
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mono-substitution of symmetric diesters: selectivity of Mycobacterium smegmatis acyltransferase variants†
Maja
Finnveden‡
 ,
Stefan
Semlitsch‡
,
Oscar
He
and
Mats
Martinelle
*
,
Stefan
Semlitsch‡
,
Oscar
He
and
Mats
Martinelle
*
KTH Royal Institute of Technology, Department of Industrial Biotechnology, School of Engineering Sciences in Chemistry, Biotechnology and Health, SE-106 91 Stockholm, Sweden. E-mail: matsm@kth.se
First published on 9th August 2019
Abstract
A method for selectively reacting one, out of two identical carboxylic esters in a symmetric diester has been developed. An esterase from Mycobacterium smegmatis (MsAcT) has a restricted active site resulting in a narrow acyl donor specificity. This constraint was used to develop a selective synthesis route from divinyl adipate (a symmetric diester) towards mixed vinyl adipate esters. To find a suitable catalyst, the wild type (wt) MsAcT and two MsAcT variants: a single point mutant (L12A) and a double point mutant (T93A/F154A), were immobilized and studied under solvent-free conditions. Out of the tested catalysts, MsAcT L12A was the most selective for mono-transesterification of divinyl adipate. When divinyl adipate was reacted with 1.5 equivalents of a hydroxyl vinyl ether full conversion of DVA was observed yielding over 95% mixed diester. Furthermore, the limitations for longer dicarboxylic esters were studied, showing that MsAcT T93A/F154A tolerated up to at least dimethyl sebacate.
Introduction
Dicarboxylic acids are essential building blocks for the production of important bifunctional molecules. Existing applications includes: synthesis of dimeric or hybrid derivatives of bioactive natural compounds; and the production of functionalised polyesters.1 Synthesis routes using dicarboxylic acids and their derivatives as monomers are generally non-selective and thus exploit both functional groups. Orthogonal approaches, where molecules containing functionalities allowing for two (or more) chemoselective reactions to operate without interference is a powerful tool in synthetic organic chemistry and polymer chemistry.2 Thus, it is desirable to selectively react dicarboxylic acids, and their derivatives, to synthesize molecules enabling such chemo-selective reactions, which is the subject of the current work (Scheme 1).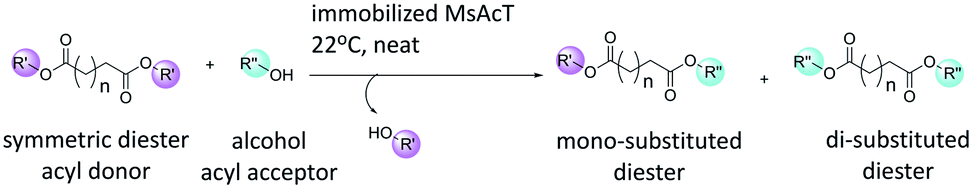 | ||
| Scheme 1 Transesterification of symmetric diesters with alcohol catalysed by Mycobacterium smegmatis esterase/acyltransferase (MsAcT). | ||
Different approaches to synthesize monoesters from aliphatic dicarboxylic esters have been reported. For acids that are able to form 5- or 6-membered cyclic anhydrides, monoacylation is possible.3,4 For other diacids both chemical and biocatalytic methods to form mono-esters have been developed.5–9 All these methods lead to dicarboxylic acid derivatives with both acid and ester functionalities, i.e. an esterification occurs on one side. However, there are applications were a selective transesterification is desirable. For example, vinyl esters have been identified as suitable alternatives to (meth)acrylates in biomedical applications due to their lower cytotoxicity and biocompatible degradation. However, there is a lack of suitable vinyl ester precursors.10 Thus, development of multifunctional vinyl ester monomers is particularly interesting.
We identified Mycobacterium smegmatis esterase/acyltransferase (MsAcT) as a potential catalyst for selective mono-substitution of dicarboxylic acid derivatives. MsAcT is an interesting enzyme in many ways: it can efficiently catalyze ester and amide formation in water;11–18 the enzyme forms an octamer in solution, also revealed in the crystal structure, which is rare for the SGNH-hydrolase family;11 and the combination of three monomers leads to a hydrophobic channel, which is believed to be the reason for its acyl transfer capabilities. MsAcT has limitations in the substrate scope especially towards the acyl donor. Previous reports have commonly used acetate esters as acyl donors with various alcohols.11–18 It has been shown that the wild type (wt) enzyme of MsAcT favors short acyl donors and has problems converting acyl donors that are longer than butyrates. Therefore, Hendil-Forssell et al. created MsAcT variants with expanded substrate scope.19 For the present investigation, we considered two of the mutants as particularly interesting: a single point mutant (L12A) and a double point mutant (T93A/F154A). As free enzyme in aqueous solutions MsAcT L12A can use methyl heptanoate as substrate, while MsAcT T93A/F154A accepts methyl nonanoate as acyl donor.19
A technique to increase enzyme stability (both during storage and reaction) is immobilization.20 Using an enzyme in an immobilized form provides convenient handling of the enzyme: protein contamination of the product and the reuse of the catalyst is possible because of easy separation. MsAcT has previously been immobilized onto carbon nanotubes and utilized for its perhydrolase activity.21 Additionally, MsAcT has been immobilized on hydrophilic carries composed of agarose, cellulose, 3-aminopropylic silica, epoxy resins22 and in siliceous monolithic microreactors.13 For both these applications the reactions were run in biphasic solution. Performing reactions under solvent-free conditions is advantageous (such as, high concentration of reactants, high product yields and simplicity of product extraction). Moreover, one of the core principals of green chemistry is to prevent waste rather than to cleanup or treat waste when produced.23
To expand the understanding of MsAcT in the current study we immobilized the wild type (MsAcT wt) and the two mutants: MsAcT L12A and MsAcT T93A/F154A on controlled pore glass particles (Ezig3). While previous studies have mainly focused on the catalytic profile of MsAcT in aqueous solutions, covering the substrate scope of the acyl acceptor the reactions reported here were performed under solvent-free conditions, focusing on the acyl donor.
