
Hybrid PDI/BiOCl heterojunction with enhanced interfacial charge transfer for a full-spectrum photocatalytic degradation of pollutants†
 *a
Xibao
Li,
b
*a
Xibao
Li,
b
Abstract
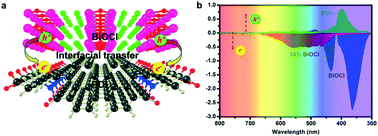
A full-spectrum (300–750 nm) responsive hybrid PDI/BiOCl photocatalyst was successfully constructed. The hybrid structure of the PDI/BiOCl photocatalyst was first examined by surface photovoltage measurements and DFT theoretical calculations. Due to the O atoms bonding to Bi atom, PDI was successfully anchored on the surface of rod-like BiOCl. Owing to the strongly coupled heterojunction interface and conjugated structure of PDI, a rapid interfacial charge transfer was allowed from PDI to BiOCl across the interface. The hybrid PDI/BiOCl photocatalyst presented the best full-spectrum photocatalytic degradation activity for organic pollutants, which was 2.2 and 1.6 times higher than that of BiOCl and PDI, respectively, for the photocatalytic degradation of phenol. The enhanced photocatalytic ability was not only due to the formation of the hybrid PDI/BiOCl composites, which could produce more active species, but also since PDI/BiOCl possessed remarkable full spectrum light-harvesting and excellent interfacial charge separation. The results of the free radical measurements showed that the main active radicals were ˙O2− and ˙OH in the full-wavelength photocatalytic oxidation degradation.
- This article is part of the themed collections: 2020 Catalysis Science & Technology Hot Articles and 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

