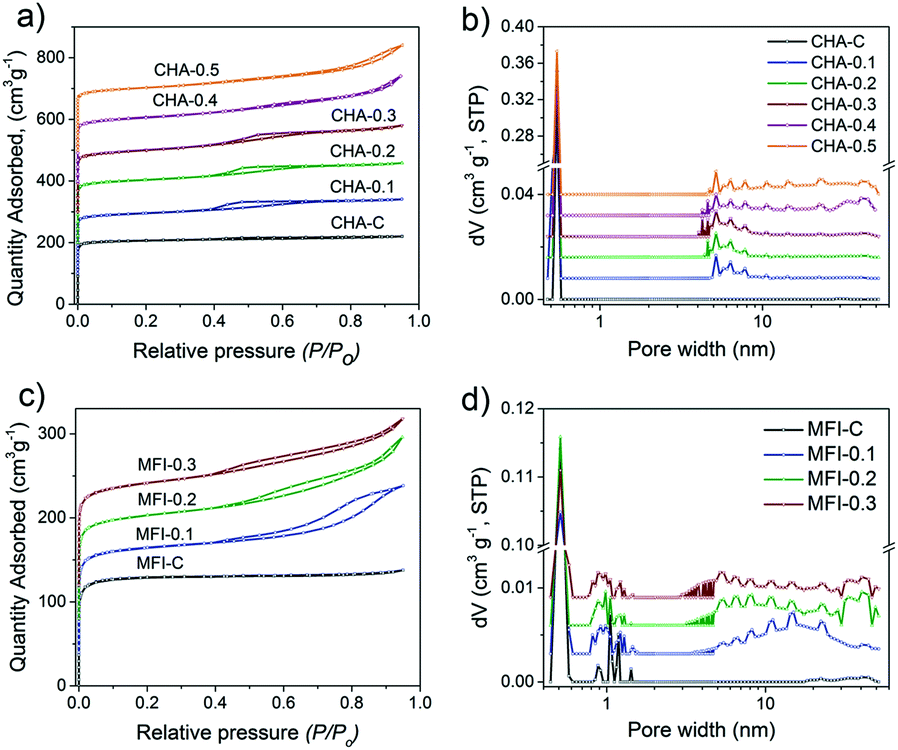 | ||
| Fig. 14 (a and c) Ar physisorption isotherm and (b and d) pore size distribution of calcined CHA and MFI samples as a function of C16NMP substitution. The isotherms were vertically offset by equal intervals of 100 cm3 g−1 (CHA) and 40 cm3 g−1 (MFI). The pore size distributions were calculated by the NLDFT method and vertically offset by equal intervals of 0.008 cm3 g−1 nm−1 and 0.003 cm3 g−1 nm−1 for CHA and MFI zeolites, respectively. | ||
The largest mesopore volume of 0.17 cm3 g−1 was obtained for MFI-0.2 (Table S4†). A further substitution of the SDA with the mesoporogen led to slightly worse textural properties. This is likely due to the formation of the amorphous phase as suggested by SEM analysis (Fig. S9†). The amount of this phase is too small to be picked up by XRD. The pore size distributions exhibited a broad mesopore range (5–45 nm) for all hierarchical MFI samples (Fig. 14d).
Based on these results, several CHA and MFI samples were chosen for further characterization. The 27Al MAS NMR spectra of these zeolites show two signals of framework (AlIV) and extraframework (AlVI) aluminum46 (Fig. 15). Table S5† shows that over 80% of the Al ended up in the zeolite framework.
