
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrosilylation of carbonyl and carboxyl groups catalysed by Mn(I) complexes bearing triazole ligands†
Oriol
Martínez-Ferraté‡
 a,
Basujit
Chatterjee‡
a,
Christophe
Werlé
*a and
Walter
Leitner
*ab
a,
Basujit
Chatterjee‡
a,
Christophe
Werlé
*a and
Walter
Leitner
*ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34–36, Mülheim an der Ruhr, 45470, Germany. E-mail: christophe.werle@cec.mpg.de; walter.leitner@cec.mpg.de
bInstitut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringer Weg 2, Aachen, 52074, Germany
First published on 17th October 2019
Abstract
Manganese(I) complexes bearing triazole ligands are reported as catalysts for the hydrosilylation of carbonyl and carboxyl compounds. The desired reaction proceeds readily at 80 °C within 3 hours at catalyst loadings as low as 0.25 to 1 mol%. Hence, good to excellent yields of alcohols could be obtained for a wide range of substrates including ketones, esters, and carboxylic acids illustrating the versatility of the metal/ligand combination.
Introduction
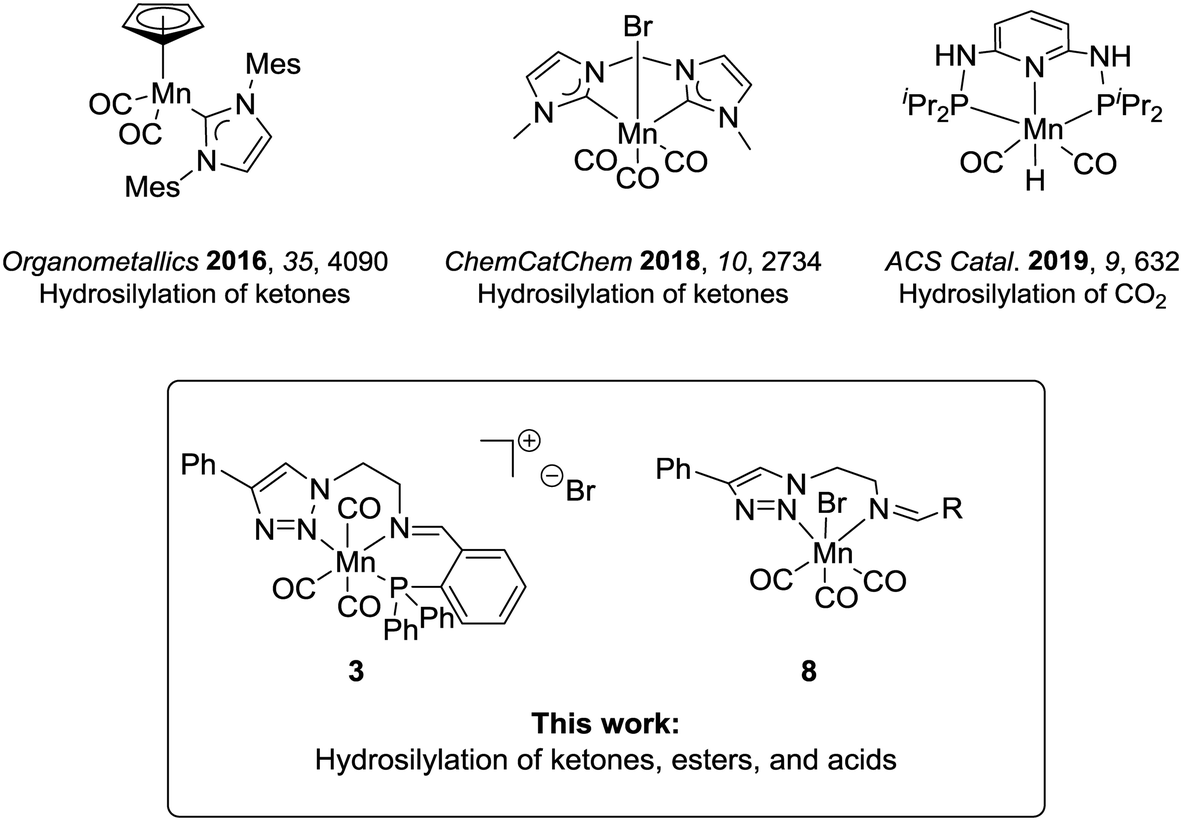
The modern chemical industry mostly relies on catalysis for the synthesis of bulk materials and fine chemicals.1 In the last three decades, industrial breakthroughs in homogeneous catalysis mainly involved catalysts based on second- and third-row transition metals, which are rare elements, whose mining generates waste, and is often associated with low abundance and high costs.2 The excellent performance of the platinum group metals has overshadowed the potential of first-row transition metals, albeit they have been widely used in academia and industry at the early days of homogeneous catalysis. This interest is currently revitalized in particular for metals which can offer potential benefits such as biocompatibility, low toxicity, and high abundance, constituting greener alternatives towards more environmentally benign processes.3 In this context, iron has arguably been the most studied candidate.4 Most recently, manganese complexes are also gaining considerable importance in homogeneous catalysis defining an active area of current research.5The hydrosilylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups is a transformation of broad synthetic utility6 and has recently been studied with manganese catalysts.7 This reaction allows the one-step synthesis of protected silyl alcohols, which can be in a second step hydrolysed to the corresponding alcohols. The protocols represent an alternative to hydrogenation reactions where easy and safe-to-handle silanes replace the use of hydrogen.8 Up to now, research in the area has focused mainly on carbonyl substrates using Mn(II) complexes,9 and only a few studies have dealt with Mn(I) or Mn(0).10 The hydrosilylation of carboxyl compounds is even less studied. For carboxylic acids, reduction with a Mn(0)-complex has been reported to yield aldehydes rather than alcohols as the preferred products.10e This lack of knowledge is surprising given that Mn(I) catalysts have already demonstrated a pronounced ability for the transfer of hydrides to substrates displaying electrophilic centres (e.g., in the case of hydrogenation and hydroboration reactions).11 In the previous studies, the Mn(I) complexes under scrutiny were composed mostly of cyclopentadienyl, carbene, or phosphine ligands (Chart 1).10a–d Nitrogen-based ligands can provide an attractive alternative, and triazole units have received attention as versatile donor units.1a,12 Their synthesis takes advantage of the modularity of copper-catalysed azide–alkyne cycloaddition (i.e., click-chemistry), which is convenient to access structurally different ligand frameworks via this atom- and step-economic robust synthetic method.13 Hence, the stereoelectronic properties of the ligand architecture can be finely adjusted. Besides, their denticity can be controlled and thus, bidentate or tridentate variations are accessible, leading to neutral or cationic complexes respectively.13d,e,14
O groups is a transformation of broad synthetic utility6 and has recently been studied with manganese catalysts.7 This reaction allows the one-step synthesis of protected silyl alcohols, which can be in a second step hydrolysed to the corresponding alcohols. The protocols represent an alternative to hydrogenation reactions where easy and safe-to-handle silanes replace the use of hydrogen.8 Up to now, research in the area has focused mainly on carbonyl substrates using Mn(II) complexes,9 and only a few studies have dealt with Mn(I) or Mn(0).10 The hydrosilylation of carboxyl compounds is even less studied. For carboxylic acids, reduction with a Mn(0)-complex has been reported to yield aldehydes rather than alcohols as the preferred products.10e This lack of knowledge is surprising given that Mn(I) catalysts have already demonstrated a pronounced ability for the transfer of hydrides to substrates displaying electrophilic centres (e.g., in the case of hydrogenation and hydroboration reactions).11 In the previous studies, the Mn(I) complexes under scrutiny were composed mostly of cyclopentadienyl, carbene, or phosphine ligands (Chart 1).10a–d Nitrogen-based ligands can provide an attractive alternative, and triazole units have received attention as versatile donor units.1a,12 Their synthesis takes advantage of the modularity of copper-catalysed azide–alkyne cycloaddition (i.e., click-chemistry), which is convenient to access structurally different ligand frameworks via this atom- and step-economic robust synthetic method.13 Hence, the stereoelectronic properties of the ligand architecture can be finely adjusted. Besides, their denticity can be controlled and thus, bidentate or tridentate variations are accessible, leading to neutral or cationic complexes respectively.13d,e,14
 | ||
| Chart 1 Examples of Mn(I) catalysts for hydrosilylation reactions. | ||
In the present study, we report the catalytic performance of Mn(I) complexes bearing triazol-based ligands for the hydrosilylation of carbonyl and in particular also for carboxyl derivatives. The new cationic complex 3 bearing a tridentate (PNN)-iminotriazole ligand and bidentate neutral complexes previously reported by our group11b were investigated. We found that the selected catalysts were able to convert a wide range of ketones to corresponding alcohols. Notably, the (PNN)-manganese(I) complex 3 showed promising results even in the reduction of ester and acid functionalities.
