
A chemical titration method for quantification of carbenes in Mo- or W-containing catalysts for metathesis of ethylene with 2-butenes: verification and application potential†

Abstract
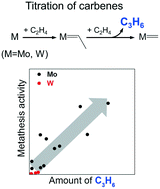
We introduce and experimentally validate a simple titration method for quantifying the number of carbenes acting as active sites in the metathesis of ethylene with 2-butenes over Mo- or W-containing catalysts. This method is based on the experimentally proven mechanistic sequence of carbene formation through the reaction of ethylene with oxidized metal oxide species: (i) reduction of M(VI) to M(IV) by ethylene, (ii) addition of ethylene to M(IV) to yield M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2H4 carbene and (iii) transformation of the latter to MCH2 upon reaction with C2H4 yielding C3H6. As MCH2 cannot produce C3H6 when reacting with C2H4, the amount of C3H6 formed in step (iii) corresponds to the number of catalytically active carbenes. The applicability of this method was supported by establishing a correlation between this number and the steady-state rate of propene formation in the metathesis of ethylene with 2-butenes.
C2H4 carbene and (iii) transformation of the latter to MCH2 upon reaction with C2H4 yielding C3H6. As MCH2 cannot produce C3H6 when reacting with C2H4, the amount of C3H6 formed in step (iii) corresponds to the number of catalytically active carbenes. The applicability of this method was supported by establishing a correlation between this number and the steady-state rate of propene formation in the metathesis of ethylene with 2-butenes.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

