
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono- and diylide-substituted phosphines (YPhos): impact of the ligand properties on the catalytic activity in gold(I)-catalysed hydroaminations†
Christopher
Schwarz
,
Jens
Handelmann
,
Daniel M.
Baier
,
Alina
Ouissa
and
Viktoria H.
Gessner
 *
*
Faculty of Chemistry and Biochemistry, Chair of Inorganic Chemistry II, Ruhr University Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: viktoria.gessner@rub.de
First published on 13th November 2019
Abstract
Understanding the impact of ligand properties on their performance in catalysis is seminal for future ligand design and catalyst improvement. In this work, the influence of the steric and electronic properties on the efficiency of a series of mono- and diylide-functionalized phosphines of type YCNPR2 and (YCN)2PR (with YCN = Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CN) and R = Ph, Cy or tBu) on their efficiency in gold(I) catalyzed haydroaminations is studied. The diylidephosphines are particularly electron-rich and sterically encumbering but led to lower conversions than expected based on their donor strength. Systematic analysis of the relation between the donor strength and the catalytic activity revealed a linear correlation for the monoylide-functionalized phosphines. Thus, the most electron-rich phosphine YCNPtBu2 gives rise to the most active catalyst with turnover numbers greater than 5000. In contrast, no correlation was found for the diylide compounds due to steric congestions which overcompensated the beneficial electronic properties. Accordingly, higher temperatures had a stronger impact on the diylide phosphine-based catalysts, thus leading to similar activities of YCNPtBu2 and (YCN)2PCy at 50 °C. Overall, these results provide valuable information for future ligand design and the general impact of steric and electronic properties on the catalytic activity.
C(CN) and R = Ph, Cy or tBu) on their efficiency in gold(I) catalyzed haydroaminations is studied. The diylidephosphines are particularly electron-rich and sterically encumbering but led to lower conversions than expected based on their donor strength. Systematic analysis of the relation between the donor strength and the catalytic activity revealed a linear correlation for the monoylide-functionalized phosphines. Thus, the most electron-rich phosphine YCNPtBu2 gives rise to the most active catalyst with turnover numbers greater than 5000. In contrast, no correlation was found for the diylide compounds due to steric congestions which overcompensated the beneficial electronic properties. Accordingly, higher temperatures had a stronger impact on the diylide phosphine-based catalysts, thus leading to similar activities of YCNPtBu2 and (YCN)2PCy at 50 °C. Overall, these results provide valuable information for future ligand design and the general impact of steric and electronic properties on the catalytic activity.
Introduction
Buchwald-type biarylphosphines are amongst the most important ligands in homogenous catalysis.1 Their success can be attributed to their electronic and steric properties, which allow the stabilization of low-coordinate as well as cationic complexes which are often the active species in catalytic transformations. In general, dialkylbiarylphosphines are usually electron-rich and sterically demanding and thus also allow for the stabilization of reactive metal species. Thereby, steric protection is further supported by a secondary interaction between the π system of the lateral arene with the metal centre (Fig. 1).2 This unique architecture was found to be particularly beneficial in palladium(0) catalysed coupling reactions3 but later also successfully applied to gold(I) catalysis.4 While first experiments focused on biaryl phosphines that were also employed in Pd-catalysed reactions,5 more recent studies addressed the adaption of the ligand design to the somewhat different requirements in gold catalysis and thus led to a further improvement in the catalyst activities and productivities.6 The customized ligand design was necessary to decrease the catalyst loading below 0.5 mol%, which is required to make gold catalysis more attractive for medium- or large-scale applications. Detailed mechanistic studies by Xu and coworkers revealed that biarylphosphines are particularly suited for Au(I)-catalysed reactions such as the hydroamination of alkynes, in which the protodeauration step is rate-determining and catalyst deactivation a serious limiting factor.7 Here, the biaryl moiety on the phosphine ligand was found to slow down the decay of the cationic gold species and thus improved catalyst stability, while the high donor strength facilitated the rate-limiting prododeauration step. Recently, this molecular design was successfully applied to other phosphines8 as well as N-heterocyclic carbenes.9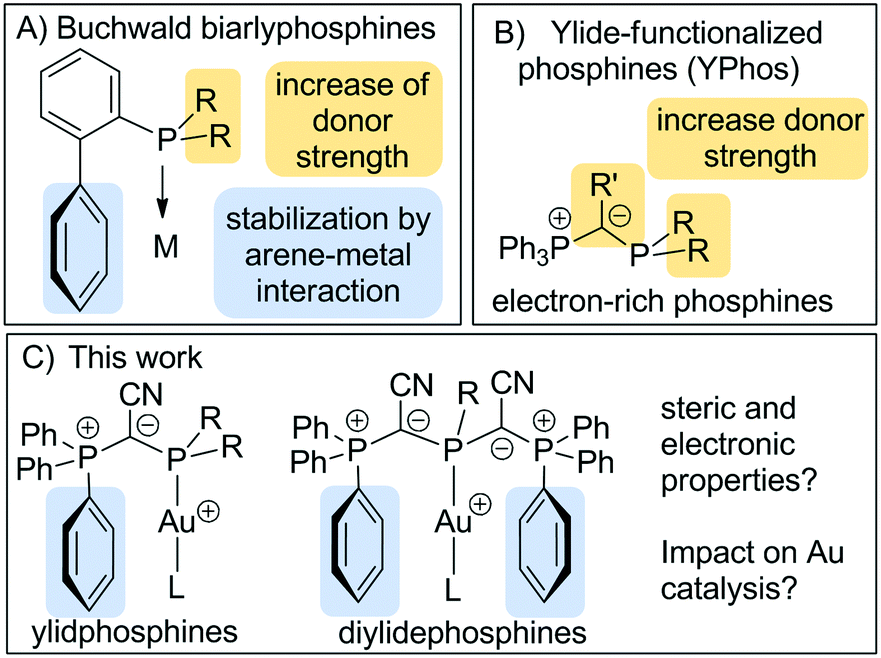 | ||
| Fig. 1 Cationic gold complexes with biaryl- and ylide-substituted phosphines. | ||
Recently, our group reported on the synthesis of ylide-substituted phosphines (YPhos) and their application in gold-catalyzed hydroamination reactions as well as palladium catalyzed Buchwald–Hartwig aminations and α-arylations.10,11 The molecular architecture of the triphenylphosphonium-substituted YPhos ligands is somewhat reminiscent to biarylphosphines due to the possible interaction of the metal with one of the phosphonium bound phenyl groups (Fig. 1). However, due to the carbanionic centre next to the phosphorus atom, these ligands possess an increased donor capacity, which – depending on the substitution pattern – may reach or even surpass that of N-heterocyclic carbenes.10 Given the efficiency of mono-ylide substituted phosphines, we now became interested in the impact of two ylide moieties on the ligand synthesis and their efficiency in catalytic applications. Owing to their enhanced donor properties as well as increased steric demand, we expected these ligands to be particularly suited for the stabilization of the cationic Au(I) species and the production of highly effective catalysts for hydroamination reactions. In order to obtain systematic information about the impact of the ligand design on the catalytic activity we addressed a comparison of the electronic and steric properties of a series of mono-, di- and triylide-substituted phosphines.