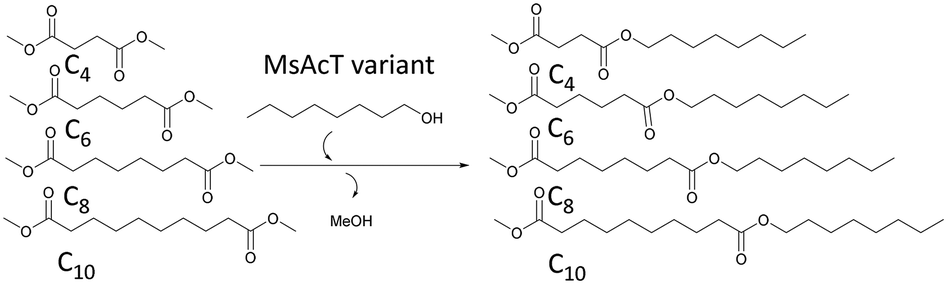
Using the limitation for MsAcT to use long acyl donors, a method for selective mono-substitution of symmetric dicarboxylic esters is presented (Scheme 1). The hypothesis builds on steric discrimination of binding the formed mono-substituted diester in the limited active site. Since the mono-substituted product will have increased size, the unreacted ester will not fit productively in the active site to achieve the di-substituted product. Divinyl adipate (DVA) was used as the acyl donor with alcohols as acyl acceptors to synthesize mixed esters. The alcohols used were two relatively large alcohols to obtain a notable change in length of the new mixed ester. MsAcT L12A was able to form mono-substituted products, with only a small fraction of di-substituted DVA. Additionally, the selectivities of MsAcT wild type and variants towards the dimethyl esters of dicarboxylic acids with varying lengths were compared.
Results and discussion
Biocatalytic route for mono-substitution of divinyl adipate
Mono-substitution of symmetric esters has previously posed a challenge. In this work, we present a method for mono-substitution of divinyl adipate (DVA). Reactions between DVA and an acyl acceptor: butanediol vinyl ether (BVE) or 1-octanol were performed under solvent-free conditions (Scheme 2). The catalysts selected for evaluation were the wild type MsAcT and the two variants MsAcT L12A and MsAcT T93A/F154A. The acyl donor DVA and the acyl acceptor were combined in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5, using an excess of alcohol to push the equilibrium towards product formation. In a first attempt 1-octanol was utilized as acyl acceptor (Fig. 1). For all three variants the main product was octyl vinyl adipate (OVA), the mono-substituted DVA. However, for both MsAcT wt and MsAcT T93A/F154A the fraction of di-substituted DVA increased with time. This was not observed when the single point mutant MsAcT L12A was used as catalyst, which almost exclusively yields the mono-substituted OVA. After 26 hours, 96% conversion of DVA was reached with 98% of the product identified as the mono-substituted product (Fig. 1). After an additional 7 hours almost no conversion of DVA or formation of the di-substituted product was observed. 1H-NMR confirmed that 50% of the vinyl groups on DVA were left intact (Fig. S2†) when MsAcT L12A was used as catalyst.
:1.5, using an excess of alcohol to push the equilibrium towards product formation. In a first attempt 1-octanol was utilized as acyl acceptor (Fig. 1). For all three variants the main product was octyl vinyl adipate (OVA), the mono-substituted DVA. However, for both MsAcT wt and MsAcT T93A/F154A the fraction of di-substituted DVA increased with time. This was not observed when the single point mutant MsAcT L12A was used as catalyst, which almost exclusively yields the mono-substituted OVA. After 26 hours, 96% conversion of DVA was reached with 98% of the product identified as the mono-substituted product (Fig. 1). After an additional 7 hours almost no conversion of DVA or formation of the di-substituted product was observed. 1H-NMR confirmed that 50% of the vinyl groups on DVA were left intact (Fig. S2†) when MsAcT L12A was used as catalyst.