Results and discussion
The synthesis of (PNN)-ligand 2 and its cationic Mn(I) complex 3 was performed as summarised in Scheme 1. Triazole 1 and the corresponding aldehyde were reacted in dry toluene at 105 °C in the presence of MgSO4 serving as a dehydrating agent. Under these reaction conditions, 2 was obtained in 99% yield. Subsequently, triazole 2 was treated with bromopentacarbonylmanganese(I) in toluene at room temperature to provide the desired complex 3 in 46% yield. High-resolution mass spectrometry confirmed the formation of the expected cationic complex with bromide as a counterion. Furthermore, 1H- and 31P{1H}-NMR spectroscopy indicated the diamagnetic nature of the complex which agrees with a d6-metal centre.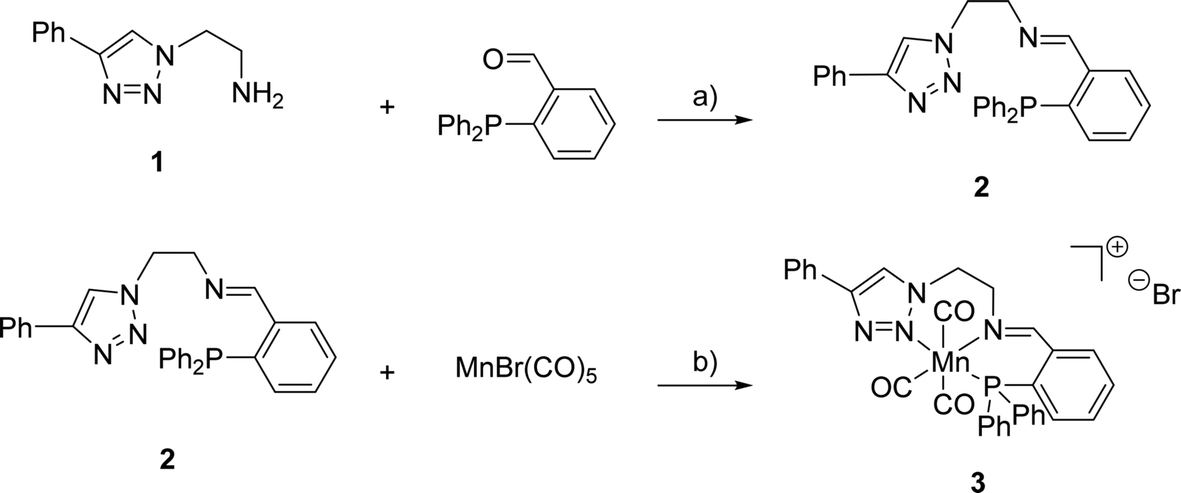 | ||
| Scheme 1 Reagents and conditions for the synthesis of 2 and 3: a) MgSO4, toluene, 105 °C, 16 h, 99%; b) toluene, r.t., 60 h, 46%. | ||
The catalytic activity of complex 3 was first studied for the hydrosilylation of ketones using acetophenone (4a) as a benchmark substrate (Table 1). Reacting 4a with one equivalent of PhSiH3 and 1 mol% of 3 under neat conditions at 80 °C for 20 h, produced alcohol 5a after hydrolysis in 85% yield. In order to improve the efficiency of the reaction, a panel of different solvents were investigated. When the reaction was carried out in tetrahydrofuran (THF) or acetonitrile (MeCN), entry 2 and 3 respectively, yields up to 99% of 5a were obtained. When the reaction time was reduced to 3 hours (entries 4–8), lower yields were obtained with apolar solvents (e.g., 15% in toluene), but yields remained high in the polar solvents (e.g., 99% in THF, 90% in MeCN).
For comparison, neutral triazole complexes 6–8 were prepared following previously described procedures,11b and tested also for the hydrosilylation of acetophenone (Fig. 1). Using 1 mol% of catalyst loading at 80 °C for 3 h, all the studied complexes were able to reduce 4a to 5a with yields up to 99%. To study more precisely the influence of the ligand, the reaction times were reduced to one hour while keeping the catalyst loading unchanged. Under these conditions, iminotriazole complexes 3 and 8 provided the best results, with yields reaching 99%. When complexes 6 and 7 were used, slightly lower yields were obtained (96%). Furthermore, when the catalyst loadings were reduced to 0.1 mol%, catalyst 8 exhibited the best performance. Finally, the activity of Mn(CO)5Br was also verified. When subjected to the standard set of conditions, 56% of conversion and 21% of the corresponding alcohol were obtained.