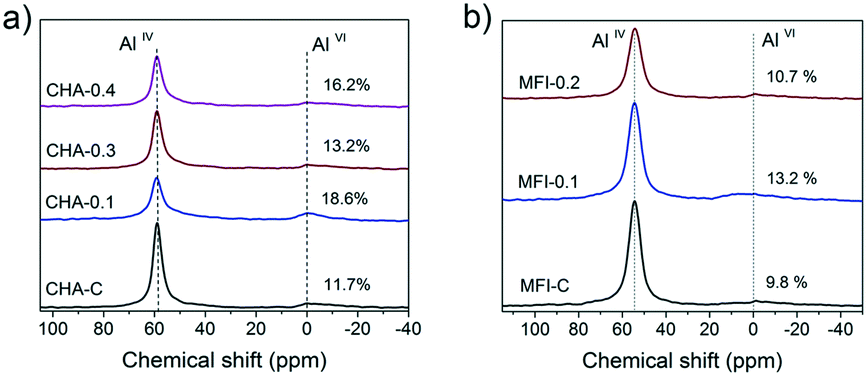 | ||
| Fig. 15 27Al MAS NMR spectra of the hydrated a) CHA and b) MFI zeolites. | ||
1H MAS NMR spectroscopy was employed to characterize the Brønsted acid sites, non-framework aluminum species and structural defects in the zeolites. This method is more suitable for characterization of CHA and MFI zeolites when compared to adsorption of probe molecules because some of these molecules cannot enter the pores. For instance, pyridine does not fit into 8-ring channels of the CHA topology.37 The 1H MAS NMR spectra contain several features that can be assigned to hydroxyl groups with different surroundings: silanol groups Si–OH at 1.3–2.3 ppm, the Al–OH group at 2.6–3.6 ppm and bridged Si–O(H)–Al groups at 3.8–6.0 ppm.68–71 Moreover, external and internal silanols groups can be distinguished, characterized by chemical shifts of 2.0 ppm and 1.8 ppm, respectively.72,73
All the mesoporous CHA and MFI zeolites contain more external silanols than the corresponding bulk reference zeolites (Fig. 16). This effect is most pronounced for MFI zeolites, where the external Si–OH probed by 1H NMR increased by 3-fold compared to MFI-C (Fig. 16d). Further analysis of 1H NMR spectra in the 3.5–8.5 ppm range revealed the presence of different Si–O(H)–Al groups: the shift of 4.1 ppm in the CHA samples is attributed to isolated groups, while the presence of hydrogen-bonded bridged hydroxyls at 5.4 ppm (ref. 71 and 74) was observed in the MFI zeolites. The total concentration of BAS was slightly lower for the mesoporous samples (Table S5†), which can be explained by a higher amount of EFAl.
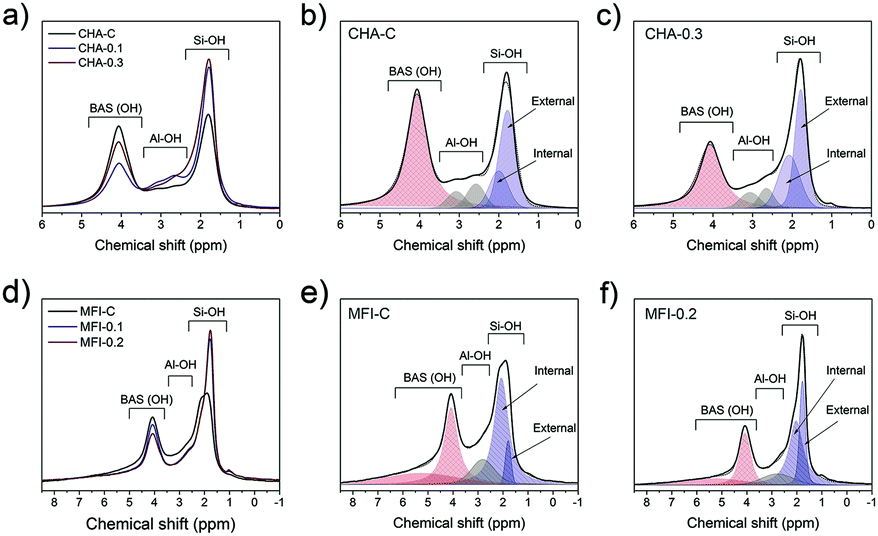 | ||
| Fig. 16 1H MAS NMR spectra of calcined a) CHA and d) MFI zeolites, and the deconvolution of the spectra of b) CHA-C, c) CHA-0.3, e) MFI-C and f) MFI-0.2 zeolites. The spectra were normalized to the sample weight. | ||
The accessibility of pores in these CHA and MFI samples was studied by following the uptake of benzene at 30 °C (Fig. 17).