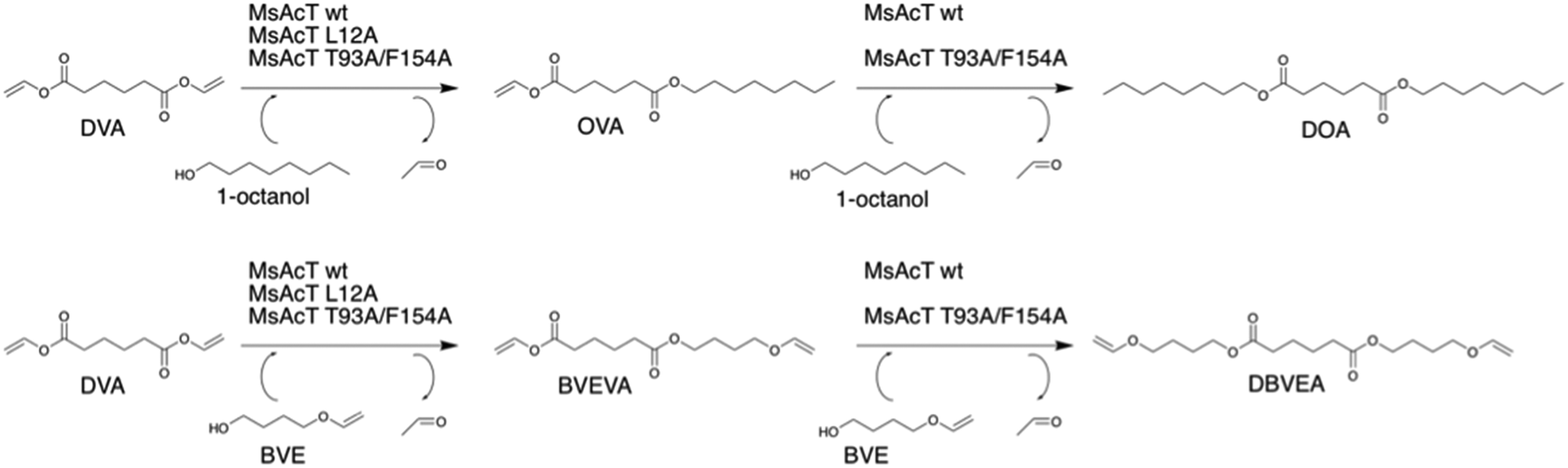 | ||
| Scheme 2 Biocatalytic route for the mono-substitution of divinyl adipate (DVA) yielding mono-substituted or di-substituted dicarboxylic esters catalysed by Mycobacterium smegmatis esterase/acyltransferase (MsAcT). MsAcT L12A was selective for mono-substitution. | ||
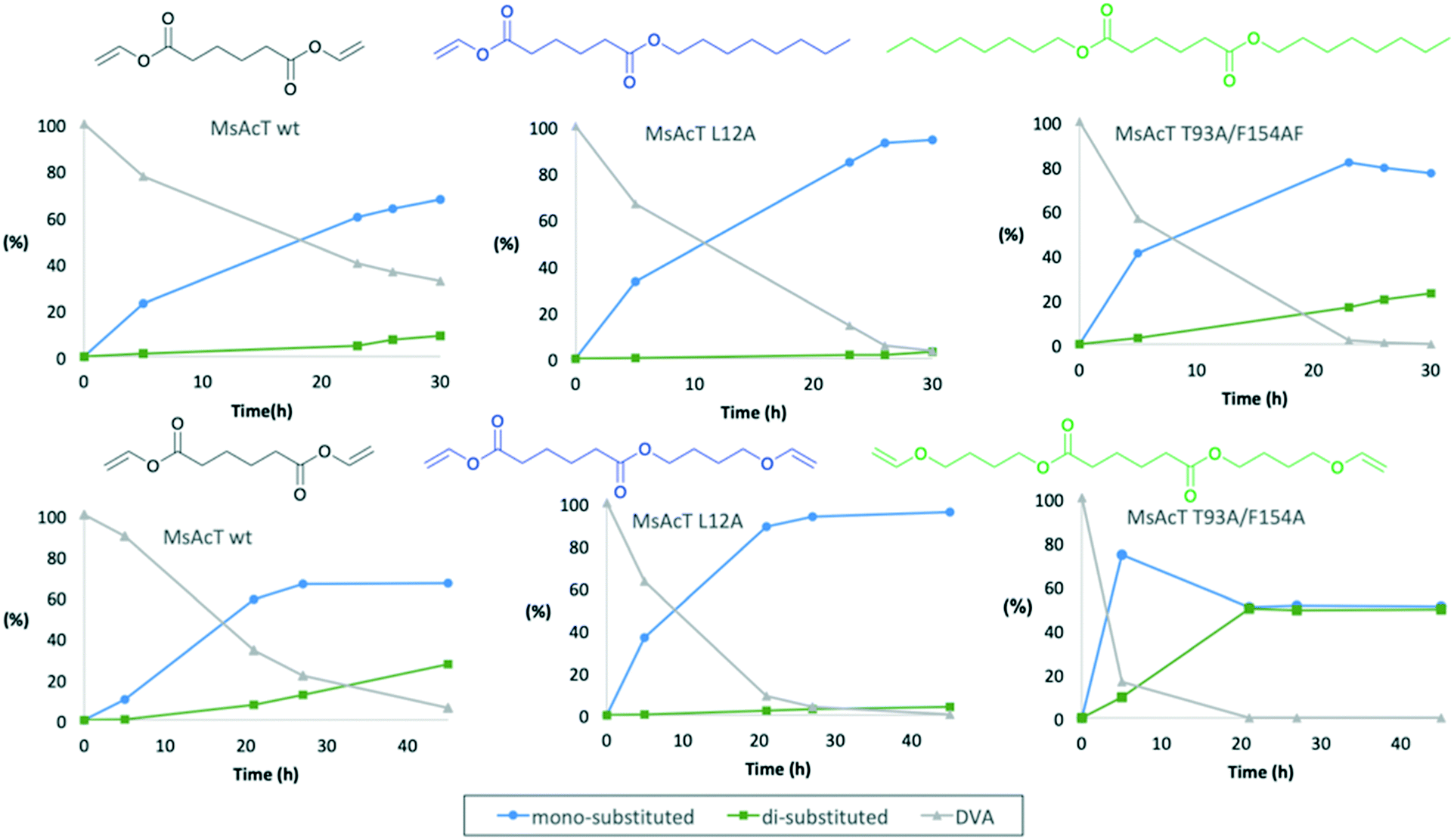 | ||
| Fig. 1
Mycobacterium smegmatis esterase/acyltransferase (MsAcT) transacylation of divinyl adipate (DVA) under solvent-free conditions. Top row using 1-octanol as acyl acceptor and bottom row using butanediol vinyl ether (BVE) as acyl acceptor. The distribution of substrate and products are shown as molar percentage as a function of time. DVA grey (Δ); mono-substituted product blue (○); di-substituted product green (□). Data points are connected by straight lines. The concentration ratio of DVA to the alcohol was 1:1.5, the reaction was run in open vessels to allow evaporation of formed acetaldehyde. | ||
To compare the MsAcT variants to a biocatalyst that has not been reported as selective towards mono-substitution of dicarboxylic esters, Candida antarctica lipase B (CalB) was added to a reaction mixture of DVA and 1-octanol. The reactants were combined in a ratio of 1:1 to favour the synthesis of the mono-substituted product. With CalB as catalyst it was not possible to selectively achieve OVA under these reaction conditions. CalB did not show any selectivity towards the formation of mono-substituted product. Within the first minute 70% of DVA was converted with a 60:40 ratio between the mono and di-substituted products.
The reaction between DVA and 1-octanol showed the potential of MsAcT L12A as a selective catalyst in the ability to achieve mono-substitution of DVA. In order to show the applicability of this system BVE was used as acyl acceptor to synthesise dual-functional adipates. The novel vinyl ester precursor with a combination of a vinyl ester and vinyl ether in the same monomer allows two different chemoselective reactions, which can be controlled by the choice of polymerization method. While vinyl esters are prone to follow radical polymerization, functional groups like vinyl ethers are typically used in cationic polymerization.24 The steric properties for BVE are similar to those of 1-octanol and thus similar reactivities for the two acyl acceptors were anticipated. The results shown in the lower row in Fig. 1 display that the same trend was observed when BVE was used as acyl acceptor. As expected, MsAcT L12A was again the most selective of the three variants, selectively building the mono-substituted 4-(vinyloxy)butyl vinyl adipate (BVEVA) (Scheme 2), with only a small fraction of the di-substituted DVA as product (Fig. 1). 