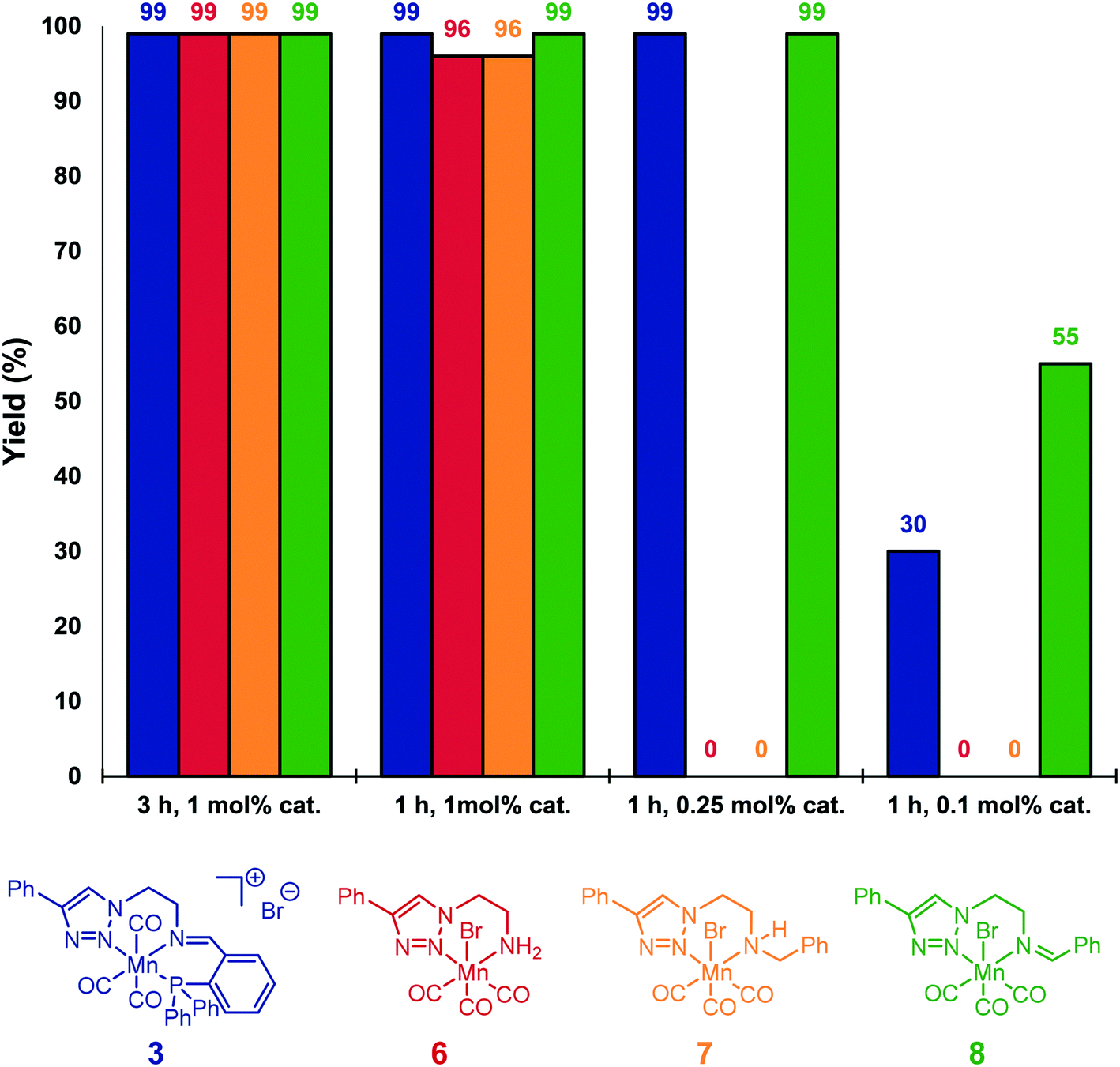 | ||
| Fig. 1 Screening of reaction times and Mn(I) complexes for the hydrosilylation of 4a. Reaction conditions: 0.5 mmol acetophenone, 0.5 mmol phenylsilane, 0.2 mL THF, 80 °C. Quantified by 1H NMR using mesitylene as internal standard. | ||
Based on the standard set of reaction conditions, we examined the substrate scope. These results are summarized in Scheme 2. For acetophenone derivatives, high yields were obtained for all studied para-substituted substrates 5b–e. Only a minor impact related to the electronic properties was observed. The yields decreased slightly in the presence of electron donating groups where 95% and 93% of 5b and 5c were obtained, respectively. Introducing electron-donating groups in meta- and para-position of substrate 4h, provided only 14% of 5h when 0.25 mol% of catalyst loading was used. However, this could be increased to 73% yield with 1 mol% of catalyst loading under otherwise identical conditions. Low yields (2% of 5f and 10% of 5g) were observed also for the ortho-substituted compounds at low catalyst loading. Again, moderate yields (49% for 5g) could be achieved when 1 mol% of catalyst 8 was used. It is conceivable that the lower yields are due to an increased steric hindrance around the carbonyl functionality for these substrates. The heteroaromatic ketone 4i were not reduced under these conditions. The naphthyl derivatives 4j and 4k could be converted with good yields showing different reactivities depending on the relative position of the ketone. In the case of 2-acetonaphtone 4j, 94% of isolated yield could be obtained with only 0.25 mol% of 8. On the other hand, 1-acetonaphthone 4k, required higher catalyst loadings (1 mol%) to furnish 5k in 77% yield. This result can be rationalized by the ortho-substituted nature of ketone 4k following the same trend as for 4f–i. Benzophenone 4l was reduced to the corresponding alcohol 5l in high yield, similar to other substrates containing unsubstituted phenyl rings (4m, n). For the aliphatic substrate cyclohexanone 4o and 1-cyclohexylethanone 4p, good yields (43% and 63% respectively) could be achieved with 1 mol% of catalyst loading. The linear aliphatic ketones 2- and 3-octanone (4q and 4r) were hydrosilylated in high yields, whereby 4q required again higher catalyst loading (1 mol%). Even the sterically congested tert-butyl methyl ketone 4s was readily reduced providing 5s in good yields (79%).
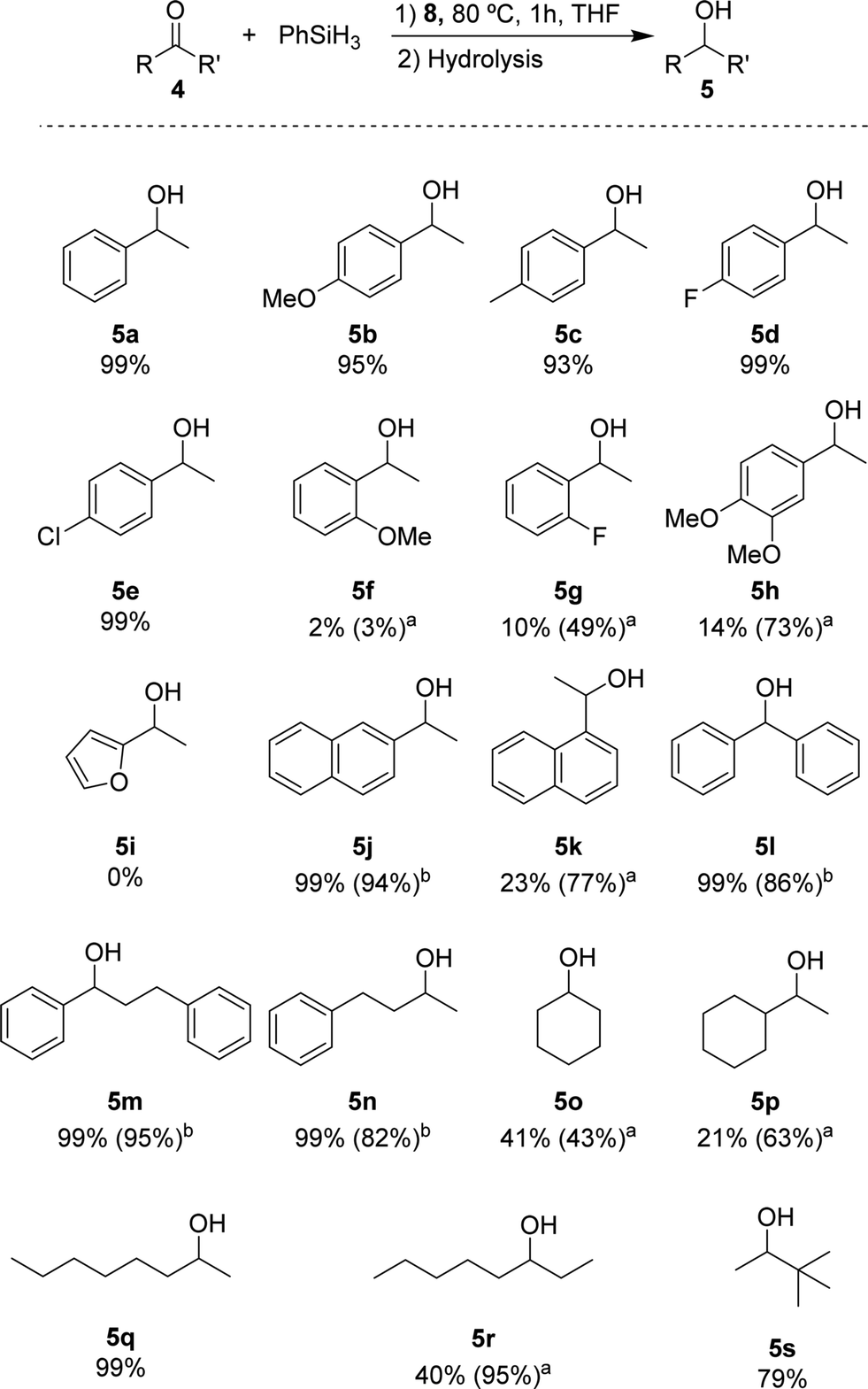 | ||
| Scheme 2 Substrate screening for the hydrosilylation of ketones catalysed by 8. Reaction conditions: 0.5 mmol substrate, 0.25 mol% of 8, 1 mmol of silane, 0.2 mL THF, 80 °C, 1 h. Quantified by 1H NMR using mesitylene or tetradecane (0.5 mmol) as the internal standard. a1 mol% catalyst loading. bIsolated yield. | ||
After the successful hydrosilylation of ketones, the catalytic competence of the Mn(I) catalysts was probed for the hydrosilylation of more challenging carboxyl groups in esters and acids. In the case of esters as substrates, the reduction and hydrolysis can lead to either the corresponding alcohols or ethers as products. Ethyl benzoate 9 was chosen as prototypical substrate for the screening of catalysts and reaction conditions (Table 2). Reacting 9 with 2 equivalents of PhSiH3 and 2 mol% of complex 8 in THF at 80 °C for 3 hours led to moderate conversions (53%), with preferential formation of ethyl benzyl ether 11 (94%) relative to benzyl alcohol 10 (6%, entry 1). Catalyst 6 gave slightly higher conversion than 8 forming a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ether and alcohol (entry 2). When complex 7 was used, only 18% conversion was obtained with alcohol 10 being the preferred product in this case (entry 3). The highest activity for reduction was observed with complex 3 that fully converted 9 to a roughly 60:40 mixture of 10 and 11 under the given conditions (entry 4).
