
Confinement of Pt nanoparticles in cage-type mesoporous silica SBA-16 as efficient catalysts for toluene oxidation: the effect of carboxylic groups on the mesopore surface†
 *ab
and
Hsien-Ming
Kao
*c
*ab
and
Hsien-Ming
Kao
*c
Abstract
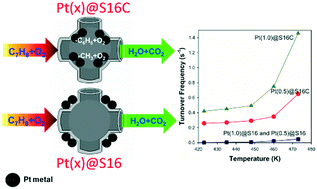
In this work, 3D cage-type mesoporous silica SBA-16, which is functionalized either with or without –COOH groups, is used to support Pt nanoparticles (NPs) and then applied for toluene oxidation. The –COOH functionalized SBA-16 exhibits higher affinity towards Pt4+ species, and as a result highly dispersed and nanosized Pt nanoparticles are formed in comparison to the case of its counterpart pure silica SBA-16 without –COOH groups. When pure SBA-16 without –COOH is used as the support, on the other hand, Pt NPs are formed outside the mesopores of SBA-16, resulting in larger Pt NPs. The catalytic activity for toluene oxidation over Pt nanoparticles deposited on SBA-16 with –COOH is significantly higher than that on pure SBA-16. The intermediate species, reaction mechanisms and active sites in the course of toluene oxidation are probed by in situ IR spectroscopy of CO and toluene adsorption. The Pt nanoparticles confined in the cage-type mesopores of SBA-16 with –COOH can induce the breakage of the strong C–C bonding between phenyl and methyl groups to form CO and carbonyl intermediates, and thus enhance the catalytic activity for toluene oxidation. The defect sites of Pt particles on SBA-16 with –COOH may play a role in the adsorption and decomposition of toluene. The low reducibility of Pt on SBA-16 results in a poor ability to degrade toluene, thus leading to a low catalytic rate for toluene oxidation.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