Results and discussion
Ligand synthesis and characterization
To access a series of mono-, di- and tri-ylide substituted phosphines for systematic studies we chose a synthetic route via the metallated ylide YCN–M (M = Na or K).12,13 We hypothesized that the cyanido group in the ylide backbone would be sufficiently small to allow for multiple ylide-functionalisations at a single phosphorus centre.14 Furthermore, YCN–M is readily available from the corresponding phosphonium salt via step-wise deprotonation with potassium or sodium hexamethyldisilazide. Thus, the corresponding phosphines are accessible in only few reaction steps from readily available compounds. The synthesis of the mono-ylide substituted phosphines 1 and 2 was previously reported via reaction of YCN–M with one equivalent of the corresponding chlorophosphine (Scheme 1).10 Analogous treatment of YCN–Na with di-tert-butylchlorophosphine, tBu2PCl, selectively produced the ylide-substituted phosphine YCNPtBu2 (3) as evidenced by 31P{1H} NMR spectroscopy. Work-up delivered the ligand as colorless solid in 46% yield. The diylide-substituted phosphines were prepared in a similar way by reaction of YCN–K with the corresponding dichloro-phosphines. Thus, (YCN)2PPh (4), (YCN)2PCy (5) and (YCN)2PtBu (6) could be obtained after work-up as colourless solids in good yields between 66 and 77%. Overall, the described reaction protocol provides facile access to a series of ligands by simple salt metatheses in the last reaction step. This versatility is certainly an advantage to many other ligand platforms.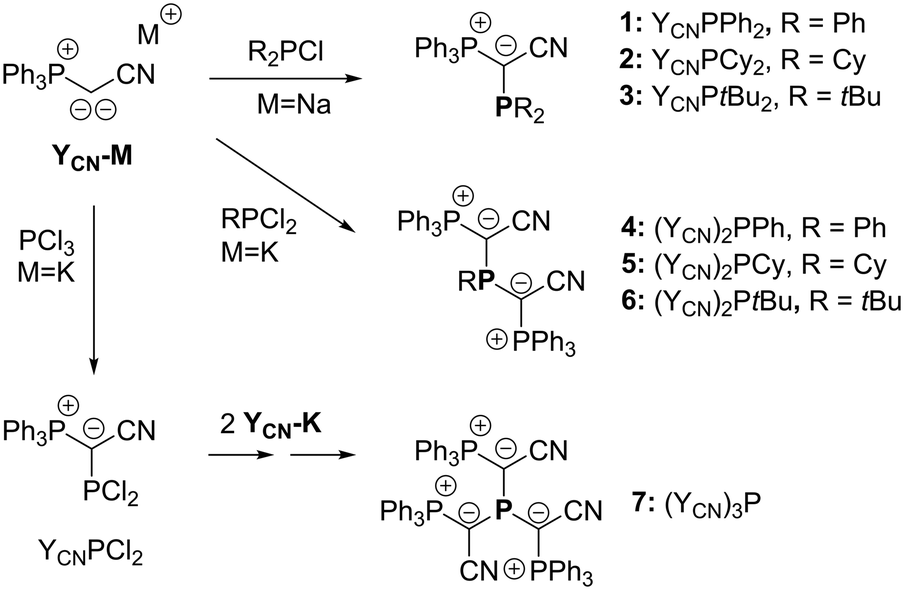 | ||
| Scheme 1 Synthesis of the mono-, di- and tri-ylide substituted phosphines. | ||
Unfortunately, employment of the same reaction protocol as outlined in Scheme 1 with phosphorus trichloride did not result in the selective formation of the desired tri-ylide substituted phosphine (YCN)3P (7). Likewise, attempts to introduce the three ylide groups in a step-wise fashion via formation of the chlorophosphines YCNPCl2 and (YCN)2PCl proofed to be unsuccessful. Only small amounts of 7 could be isolated in form of yellow crystals, which confirmed the identity of 7 (Fig. 2). However, this reaction protocol did not deliver sufficient amounts of the triylidephosphine for further investigations.
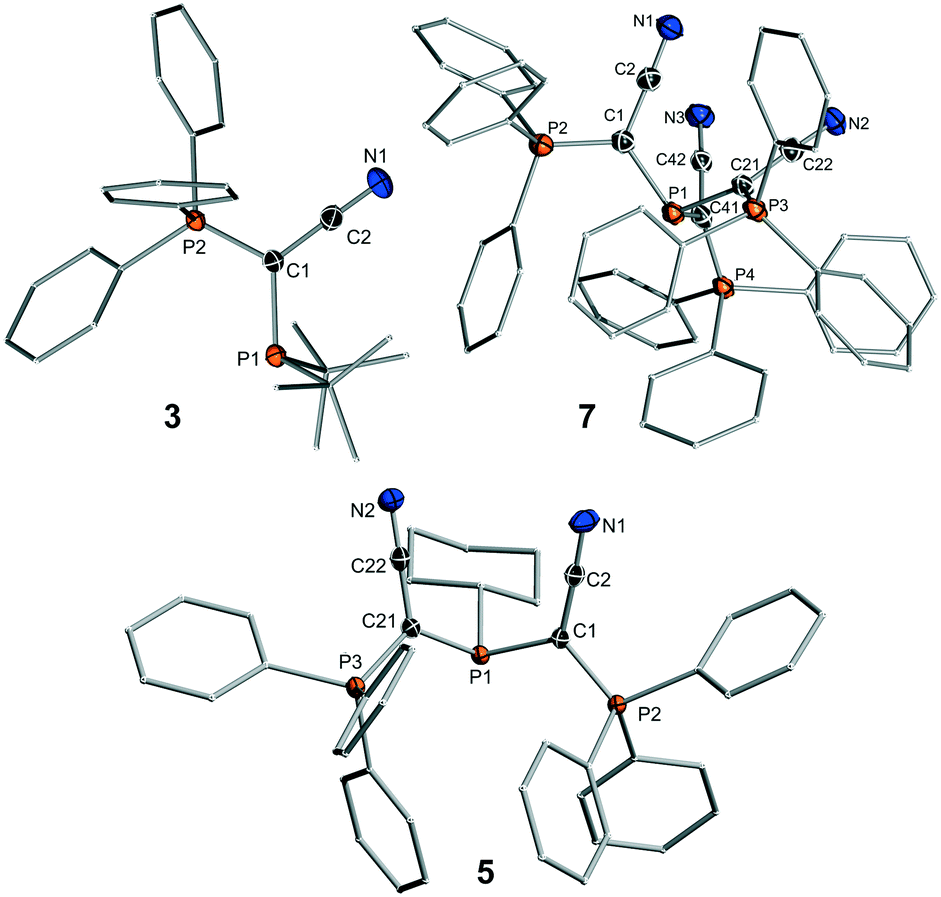 | ||
| Fig. 2 Molecular structures of YCNPtBu2 (3), (YCN)2PCy (5) and (YCN)3P (7). Thermal ellipsoids drawn at the 50% probability level. Structures of the other ligands are given in the SI, important bond lengths and angles in Table 1. | ||
All mono- and diylidephosphine ligands were characterized by NMR and IR spectroscopy as well as elemental and single-crystal X-ray diffraction analysis.15 Particularly, the NMR data of these ligand systems are highly meaningful (Table 1). The mono-ylide-substituted phosphines exhibit two doublets with 2JPP coupling constants between 135 and 150 Hz. In contrast, the diylide-functionalized compounds Y2PR show AX2 coupling patterns with slightly larger coupling constants than their monoylide analogues. This is probably due to the increased steric bulk, which results in larger P–C–P angles and thus larger coupling constants. For example, in case of the phenylphosphines 1 and 4 an increase of 13.4 Hz was observed. It is also worth mentioning, that the introduction of the second ylide moiety is also accompanied by a high-field shift of the signal of the phosphine phosphorus atom. For example, the signal of the phosphine moiety appears at δP 17.0 ppm in YCNPtBu2 (3), while it is shifted to δP −24.4 ppm in (YCN)2PtBu (6).
