
Ceria imparts superior low temperature activity to nickel catalysts for CO2 methanation

Abstract
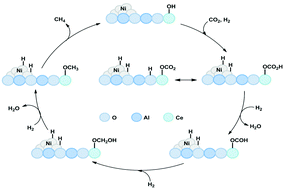
Carbon dioxide methanation is well known to achieve the catalytic conversion of the CO2 molecule into value-added chemicals. At present, the important challenge is the development of efficient methanation catalysts at low reaction temperature. In this study, Ni–Al layered double hydroxides (LDHs) were used as catalyst precursors and modified by CeO2 for CO2 methanation. By the optimization of CeO2 loading, the reaction activity was improved, especially at low temperature. Among the prepared catalysts, the NiAl-MO/CeO2-5 catalyst exhibited the highest catalytic activity with a CO2 conversion of 91% at 250 °C. The addition of CeO2 was beneficial to the formation of small particles with good dispersion and provided appropriate basic sites and oxygen vacancies, which were conducive to the catalytic performance. DFT calculation and in situ DRIFTS results further verified that the catalyst with an appropriate amount of CeO2 was favorable for CO2 adsorption and conversion. The reaction mechanism results demonstrated that the high amount of formate intermediate species can accelerate the reaction and a certain amount of ceria in the catalyst could provide a suitable metal–support interaction, which was beneficial for CO2 methanation. The LDH as a catalyst precursor integrated with CeO2 shows a promising effect on CO2 methanation at low temperature.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