:1 mixture of ether and alcohol (entry 2). When complex 7 was used, only 18% conversion was obtained with alcohol 10 being the preferred product in this case (entry 3). The highest activity for reduction was observed with complex 3 that fully converted 9 to a roughly 60:40 mixture of 10 and 11 under the given conditions (entry 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | t (°C) | Cat. (mol%) | Conv.a (%) | Sel. 10a (%) | Sel. 11a (%) |
| 0.5 mmol ethyl benzoate, 1 mmol phenylsilane, 3 h, 0.2 mL of THF. a Quantified by 1H NMR using tetradecane as an internal standard. b Slow addition of phenyl silane, 0.33 mmol per hour. c Hydrolysis with Me4NF, and further addition of 10 equivalents of sodium hydride and 5 equivalents of ethyl bromide. d Yield after workup. | |||||
| 1 | 80 | 8 (2) | 53 | 6 | 94 |
| 2 | 80 | 6 (2) | 67 | 48 | 52 |
| 3 | 80 | 7 (2) | 18 | 67 | 33 |
| 4 | 80 | 3 (2) | 100 | 62 | 38 |
| 5 | 105 | 8 (2) | 88 | 35 | 65 |
| 6 | 120 | 8 (2) | 100 | 32 | 68 |
| 7 | 140 | 8 (2) | 100 | 25 | 75 |
| 8 | r.t. | 3 (2) | 18 | 83 | 17 |
| 9 | 60 | 3 (2) | 64 | 77 | 23 |
| 10 | 100 | 3 (2) | 100 | 57 | 43 |
| 11b | 80 | 3 (2) | 81 | 84 | 16 |
| 12c | 80 | 3 (2) | 78d | — | 100 |
To investigate whether the two products 10 and 11 are interconverted under the given reaction conditions, two control experiments were carried out (Scheme S1†). Firstly, reductive ether cleavage was investigated. Only 1% of 11 was converted to alcohol 10 under the optimized reaction conditions established for the hydrosilylation of 9 in presence of excess phenylsilane. Similarly, no etherification was observed when alcohol 10 was reacted with ethanol in the presence of 3 and phenylsilane. These results indicate that the selectivity is controlled through competing pathways, presumable branching from a common intermediate RC(OR′)(OSiH2Ph), rather than by secondary interconversion.
Having established the principle ability for carboxyl reduction, the variation of reaction conditions was investigated (Table 2). The rate of reduction increased at elevated temperatures reaching full conversion above 120 °C for catalysts 8 (entries 1, 5–7). The ether 11 remained the preferred product, but selectivity decreased from 94% to ca. 70% at higher temperature. For catalyst 3, the selectivity towards the alcohol product 10 also increased with decreasing temperature, albeit at the expense of conversion (entry 4, 8–10). The reaction was not very solvent dependent and toluene, as well as neat conditions, provide potential alternatives to THF (Fig. S1†). The reactivity of silanes followed the hydricity strength in the order PhSiH3 > Ph2SiH2 ≫ Ph3SiH with only moderate influence on product distribution (Table S1†). A significant improvement could be achieved when PhSiH3 was added slowly (0.33 mmol per hour) to the reaction mixture at 80 °C. This protocol combined high conversion (81%) with good selectivity towards the alcohol 10 (84%) (entry 11).
Examples for the synthesis of ethers via hydrosilylation of esters are limited and ample scope prevails for further development.10e,g We, therefore, investigated the possibility to combine the reduction step directly with a workup under etherification conditions to provide access to 11 from 9. After reduction of 9 with PhSiH3 using catalyst 3 under standard conditions, the reaction mixture was treated with tetramethylammonium fluoride to remove the silyl group, followed by addition of ethyl bromide and base. The ether 11 was isolated in 78% yield directly from this method after standard workup procedure (Table 2, entry 12).