| Ligand | δ(31P(1)) [ppm], 2JPP [Hz] | C–P(1) | C–CCN | C–N | P–C–P |
|---|---|---|---|---|---|
| a Two molecules in the asymmetric unit; values are average values. | |||||
| 1 | −16.3, 146.8 | 1.800(1) | 1.407(2) | 1.160(2) | 118.1(1) |
| 2 | −11.3, 135.7 | 1.819(1) | 1.398(2) | 1.161(2) | 119.4(1) |
| 3 | 17.0, 149.7 | 1.813(2) | 1.404(3) | 1.163(3) | 119.7(1) |
| 4 | −26.4, 160.2 | 1.809(3) | 1.407(4) | 1.157(4) | 115.2(2) |
| 1.792(3) | 1.412(4) | 1.160(4) | 120.1(2) | ||
| 5 | −31.2, 140.5 | 1.809(2) | 1.404(3) | 1.163(3) | 120.3(1) |
| 1.818(2) | 1.392(3) | 1.160(3) | 118.8(1) | ||
| 6 | −24.4, 161.2 | 1.808(2) | 1.410(3) | 1.158(3) | 119.5(1) |
| 1.814(2) | 1.400(3) | 1.161(3) | 114.1(1) | ||
The molecular structures of 3, 5 and 7 are shown in Fig. 2, those of the other ligands are provided in the ESI.† Interestingly, in all structures of the diylidephosphines and even in that of (YCN)3P, the cyano moieties point to the same side of the molecule, that is the opposite side of the phosphine lone pair. This is particularly noteworthy for the triylide-substituted ligand, where all bulky triphenylphosphonium moieties still prefer to arrange on the same side. We assume that this arrangement is due to electronic effects and the stabilization of the lone pair by interaction with antibonding CN orbitals or the minimization of repulsion between the phosphine lone pair and the π orbitals of the CN moiety. The P(1)–C bonds are all longer than the corresponding P–C bonds to the phosphonium moieties and only change slightly within the whole ligand series. The same holds true for the C–CCN and C–N bonds in the ligand backbone. It is also noteworthy, that the P–C–P angles are similar in all structures despite the different steric demand of the ligands. This suggests that despite the bulk of the PPh3 groups the ligands are not overcrowded.
Determination of the steric and electronic ligand properties
To estimate the donor strengths of the phosphines 1–6 we set out to measure the corresponding Tolman electronic parameter (TEP). Since the classical procedure via synthesis of the LNi(CO)3 complexes was found to be no reliable method for the bulky YPhos ligands due to the often observed displacement of two CO ligands,16 the [(L)Rh(acac)CO] complexes (acac = acetylacetonate) were used as alternative.17 The TEP value can be calculated from the linear relationship of the CO stretching frequencies in both complexes. In case of the diylidephosphine (YCN)2PCy, we were also able to isolate the rhodium complex in crystalline form to verify the identity of the complex. X-ray crystallography (Fig. 3) unambiguously proved the formation of the expected rhodium complex with one YPhos and one CO ligand coordinating to the metal. The TEP values obtained by IR spectroscopy are given in Table 2. Unfortunately, attempts to determine the TEP value of the bulkiest ligand 6 by this method repeatedly failed. Thus, the corresponding [(L)Ir(CO)2Cl] complex of 6 was synthesized and the TEP value determined according to literature reports.18 The obtained data clearly reflect the expected trends in the donor strengths, i.e. YPPh2 < YPCy2 < YPtBu2 and YPR2 < Y2PR. Thus, (YCN)2PtBu is the strongest donor in the ligand series. With a TEP value of 2054.7 cm−1, the donor capacity of 6 is somewhat larger than that of PtBu3 (TEP(PtBu3) = 2056.1 cm−1).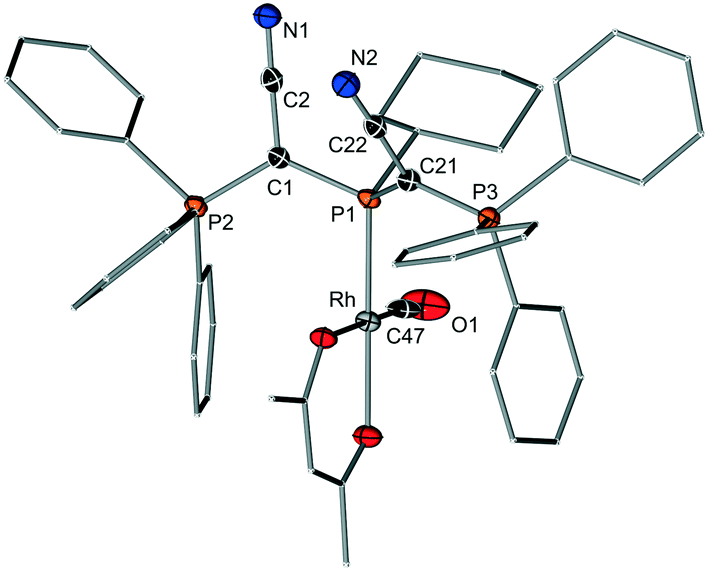 | ||
| Fig. 3 Molecular structure of [5·Rh(CO)acac]. | ||
| Ligand | ν CO(Rh)a [cm−1] | TEPb [cm−1] | % Vburc |
|---|---|---|---|
| a CO stretching frequency in the [(L)Rh(CO)acac] complexes. b Determined by the relationship between νCO for [(L)Ni(CO)3] and [(L)Rh(acac)(CO)].19 c Calculated using the SambVca 2.0 program for the LAuCl complexes with a P–M distance of 2.28 Å including H atoms.20 d Determined by the relationship between νCO for [(L)Ni(CO)3] and [(L)Ir(CO)2Cl].18 | |||
| 1 | 1973.8 | 2066.7 | 44.3 |
| 2 | 1958.3 | 2057.8 | 45.4 |
| 3 | 1954.1 | 2055.4 | 50.8 |
| 4 | 1963.6 | 2060.9 | 57.2 |
| 5 | 1955.0 | 2055.9 | 55.2 |
| 6 | — | 2054.7d | 62.7 |
Next, the steric properties of the ligands were studied by determination of their buried volume (% Vbur). To this end, the gold(I) chloride complexes of all ligands were prepared by treatment of the phosphines with (THT)AuCl. All complexes could be isolated as colourless solids in good yields higher than 80%, except for 6·AuCl which was only isolable in 33% yield. In the 31P{1H} NMR spectra, all complexes exhibit a distinct down-field shift of the signal of the phosphine phosphorus atom as expected for metal coordination. Single crystals were grown to determine the structure and the buried volume of the ligands. The structures of the AuCl complexes with 3 and 5 as ligands are exemplarily depicted in Fig. 4, the others are given in the ESI.† In the molecular structures of the gold complexes, the phosphines retain the conformation observed in the free ligands with the CN moieties pointing away from the phosphine lone pair. Thus, the metal is always shielded by one or two PPh3 moieties. All ligands – particularly the diylide-substituted compounds – are extremely bulky showing a buried volume between 42 (for Y2PCy) and 63% (Y2PtBu). The diylidephosphines are thus more encumbering than classical phosphine ligands such as PtBu3 (% Vbur = 38.1 (ref. 20)) or PAd3 (% Vbur = 40.5 (ref. 19)) and in the range of biaryl phosphines (e.g. (% Vbur(CyJohnPhos) = 46.7 (ref. 20)). Hence, they should be capable to efficiently sterically protect a cationic gold species formed after halide abstraction. Overall, the analysis of the steric and electronic properties of the YPhos ligands showed that the ylide substituent YCN can be regarded as a slightly more electron-releasing and considerably more sterically demanding tert-butyl group.