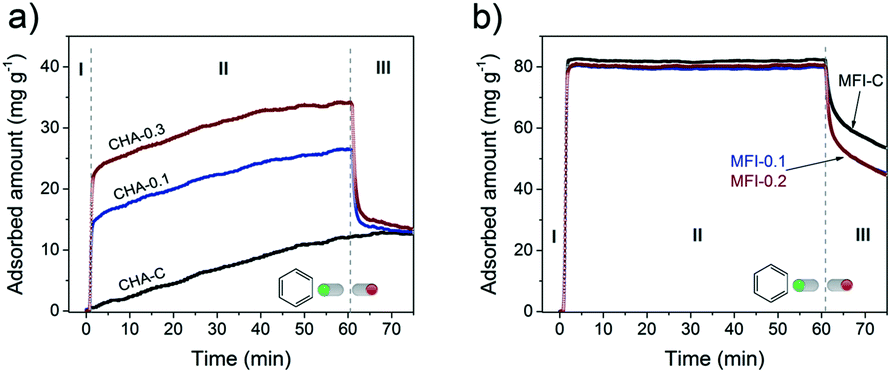 | ||
| Fig. 17 Benzene uptake experiments performed at 30 °C on a) CHA and b) MFI zeolites. The samples were dehydrated before the adsorption. | ||
Benzene fits in the 10MR channels of the MFI structure,75,76 but its diffusion through small 8MR pores of the CHA framework is restricted. This difference can explain the slow uptake to a low level of ∼10 mg g−1 for microporous CHA (Fig. 17a, stage I and II) and the much faster uptake of benzene in the MFI sample to a higher adsorption capacity of ∼80 mg g−1 (Fig. 17b, stage I). Although benzene should in principle not be able to adsorb in CHA, slow adsorption of molecules inside the CHA zeolite with a larger kinetic diameter than the 8MR pore windows (3.8 × 3.8 Å) has been reported. For instance, n-butane (kinetic diameter of 4.3 Å)77 and n- and iso-butanol (kinetic diameter of >5.0 Å)54,78 could adsorb in small amounts (up to ∼10% of total pore volume) in CHA zeolite.
The introduction of secondary porosity in CHA zeolites substantially changed the uptake behavior: (I) rapid adsorption, (II) slow diffusion and (III) partial desorption obtaining the same uptake level as for the CHA-C reference (Fig. 17a). The smaller crystallites of the hierarchical SSZ-13 zeolites led to a higher accessibility, explaining the higher initial uptake of benzene, which is indeed proportional to the external surface area (Fig. S5†). A very slow adsorption during stage II with similar rates for all the samples can be ascribed to very slow diffusion in SSZ-13 micropores. After exposure to benzene for 1 h, the maximum capacity values reached 5% (CHA-C), 11% (CHA-0.1) and 19% (CHA-0.3) of the respective micropore volumes (Table S4†). Quick benzene desorption (stage III) from the hierarchical CHA samples to the same residual benzene amounts as bulk CHA-C indicates that some benzene molecules remain in the micropores.
The uptake curves of hierarchical and bulk MFI zeolites behaved very similarly (Fig. 17b). The presence of mesopores did not significantly change the total amount of adsorbed benzene of 0.09 cm3 g−1 and 0.10 cm3 g−1 for MFI-0.1/0.2 and MFI-C, corresponding to a high degree of saturation of MFI micropores (63–82%). Moreover, the uptake rates are very similar which can be related to a high diffusion rate of benzene through the 10MR pores. However, the rate of desorption from the nearly saturated micropores in the hierarchical samples was higher than that in the microporous sample (Fig. 17b stage III).
These CHA and MFI zeolites were evaluated for their performance in the methanol to hydrocarbons (MTH) reaction, an important chemical process to produce light olefins from methanol.79–83 The activity tests were carried out at a temperature of 350 °C and a WHSV of 0.93 h−1 for CHA and a temperature of 400 °C and a WHSV of 6.0 h−1 for MFI samples. Fig. 18 shows the methanol conversion as a function of time on stream. We defined the catalyst lifetime as the time to reach a methanol conversion of 50% (t50). The resulting activity and selectivity data are given in Tables S6 and S7.† All the tested zeolites completely converted the methanol feed at the start of the reaction (Fig. 18). While the CHA-C zeolite showed a rapid decrease in methanol conversion after 4 h, deactivation of mesoporous CHA-0.1 and CHA-0.3 was more gradual and occurred after a longer time on stream (Fig. 18a). This difference is due to a higher crystal utilization degree due to the shortened microporous domains and the improved transport of reactants and products to and from the active sites.84 As a result, less coke was formed in hierarchical CHA samples during the methanol conversion reaction (Fig. S11†). We cannot