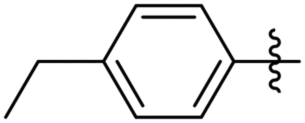
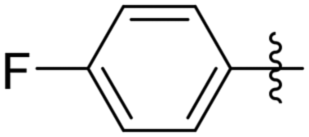
Subsequently, various esters were hydrosilylated using cationic catalyst 3 under the standard set of reaction conditions to assess the scope and limitation of the reduction (Table 3). When benzoates were used as substrates, comparable results in yield and selectivity for methyl-, ethyl- and benzyl-benzoates were observed (entry 1–3, Table 3). For the bulky tert-butyl ester a low conversion was obtained (14%) with the selectivity favouring formation of ether-type product 11 over the alcohol 10 (60% versus 40%, entry 4). Interestingly, phenyl benzoate (entry 5), provided satisfactory results with 80% conversion and high selectivity 82% for corresponding benzyl alcohol. When ethyl heptanoate was used as substrate we observed a full reduction of the ester, with the ether being the major product (60%, entry 6). Finally, para-substituted methyl-benzoates substrates could be fully converted to the alcohols as the major product (entry 7–8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | Conv.a (%) | Sel. 10a (%) | Sel. 11a (%) |
| 0.5 mmol ethyl benzoate, 2 mol% of 3, 1 mmol phenylsilane, 0.2 mL of THF. a Quantified by 1H NMR using tetradecane or mesitylene as an internal standard. | |||||
| 1 | Ph | Me | 97 | 69 | 31 |
| 2 | Ph | Et | 100 | 62 | 38 |
| 3 | Ph | Bn | 100 | 70 | 30 |
| 4 | Ph | t Bu | 14 | 40 | 60 |
| 5 | Ph | Ph | 80 | 82 | 18 |
| 6 | C6H13 | Et | 100 | 40 | 60 |
| 7 |
|
Me | 100 | 73 | 27 |
| 8 |
|
Me | 100 | 78 | 22 |
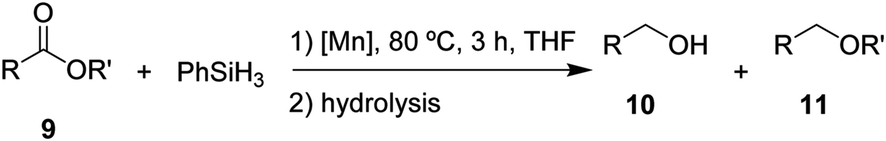
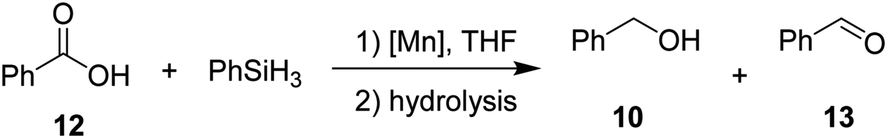
Then, we explored the hydrosilylation of acids with leading complexes 3 and 8 and benzoic acid as the benchmark substrate (Table 4). Gratifyingly, conversion of 50% with 98% selectivity towards the corresponding alcohol was observed with catalyst 3 at 80 °C (entry 2). At lower (60 °C, entry 1) as well as higher temperatures (140 °C, entry 3) the conversions towards 10 decreased to 26% and 31% respectively. At 60 °C (entry 1), significant amounts of aldehyde 13 were formed (38%). Control experiments at 80 °C for 2 hours in absence of catalyst 3 in solution (entry 8) and neat (entry 9) revealed no conversion. The neutral complex 8 showed lower catalytic activity than 3 (entries 4–6). When catalyst loading for 3 was increased to 2 mol%, 94% conversion (entry 7) with nearly perfect selectivity for the alcohol could be obtained even with lower reaction time of 2 h, setting these conditions as standard conditions for exploring the substrate scope.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | t (°C) | Time (h) | Cat. (mol%) | Conv.a (%) | Sel. 10a (%) | Sel. 13a (%) |
| 0.5 mmol acid, 1 mmol phenylsilane, 0.2 mL of THF. a Quantified by 1H NMR using mesitylene or tetradecane as an internal standard. b Reaction was performed in the absence of catalyst. c Reaction was performed in absence of catalyst under neat condition. | ||||||
| 1 | 60 | 3 | 3 (1) | 26 | 62 | 38 |
| 2 | 80 | 3 | 3 (1) | 50 | 98 | <1 |
| 3 | 140 | 3 | 3 (1) | 31 | 100 | — |
| 4 | 80 | 3 | 8 (1) | 28 | 100 | — |
| 5 | 120 | 3 | 8 (1) | 10 | 80 | 20 |
| 6 | 140 | 3 | 8 (1) | 2 | 100 | — |
| 7 | 80 | 2 | 3 (2) | 94 | 100 | — |
| 8b | 80 | 2 | — | 0 | — | — |
| 9c | 80 | 2 | — | 0 | — | — |
Based on the standard set of reaction conditions, we examined the substrate scope of carboxylic acids as reported in Scheme 3. It is shown that the para-substituted substrates 15b and 15c could be converted in high yields, albeit with a noticeable influence of the electron-withdrawing substituents. Introducing a NO2 group in the meta-position in substrate 14d, reduced the yield to 12% for 15d. The aliphatic substrates 14e–h could be converted in good yields (67% and 66% for 14e and 14h, respectively) to excellent yields (95% and 86% for 14f and 14g, respectively). In addition, oxalic acid 14i remarkably provided the corresponding diol (15i) with a high yield of 91%. These results indicate a broad portfolio of potential target substrates.
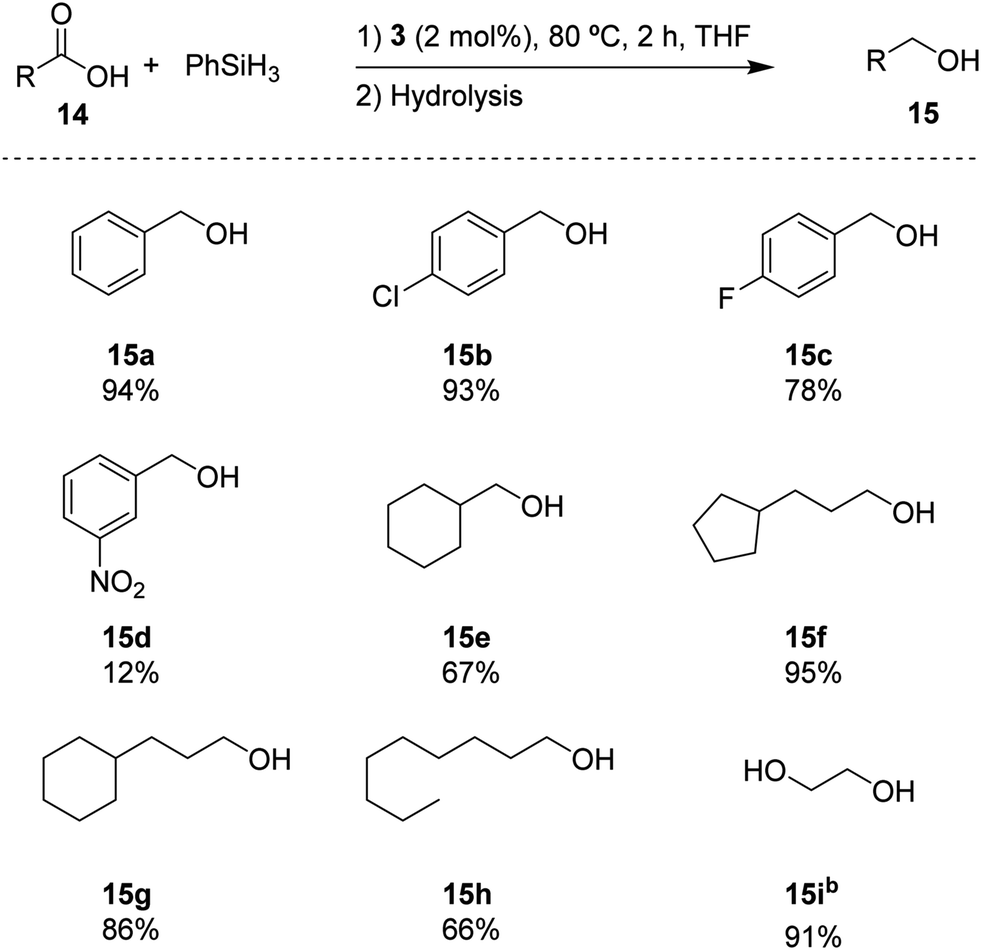 | ||
| Scheme 3 Substrate screening for the hydrosilylation of carboxylic acids catalysed by 3. Reaction conditions: 0.5 mmol substrate, 2 mol% of 3, 1 mmol of silane, 0.2 mL THF, 80 °C, 2 h. Quantified by 1H NMR using mesitylene as an internal standard. b2 mmol of silane. | ||
On the basis of the experimental observations and current reports in the literature,7b,15 a tentative mechanism for the hydrosilylation of the carboxylic CO units using Mn(I) complexes can be proposed (Scheme 4). In the presence of phenylsilane, a catalytically active neutral hydride complex [Mn-H] may be formed under concomitant formation of a silyl cation.16 This mode of activation is supported by ex situ reaction of complex 3 with PhSiH3. Analysis of the reaction mixture revealed the formation of a hydride complex 16 based on HR-MS data indicating a molecular formula [C31H25MnN4O2P]+ and a hydride signal in the 1H-NMR spectrum at −8.56 ppm (see ESI† for further details). The [Mn-H] species are expected to catalyse hydride transfer from PhSiH3 to the silyl-activated substrate. The resulting intermediate A can be converted by hydrolysis or desilylation to aldehyde B or it is reduced further via a second Mn-catalysed hydride transfer to the alkyl- or silyl-ether derivatives affording after hydrolysis the corresponding organic product alcohol C or ether D. While the exact nature of the catalytically active [Mn-H] species and how it enables the hydride transfer still needs to be elucidated, the proposed pathway is in line with the currently available experimental data and may thus serve as a working hypothesis for future studies.