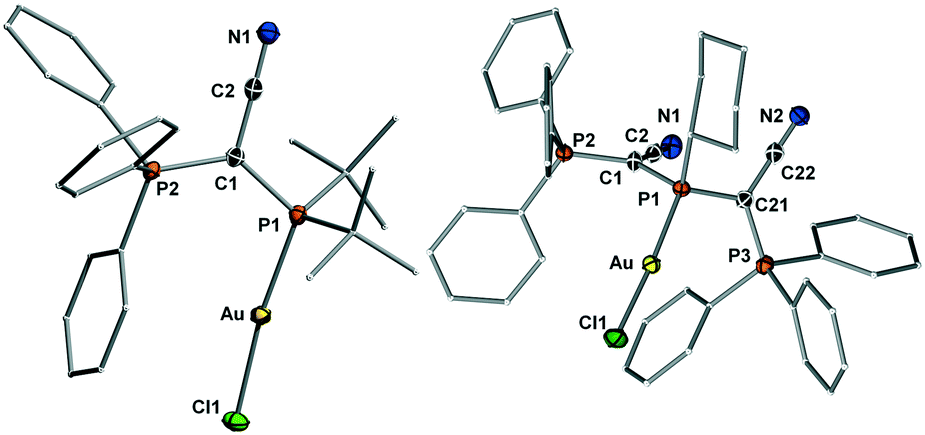 | ||
| Fig. 4 Molecular structures of the gold complexes of YCNPtBu2 (3) and (YCN)2PCy (5). Thermal ellipsoids drawn at the 50% probability level. | ||
Catalytic activity
With the AuCl complexes in hand we turned our attention towards catalytic applications. As a preliminary test reaction to determine the relative activities of 1–6 we compared their performance in the hydroamination of phenyl acetylene with aniline. The performance was tested with NaBArF4 for halide abstraction and a low catalyst loading of only 0.1 mol%. First experiments were conducted at room temperature to examine, whether the catalysts are already active at these mild conditions. The conversion over time plots are depicted in Fig. 5 (top). To our delight, all gold complexes exhibited a remarkable activity already at room temperature with only 0.1 mol% of catalyst loading. Typically, higher catalyst loadings as well as harsher reaction conditions are required in gold catalysis to obtain high conversions in hydroamination reactions.21 However, all catalysts based on L1–L6 delivered more than 80% conversion after 48 h. The highest activity was observed with the most electron-rich mono-ylide substituted phosphine YCNPtBu2, which delivers 98% of product after 24 h. With 0.5 mol% of catalyst loading the reaction was complete within only 3 h reaction time, which is certainly a remarkable activity for room temperature Au(I) catalysis and has rarely been observed for any other catalytic system.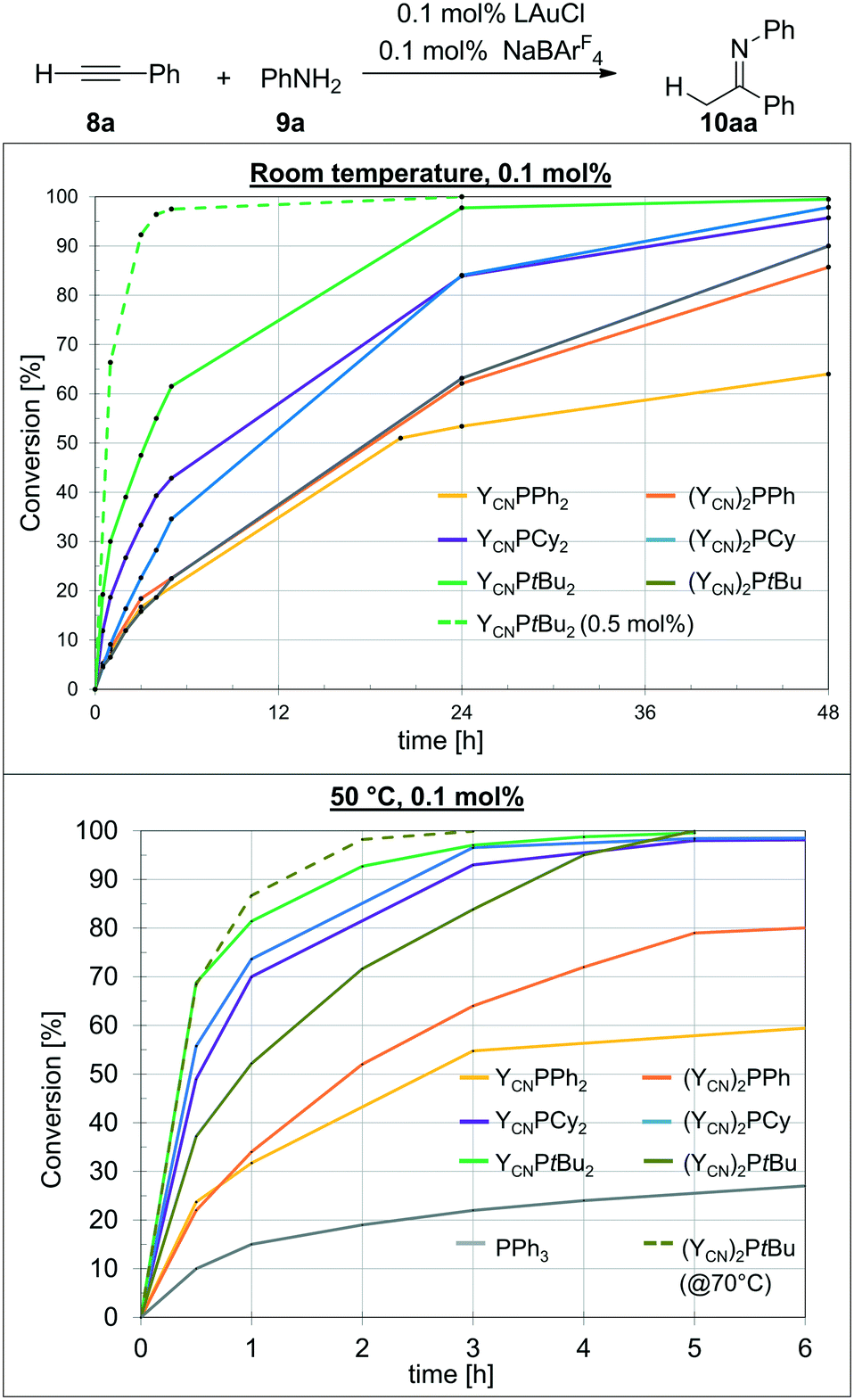 | ||
| Fig. 5 Conversion over time plots of the hydroamination of phenyl acetylene with aniline at (top) room temperature and (bottom) 50 °C. NMR yields determined by direct integration of the peak for the imine product with respect to the peak for the alkyne starting material. Values are average values of at least two runs. | ||
The monoylide phosphines YCNPR2 show a clear increase in activity with increasing donor strength, i.e. YCNPPh2 < YCNPCy2 < YCNPtBu2. This is expected, since previous studies have shown that more electron-rich ligands usually facilitate the rate-limiting protodeauration step and give way to more active hydroamination catalysts.6 However, this contrasts the observations for the diylide phosphines. For example, the most electron-rich diylidephosphine (YCN)2PtBu gave lower yields than its monoylide-substituted analogue and comparable yields than YCNPPh2. In fact, all three diylidephosphines showed lower activities than expected based on their donor strength. This indicated that either the beneficial electronic effects of the diylide systems are overcompensated by steric crowding or that these ligands become too electron-rich, thus resulting in a less effective alkyne activation and hence in a shift of the rate-limiting step to an earlier point in the catalytic cycle. However, since for example (YCN)2PCy is according to its TEP value (Table 2) a similar strong donor than YCNPtBu2 we believe that steric hinderance is the main factor leading to different activation enthalpies for the gold mono- and diylidephosphine-catalysed transformations. Accordingly, we turned our attention toward catalysis studies at 50 °C to examine whether at those temperatures the diylide systems perform superior to their monoylide congeners.