1H-NMR confirmed these results (Fig. S6†). Noteworthy, the double point mutant showed 85% conversion of DVA within 5 hours with 9% of the di-substituted product. However, as the reaction progressed, full conversion of both acyl donor and acyl acceptor (DVA and BVE) was reached, yielding a 50:50 distribution of the mono and di-substituted products. The results were confirmed by 1H-NMR (Fig. S7 in ESI†) with 25% remaining vinyl groups.
The acylation of DVA with 1-octanol was modelled by building the second tetrahedral intermediate of the reaction, where the acyl acceptor 1-octanol makes a nucleophilic attack on the acyl-enzyme (see Fig. 2 and 3). During the molecular dynamics (MD) simulations the important hydrogen bonds in the tetrahedral intermediate necessary for the catalytic activity of MsAcT were kept intact (for MsAcT wt and the two variants MsAcT L12A and MsAcT T93A/F154A). The simulations give a model for the binding position of the substrates in the wild type MsAcT and the two variants (Fig. 3). A close up of the surface around the active site of one subunit for each MsAcT variant is shown in Fig. 2. The structures in the second row show the tetrahedral intermediate of the deacylation step. As indicated by the white arrow in the top middle frame in Fig. 2, MsAcT L12A acquires a deeper binding-site behind the side chain. Due to this MsAcT L12A has an extended substrate scope on the acyl donor side. DVA seemingly continues inwards into the enlarged pocket, indicated by the white arrow in the top row in Fig. 2. Fig. 3B shows a different orientation of the active site with the surface of a few selected amino acids revealed. Fig. 3B shows the new orientation of DVA (magenta) while 1-octanol (yellow) still points towards the entrance of the active site (black arrow). When the product OVA has been released, this mono-substituted product is too long to fit into the restricted space generated from mutating leucin to alanine (mutation shown in cyan in the Fig. 3B). In the MsAcT wt, DVA must accommodate in the narrow entrance of the active-site and, as can be seen from the modelling shown in Fig. 3A, DVA points toward the entrance of the binding-site in the vicinity of the alcohol binding area. The restricted space, caused by F154, might be the explanation for the lower conversion of DVA when MsAcT wt was used as catalyst compared to the two mutants (Fig. 1).
