
The control effects of different scaffolds in chiral phosphoric acids: a case study of enantioselective asymmetric arylation†

Abstract
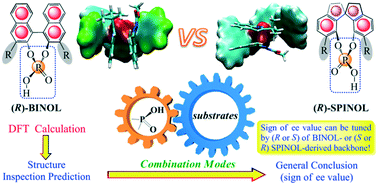
Chiral phosphoric acid (CPA) catalysts with BINOL or SPINOL backbones have attracted immense attention in recent years. Despite a number of successful studies, the theoretical elucidation on the role of the CPA scaffold has rarely been considered, and its effects on enantioselectivity are still far from being understood. Structure inspection shows that axially chiral scaffolds induce a stereogenic central phosphate, and the phosphoric acid functional groups in the (R/S)-BINOL skeleton are identical to those in the (S/R)-SPINOL skeleton. Therefore, a hypothesis is proposed that the orientation of each phosphoric acid functional group regulated by different (R/S) BINOL- and SPINOL-derived scaffolds may control the signs of enantioselectivity (positive or negative ee value) by changing the combination modes of the substrates. In other words, the sign of enantioselectivity (positive or negative ee value) can be tuned with an (R or S) BINOL-derived backbone or an (S or R) SPINOL-derived backbone, respectively. Thus, we elaborate an in-depth mechanistic case study of newly reported CPA-catalyzed enantioselective asymmetric arylation reactions (a–c) catalyzed by CPAs with two different backbones. We found that although the origin of the high enantioselectivity of these three case reactions a–c can be ascribed to favorable C–H⋯O interactions, catalyst and substrate distortion interactions, and electrostatic interactions in the preferential TSs leading to the major products, the signs of enantioselectivity still follow our hypothesis. Our general applicability finding encourages the further development of more effective CPA catalysts and can be used to guide the strategic choice of CPA skeletons.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