At 50 °C, the catalysis becomes in general faster (Fig. 5, bottom). However, this particularly accounts for the diylidephosphines. While YCNPtBu2 still yields the most active catalyst, (YCN)2PCy now shows a similar activity, giving more than 95% yield after 3 h reaction time with 0.1 mol% catalyst loading. The gold catalysts based on YCNPCy2 (2) and (YCN)2PtBu (6) are somewhat slower, but also allow for full conversion to the imine within 5 h reaction time. For comparison, PPh3 only delivers approx. 30% conversion after 24 h under the same reaction conditions.10 At higher temperatures (70 °C), the reaction with 6 as catalyst is also completed within less than 3 h reaction time. Overall, YCNPtBu2 and (YCN)2PCy are the most active catalysts and comparable to CyJohnPhos under these reaction conditions (see the ESI,† Table S1).
It has to be noted that the temperature dependency of the catalytic activity is more pronounced for the diylide-substituted phosphines than for their monoylide analogues. For example, the most demanding phosphine (YCN)2PtBu (6), which performed equally than YCNPPh2 at room temperature, is much more active at 50 °C and gives – in contrast to YCNPPh2 – full conversion within 5 h. Likewise, (YCN)2Cy and (YCN)2Ph now perform superior than their mono-ylide congeners at higher temperatures. This observation supports our hypothesis that effective room-temperature catalysis with the diylide phosphines is hampered by steric effects. Spatial limitations are presumably reached in case of the tert-butyl system. Here, the monoylide-substituted phosphine is still more active than its diylide analogue at 50 °C.
To explain the different activities of our catalysts we looked for any structure–activity relationship as guideline for future ligand design. Since the rate-limiting protodeauration step is facilitated by more electron-rich ligands,6 one could expect a dependency between the TEP value and the catalytic activity. Due to the already noted different performance of the mono and diylide phosphines at room temperature no correlation between the TEP and the catalytic performance exists for the whole ligand set. However, a linear correlation between the TEP value and the initial reaction rates22 is observed for the monoylide phosphines where presumably no or only little steric hindrance is present (green and blue lines in Fig. 6, top, Table S4, Fig. S3†). In contrast, no such correlation is seen for the diylide-substituted phosphines (orange and yellow line). Here, at first an increase in activity with an increase of donor strength is observed when going from (YCN)2PPh to (YCN)2PCy, but a clear drop in activity is found when moving to (YCN)2PtBu. This further supports that steric limits are reached with (YCN)2PtBu.
