
Electrocatalytic conversion of carbon dioxide to formic acid over nanosized Cu6Sn5 intermetallic compounds with a SnO2 shell layer†

Abstract
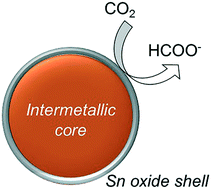
A novel ordered intermetallic compound of carbon-black-supported Cu6Sn5 nanoparticles (Cu6Sn5 NP/CB) in which Cu6Sn5 has a NiAs-type structure was successfully prepared through a wet chemical method using lithium triethylborohydride as a reducing agent. The prepared ordered intermetallic compound was characterized using X-ray diffraction (XRD), transmission electron microscopy, X-ray photoelectron spectroscopy (XPS), scanning transmission electron microscopy (STEM), and X-ray absorption fine structure spectroscopy (XAFS). The XRD measurements confirm the formation of the NiAs-type ordered intermetallic Cu6Sn5. XPS and STEM-X-ray energy dispersive spectroscopy measurements allowed us to confirm the Cu6Sn5 structure. The surface of the intermetallic Cu6Sn5 was found to be covered by SnO2, indicating that a core–shell structured intermetallic compound (i.e., Cu6Sn5 core/SnO2 shell) had formed. The Cu6Sn5 NP/CB material exhibited a faradaic efficiency of 65.3% at −0.6 V for HCOO− formation via electrochemical CO2 reduction, which is superior to those of the non-intermetallic Cu NP/CB and Sn NP/CB samples. From the XAFS measurements, we determined the Sn–Sn distance in the SnO2 on the surface of the Cu6Sn5 NPs, and the key factor affecting the high selectivity was found to be the 4.9% compressive strain of the SnO2 shell layers on the Cu6Sn5 compared to that of the Sn NP/CB sample.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