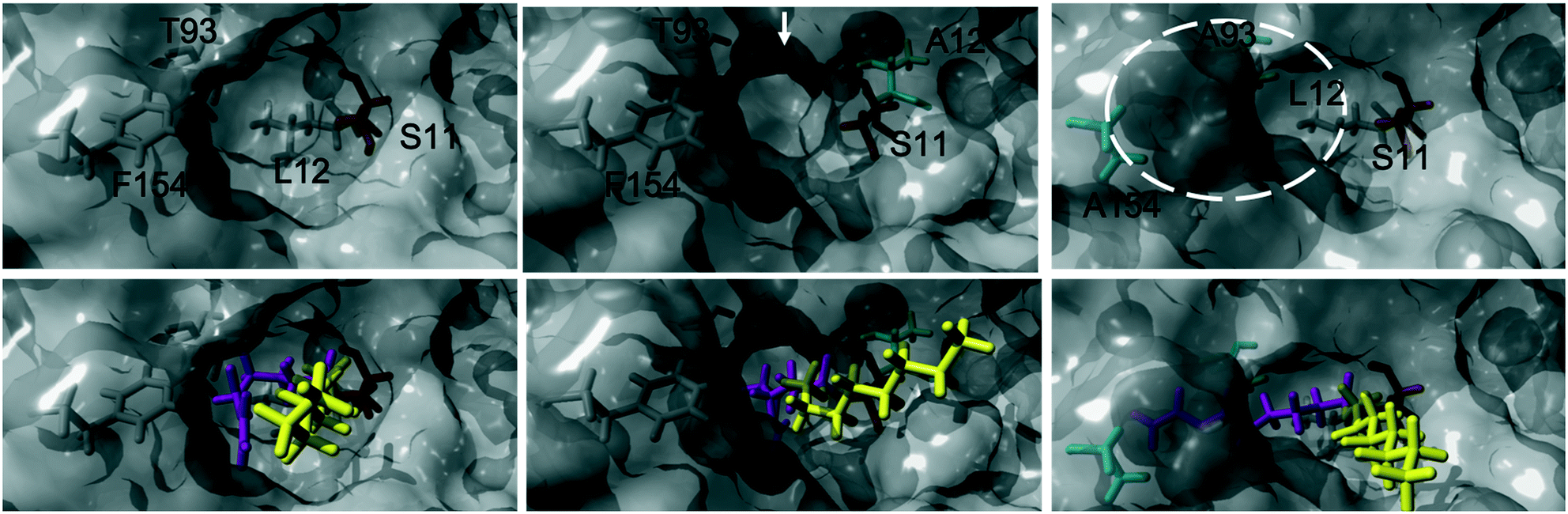 | ||
| Fig. 2 Close up of the surface around the active site of one subunit for each Mycobacterium smegmatis esterase/acyltransferase (MsAcT) variants (wt, L12A, T93A/F154A from left to right column). Mutated amino acids are shown in cyan. Amino acids shown in grey are conserved and the catalytic serine is shown in orange. Acyl donor (DVA) in magenta and acyl acceptor (1-octanol) in yellow. The model represents the tetrahedral intermediate formed during the deacylation with 1-octanol. The white arrow and dotted circle indicates the space formed from the mutation in MsAcT L12A and MsAcT T93A/F154A, respectively. MsAcT wt has a narrow entrance pocket forcing the acyl donor and acceptor close together. The L12A mutation creates a passage to a small empty pocket deeper into the active site, which fits DVA well. T93A/F154A widens the entrance enabling higher degree of flexibility for the substrates. Figure made in YASARA and modelled based on (PDB ID: 2Q0S). | ||
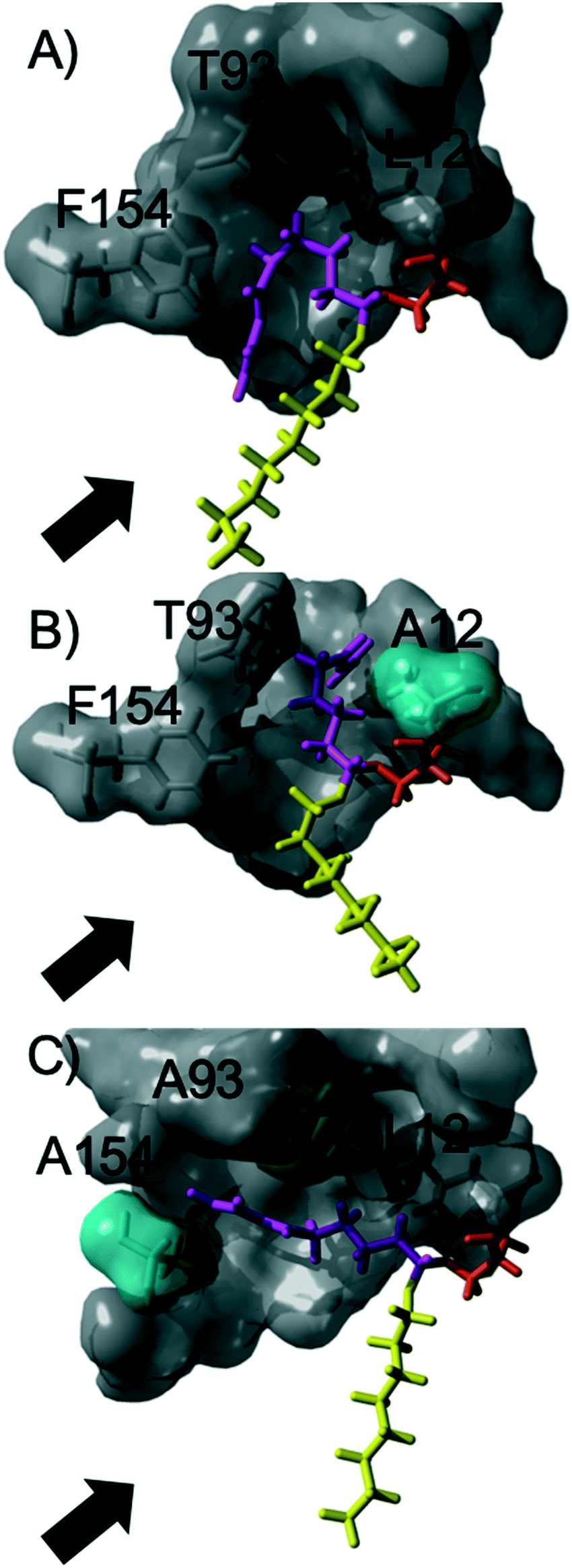 | ||
| Fig. 3 A close up of the active sites displaying the difference in orientation of the acyl donor (DVA) in MsAcT wild type and variants: wt (A), L12A (B) and T93A/F154A (C). The surfaces of selected amino acids are shown. Grey surface indicate conserved amino acids and surfaces in cyan show mutated amino acids. The catalytic serine is shown in orange and the black arrow indicates the entrance. | ||
The new space generated in the double point mutant MsAcT T93A/F154A is indicated by a white circle in the top right frame in Fig. 2 (mutations shown in cyan). The modelling showed that mutating T93 and F154 to alanine facilitated a relief in steric hindrance by generating a volume allowing the acyl donor to bend out towards the entrance of the active site. The new space separated the acyl donor (magenta) from the acyl acceptor (yellow), as can be seen in Fig. 3C. The experimental results presented in Fig. 1 show the catalytic potential of MsAcT T93A/F1545A to produce a highly enriched amount of di-substituted ester relative to the two other MsAcT variants. Interestingly, it can be concluded that the immobilized MsAcT wt and MsAcT L12 have extended substrate scopes towards the acyl donor. Previous reported results where the free enzymes were used in aqueous solutions show that the limiting length for the acyl donor is: methyl butyrate for MsAcT wt and methyl heptanoate for MsAcT L12A.19 Here we report that both these catalyst can use DVA as acyl donor. 