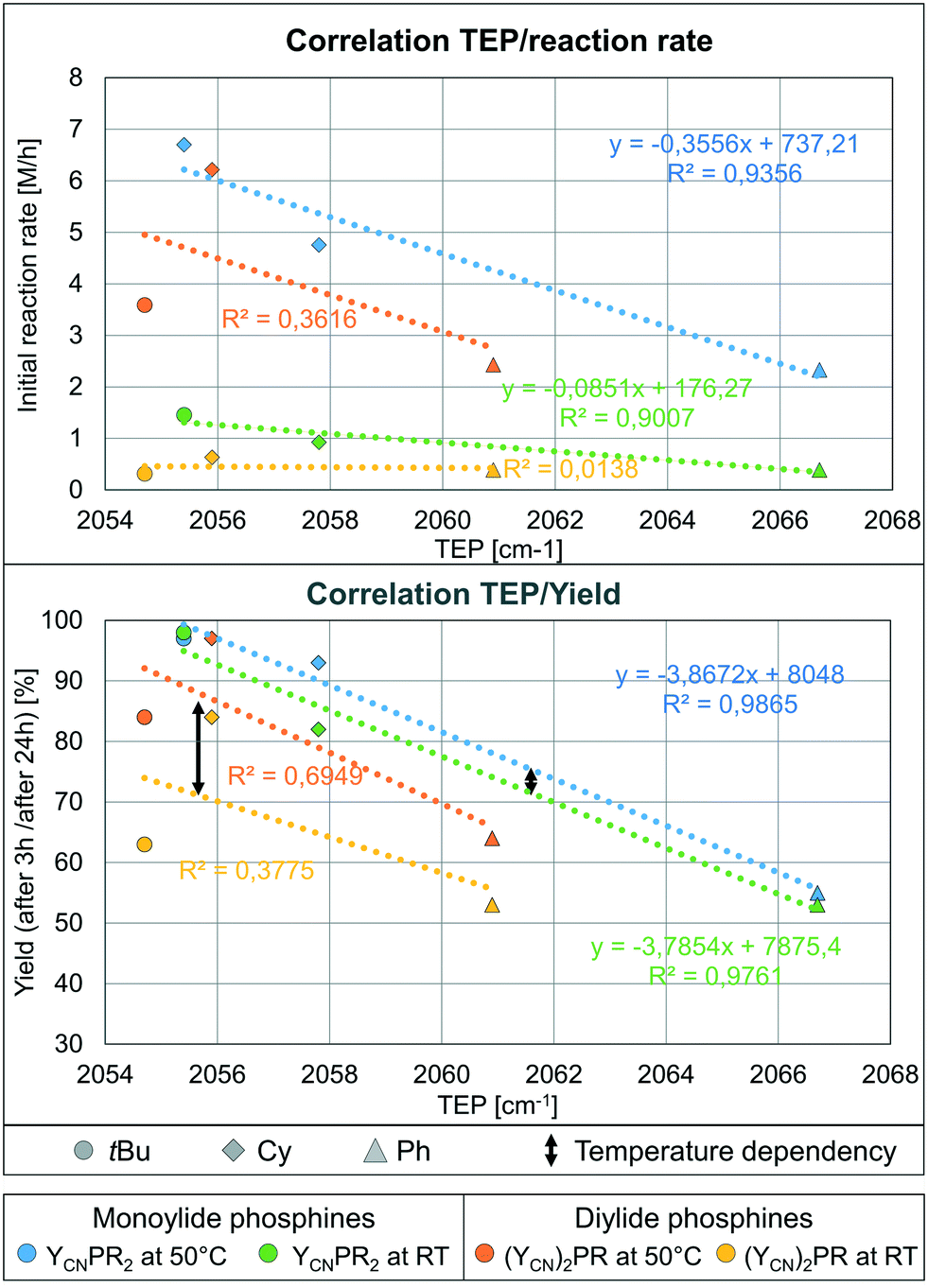 | ||
| Fig. 6 Correlation between the TEP value and the catalytic performance for the mono and diylide-substituted phosphines at rt and 50 °C: (top) with the initial reaction rate (in the first 30 min at 50 °C and in the 1st h at RT) and (below) the yield of the reaction after 3 h (50 °C) and 24 h (RT). | ||
The same observations can be made when correlating the TEP with the yields obtained after 3 and 24 h, respectively (Fig. 6, bottom). This suggests that besides facilitating the rate-limiting protodeauration step (kinetic effect), the stronger donors also stabilize the active cationic gold species more efficiently and thus lead to higher yields (thermodynamic effect). This is well in line with studies by Xu and coworkers showing that the protodeauration determines the reaction rate of the hydroamination reaction and that catalyst deactivation significantly affects the final yield of the reaction.7 Both processes are positively influenced by strong donor ligands.
The correlation between the TEP and the yield at different temperatures (black arrow, Fig. 6) nicely illustrates the stronger impact of the temperature on the activity for the diylidephosphine-based catalysts (cf. yellow line and orange line). This supports the conclusion that steric crowding in the diylide ligands hampers fast catalysis at room temperature. It should be noted that there is neither a direct correlation between the yield/rate and the buried volume nor a correlation between the difference between the observed and expected rate (calculated based on the linear regression obtained for the monoylide phosphines) and the % Vbur (see Fig. S1 and S2†). This is of course expected, since on the one hand steric protection is required for the stabilization of LAu+, but on the other hand seems to increase the activation enthalpy of the rate-limiting step. The fact that we still observe a correlation between the TEP and the reaction rates indicates that the performance of the complexes based on the “smaller” monoylide phosphines is predominately determined by the different electronic effects. This is also supported by the fact that the results for the smallest diylidephosphine (YCN)2PCy best fit into the linear regressions obtained for the monoylide catalysts (orange and yellow diamond in Fig. 6). Overall, the findings clearly show that the catalytic activity in the hydroamination nicely correlates with the donor strength of the YPhos ligands, but that steric congestions might lead to restrictions in the performance.
Catalyst productivity and scope of catalytic applications
Motivated by the excellent performance of the gold complexes based on YCNPtBu2 and (YCN)2PCy we examined their actual productivity. Overall, the catalysts provide TOFs of more than 500 h−1 with only 0.1 mol% of catalyst loading. A further decrease of the catalyst loading to 0.05 mol% still gave good yields of greater than 85% within 5 h when using YCNPtBu2 and (YCN)2PCy as ligands (Table 3). Without further optimization of the reaction conditions (e.g. impact of additives or further increase of temperature) these ligands allowed for turnover numbers of 5000 and more at 50 °C. Thus, the ylide and diylidephosphines render remarkably active catalysts, which are amongst the very few catalytic systems that reach TON of 5000 in Au(I) catalysis. Notable examples include, catalysts based on specifically functionalized biarylphosphines,6 a carbanionic phosphine decorated with a chlorinated carba-closo-dodecaborate23 or specifically designed NHC ligands.9a The activity of our YPhos-based catalysts is certainly remarkable particularly due to their facile synthesis.| Entry | Ligand | Mol% LAuCl | Reaction time [h] | Yield [%] | TON |
|---|---|---|---|---|---|
| a 5.0 mmol phenylacetylene, 5.05 mmol aniline, 50 °C, NMR yields. b After 2 h reaction time with 0.1 mol% catalyst loading a further portion of reagents was added. This was repeated 6 times. c Reaction temperature: 70 °C; 6 portions of reagents were added after 1 h reaction time each to 0.1 mol% catalyst. | |||||
| 1 | YCNPCy2 | 0.05 | 5 | 68 | 1360 |
| 2 | YCNPtBu2 | 0.05 | 5 | 90 | 1800 |
| 3 | (YCN)2PCy | 0.05 | 5 | 87 | 1740 |
| 4 | YCNPtBu2 | 0.01 | 48 | 58 | 5800 |
| 5 | YCNPtBu2 | 0.01 | 72 | 62 | 6200 |
| 6 | (YCN)2PCy | 0.01 | 48 | 48 | 4800 |
| 7 | YCNPtBu2 | 0.1b | 72 | 92 | 7360 |
| 8 | (YCN)2PCy | 0.1b | 72 | 73 | 5840 |
| 9 | (YCN)2PtBu | 0.1c | 48 | 49 | 3920 |
Next, we addressed the substrate scope of the amination reaction with both ligands YCNPtBu2 (L3) and (YCN)2PCy (L5). Fortunately, high conversions were observed with both gold complexes at 50 or 80 °C for a series of different amines and alkynes (Fig. 7). Electron-rich as well as electron-poor aryl amines as well as secondary amines could be coupled as well as internal alkynes. In general, both phosphines performed equally well, albeit in case of the secondary amine (N-methyl aniline) L5 gave only poor yields, presumably due to steric congestions. Accordingly. the amination with sterically even more demanding amines (e.g. H2N-Mes, Mes = 1,3,5-trimethylphenyl) failed, thus further supporting that steric limitations are reached with these ligands. It is also interesting to note that in case of the amination of methyl phenyl acetylene to 10ea both ligands prefer the formation of the opposite regioisomers. While L3 results in the preferred formation of the Markovnikov product, L5 favored the anti-Markovnikov product, albeit with only low selectivities (1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Nonetheless, this suggests that tailoring of the steric properties of these ligands can also allow for the regioselective amination reactions of unsymmetrical alkynes. We also attempted the amination with alkyl amines. However, these compounds showed no conversion under the reaction conditions.
