
Homogenous electrochemical water oxidation by a nickel(ii) complex based on a macrocyclic N-heterocyclic carbene/pyridine hybrid ligand†
 abc
Francis
Verpoort
*abgh
abc
Francis
Verpoort
*abgh
Abstract
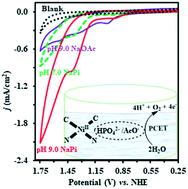
Water-soluble homogeneous nickel catalysts have been rarely investigated for catalytic water oxidation as compared to their heterogeneous counterparts. Herein, we report homogenous electrochemical water oxidation by a nickel(II) complex, ([NiL](PF6)2 (L = bis(2-pyridyl-methylimidazolylidene)methane), based on a macrocyclic N-heterocyclic carbene/pyridine hybrid ligand under neutral and alkaline conditions. The catalyst displayed the stable catalytic current of 0.65 mA cm−2 at the overpotential of 0.80 V (∼0.55 V at GCE for CV) with a ∼93% Faradaic efficiency at pH 9.0 for oxygen evolution in long-term bulk electrolysis. The CV, UV-vis, ESI-MS, SEM, and EDX results demonstrated that the catalyst was impressively stable even after long-term controlled potential electrolysis (CPE) (11 h) and homogeneous in nature. The synthesis of this catalyst is straightforward, and its complex is air and moisture stable. To the best of our knowledge, this is the first study on the investigation of a Ni–NHC complex for water oxidation under aqueous conditions (acetate/phosphate). According to the literature, the role of the phosphate ion in homogenous nickel-catalysed water oxidation was found to vary from catalyst poisoning to activation. Interestingly, the catalytic activity of our catalyst in phosphate buffer was significantly higher than that with acetate ions at the same pH value; this might indicate the key role of phosphate ions as proton acceptors, which boosted the catalyst activity via enhanced PCET during catalysis.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