1H-NMR spectra from end-point samples for the reactions presented in Fig. 1 can be found in ESI† (Fig. S1–S7).
Enzyme selectivity towards dicarboxylic esters
To further investigate the applicability of the three MsAcT variants, selectivities towards dimethyl esters with lengths varying from succinate to sebacate as acyl donors were studied in bulk. The selectivity was calculated by the relative specificities (kcat/KM) towards the dimethyl esters when incubated in a mixture. The relative specificities were obtained from the initial relative reaction velocities (Fig. S8 to S10 in ESI†).The selectivities of MsAcT wild type and variants displayed in Table 1 support the results in the previous section. MsAcT T93A/F154A, which was the MsAcT variant with the highest activity towards di-substitution of DVA (Fig. 1), was the only tested variant where the selectivity could be determined for the acyl donors longer than adipate. MsAcT T93/F154 was able to use dimethyl sebacate as acyl donor, although the variant showed 20 times higher selectivity towards dimethyl succinate. The increased substrate scope further strengthens the models shown in Fig. 2 and 3 displaying a widened entrance to the active site allowing the acyl donor to bend out towards the entrance. For the reaction catalysed by MsAcT wt product traces were observed at the correct retention time for the mono-substituted suberate on GC, but the integrals of the peaks were not quantifiable, due to the background noise. A substance (peak) was defined as detected if the signal was more than three times the noise of the surrounding baseline. A peak was quantified if it was larger than seven times the noise of the surrounding baseline. Since the MsAcT wt is thought to have a similar positioning of the substrate as MsAcT T93A/F154A (pointing toward the entrance of the binding-site) but with less space for the substrates, the product can be formed but the catalyst is considerably less active compared to the other two.