exclude that the difference in catalytic performance of the CHA-0.1 sample is also caused by its lower Brønsted acidity (BAS 0.4 vs. 0.6–0.7 mmol g−1). A higher density of acid sites can lead to the fast formation of aromatic intermediates, which not only act as reaction intermediates but also as precursors in coke formation.85 A previous study revealed, however, that coke deposition decreased by only ∼10% when the number of BAS was decreased by a factor of 2 in ZSM-5 zeolite.86 Thus, we suspect that the influence of acidity differences on the coking behavior of the CHA samples is also less pronounced than the influence of pore hierarchization. Similar results were observed for the MFI zeolites (Fig. 18b). Mesoporous MFI-0.1 and MFI-0.2 samples have lifetimes of 75.9 h and 63.7 h, respectively, which are about three times higher than that of bulk MFI-C (t50 = 26.0 h). The MFI catalysts are selective to a fraction of C4+ hydrocarbons (>50%), which together with propylene constitutes more than 85% of the total product yield (Table S7†). The small-pore CHA zeolites, on the other hand, are very selective to light olefins, mainly ethylene and propylene (Table S6†). The introduction of mesopores does not significantly influence the product distribution (propylene/ethylene ratio ∼0.9). This latter finding emphasizes that the main methanol conversion reaction takes place in the micropores, where the arene cycle dominates.87
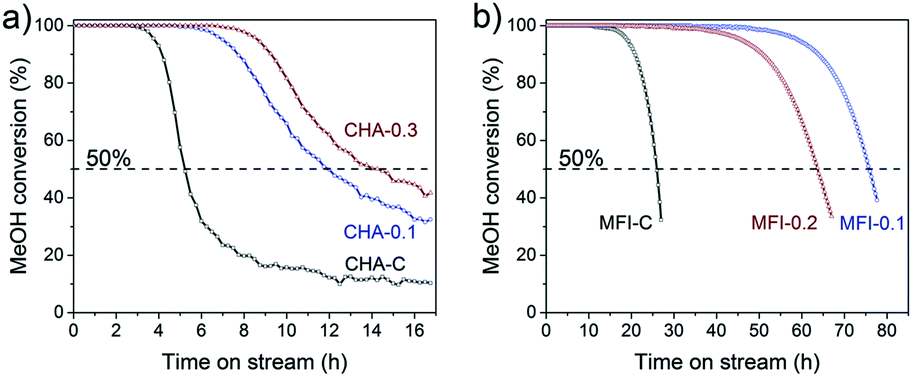 | ||
| Fig. 18 Methanol conversion as a function of time-on-stream: a) CHA zeolites (reaction conditions: WHSV = 0.93 h−1, 350 °C) and b) MFI zeolites (reaction conditions: WHSV = 6.0 h−1, 400 °C). | ||
4. Conclusions
In this work, we showed that a dual-template approach in the presence of inexpensive C16NMP as a mesoporogen is an efficient method for obtaining hierarchically porous zeolites with FER, CHA and MFI topologies. Varying substitution levels of the zeolite-providing SDA with C16NMP (10–30 mol%) allows the morphology and textural properties of the final materials to be controlled. All the prepared zeolites are highly crystalline, and contain mesopores and strong Brønsted acid sites. The significantly improved catalytic performance of FER and CHA/MFI in the direct conversion of 1-butanol to iso-butene and MTH reaction, respectively, can be attributed to the shorter diffusion lengths in the microporous domains, resulting in more effective utilization of the microporous space in comparison with the bulk counterparts. Current results combined with our previous work, where MOR nanorods were also synthesized using the same mesoporogen, confirm that the relatively inexpensive C16NMP template is a promising agent for the synthesis of various hierarchically porous zeolites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Impuls program of the Eindhoven University of Technology for financial support.Notes and references
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Adv. Mater., 2017, 29, 1701139 CrossRef PubMed.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–701 CrossRef CAS PubMed.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- L. B. McCusker, D. H. Olson and C. Baerlocher, Atlas of Zeolite Framework Types, 2007 Search PubMed.
- J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- J. Coronas, Chem. Eng. J., 2010, 156, 236–242 CrossRef CAS.
- S. F. Abdo and S. T. Wilson, Zeolites in Catalysis: Properties and Applications, 2017, pp. 310–350 Search PubMed.
- V. Valtchev and S. Mintova, MRS Bull., 2016, 41, 689–693 CrossRef CAS.
- D. P. Serrano, J. M. Escola and P. Pizarro, in Mesoporous Zeolites: Preparation, Characterization and Applications, 2015, pp. 157–198 Search PubMed.
