
Enhancement mechanism of sulfur dopants on the catalytic activity of N and P co-doped three-dimensional hierarchically porous carbon as a metal-free oxygen reduction electrocatalyst†
 *ab
*ab
Abstract
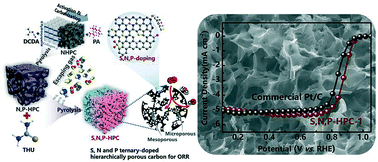
Doping carbon networks with multi-heteroatoms is one of the most effective ways to achieve a carbon-based metal-free electrocatalyst with a superior activity for oxygen reduction reaction (ORR). Herein, multi-heteroatom doped three-dimensional hierarchically micro/mesoporous carbon was synthesized as an efficient metal-free electrocatalyst towards ORR by pyrolyzing thiourea (THU) with nitrogen- and phosphorus-doped (N- and P-doped) hierarchically porous carbon networks derived from cattle bone. The obtained sulfur, nitrogen and phosphorus ternary-doped (S, N and P ternary-doped) carbon material has both a high porosity for effective mass transport of ORR reactants and abundant surface active sites for efficient ORR. As identified by X-ray absorption near edge structure spectroscopy, the S dopant in the reduced form (SRed) plays a major role during the ORR catalytic process. When used as an ORR electrocatalyst, the S, N and P ternary-doped carbon with the highest content of SRed outperformed the commercial Pt/C electrocatalyst in alkaline electrolytes with a superior electrocatalytic activity (∼40 mV higher half-wave potential), outstanding electrochemical durability and a good anti-poisoning capability. This work presents an effective approach for the design and synthesis of efficient metal-free ORR electrocatalysts from earth-abundant cattle bone and analogues.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