|
|
|||
|---|---|---|---|
| Substratea | MsAcT variant | ||
| wt | L12A | T93A/F154A | |
| a C4, C6, C8, and C10 correspond to dimethyl succinate, dimethyl adipate, dimethyl suberate, and dimethyl sebacate, respectively. b Below limit of quantification, some product was formed, but the curve fitting was not reliable. c Below detection limit | |||
| C 4 | 1 | 1 | 1 |
| C 6 | 0.3 | 0.4 | 0.4 |
| C 8 | blqb | bdlc | 0.2 |
| C 10 | bdlc | bdlc | 0.05 |
A noteworthy observation was that MsAcT L12A had over 5 times higher initial reaction velocity towards succinate compared to MsAcT wt (Fig. S8–S10 in ESI†). Even though, without active-site titration data, the experimental results provide primarily the relative substrate selectivities for each enzyme this indicates an improved activity of the MsAcT L12A variant compared to wt. MsAcT L12A is postulated to bind the substrate inwards into the enlarged binding-site and, as was shown in the previous section, MsAcT L12A was able to utilise adipate as acyl donor. However, when increasing the chain length to suberate no product formation was detected.
Experimental
Materials and methods
All chemicals were used as received. The carriers for the enzyme immobilization EziG3 were a kind gift from EnginZyme (Sweden). 1-Octanol, 1,4-butanediol vinyl ether (4-(vinyloxy)butanol), dodecane, dimethyl succinate, dimethyl adipate, dimethyl suberate, dimethyl sebacate and Candida antarctica lipase B (CalB) immobilised on acrylic carriers (Novozym® 435) were purchased from Sigma Aldrich. Divinyl adipate was bought from TCI chemicals Acetonitrile and chemicals for enzyme production were purchased from VWR.000g and 4 °C. The supernatant was collected and filtered (0.2 μM). Protein purification was performed by using an ÄKTA explorer (GE Healthcare) by IMAC using HisTrap™ HP-column (GE Healthcare). Fractions giving rise to an absorbance signal at 280 nm were pooled and a concentration and buffer change (potassium phosphate buffer; 20 mM, pH 7.2) was performed using Centricon centrifugal filters with a MWCO of 30 kDa (Millipore, MA, USA). The flow-through was tested for residual hydrolytic activity by hydrolysis of p-nitrophenyl acetate. The enzyme solution was stored at −20 °C.
MsAcT L12A forward primer:
GTGACTCCgctACCTGGGGTTG
MsAcT L12A reverse primer:
CCCAGGTagcGGAGTCACCAAAG
MsAcT T93A forward primer:
CATGCTGGGCgctAACGACACGAAAGC
MsAcT T93A reverse primer:
CGTGTCGTTagcGCCCAGCATGATAATAACC
MsAcT F154A forward primer:
CAGCTGATCgctGAAGGCGGTGAAC
MsAcT F154A reverse primer:
CGCCTTCagcGATCAGCTGAAACC
000 M−1 cm−1). 4.4 mg enzyme solution (wt, L12A or T93A/F153A) were mixed with 50 mg Ezig 3 (Enginzyme). This was incubated at room temperature for three hours and 230 rpm. The enzyme activity of the supernatant decreased by 85–90% compared to the enzyme solution before the immobilization. The supernatant was removed by vacuum filtration and the beads were washed with HEPES buffer followed by Milli-Q-H2O. Finally the beads were dried in a vacuum oven and the immobilized enzyme was stored at 4 °C. Theoretical amount of immobilized enzyme: 80 ± 8 μgMsAcT/mgEzig3.
600 M−1 cm−1). The reaction system contained 100 mM potassium phosphate buffer, pH 8.0, 0.1 mM p-nitrophenyl acetate and 0.1% enzyme solution. To assess the activity of the immobilized enzymes the same theoretical amount of immobilized enzyme (amount), (calculated based on each individual bead saturation percentage), was analysed using the hydrolysis activity assay described above. The absorbance of the product p-nitrophenolate was measured every 1 min for 15 min at 405 nm. A reference from the solution (without beads) was measured every 5 min for 30 min to adjust for spontaneous hydrolysis of p-nitrophenyl acetate.