:1). Nonetheless, this suggests that tailoring of the steric properties of these ligands can also allow for the regioselective amination reactions of unsymmetrical alkynes. We also attempted the amination with alkyl amines. However, these compounds showed no conversion under the reaction conditions.
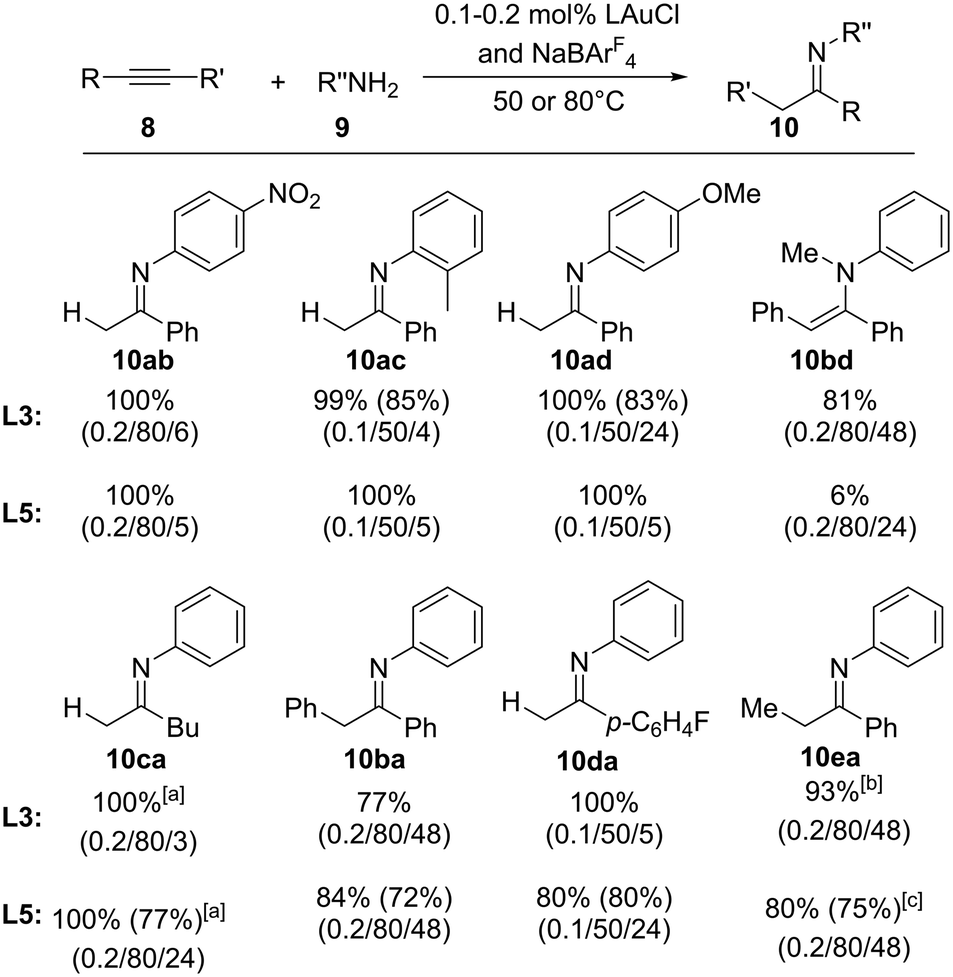 | ||
| Fig. 7 Scope of the amination of alkynes with YCNPtBu2·AuCl and (YCN)2PCy·AuCl as precatalysts. NMR yields (yields in brackets are isolated yields). Reaction conditions are given in brackets as follows (mol% catalyst/temperature/reaction time). [a] 2.6:1 mixture of the E and Z isomers. [b] 1.5:1 mixture of both isomers. [c] 1:1.5 mixture of both isomers (N,1-diphenylpropan-1-imine: N,1-diphenylpropan-2-imine). | ||
Conclusions
In summary, we have prepared a series of mono and diylide-substituted phosphines and examined their performance in the gold(I) catalysed hydroamination of alkynes. Thereby, a clear dependency between the activity of the gold catalysts and the donor strength of the phosphine ligands was found for the monoylide-substituted systems YCNPR2, which showed a linear correlation between the determined TEP value and the initial reaction rates. This correlation is not observed for the more electron-rich, but bulkier diylide compounds (YCN)2PR. Here, steric congestion leads to lower activities than expected based on the donor strength of the ligands. Accordingly, higher temperatures led to a more pronounced increase in activity of the diylide-substituted phosphines, so that the gold complex based on (YCN)2PCy (L5) becomes similarly active than the one of the most electron-rich monoylide phosphine YCNPtBu2 (L3) at 50 °C. Both, L3 and L5 provide highly productive catalysts, which give rise to turnover numbers greater than 5000.Overall, these studies demonstrate the beneficial effect of strong donor properties on the activity of gold(I) catalysts in alkyne hydroaminations. The increase of activity appears to be almost linear to the TEP value of the YPhos ligands. This suggests that the catalytic efficiency can be reliably estimated based on the donor strength of a ligand. This predictability however is complicated by steric congestions (as well as other stabilizing effects on the catalytically active gold(I) species). Our data implicate that a buried volume greater than 55% can lead to detrimental steric encumbrance which might lead to an overcompensation of beneficial electronic effects at mild conditions. Since phosphines are ubiquitous ligands in homogenous catalysis, we are currently studying the impact of the steric and electronic properties of our ligands on their catalytic efficiency to further facilitate parametrization and ligand design in the future.
Conflicts of interest
The authors have filed patent WO2019030304 covering the YPhos ligands discussed, which is held by UMICORE and products will be made commercially available.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy-EXC-2033-Projektnummer 390677874 and the European Research Council (Starting Grant: YlideLigands 677749). We thank UMICORE for donating chemicals.Notes and references
- For review articles: (a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed; (b) S. L. Buchwald, C. Mauger, G. Mignani and U. Scholz, Adv. Synth. Catal., 2006, 348, 23–39 CrossRef CAS; (c) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed; (d) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 12003–12010 CrossRef CAS PubMed.