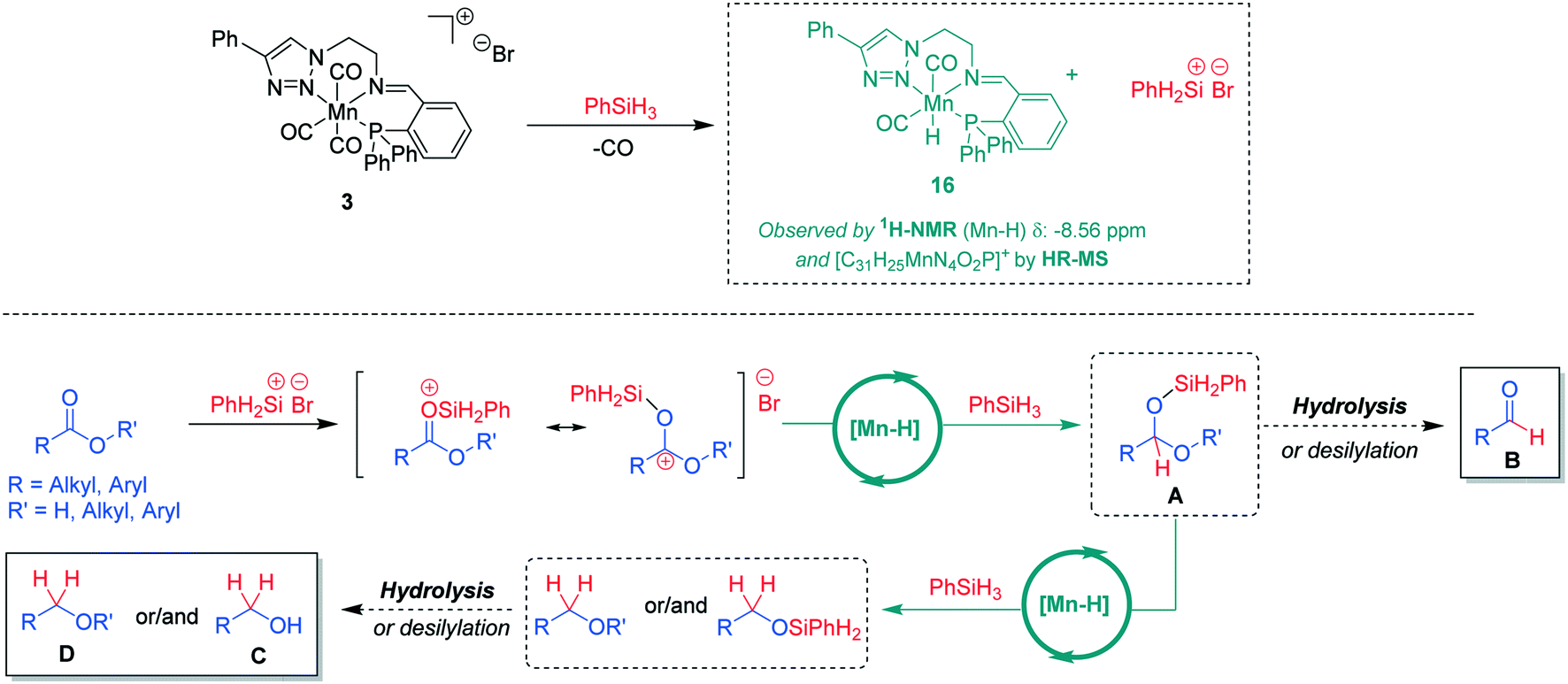 | ||
| Scheme 4 A tentative reaction mechanism for Mn(I)-catalysed hydrosilylation of carboxylic groups in acids or esters. | ||
In summary, we have shown that manganese(I) complexes based on iminotriazole ligands constitute efficient catalysts for the hydrosilylation of carbonyl and carboxyl groups. Aromatic and aliphatic ketones can be reduced with good to excellent yields at low catalyst loadings within 1 h reaction time, using either neutral or cationic complexes comprising bidentate or tridentated ligands, respectively. Extending the reaction to carboxyl groups, esters were reduced with very high conversions, whereby the bidentate-ligated complex 8 favoured formation of ether products while in complex 3 bearing a tridentate (PNN)-iminotriazole ligand lead preferentially to alcohols. Most significantly, complex 3 provides the first example for effective manganese(I) catalysed hydrosilylation of carboxylic acids to alcohols, we believe that the insights provided herein will encourage further investigations into the dynamic field of manganese(I) catalysis in the prospect of finding greener alternatives to existing catalytic methodologies.
Experimental section
General procedure for the catalytic hydrosilylation of carbonyl and carboxyl substrates
Selected ketone/ester/acid (0.5 mmol), phenylsilane (0.5 mmol for ketone and 1–2 mmol for ester/acid), and mesitylene or tetradecane (0.5 mmol) were added to a stock solution (0.2 mL) of the Mn(I) catalyst. The reaction mixture was stirred at 80 °C for the required time (1 to 3 h). After this time, the reaction was cooled to room temperature and the corresponding hydrolysis was performed (see ESI†). After hydrolysis, the sample was diluted with CDCl3 (0.6 mL), and subjected to 1H-NMR spectroscopy to determine the yield of the product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge generous financial support by the Max Planck Society and the RWTH Aachen University. We thank all analytical departments involved in this project for their excellent support. This work was supported by the Bundesministerium für Bildung und Forschung as part of the project “MANGAN” (03SF0508). Additional support by the Cluster of Excellence “The Fuel Science Center” is gratefully acknowledged. Open Access funding provided by the Max Planck Society.References
- (a) A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J. B. Sortais, Org. Lett., 2017, 19, 3656–3659 CrossRef CAS PubMed; (b) J. Hagen, Industrial Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed; (c) J. G. de Vries and S. D. Jackson, Catal. Sci. Technol., 2012, 2, 2009 RSC; (d) J. Heveling, J. Chem. Educ., 2012, 89, 1530–1536 CrossRef CAS.