- K. Zhang and M. L. Ostraat, Catal. Today, 2016, 264, 3–15 CrossRef CAS.
- W. Schwieger, A. G. Machoke, B. Reiprich, T. Weissenberger, T. Selvam and M. Hartmann, Zeolites in Catalysis: Properties and Applications, 2017, pp. 103–145 Search PubMed.
- T. Prasomsri, W. Jiao, S. Z. Weng and J. Garcia Martinez, Chem. Commun., 2015, 51, 8900–8911 RSC.
- A. Feliczak-Guzik, Microporous Mesoporous Mater., 2018, 259, 33–45 CrossRef CAS.
- D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC.
- H. K. Beyer, Molecular Sieves: Science and Technology, 2007, pp. 203–255 Search PubMed.
- R. Millini and G. Bellussi, in Mesoporous Zeolites: Preparation, Characterization and Applications, 2015, pp. 541–564 Search PubMed.
- R. Chal, C. Gérardin, M. Bulut and S. VanDonk, ChemCatChem, 2011, 3, 67–81 CrossRef CAS.
- W. Schwieger, A. G. Machoke, T. Weissenberger, A. Inayat, T. Selvam, M. Klumpp and A. Inayat, Chem. Soc. Rev., 2016, 45, 3353–3376 RSC.
- Y. Zhu, Z. Hua, J. Zhou, L. Wang, J. Zhao, Y. Gong, W. Wu, M. Ruan and J. Shi, Chem. – Eur. J., 2011, 17, 14618–14627 CrossRef CAS PubMed.
- K. Zhang, C. Li, Z. Liu, M. Wang, X. Yan and H. Xi, Chem. – Asian J., 2017, 12, 2711–2719 CrossRef CAS PubMed.
- F. S. Xiao, L. Wang, C. Yin, K. Lin, Y. Di, J. Li, R. Xu, D. S. Su, R. Schlögl, T. Yokoi and T. Tatsumi, Angew. Chem., Int. Ed., 2006, 45, 3090–3093 CrossRef CAS PubMed.
- F. Liu, T. Willhammar, L. Wang, L. Zhu, Q. Sun, X. Meng, W. Carrillo-Cabrera, X. Zou and F. S. Xiao, J. Am. Chem. Soc., 2012, 134, 4557–4560 CrossRef CAS PubMed.
- A. Sachse and J. García-Martínez, Chem. Mater., 2017, 29, 3827–3853 CrossRef CAS.
- A. J. J. Koekkoek, C. H. L. Tempelman, V. Degirmenci, M. Guo, Z. Feng, C. Li and E. J. M. Hensen, Catal. Today, 2011, 168, 96–111 CrossRef CAS.
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D. H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718–723 CrossRef CAS PubMed.
- C. H. L. Tempelman, M. T. Portilla, M. E. Martínez-Armero, B. Mezari, N. G. R. De Caluwé, C. Martínez and E. J. M. Hensen, Microporous Mesoporous Mater., 2016, 220, 28–38 CrossRef CAS.
- K. Cho, H. S. Cho, L. C. De Ménorval and R. Ryoo, Chem. Mater., 2009, 21, 5664–5673 CrossRef CAS.
- A. Inayat, I. Knoke, E. Spiecker and W. Schwieger, Angew. Chem., Int. Ed., 2012, 51, 1962–1965 CrossRef CAS PubMed.
- X. F. Li, R. Prins and J. A. van Bokhoven, J. Catal., 2009, 262, 257–265 CrossRef CAS.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- F. M. Menger and C. A. Littau, J. Am. Chem. Soc., 1991, 113, 1451–1452 CrossRef CAS.
- C. Jo, J. Jung, H. S. Shin, J. Kim and R. Ryoo, Angew. Chem., Int. Ed., 2013, 52, 10014–10017 CrossRef CAS PubMed.
- W. Kim, J. C. Kim, J. Kim, Y. Seo and R. Ryoo, ACS Catal., 2013, 3, 192–195 CrossRef CAS.
- D. Xu, Y. Ma, Z. Jing, L. Han, B. Singh, J. Feng, X. Shen, F. Cao, P. Oleynikov, H. Sun, O. Terasaki and S. Che, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- Z. Liu, Y. Hua, J. Wang, X. Dong, Q. Tian and Y. Han, Mater. Chem. Front., 2017, 1, 2195–2212 RSC.
- B. K. Singh, D. Xu, L. Han, J. Ding, Y. Wang and S. Che, Chem. Mater., 2014, 26, 7183–7188 CrossRef CAS.