- For examples: (a) J. M. Dennis, N. A. White, R. Y. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 4721–4725 CrossRef CAS PubMed; (b) N. H. Park, E. V. Vinogradova, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 8259–8262 CrossRef CAS PubMed; (c) P. Ruiz-Castillo, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 3085–3092 CrossRef CAS PubMed.
- Selected recent reviews: (a) A. M. Asiri and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471 RSC; (b) A. Fürstner, Angew. Chem., Int. Ed., 2018, 57, 4215 CrossRef PubMed; (c) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (d) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232–5241 CrossRef CAS PubMed.
- For examples: (a) C. Nieto-Oberhuber, S. Lopez and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178–6179 CrossRef CAS PubMed; (b) L. Ye, W. He and L. A. Zhang, Angew. Chem., Int. Ed., 2011, 50, 3236–3239 CrossRef CAS PubMed; (c) V. López-Carrillo and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 9292–9294 CrossRef PubMed.
- (a) Y. Wang, Z. Wang, L. Yuxue, G. Wu, Z. Cao and L. A. Zhang, Nat. Commun., 2014, 5, 3470 CrossRef PubMed; (b) D. Malhotra, M. S. Mashuta, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 4456 CrossRef CAS PubMed; (c) S. Liao, A. Porta, X. Cheng, X. Ma, G. Zanoni and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8250–8254 CrossRef CAS PubMed.
- W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697–5705 CrossRef CAS PubMed.
- (a) Z. Wang, C. Nicolin, C. Hervieu, Y.-F. Wong, G. Zanoni and L. Zhang, J. Am. Chem. Soc., 2017, 139, 16064–16067 CrossRef CAS PubMed; (b) H. Tinnermann, L. D. M. Nicholls, T. Johannsen, C. Wille, C. Golz, R. Goddard and M. Alcarazo, ACS Catal., 2018, 8, 10457–10463 CrossRef CAS; (c) L. Noel-Duchesneau, N. Lugan, G. Lavigne, A. Labande and V. César, Organometallics, 2014, 33, 5085 CrossRef CAS; (d) H. Tinnermann, C. Wille and M. Alcarazo, Angew. Chem., Int. Ed., 2014, 53, 8732 CrossRef CAS PubMed; (e) E. Deck, H. E. Wagner, J. Paradies and F. Breher, Chem. Commun., 2019, 55, 5323–5325 RSC; (f) T. Witteler, H. Darmandeh, P Mehlmann and F. Dielmann, Organometallics, 2018, 37, 3064–3072 CrossRef CAS; (g) K. D. Hesp and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 18026–18029 CrossRef CAS PubMed; (h) N. L. Rotta-Loria, A. J. Chisholm, P. M. MacQueen, R. McDonald, M. J. Ferguson and M. Stradiotto, Organometallics, 2017, 36, 2470–2475 CrossRef CAS; (i) X. Cheng, Z. Wang, C. D. Quintanilla and L. Zhang, J. Am. Chem. Soc., 2019, 141, 3787–3791 CrossRef CAS PubMed.
- (a) Y. Tang, I. Benaissa, M. Huynh, L. Vendier, N. Lugan, S. Bastin, P. Belmont, V. César and V. Michelet, Angew. Chem., Int. Ed., 2019, 58, 7977–7981 CrossRef CAS PubMed; (b) M. Teci, D. Hueber, P. Pale, L. Toupet, A. Blanc, E. Brenner and D. Matt, Chem. – Eur. J., 2017, 23, 7809–7818 CrossRef CAS PubMed; (c) I. Varela, H. Faustino, E. Díez, J. Iglesias-Siguenza, F. Grande-Carmona, R. Fernández, J. M. Lassaletta, J. L. Mascareñas and F. López, ACS Catal., 2017, 7, 2397–2402 CrossRef CAS; (d) J. Francos, F. Grande-Carmona, H. Faustino, J. Iglesias-Siguenza, E. Díez, I. Alonso, R. Fernández, J. M. Lassaletta, F. López and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322–14325 CrossRef CAS PubMed; (e) M. Alcarazo, T. Stork, A. Anoop, W. Thiel and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 2542–2546 CrossRef CAS PubMed.
- T. Scherpf, C. Schwarz, L. T. Scharf, J.-A. Zur, A. Helbig and V. H. Gessner, Angew. Chem., Int. Ed., 2018, 57, 12859–12864 CrossRef CAS PubMed.
- (a) P. Weber, T. Scherpf, I. Rodstein, D. Lichte, L. T. Scharf, L. J. Gooßen and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 3203–3207 CrossRef CAS PubMed; (b) X.-Q. Hu, D. Lichte, I. Rodstein, P. Weber, A.-K. Seitz, T. Scherpf, V. H. Gessner and L. J. Gooßen, Org. Lett., 2019, 21, 7558–7562 CrossRef CAS PubMed.
- T. Scherpf, R. Wirth, S. Molitor, K.-S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2015, 54, 8542–8546 CrossRef CAS PubMed.
- C. Schwarz, L. T. Scharf, T. Scherpf, J. Weismann and V. H. Gessner, Chem. – Eur. J., 2019, 25, 2793 CrossRef CAS PubMed.
- (a) C. Schwarz, T. Scherpf, I. Rodstein, J. Weismann, K. Feichtner and V. H. Gessner, ChemistryOpen, 2019, 8, 621–626 CrossRef CAS PubMed; (b) L. T. Scharf and V. H. Gessner, Inorg. Chem., 2017, 56, 8599–8607 CrossRef CAS PubMed.
- Crystallographic data (including structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-1943517–1943526.
- (a) R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2489 CrossRef PubMed.
- S. Serron, J. Huang and S. P. Nolan, Organometallics, 1998, 17, 534–539 CrossRef CAS.
- (a) R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef; (b) O. Diebolt, G. C. Fortman, H. Clavier, A. M. Z. Slawin, E. C. Escudero-Adán, J. Benet-Buchholz and S. P. Nolan, Organometallics, 2011, 30, 1668–1676 CrossRef CAS.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392–6395 CrossRef CAS PubMed.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- For recent general reviews on gold catalysis: (a) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536–6544 RSC; (b) D. J. Gorin, B. D. Sherry and F. D. Toste, Rev., 2008, 108, 3351–3378 CAS; (c) E. Jiménez-Núnez and A. M. Echavarren, Chem. Commun., 2007, 333–346 RSC; (d) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed; (e) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- The reaction rates were calculated after 0.5 h for the reactions at 50°C and 1h at RT.
- V. Lavallo, J. H. Wright and F. S. Tham, Angew. Chem., Int. Ed., 2013, 52, 3172–3176 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1943517–1943526. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cy01861a |
| This journal is © The Royal Society of Chemistry 2019 |