- (a) B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Applied homogeneous catalysis with organometallic compounds: a comprehensive handbook in four volumes, 2018 Search PubMed; (b) J. F. Hartwig, Organotransition metal chemistry: from bonding to catalysis, University Science Books, Sausalito, 2010 Search PubMed; (c) P. W. N. M. van Leeuwen, Homogeneous Catalysis, Springer Netherlands, Dordrecht, 2004 CrossRef.
- (a) A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS; (b) S. Bezzenine-Lafollee, R. Gil, D. Prim and J. Hannedouche, Molecules, 2017, 22, 1901 CrossRef PubMed; (c) J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680–11752 CrossRef CAS PubMed; (d) P. Gandeepan and C. H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed; (e) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (f) K. Junge, K. Schroder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC; (g) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- (a) S. Chakraborty, G. Leitus and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 2074–2078 CrossRef CAS PubMed; (b) O. Martinez-Ferrate, J. M. Lopez-Valbuena, M. M. Belmonte, A. J. White, J. Benet-Buchholz, G. J. Britovsek, C. Claver and P. W. van Leeuwen, Dalton Trans., 2016, 45, 3564–3576 RSC; (c) T. Dombray, C. G. Werncke, S. Jiang, M. Grellier, L. Vendier, S. Bontemps, J. B. Sortais, S. Sabo-Etienne and C. Darcel, J. Am. Chem. Soc., 2015, 137, 4062–4065 CrossRef CAS PubMed; (d) L. C. Misal Castro, H. Li, J.-B. Sortais and C. Darcel, Green Chem., 2015, 17, 2283–2303 RSC; (e) Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC; (f) S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed; (g) A. Correa, O. Garcia Mancheno and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (h) A. M. Tondreau, E. Lobkovsky and P. J. Chirik, Org. Lett., 2008, 10, 2789–2792 CrossRef CAS PubMed; (i) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed; (j) C. Pavan, J. Legros and C. Bolm, Adv. Synth. Catal., 2005, 347, 703–705 CrossRef CAS; (k) M. Costas, K. Chen and L. Que, Coord. Chem. Rev., 2000, 200, 517–544 CrossRef.
- (a) A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS PubMed; (b) V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS PubMed; (c) K. Z. Demmans, M. E. Olson and R. H. Morris, Organometallics, 2018, 37, 4608–4618 CrossRef CAS; (d) U. Chakraborty, E. Reyes-Rodriguez, S. Demeshko, F. Meyer and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2018, 57, 4970–4975 CrossRef CAS PubMed; (e) F. Bertini, M. Glatz, N. Gorgas, B. Stoger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Chem. Sci., 2017, 8, 5024–5029 RSC; (f) S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS; (g) J. Neumann, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Chem. – Eur. J., 2017, 23, 5410–5413 CrossRef CAS PubMed; (h) M. Perez, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, ChemSusChem, 2017, 10, 83–86 CrossRef CAS PubMed; (i) F. Kallmeier, B. Dudziec, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 7261–7265 CrossRef CAS PubMed; (j) N. Deibl and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 1663–1666 CrossRef CAS PubMed; (k) N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CrossRef CAS PubMed; (l) M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS PubMed; (m) A. Zirakzadeh, S. R. M. M. de Aguiar, B. Stoger, M. Widhalm and K. Kirchner, ChemCatChem, 2017, 9, 1744–1748 CrossRef CAS; (n) A. Mukherjee, A. Nerush, G. Leitus, L. J. Shimon, Y. Ben David, N. A. Espinosa Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed; (o) S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed; (p) S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- (a) C. H. Schiwek, V. Vasilenko, H. Wadepohl and L. H. Gade, Chem. Commun., 2018, 54, 9139–9142 RSC; (b) M. Zhang, N. Li, X. Tao, R. Ruzi, S. Yu and C. Zhu, Chem. Commun., 2017, 53, 10228–10231 RSC; (c) T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2015, 137, 2456–2459 CrossRef CAS PubMed; (d) D. Bezier, S. Park and M. Brookhart, Org. Lett., 2013, 15, 496–499 CrossRef CAS PubMed; (e) D. C. Sauer, H. Wadepohl and L. H. Gade, Inorg. Chem., 2012, 51, 12948–12958 CrossRef CAS PubMed; (f) C. Cheng and M. Brookhart, Angew. Chem., Int. Ed., 2012, 51, 9422–9424 CrossRef CAS PubMed; (g) N. Sakai, K. Kawana, R. Ikeda, Y. Nakaike and T. Konakahara, Eur. J. Org. Chem., 2011, 3178–3183 CrossRef CAS; (h) S. Park and M. Brookhart, Organometallics, 2010, 29, 6057–6064 CrossRef CAS PubMed; (i) N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Angew. Chem., Int. Ed., 2009, 48, 1609–1613 CrossRef CAS PubMed; (j) N. Schneider, M. Kruck, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2009, 2009, 493–500 CrossRef; (k) A. Furuta and H. Nishiyama, Tetrahedron Lett., 2008, 49, 110–113 CrossRef CAS; (l) J. K. Kassube, H. Wadepohl and L. H. Gade, Adv. Synth. Catal., 2008, 350, 1155–1162 CrossRef CAS; (m) H. Nishiyama and A. Furuta, Chem. Commun., 2007, 760–762 RSC; (n) L. H. Gade, V. Cesar and S. Bellemin-Laponnaz, Angew. Chem., Int. Ed., 2004, 43, 1014–1017 CrossRef CAS PubMed; (o) H. Brunner and C. Henrichs, Tetrahedron: Asymmetry, 1995, 6, 653–656 CrossRef; (p) H. Brunner and P. Brandl, Tetrahedron: Asymmetry, 1991, 2, 919–930 CrossRef CAS; (q) H. Brunner and K. Fisch, Angew. Chem., Int. Ed., 1990, 29, 1131–1132 CrossRef; (r) H. Brunner and A. Kürzinger, J. Organomet. Chem., 1988, 346, 413–424 CrossRef CAS.
- (a) X. Yang and C. Wang, Chem. – Asian J., 2018, 13, 2307–2315 CrossRef CAS PubMed; (b) R. J. Trovitch, Acc. Chem. Res., 2017, 50, 2842–2852 CrossRef CAS PubMed; (c) J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Org. Chem., 2016, 3912–3929 CrossRef CAS.
- (a) D. Bézier, G. T. Venkanna, L. C. M. Castro, J. Zheng, T. Roisnel, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2012, 354, 1879–1884 CrossRef; (b) L. C. Misal Castro, H. Li, J. B. Sortais and C. Darcel, Chem. Commun., 2012, 48, 10514–10516 RSC; (c) S. J. Clarson, Silicon, 2009, 1, 57–58 CrossRef CAS; (d) B. M. Trost, I. Fleming, Comprehensive organic synthesis selectivity, strategy, and efficiency in modern organic chemistry, Pergamon Press, Oxford, England, New York, 2007 Search PubMed.