- X. Zhu, R. Rohling, G. Filonenko, B. Mezari, J. P. Hofmann, S. Asahina and E. J. M. Hensen, Chem. Commun., 2014, 50, 14658–14661 RSC.
- L. Meng, X. Zhu, W. Wannapakdee, R. Pestman, M. G. Goesten, L. Gao, A. J. F. van Hoof and E. J. M. Hensen, J. Catal., 2018, 361, 135–142 CrossRef CAS.
- V. J. Margarit, M. R. Díaz-Rey, M. T. Navarro, C. Martínez and A. Corma, Angew. Chem., Int. Ed., 2018, 57, 3459–3463 CrossRef CAS PubMed.
- A. Bolshakov, D. E. Romero Hidalgo, A. J. F. van Hoof, N. Kosinov and E. J. M. Hensen, ChemCatChem, 2019, 11, 2803–2811 CrossRef CAS.
- L. Meng, B. Mezari, M. G. Goesten and E. J. M. Hensen, Chem. Mater., 2017, 29, 4091–4096 CrossRef CAS PubMed.
- V. R. R. Marthala, M. Hunger, F. Kettner, H. Krautscheid, C. Chmelik, J. Kärger and J. Weitkamp, Chem. Mater., 2011, 23, 2521–2528 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459–8467 CrossRef CAS PubMed.
- S. Storck, H. Bretinger and W. F. Maier, Appl. Catal., A, 1998, 174, 137–146 CrossRef CAS.
- J. Shi, M. W. Anderson and S. W. Carr, Chem. Mater., 1996, 8, 369–375 CrossRef CAS.
- Y. Lee, M. B. Park, P. S. Kim, A. Vicente, C. Fernandez, I. S. Nam and S. B. Hong, ACS Catal., 2013, 3, 617–621 CrossRef CAS.
- V. L. Zholobenko, D. B. Lukyanov, J. Dwyer and W. J. Smith, J. Phys. Chem. B, 2002, 102, 2715–2721 CrossRef.
- A. B. Pinar, C. Márquez-Álvarez, M. Grande-Casas and J. Pérez-Pariente, J. Catal., 2009, 263, 258–265 CrossRef CAS.
- G. Onyestyák, Microporous Mesoporous Mater., 2007, 104, 192–198 CrossRef.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Petrini, G. Leofanti, M. Padovan and C. O. Areàn, J. Chem. Soc., Faraday Trans., 1992, 88, 2959–2969 RSC.
- C. Márquez-Alvarez, A. B. Pinar, R. García, M. Grande-Casas and J. Pérez-Pariente, Top. Catal., 2009, 52, 1281–1291 CrossRef.
- J. N. Kondo, E. Yoda, H. Ishikawa, F. Wakabayashi and K. Domen, J. Catal., 2000, 191, 275–281 CrossRef CAS.
- X. Zhu, J. P. Hofmann, B. Mezari, N. Kosinov, L. Wu, Q. Qian, B. M. Weckhuysen, S. Asahina, J. Ruiz-Martínez and E. J. M. Hensen, ACS Catal., 2016, 6, 2163–2177 CrossRef CAS.
- M. S. Holm, S. Svelle, F. Joensen, P. Beato, C. H. Christensen, S. Bordiga and M. Bjørgen, Appl. Catal., A, 2009, 356, 23–30 CrossRef CAS.
- G. Busca, Microporous Mesoporous Mater., 2017, 254, 3–16 CrossRef CAS.
- B. Bonelli, B. Onida, J. D. Chen, A. Galarneau, F. Di Renzo, F. Fajula and E. Garrone, Microporous Mesoporous Mater., 2004, 67, 95–106 CrossRef CAS.
- B. Onida, L. Borello, B. Bonelli, F. Geobaldo and E. Garrone, J. Catal., 2003, 214, 191–199 CrossRef CAS.
- W. Schirmer, Molecular Transport and Reaction in Zeolites: Design and Application of Shape Selective Catalysis, 1995, vol. 191, pp. 55–66 Search PubMed.
- C. D. Baertsch, H. H. Funke, J. L. Falconer and R. D. Noble, J. Phys. Chem., 1996, 100, 7676–7679 CrossRef CAS.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS PubMed.
- B. de Ménorval, P. Ayrault, N. S. Gnep and M. Guisnet, Appl. Catal., A, 2006, 304, 1–13 CrossRef.
- D. Zhang, R. Al-Hajri, S. a I. Barri and D. Chadwick, Chem. Commun., 2010, 46, 4088–4090 RSC.
- T. Takata, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2016, 225, 524–533 CrossRef CAS.
- D. H. Olson, G. T. Kokotailo, S. L. Lawton and W. M. Meier, J. Phys. Chem., 1981, 85, 2238–2243 CrossRef CAS.
- H. Robson, Verified Syntheses of Zeolitic Materials, 2016 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- D. Freude, M. Hunger, H. Pfeifer and W. Schwieger, Chem. Phys. Lett., 1986, 128, 62–66 CrossRef CAS.
- F. Deng, Y. Yue and C. Ye, J. Phys. Chem. B, 2002, 102, 5252–5256 CrossRef.
- J. Huang, Y. Jiang, V. R. R. Marthala, B. Thomas, E. Romanova and M. Hunger, J. Phys. Chem. C, 2008, 112, 3811–3818 CrossRef CAS.
- A. A. Gabrienko, I. G. Danilova, S. S. Arzumanov, L. V. Pirutko, D. Freude and A. G. Stepanov, J. Phys. Chem. C, 2018, 122, 25386–25395 CrossRef CAS.
- H. Sachsenröder, E. Brunner, M. Koch, H. Pfeifer and B. Staudte, Microporous Mater., 1996, 6, 341–347 CrossRef.
- E. Brunner, J. Mol. Struct., 1995, 355, 61–85 CrossRef CAS.
- A. A. Gabrienko, I. G. Danilova, S. S. Arzumanov, A. V. Toktarev, D. Freude and A. G. Stepanov, Microporous Mesoporous Mater., 2010, 131, 210–216 CrossRef CAS.
- L. Song, Z. L. Sun, H. Y. Ban, M. Dai and L. V. C. Rees, Adsorption, 2005, 11, 325–339 CrossRef CAS.
- H. Ban, J. Gui, X. Zhang, M. Dai, L. Song, Z. Sun and L. V. C. Rees, Thermochim. Acta, 2005, 439, 121–126 CrossRef CAS.
- F. Schmidt, S. Paasch, E. Brunner and S. Kaskel, Microporous Mesoporous Mater., 2012, 164, 214–221 CrossRef CAS.
- T. Remy, J. Cousin Saint Remi, R. Singh, P. A. Webley, G. V. Baron and J. F. M. Denayer, J. Phys. Chem. C, 2011, 115, 8117–8125 CrossRef CAS.
- W. Dai, G. Wu, L. Li, N. Guan and M. Hunger, ACS Catal., 2013, 3, 588–596 CrossRef CAS.
- D. Mores, E. Stavitski, M. H. F. Kox, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Chem. – Eur. J., 2008, 14, 11320–11327 CrossRef CAS PubMed.
- M. Bertau, H. Offermanns, L. Plass, F. Schmidt and H. J. Wernicke, Methanol: The basic chemical and energy feedstock of the future, 2014, pp. 423–489 Search PubMed.
- D. Fan, P. Tian, X. Su, Y. Yuan, D. Wang, C. Wang, M. Yang, L. Wang, S. Xu and Z. Liu, J. Mater. Chem. A, 2013, 1, 14206–14213 RSC.
- B. Vora, J. Q. Chen, A. Bozzano, B. Glover and P. Barger, Catal. Today, 2009, 141, 77–83 CrossRef CAS.
- X. Zhu, N. Kosinov, A. V. Kubarev, A. Bolshakov, B. Mezari, I. Valastyan, J. P. Hofmann, M. B. J. Roeffaers, E. Sarkadi-Pribóczki and E. J. M. Hensen, ChemCatChem, 2017, 9, 3470–3477 CrossRef CAS.
- L. Qi, J. Li, Y. Wei, Y. He, L. Xu and Z. Liu, RSC Adv., 2016, 6, 52284–52291 RSC.
- M. Dyballa, U. Obenaus, M. Blum and W. Dai, Catal. Sci. Technol., 2018, 8, 4440–4449 RSC.
- B. Arstad and S. Kolboe, J. Am. Chem. Soc., 2001, 123, 8137–8138 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional IR spectra, TGA, Ar adsorption and FTIR results, and SEM images. See DOI: 10.1039/c9cy02001b |
| This journal is © The Royal Society of Chemistry 2019 |