
The effects of H2O on a vanadium-based catalyst for NH3-SCR at low temperatures: a quantitative study of the reaction pathway and active sites
 *c
*c
Abstract
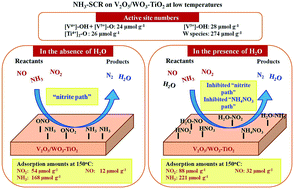
The influences of H2O on the adsorption amounts of NO, NO2, and NH3 on V2O5/WO3–TiO2 and the reaction pathway at low temperatures for selective catalytic reduction of nitrogen oxides by NH3 (NH3-SCR) were quantitatively obtained using the temperature programmed surface reaction as well as the transient response method (TRM). The active site distribution on V2O5/WO3–TiO2 was also clarified quantitatively. NOx adsorption took place on [V4+]–OH + [V5+]–O, [V5+]–OH, and [Ti4+]2–O, the surface concentrations of which were 24, 28 and 26 μmol g−1, respectively, whereas NH3 was activated on [V4+]–OH + [V5+]–O, [V5+]–OH, [Ti4+]2–O and W species, with concentrations of 24, 28, 26 and 274 μmol g−1, respectively. The V and W species did not distribute as a monolayer. The “nitrite path”, referring to the reaction between adsorbed NH3 and nitrite species to produce N2 and water (H2O), contributed to standard and fast SCR mechanisms, but the “NH4NO3 path”, referring to the reaction between the NO gas and NH4NO3 formed from the surface nitrates and adsorbed NH3 species to emit NO2, H2O and N2, did not occur at low temperatures without H2O. Both the “NH4NO3 path” and the “nitrite path” contributed to NH3-SCR with H2O in the gas feed. The presence of H2O increased the amounts of the adsorbed nitrates and ammonia at 150 °C, but hindered the reaction between the adsorbed NH3 and NO gas, lowering the SCR activity.
 Please wait while we load your content...
Please wait while we load your content...