- (a) R. Rahman, V. Uahengo and D. Likius, Glob. Drugs Ther., 2017, 2, 1–6 Search PubMed; (b) T. K. Mukhopadhyay, C. L. Rock, M. Hong, D. C. Ashley, T. L. Groy, M. H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2017, 139, 4901–4915 CrossRef CAS PubMed; (c) T. K. Mukhopadhyay, C. Ghosh, M. Flores, T. L. Groy and R. J. Trovitch, Organometallics, 2017, 36, 3477–3483 CrossRef CAS; (d) C. M. Kelly, R. McDonald, O. L. Sydora, M. Stradiotto and L. Turculet, Angew. Chem., Int. Ed., 2017, 56, 15901–15904 CrossRef CAS PubMed; (e) C. Ghosh, T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, Inorg. Chem., 2015, 54, 10398–10406 CrossRef CAS PubMed; (f) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, J. Am. Chem. Soc., 2014, 136, 882–885 CrossRef CAS PubMed; (g) V. K. Chidara and G. D. Du, Organometallics, 2013, 32, 5034–5037 CrossRef CAS; (h) P. Magnus and M. R. Fielding, Tetrahedron Lett., 2001, 42, 6633–6636 CrossRef CAS.
- (a) F. Bertini, M. Glatz, B. Stoger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, ACS Catal., 2019, 9, 632–639 CrossRef CAS; (b) M. Pinto, S. Friaes, F. Franco, J. Lloret-Fillol and B. Royo, ChemCatChem, 2018, 10, 2734–2740 CrossRef CAS; (c) D. A. Valyaev, D. Wei, S. Elangovan, M. Cavailles, V. Dorcet, J. B. Sortais, C. Darcel and N. Lugan, Organometallics, 2016, 35, 4090–4098 CrossRef CAS; (d) J. Zheng, S. Elangovan, D. A. Valyaev, R. Brousses, V. César, J.-B. Sortais, C. Darcel, N. Lugan and G. Lavigne, Adv. Synth. Catal., 2014, 356, 1093–1097 CrossRef CAS; (e) J. Zheng, S. Chevance, C. Darcel and J. B. Sortais, Chem. Commun., 2013, 49, 10010–10012 RSC; (f) B. T. Gregg and A. R. Cutler, J. Am. Chem. Soc., 1996, 118, 10069–10084 CrossRef CAS; (g) Z. Mao, B. T. Gregg and A. R. Cutler, J. Am. Chem. Soc., 1995, 117, 10139–10140 CrossRef CAS; (h) M. DiBiase Cavanaugh, B. T. Gregg and A. R. Cutler, Organometallics, 1996, 15, 2764–2769 CrossRef; (i) B. T. Gregg, P. K. Hanna, E. J. Crawford and A. R. Cutler, J. Am. Chem. Soc., 1991, 113, 384–385 CrossRef CAS; (j) P. K. Hanna, B. T. Gregg and A. R. Cutler, Organometallics, 1991, 10, 31–33 CrossRef CAS; (k) R. L. Yates, J. Catal., 1982, 78, 111–115 CrossRef CAS.
- (a) D. Wei, A. Bruneau-Voisine, T. Chauvin, V. Dorcet, T. Roisnel, D. A. Valyaev, N. Lugan and J. B. Sortais, Adv. Synth. Catal., 2018, 360, 676–681 CrossRef CAS; (b) O. Martinez-Ferrate, C. Werle, G. Francio and W. Leitner, ChemCatChem, 2018, 10, 4514–4518 CrossRef CAS PubMed; (c) C. Erken, A. Kaithal, S. Sen, T. Weyhermuller, M. Holscher, C. Werle and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef PubMed; (d) A. Kaithal, M. Holscher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449–13453 CrossRef CAS PubMed; (e) R. van Putten, E. A. Uslamin, M. Garbe, C. Liu, A. Gonzalez-de-Castro, M. Lutz, K. Junge, E. J. M. Hensen, M. Beller, L. Lefort and E. A. Pidko, Angew. Chem., Int. Ed., 2017, 56, 7531–7534 CrossRef CAS PubMed; (f) M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed; (g) F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed.
- (a) D. Wang, A. Bruneau-Voisine and J. B. Sortais, Catal. Commun., 2018, 105, 31–36 CrossRef CAS; (b) C. A. Caputo and N. D. Jones, Dalton Trans., 2007, 4627–4640 RSC; (c) R. ter Halle, A. Bréhéret, E. Schulz, C. Pinel and M. Lemaire, Tetrahedron: Asymmetry, 1997, 8, 2101–2108 CrossRef CAS.
- (a) D. L. Broere, R. Plessius, J. Tory, S. Demeshko, B. de Bruin, M. A. Siegler, F. Hartl and J. I. van der Vlugt, Chem. – Eur. J., 2016, 22, 13965–13975 CrossRef CAS PubMed; (b) S. Paganelli, M. M. Alam, V. Beghetto, A. Scrivanti, E. Amadio, M. Bertoldini and U. Matteoli, Appl. Catal., A, 2015, 503, 20–25 CrossRef CAS; (c) K. Q. Vuong, M. G. Timerbulatova, M. B. Peterson, M. Bhadbhade and B. A. Messerle, Dalton Trans., 2013, 42, 14298–14308 RSC; (d) E. M. Schuster, M. Botoshansky and M. Gandelman, Organometallics, 2009, 28, 7001–7005 CrossRef CAS; (e) R. J. Detz, S. A. Heras, R. de Gelder, P. W. van Leeuwen, H. Hiemstra, J. N. Reek and J. H. van Maarseveen, Org. Lett., 2006, 8, 3227–3230 CrossRef CAS PubMed.
- (a) D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Z. Anorg. Allg. Chem., 2017, 643, 554–584 CrossRef CAS; (b) J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 612–623 RSC.
- (a) D. H. Binh, M. Hamdaoui, D. Fischer-Krauser, L. Karmazin, C. Bailly and J. P. Djukic, Chem. – Eur. J., 2018, 24, 17577–17589 CrossRef CAS PubMed; (b) D. H. Binh, M. Milovanovic, J. Puertes-Mico, M. Hamdaoui, S. D. Zaric and J. P. Djukic, Chem. – Eur. J., 2017, 23, 17058–17069 CrossRef CAS PubMed; (c) Y. Corre, V. Rysak, X. Trivelli, F. Agbossou-Niedercorn and C. Michon, Eur. J. Org. Chem., 2017, 4820–4826 CrossRef CAS; (d) M. Hamdaoui, M. Ney, V. Sarda, L. Karmazin, C. Bailly, N. Sieffert, S. Dohm, A. Hansen, S. Grimme and J. P. Djukic, Organometallics, 2016, 35, 2207–2223 CrossRef CAS.
- (a) S. J. Connelly, W. Kaminsky and D. M. Heinekey, Organometallics, 2013, 32, 7478–7481 CrossRef CAS; (b) M. Nava and C. A. Reed, Organometallics, 2011, 30, 4798–4800 CrossRef CAS PubMed; (c) S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767–769 RSC; (d) J. B. Lambert, S. Zhang, C. L. Stern and J. C. Huffman, Science, 1993, 260, 1917 CrossRef CAS PubMed; (e) P. A. McCusker and E. L. Reilly, J. Am. Chem. Soc., 1953, 75, 1583–1585 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01738k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |