
Coordination chemistry and catalysis with secondary phosphine oxides†

Abstract
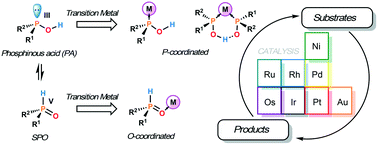
Secondary phosphine oxides present tautomeric equilibrium between the pentavalent oxide form (SPO) and the trivalent phosphinous acid (PA). This dichotomy is the origin of the rich coordination chemistry of this class of compounds. As the pentavalent oxide form usually predominates, SPOs are air-stable but at the same time metal coordination can shift the tautomerism towards the PA form, making the ligand act as an ordinary trivalent phosphine. For this reason, this class of ligands has found application in numerous homogeneously catalysed reactions, including some enantioselective transformations. This review aims to give an up-to-date account on the synthesis, coordination chemistry and homogeneous catalysis of SPOs.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