Analytic methods
Conclusions
A novel synthesis route for mono-substitution of symmetric diesters utilising the single point mutant MsAcT L12A as catalyst was developed. The combined results show that the immobilized MsAcT variants yields catalyst for different applications. MsAcT L12A is a selective catalyst capable of reacting one group out of two identical chemical groups. In contrast, the double point mutant MsAcT T93A/F154A poses as a more versatile catalyst. It can accommodate dicarboxylic esters of varying lengths and is able to catalyse di-substituted adipate. Furthermore, immobilising the MsAcT enabled the catalyst to work under solvent-free conditions and therefore it could be recycled. As MsAcT accepts amines as acyl acceptor interesting amide derivatives might extend the synthetic scope.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study has been funded by Formas, grant No. 211-2013-70 and European Union's Seventh Framework Program for research, technological development and demonstration under grant No. 289253. EnginZyme (Sweden) are acknowledged for the EziG3 carriers. Professor emeritus Karl Hult is acknowledged for valuable discussions and general input.References
- I. Bassanini, K. Hult and S. Riva, Beilstein J. Org. Chem., 2015, 11, 1583–1595 CrossRef CAS PubMed.
- C.-H. Wong and S. C. Zimmerman, Chem. Commun., 2013, 49, 1679–1695 RSC.
- S.-Y. Baek, Y.-W. Kim, K. Chung, S.-H. Yoo, N. K. Kim and Y.-J. Kim, Ind. Eng. Chem. Res., 2012, 51, 3564–3568 CrossRef CAS.
- H. Bart, J. Reidetschläger, K. Schatka and A. Lehmann, Int. J. Chem. Kinet., 1994, 26, 1013–1021 CrossRef CAS.
- A. Sharma, S. Chattopadhyay and V. R. Mamdapur, Biotechnol. Lett., 1995, 17, 939–942 CrossRef CAS.
- R. Shelkov, M. Nahmany and A. Melman, J. Org. Chem., 2002, 67, 8975–8982 CrossRef CAS PubMed.
- H. Ogawa, T. Chihara and K. Taya, J. Am. Chem. Soc., 1985, 107, 1365–1369 CrossRef CAS.
- V. Santacroce, F. Bigi, A. Casnati, R. Maggi, L. Storaro, E. Moretti, L. Vaccaro and G. Maestri, Green Chem., 2016, 18, 5764–5768 RSC.
- M. Lobell and M. P. Schneider, J. Chem. Soc., Perkin Trans. 1, 1993, 1713–1714 RSC.
- A. Mautner, B. Steinbauer, S. Orman, G. Russmüller, K. Macfelda, T. Koch, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1987–1997 CrossRef CAS.
- I. Mathews, M. Soltis, M. Saldajeno, G. Ganshaw, R. Sala, W. Weyler, M. A. Cervin, G. Whited and R. Bott, Biochemistry, 2007, 46, 8969–8979 CrossRef CAS PubMed.
- L. Wiermans, S. Hofzumahaus, C. Schotten, L. Weigand, M. Schallmey, A. Schallmey and P. Domínguez de María, ChemCatChem, 2013, 5, 3719–3724 CrossRef CAS.
- K. Szymańska, K. Odrozek, A. Zniszczoł, G. Torrelo, V. Resch, U. Hanefeld and A. B. Jarzębski, Catal. Sci. Technol., 2016, 6, 4882–4888 RSC.
- N. de Leeuw, G. Torrelo, C. Bisterfeld, V. Resch, L. Mestrom, E. Straulino, L. van der Weel and U. Hanefeld, Adv. Synth. Catal., 2018, 360, 242–249 CrossRef CAS.
- H. Land, P. Hendil-Forssell, M. Martinelle and P. Berglund, Catal. Sci. Technol., 2016, 6, 2897–2900 RSC.
- M. L. Contente, A. Pinto, F. Molinari and F. Paradisi, Adv. Synth. Catal., 2018, 360, 4814–4819 CrossRef CAS.
- L. Mestrom, J. G. Claessen and U. Hanefeld, ChemCatChem, 2019, 11, 2004–2010 CrossRef CAS.
- I. C. Perdomo, S. Gianolio, A. Pinto, D. Romano, M. L. Contente, F. Paradisi and F. Molinari, J. Agric. Food Chem., 2019, 67, 6517–6522 CrossRef CAS PubMed.
- P. Hendil-Forssell, Rational engineering of esterases for improved amidase specificity in amide synthesis and hydrolysis, Ph.D. Dissertation, KTH Royal Institute of Technology, 2016 Search PubMed.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- C. Z. Dinu, G. Zhu, S. S. Bale, G. Anand, P. J. Reeder, K. Sanford, G. Whited, R. S. Kane and J. S. Dordick, Adv. Funct. Mater., 2010, 20, 392–398 CrossRef CAS.
- M. Contente, S. Farris, L. Tamborini, F. Molinari and F. Paradisi, Green Chem., 2019, 21, 3263–3266 RSC.
- S. Y. Tang, R. A. Bourne, R. L. Smith and M. Poliakoff, Green Chem., 2008, 10, 268–269 RSC.
- Q. Michaudel, V. Kottisch and B. P. Fors, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS PubMed.
- L. Zheng, U. Baumann and J.-L. Reymond, Nucleic Acids Res., 2004, 32, e115 CrossRef PubMed.
- J. T. Scanlon and D. E. Willis, J. Chromatogr. Sci., 1985, 23, 333–340 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01181a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |