
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBi-functional Ru/Ca3Al2O6–CaO catalyst-CO2 sorbent for the production of high purity hydrogen via sorption-enhanced steam methane reforming†
Sung Min
Kim
 a,
Paula M.
Abdala
a,
Davood
Hosseini
a,
Andac
Armutlulu
a,
Tigran
Margossian
b,
Christophe
Copéret
b and
Christoph
Müller
*a
a,
Paula M.
Abdala
a,
Davood
Hosseini
a,
Andac
Armutlulu
a,
Tigran
Margossian
b,
Christophe
Copéret
b and
Christoph
Müller
*a
aDepartment of Mechanical and Process Engineering, ETH Zurich, Leonhardstrasse 21, 8092 Zurich, Switzerland. E-mail: muelchri@ethz.ch
bDepartment of Chemistry and Applied Sciences, ETH Zurich, Vladimir Prelog Weg 1-5, 8093 Zurich, Switzerland
First published on 9th September 2019
Abstract
Sorption-enhanced steam methane reforming (SE-SMR) combines steam methane reforming and a CO2 abstraction reaction to yield high purity hydrogen. In this work, we report on the development of a bi-functional catalyst–sorbent containing Ru as the reforming catalyst and CaO as the solid CO2 sorbent via a citrate sol–gel route. The material contains CaO, a structural stabilizer (Ca3Al2O6) and Ru nanoparticles (∼5 nm, 3 wt%) that are formed upon reduction in H2. This new material was found to outperform significantly the benchmarks Ni–CaO and Ru/limestone in terms of yield of high-purity hydrogen and coke resistance. Using highly active Ru nanoparticles for the SMR allowed to maximize the weight fraction of the CO2 sorbent CaO, hence increasing significantly the CO2 capture capacity of the material. This favorable characteristic of the material led to an appreciably extended pre-breakthrough duration. In addition, we demonstrate that the material developed was very stable over multiple SE-SMR/regeneration cycles. The excellent cyclic stability is ascribed to the presence of Ca3Al2O6 that stabilized effectively the porous structure of the material against sintering.
1. Introduction
Hydrogen is a carbon-free energy carrier that features a high energy density (120–142 MJ kg−1).1,2 In addition, hydrogen is also an important chemical feedstock for a number of large scale processes such as ammonia synthesis or the refining of crude oil.3 Although its combustion does not yield any CO2 emissions, the large scale processes through which it is currently produced, e.g. the steam reforming of natural gas (SMR), are energy intensive and emit large quantities of CO2 as a side product.4 For example, the conventional SMR process (CH4 + H2O ⇆ 3H2 + CO, ΔH0298 K = 206 kJ mol−1) produces 7 kg CO2 per kg H2.5 Besides the emission of CO2, SMR is also an equilibrium limited process, i.e. at the SMR working a gas mixture containing approximately 76% H2, 17% CO2, 3% CO and 4% CH4 is obtained.6,7 CO is converted further with steam via the water gas shift reaction (CO + H2O ⇄ CO2 + H2, ΔH0298 K = −41 kJ mol−1) to CO2, yielding an additional mole of H2 per mole CO. Unconverted CO (after the high and low temperature WGS steps) is selectively oxidized to CO2 which is subsequently removed from the hydrogen stream by pressure swing adsorption (PSA) or amine scrubbing.8The sorption-enhanced steam methane reforming (SE-SMR: CH4 + 2H2O + sorbent ⇆ sorbent-CO2 + 4H2) is a promising approach to overcome the drawbacks of the traditional SMR.9 In this process the SMR and WGS reactions proceed in the presence of a solid CO2 sorbent. The abstraction of the product CO2 shifts the equilibrium to the product side (Le Chatelier's principle) yielding high-purity H2 in a single step. This allows the SE-SMR reaction to be operated also at a lower temperature of ≈550 °C compared to the conventional SMR (800–1000 °C), potentially reducing the rate of catalyst deactivation by thermal sintering.
A key component of the SE-SMR process is a CO2 sorbent that should possess a high CO2 uptake in the temperature range of interest (500–900 °C). In this context, CaO is a promising candidate due to (i) its high theoretical CO2 uptake (0.78 g CO2/g sorbent), (ii) the high abundance of its natural precursors, e.g., limestone, (iii) its low CO2 capture costs ($9–11 per ton of CO2 captured),10 and (iv) its fast kinetics for CO2 uptake and release (CaO + CO2 ⇆ CaCO3, ΔH0298 K = −178 kJ mol−1). It is important to note that the incorporation of a CaO-based CO2 abstraction reaction turns the highly endothermic SMR reaction into an exothermic reaction (CH4 + 2H2O + CaO ⇆ 4H2 + CaCO3, ΔH0298 K = −13 kJ mol−1).
However, the cyclic performance of CaO-based CO2 sorbents is hampered by the low Tammann temperature of CaCO3 (TT ≈ 530 °C) leading to sintering and in turn a loss in porosity. A high porosity is, however, required as the molar volume of the reaction product (CaCO3) is almost twice as high as that of the reactant (CaO).11,12 Therefore, to avoid that the CO2 uptake proceeds in a slow, diffusion-limit regime (the diffusivity of CO2 in CaCO3, DCaCO3 = 0.003 cm2 s−1, is two orders of magnitude lower than that in CaO, DCaO = 0.3 cm2 s−1), a high porosity of the sorbent is critical. To counteract sintering, attempts have been made to stabilize the CaCO3/CaO structure with high Tammann temperature metal oxides such as Al2O3, MgO, TiO2, SiO2, and ZrO2.13–16 As the reaction becomes diffusion-limited at a CaCO3 thickness of ∼50 nm,17 also nano-structuring of the sorbent becomes an important aspect to minimize diffusion lengths.
Besides requiring a solid CO2 sorbent, the SE-SMR relies on a SMR catalyst, typically a transition metal such as Pt, Ru, Rh, Pd, Ir, Co or Ni.18 An additional requirement for the catalyst in the SE-SMR process is its stability in cyclic SE-SMR–regeneration. Conceptually, it is possible to simply mix a SMR catalyst with a CO2 sorbent. However, the availability of a bi-functional material that contains both the CO2 sorbent and the catalyst would have a number of advantages, such as enhancing heat and mass transfer and reducing the quantity of inactive support.19,20 However, the design and manufacture of highly active and stable bi-functional materials is challenging as there is a potential for an irreversible interaction (e.g. formation of inactive solid solutions) between the CO2 sorbent, the catalyst and the support.21–23 In addition, material migration during CO2 uptake and regeneration may block the active sites of the catalyst. In order to overcome the sintering-induced deactivation, approaches that incorporate an structural stabilizer such as MgxAlyOz,24 CaZrO3,25 Ca12Al14O33,26–29 Ca9Al6O18,30 Ca5Al6O14,31 CaAl2O6 (ref. 32) have been applied to develop more effective bi-functional catalyst–sorbents. However, high metal loadings of the catalyst (15–50 wt% Ni or Co)24,25,33 have been typically required to obtain materials with acceptable SMR activity. However, such high catalyst loadings lead to a reduced CO2 uptake capacity per gram of material, thus, affecting negatively the economics of the SE-SMR process.
In this work, we report the development of a bi-functional material that contains Ru as the reforming catalyst and CaO as the solid CO2 sorbent. The materials were synthesized via a scalable, one-pot synthesis route (citrate sol–gel method). The synthesized materials (Ru/CaO and Ru/Ca3Al2O6–CaO) were compared to the following benchmark materials: Ru impregnated on limestone-derived CaO (Ru/lime) and a bi-functional Ni-based material (Ca–Ni-ex-Htlc).24 The SE-SMR performance of the synthesized bi-functional materials was tested at 550 °C under equilibrium limitations (8.4 L gcat−1 h−1 of GHSV, CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:N2 = 1:4:9). The yield of high purity hydrogen was evaluated over ten repeated cycles of SE-SMR and regeneration (at 750 °C). The stability of the materials for SE-SMR was also determined under kinetic control at 550 °C (84 L gcat−1 h−1 of GHSV, CH4:H2O:N2 = 1:4:9). The bi-functional material Ru/Ca3Al2O6 yielded a remarkable quantity of high purity hydrogen (12–13 mmol) in the pre-breakthrough regime, outperforming the benchmark Ca–Ni-ex-Htlc and Ru/lime by more than 590%. Differences in the SE-SMR performance of the materials tested, are discussed in view of a detailed characterization of the as-synthesized and reacted materials using X-ray absorption spectroscopy (XAS), electron microscopy, X-ray powder diffraction (XRD) and N2 physisorption.
:H2O:N2 = 1:4:9). The yield of high purity hydrogen was evaluated over ten repeated cycles of SE-SMR and regeneration (at 750 °C). The stability of the materials for SE-SMR was also determined under kinetic control at 550 °C (84 L gcat−1 h−1 of GHSV, CH4:H2O:N2 = 1:4:9). The bi-functional material Ru/Ca3Al2O6 yielded a remarkable quantity of high purity hydrogen (12–13 mmol) in the pre-breakthrough regime, outperforming the benchmark Ca–Ni-ex-Htlc and Ru/lime by more than 590%. Differences in the SE-SMR performance of the materials tested, are discussed in view of a detailed characterization of the as-synthesized and reacted materials using X-ray absorption spectroscopy (XAS), electron microscopy, X-ray powder diffraction (XRD) and N2 physisorption.
2. Experimental
2.1. Material preparation
:Al3+ in Ru/Ca3Al2O6–CaO was 9:1 (as determined by ICP-OES). The solution was heated up to 110 °C under vigorous stirring until gel formation occurred. This was followed by drying at 130 °C in an oven. Subsequently, the dried materials were calcined at 850 °C (5 °C min−1 heating rate) for 2 h and sequentially ground and sieved to 100–300 μm. The bi-functional Ru/lime (3 wt% Ru) was prepared by wet impregnation. 1 g of Rheinkalk limestone calcined at 800 °C was added to 100 ml of an aqueous solution of ruthenium nitrosyl nitrate (3.0 mM) under vigorous stirring for 5 h. The water was evaporated subsequently in a rotary evaporator (Heidolph) at 80 °C (300 mbar). The materials were dried at 110 °C in an oven overnight, followed by calcination at 850 °C. A bi-functional material containing a Ni catalyst (45 wt% Ni, Ca–Ni-ex-Htlc) was prepared by co-precipitation and contained the following phases (after calcination): NiO, CaO and MgAl2O4. Details of its physical–chemical properties have been reported elsewhere.24
:1 molar ratio; in total 10 mmol of metal precursors), and (ii) Ru (3 wt%) supported on γ-Al2O3 (Alfa Aesar) was prepared by impregnation using Ru(NO)(NO3)3 (Acros Organics) and it is referred as Ru/Al2O3. The reference materials were calcined at 850 °C (5 °C min−1) for 2 h. Additionally, RuO2 (Sigma-Aldrich) with a rutile structure was used as a reference material.
2.2. Material characterization
The elemental composition of the calcined materials was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Agilent 5100 VDV).The surface area and pore volume of the freshly calcined and reacted materials were determined using a Quantachrome (NOVA 4000e) N2 adsorption analyzer. Prior to N2 adsorption, each sample was degassed at 250 °C for 3 h under vacuum (10−3 mbar). The Brunauer et al. (BET)35 and the Barrett et al. (BJH) models36 were used to calculate the surface area and pore size distribution, respectively.
Temperature-programmed reduction in H2 (H2-TPR) was conducted in an Autochem 2920 equipped with a thermal conductivity detector. For these measurements, 30–50 mg of the calcined catalyst was placed in a quartz reactor and heated to 300 °C in an argon atmosphere (50 mL min−1) to dehydrate the sample. Next, the materials were cooled down to 50 °C and the gas was switched to 5 vol% H2/Ar (50 mL min−1). Subsequently, the temperature was increased at a rate of 5 °C min−1.
Temperature-programmed oxidation (TPO) was used to determine the quantity of coke deposited on the spent catalyst. TPO experiments were performed in a thermogravimetric analyzer (TGA, Mettler Toledo TGA/DSC 3). Here, approximately 20 mg of the spent catalyst was loaded into an alumina crucible. Prior to TPO, the sample was heated to 100 °C with a heating rate of 10 °C min−1 in 100 mL min−1 of air. After a holding time of 1 h, the sample was heated further to 1000 °C (10 °C min−1 in 100 ml min−1 of air). The weight loss of the sample was continuously recorded and used to determine the mass of coke deposited.
H2 chemisorption was carried out in an Autochem 2920 apparatus. 50–60 mg of a material was reduced in a quartz reactor at 500 °C for 1 h in 5 vol% H2/Ar (50 mL min−1). The sample was then cooled down to 400 °C and purged with Ar (50 mL min−1) for 30 min. After cooling down further to 50 °C in Ar, the quantity of chemisorbed H2 was determined by repeatedly injecting pulses of 5 vol% H2/Ar into the reduced catalyst bed. The stoichiometry factor of dissociated H2 to Ru was set to 1.0 (H/Ru).37–39
The crystalline phases in the materials were studied using an Empyrean (PANalytical) powder X-ray diffractometer operated at 40 mA and 40 kV using Cu Kα (λ = 0.1541 nm) radiation. The diffraction patterns were recorded in the range 2θ = 5–90° with 0.016° of step size.
The morphology of the samples was studied by electron microscopy (viz. scanning electron microscopy, SEM, FEI Magellan 400 FEG, transmission electron microscopy, TEM, Philips CM12 and FEI Talos F200X and a scanning transmission electron microscopy, STEM, Hitachi HD-2700) equipped also with an energy-dispersive X-ray (EDX) detector.
In situ X-ray absorption spectroscopy (XAS) experiments (Ru K-edge, 22.1 keV) were performed at the Swiss-Norwegian Beamlines (SNBL, BM31) of the European Synchrotron Radiation Facility (ESRF). Here, the calcined material, approximately 2 mg, was loaded in a capillary quartz reactor (1.0 mm of outer diameter) and heated up to 500 °C, with a ramp rate of 10 °C min−1 in 5 vol% H2/He (10 ml min−1). Ex situ XAS measurement were also performed on the as-synthesized materials (bi-functional materials and references). The materials were mixed with cellulose (ratio chosen such that XAS measurements in transmission mode were optimized) and ground to a fine powder. The pelletized samples were measured in transmission mode. XAS spectra were collected at the Ru K-edge using a double-crystal Si (111) monochromator (continuous scanning mode) and the data were analyzed using the Athena and Arthemis software.40
2.3. Cyclic CO2 capture experiments
Cyclic CO2 capture and regeneration experiments were performed in a TGA (Mettler Toledo TGA/DSC 3). Approximately 10 mg of the calcined material were heated to 900 °C at a heating rate of 50 °C min−1 under a flow of N2 (total flow rate of 120 mL min−1 including a constant purge flow of N2, 25 mL min−1, over the microbalance). When the temperature of 900 °C was reached a holding time of 4 min was applied to ensure complete calcination. This was followed by a reduction of the temperature to 650 °C. At 650 °C, the CO2 capture step was performed (20 min under 20 vol% of CO2 in N2, total flow rate: 150 ml min−1). After carbonation, the CO2 sorbent was regenerated at 900 °C for 10 min in a CO2 atmosphere (30 mL min−1). 10 cycles of repeated carbonation and calcination steps were performed.The cyclic CO2 capture performance was tested further under SE-SMR-mimicking conditions (carbonation: 550 °C using 50 ml min−1 of 20% CO2/N2, 2 h, and regeneration: 750 °C, 50 ml min−1 of N2, 15 min). Prior to CO2 capture, the calcined material was heated to 800 °C (10 °C min−1) under N2 (50 ml min−1) followed by reduction at 500 °C (10 °C min−1) for 2 h in 10 vol% H2/N2 (50 ml min−1). Ten repeated cycles of carbonation (550 °C, 50 ml min−1 of 20 vol% CO2/N2) and regeneration (750 °C, 50 ml min−1 of N2) were performed.
2.4. Cyclic SE-SMR experiments
The cyclic SE-SMR reaction was carried out in a fixed-bed quartz reactor (12.6 mm internal diameter and 400 mm length). 100 mg of the calcined bi-functional material (diluted with SiC with a weight ratio of 1:10) was used. Prior to the activity test, the bed was heated up to 800 °C (10 °C min−1). A holding time of 1 h (N2 with 100 mL min−1) ensured the complete calcination of CaCO3 to CaO. Subsequently, the bed temperature was reduced to 500 °C. At 500 °C the bed was treated for 2 h in 10 vol% H2/N2 (100 mL min−1). After reduction, the SE-SMR was performed at 550 °C using a total flow rate of 14 mL min−1 (8.4 L gcat−1 h−1 of GHSV and CH4:H2O:N2 = 1:4:9). After 2 h of the SE-SMR, the bed was heated up to 750 °C (20 °C min−1) in N2 (50 ml min−1) for regeneration (15 min). In total 10 cycles of SE-SMR/regeneration were performed. The composition of the off-gas (after condensation of unreacted steam) was analyzed using a micro-GC (C2V-200, Thermo Scientific) equipped with a thermal conductivity detector (TCD) and molecular sieve 5A and U-plot column cartridges.
3. Results and discussion
3.1. Physical–chemical and textural properties
The elemental composition determined by ICP-OES and the textural characteristics of the Ru–CaO-based materials (Ru/CaO, Ru/Ca3Al2O6–CaO and Ru/lime) after calcination (850 °C in air) and reduction (500 °C under 10 vol% H2/N2) are summarized in Table 1. The elemental composition of the calcined catalysts shows a very similar loading of Ru in the materials studied here (1.6–1.7 mol% corresponding to 2.8–2.9 wt%) and is in good agreement with the desired composition. The N2 isotherms of all of the calcined materials tested is represented well by a type III isotherm and a H3 type hysteresis loop at 0.2–0.9 of p/p0, indicative of a randomly distributed macro-porosity (Fig. S1a†). The corresponding BJH pore size distributions show a bimodal porosity distribution with peaks located at 1.5–2.1 nm and 27.4–28.5 nm (Fig. S1b†). Overall, Ru/Ca3Al2O6–CaO features a higher surface area and pore volume than Ru/lime and Ru/CaO (Table 1).| Materiala | Elemental compositionb | N2 physisorptionc | Crystallite sized [nm] | Ru particle sizee [nm] | H2 uptakef [μmolRu gcat−1] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ru [mol%] | Ca [mol%] | Al [mol%] | S BET [m2 g−1] | V Pore [cm3 g−1] | D pore [nm] | CaO | Ru | ||||
| a The bi-functional Ru–CaO catalyst–sorbent was calcined at 800 °C in static air and reduced in 10 vol% H2/N2. b The normalized metal mol% of Ru, Ca an Al in the calcined materials were obtained by ICP-OES. c Textural properties of the calcined materials. d Average crystallite size of CaO and Ru (in the reduced materials) determined by Scherrer's equation.41 e Average Ru particle size of the reduced materials determined by TEM. f The quantity of surface Ru in the reduced materials as determined by H2 chemisorption using a stoichiometry factor of 1.0 of H/Ru,37–39 parenthesis represents Ru dispersion (%) in the reduced catalysts. | |||||||||||
| Ru/lime | 1.7 | 97.3 | — | 12 | 0.11 | 2.2 | 70 | 18 | 22.6 ± 2.6 | 4.8 | (1.6%) |
| Ru/CaO | 1.6 | 98.4 | — | 17 | 0.19 | 1.8 | 36 | 10 | 11.2 ± 2.2 | 18.4 | (6.2%) |
| Ru/Ca3Al2O6–CaO | 1.7 | 88.6 | 9.7 | 23 | 0.26 | 1.8 | 27 | 5 | 5.3 ± 1.3 | 37.4 | (12.7%) |
3.2. Structure of the calcined catalyst–sorbents and evolution upon reductive treatment
HAADF STEM images (with EDX analysis) of calcined Ru/Ca3Al2O6–CaO showed a homogenous distribution of Ru and Al in CaO matrix (Fig. S2†).The crystalline phases of the bifunctional catalyst–sorbents (calcined and after reduction) were characterized by XRD. All of the calcined and reduced materials contain cubic CaO (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group), while Ru/Ca3Al2O6–CaO contains an additional Ca3Al2O6 (Pa) phase (Fig. 1a). Some small amounts of Ca(OH)2 are also present due to the hygroscopic nature of CaO, (Ca(OH)2 is formed after exposing the materials to humid air). The diffraction patterns of Ru/CaO and Ru/Ca3Al2O6–CaO feature relatively broad peaks of CaO, characteristic of a nanostructured material. Indeed, the average crystallite sizes of CaO, calculated by the Scherrer equation,41 are ∼36 nm and ∼27 nm for Ru/CaO and Ru/Ca3Al2O6–CaO, respectively (Table 1). These values are considerably smaller than that determined for Ru/lime (∼70 nm). In addition, for all of the calcined, Ru–CaO containing materials we observe additional, weak peaks that are likely due to the presence of a perovskite CaRuO3 phase (Pnma space group, inset in Fig. 1a). However, due to the low intensity of these peaks (low Ru loading) and the overlap with other phases present, XRD provides only limited information of the Ru containing phases. Therefore, the materials were characterized further by Ru K-edge XAS to understand better the local environment of Ru after calcination and reduction (Fig. 1b) and in particular the interaction with the CO2 capture active phase CaO.
m space group), while Ru/Ca3Al2O6–CaO contains an additional Ca3Al2O6 (Pa) phase (Fig. 1a). Some small amounts of Ca(OH)2 are also present due to the hygroscopic nature of CaO, (Ca(OH)2 is formed after exposing the materials to humid air). The diffraction patterns of Ru/CaO and Ru/Ca3Al2O6–CaO feature relatively broad peaks of CaO, characteristic of a nanostructured material. Indeed, the average crystallite sizes of CaO, calculated by the Scherrer equation,41 are ∼36 nm and ∼27 nm for Ru/CaO and Ru/Ca3Al2O6–CaO, respectively (Table 1). These values are considerably smaller than that determined for Ru/lime (∼70 nm). In addition, for all of the calcined, Ru–CaO containing materials we observe additional, weak peaks that are likely due to the presence of a perovskite CaRuO3 phase (Pnma space group, inset in Fig. 1a). However, due to the low intensity of these peaks (low Ru loading) and the overlap with other phases present, XRD provides only limited information of the Ru containing phases. Therefore, the materials were characterized further by Ru K-edge XAS to understand better the local environment of Ru after calcination and reduction (Fig. 1b) and in particular the interaction with the CO2 capture active phase CaO.
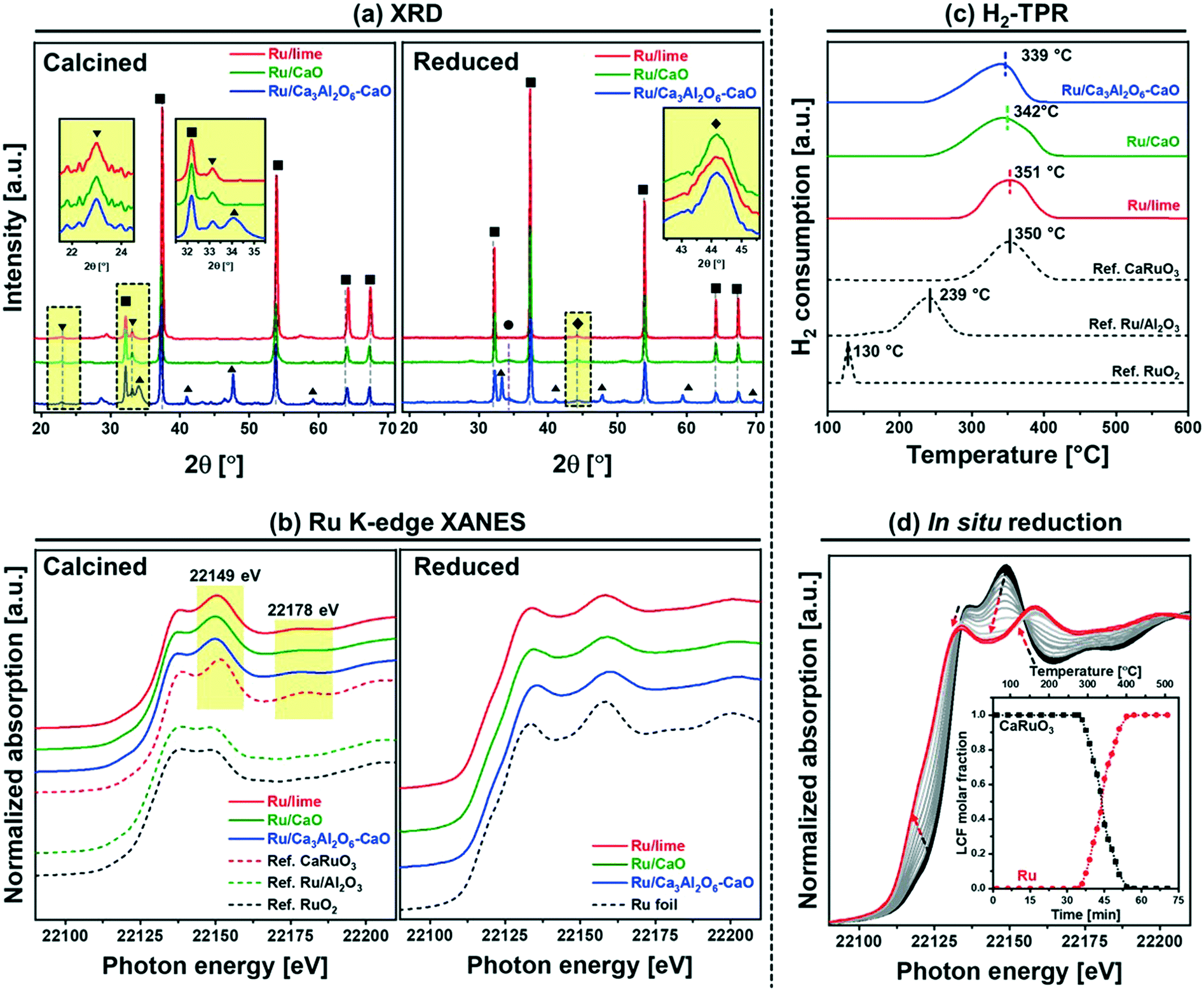 | ||
| Fig. 1 Structural characterization of the calcined and reduced materials: (a) XRD of the calcined and reduced bifunctional materials. The insets show Bragg reflections due to CaRuO3 and Ru, respectively. The symbols represent the following phases: (■) CaO, (▲) Ca3Al2O6, (●) Ca(OH)2, (▼) CaRuO3 and (♦) Ru. (c) Ru K-edge, XANES of bifunctional materials after calcination in air at 850 °C air and reduction in 10 vol% H2/N2 at 500 °C. (c) H2-TPR of bifunctional materials and references. (d) In situ Ru K-edge XANES spectra of Ru/Ca3Al2O6–CaO, during temperature programed reduction in 5 vol% H2/He. The arrows indicate the direction of change with time and temperature. The inset shows the results of the LCF analysis. | ||
For XAS analysis, the following reference materials were measured, and also characterized by XRD (Fig. S3†) prior to XAS analysis: (i) bulk (rutile) RuO2 and (ii) CaRuO3 which contains a pure orthorhombic perovskite phase (Pnma space group). In CaRuO3, Ru is in an octahedral coordination, and in rutile RuO2 in an axially elongated octahedral coordination. The X-ray absorption near edge structure (XANES) measurements acquired (Fig. 1b) show very distinct features for the CaRuO3 and RuO2 references. Both RuO2 and CaRuO3 compounds exhibit two peaks: at 22136 and 22149 eV, with the latter being more intense for CaRuO3. Additionally, CaRuO3 exhibits a feature at 22178 eV which is absent in RuO2. Owing to these features in the XANES signal we can distinguish between a RuO2 or CaRuO3-like local environment of Ru. For instance, in agreement with XRD, the Ru/Al2O3 reference shows the same XANES features as RuO2. Turning to the bi-functional materials, the presence of features at 22149 and 22178 eV in Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO indicates that in these materials Ru is in a CaRuO3-like local environment. To provide further insight into the local environment of Ru in the calcined materials, the extended X-ray absorption fine structure (EXAFS) data of the CaRuO3 reference, Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO were fitted (Table S1 and Fig. S4†). In line with the atomic arrangement of a CaRuO3 perovskite according to crystallographic data,42 the CaRuO3 reference data were fitted with a Ru–O shell at 2.0 Å, four Ru–Ca shells with interatomic distances in the range of 3.14–3.61 Å and one Ru–Ru shell at 3.84 Å (and with coordination numbers set to the crystallographic values). The same model of the local structure of Ru was applied to Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO. All three Ru–CaO-based sorbents showed a Ru–O coordination sphere at interatomic distances in the range of 1.95–1.97 Å, four Ru–Ca shells with interatomic distances in the range of 3.10–3.47 Å and a Ru–Ru shell at 3.76–3.87 Å. These results are in line with the XANES observation that suggests Ru has a local environment consistent with CaRuO3. On the other hand, the Debye Waller factors observed for the synthetic materials were larger than for the reference crystalline perovskite. This can be explained by a higher disorder of the local environmental of Ru in a highly dispersed CaRuO3-phase within the CaO or CaO–Ca3Al2O6 matrix.
The reduction characteristics of the different materials (bi-functional materials and references) were assessed by H2-TPR experiments (Fig. 1c) after calcination (in air) at 800 °C. The diffractogram of the calcined materials (Fig. 1a) confirmed that there was no CaCO3 in the samples. In addition, all H2-TPR profiles showed a stable baseline between 600–1000 °C (without any further H2 consumption). The TPR profile of the reference bulk RuO2 shows a reduction peak at 130 °C, in agreement with literature.43 On the other hand, Ru/Al2O3 show higher reduction temperatures, viz. 239 °C. In agreement with literature.44,45 The reference CaRuO3 shows a reduction peak at 350 °C, according to: CaRuO3 + 2H2 → Ru0 + CaO + 2H2O. All the Ru–CaO-containing materials (Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO) show a reduction peak in the temperature range 339–351 °C (similar to CaRuO3) indicating that Ru0 is obtained through the reduction of a CaRuO3 phase. The TPR experiments were complemented with in situ Ru K-edge XANES (Fig. 1d). Linear combination fitting (LCF) using Ru foil, RuO2 and CaRuO3 as references allowed us to quantify the evolution of these phases in Ru/Ca3Al2O6–CaO (Fig. 1d) during in situ XANES TPR. Our XANES analysis shows that CaRuO3 reduces to Ru0 between 260–400 °C. We could not detect any intermediate phases or segregation of RuO2.
Additionally, using ex situ XRD analysis, the formation of metallic Ru particles after reduction at 500 °C in 10 vol% H2/N2 is evidenced by the appearance of, albeit weak, (101) reflections due to hexagonal Ru (P63/mmc space group) at ∼44° (Fig. 1a). This Bragg reflection is observed in all of the Ru containing materials. Using Scherrer's equation, the average Ru crystallite sizes were estimated as: Ru/lime (18 nm) > Ru/CaO (10 nm) > Ru/Ca3Al2O6–CaO (5 nm).
H2 chemisorption was employed to quantify the amount of surface Ru (Table 1). Considering a stoichiometry factor of 1.0 of H/Ru,37–39 the quantity of surface Ru and its dispersion were determined as follows: 4.8 μmolRu gcat−1 and 1.6% (Ru/lime) < 18.4 μmolRu gcat−1 and 6.2% (Ru/CaO) < 37.4 μmolRu gcat−1 and 12.7% (Ru/Ca3Al2O6–CaO).
HR-SEM and HAADF-STEM images of the reduced materials are shown in Fig. 2. In HR-SEM (Fig. 2a), reduced Ru/Ca3Al2O6–CaO exhibits particles of CaO or Ca3Al2O6 with an average size of 90 ± 10 nm, which is a factor of 1.6–2.0 smaller than the size observed in Ru/CaO (140 ± 20 nm) and Ru/lime (170 ± 40 nm). Ru nanoparticle were observed by HAADF STEM in all of the materials (Fig. 2b and c) with average sizes determined as: 22.6 ± 2.6 nm (Ru/lime) > 11.2 ± 2.2 nm (Ru/CaO) > 5.3 ± 1.3 nm (Ru/Ca3Al2O6–CaO). Hence electron microscopy, in combination with H2 chemisorption measurements, show that the one-pot, citrate sol–gel method (Ru/CaO and Ru/Ca3Al2O6–CaO) leads to smaller Ru nanoparticles compared to impregnation (Ru/lime), yielding in turn a higher quantity of Ru surface sites.
 | ||
| Fig. 2 Electron microscopy based characterization of the reduced materials: (a) HR-SEM and (b) HAADF-STEM of reduced Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO. (c) HR-TEM and STEM EDX mapping for reduced Ru/Ca3Al2O6–CaO. | ||
3.3. CO2 capture performance
The cyclic CO2 capture performance of the bi-functional catalyst–sorbents (Ru/CaO, Ru/Ca3Al2O6–CaO and the benchmarks Ru/lime and Ca–Ni-ex-Htlc) was assessed in a TGA (carbonation was performed in 20% CO2/N2 at 650 °C and calcination in CO2 at 900 °C, Fig. 3a). The CO2 uptake in the 1st cycle decreases in the following order: Ru/CaO (0.57 gCO2/g) > Ru/Ca3Al2O6–CaO (0.54 gCO2/g) > Ru/lime (0.45 gCO2/g) ≫ Ca–Ni-ex-Htlc (0.11 gCO2/g). The reference material Ca–Ni-ex-Htlc has a comparatively low CO2 uptake, owing to the high content of Ni (45 wt%). After 10 cycles of repeated carbonation–calcination, the CO2 uptake of Ru/CaO and Ru/lime has reduced appreciably to 0.16 gCO2 gcat−1 and 0.15 gCO2 gcat−1, corresponding to a capacity retention of 31% and 33%, respectively. On the other hand, a significantly higher cyclic stability was observed for Ru/Ca3Al2O6–CaO and Ca–Ni-ex-Htlc, yielding a CO2 uptake of 0.46 gCO2/g and 0.09 gCO2/g, respectively (corresponding to capacity retentions of respectively, 92% and 82% after 10 cycles). These findings indicate that the presence of a high Tammann temperature stabilizer, viz. Ca3Al2O6 (TT = 771 °C) or MgAl2O4 (TT = 1065 °C) in, respectively, Ru/Ca3Al2O6–CaO and Ca–Ni-ex-Htlc, improves the cyclic stability over ten cycles of the CO2 uptake in these materials (although the CaO content in Ca–Ni-ex-Htlc is rather low, i.e. 21 wt%), in agreement with previous studies.13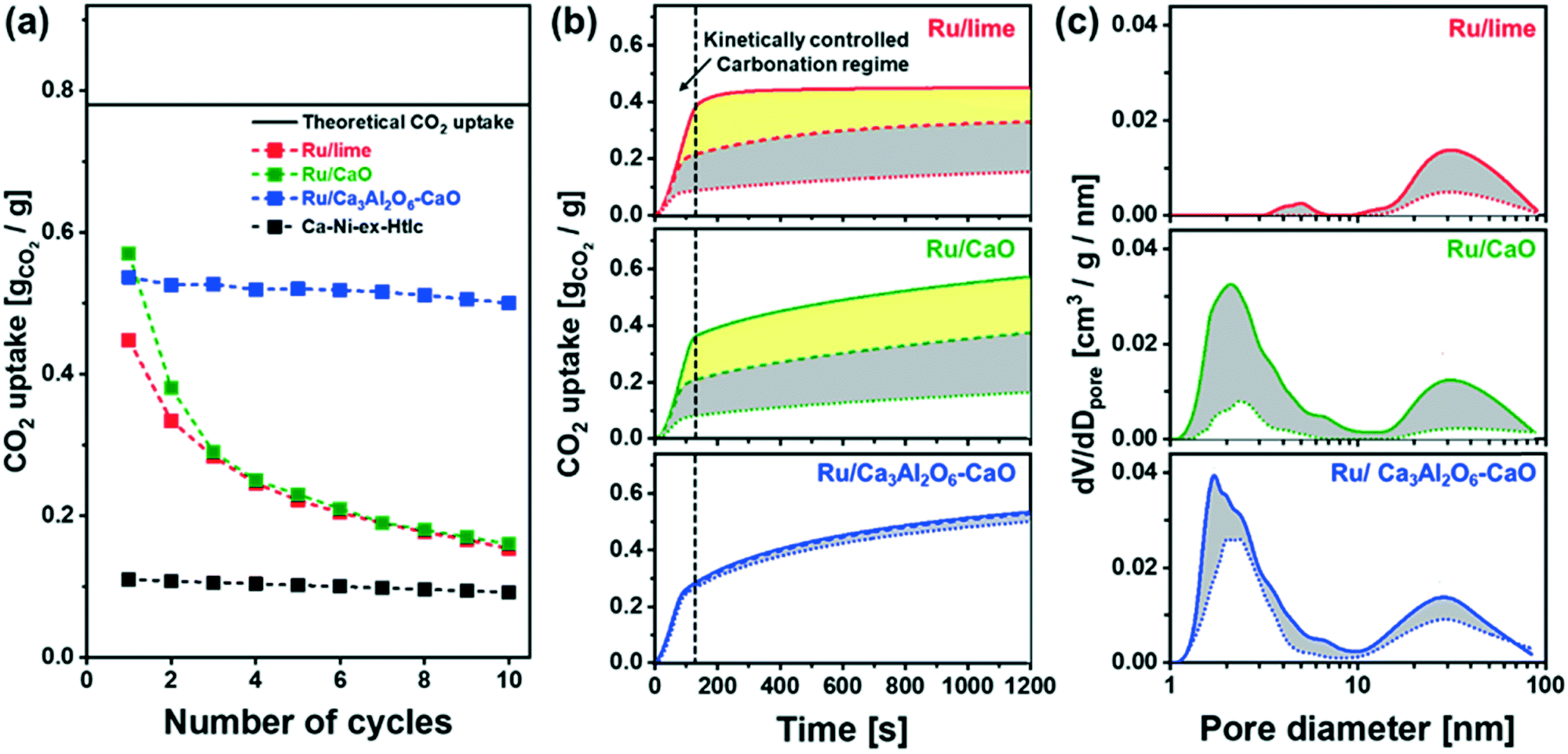 | ||
| Fig. 3 CO2 capture: (a) CO2 uptake as a function of number of carbonation–regeneration cycles. The solid line is the theoretical CO2 uptake of pure CaO, i.e., 0.78 gCO2/g. (b) CO2 uptake vs. carbonation time. The solid dashed and dotted lines represent the 1st, 2nd and 10th carbonation cycle, respectively. The reduction in the CO2 uptake is highlighted by grey and yellow fills. (c) BJH pore size distribution of freshly calcined (dash line) materials and materials that have undergone 10 cycles (dotted line) of carbonation and regeneration (calcined form). The reduction in pore volume over 10 cycles is highlighted by a grey fill. | ||
Temporally resolved carbonation profiles were acquired for each material to elucidate the origin of deactivation upon cyclic operation. Fig. 3b plots the CO2 uptake of a series of materials in the 1st and 10th carbonation cycle as a function of time. The carbonation of CaO is known to occurs in two regimes:17,46 i) a reaction stage that is kinetically controlled and in which CaCO3 fills the volume available in small pores (dpore < 100 nm) and ii) a diffusion-limited reaction stage that is controlled by the slow diffusion of CO2 through the CaCO3 product layer. The diffusivity of CO2 through CaCO3 (DCaCO3 = 0.003 cm2 s−1) is significantly smaller than its diffusion through CaO (DCaO = 0.3 cm2 s−1).47 Here, we determine the transition between these two reaction stages as the intersection of two lines that fit linearly the CO2 uptake with time in the two carbonation regimes. Owing to the large difference in the rate of carbonation in the two reaction regimes, a large fraction of the overall CO2 uptake occurs in the kinetically-controlled regime, viz. 0.40 gCO2/g, 0.35 gCO2/g and 0.33 gCO2/g for, respectively, Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO, in 1st cycle (contributing to 88%, 62% and 63% of the respective overall CO2 uptake). However, after two cycles, the CO2 uptake of Ru/CaO and Ru/lime in the kinetically-controlled reaction stage decreased significantly to 0.19 gCO2/g and 0.21 gCO2/g, i.e., a reduction by 45% and 48%, respectively (these values reduced to 0.10 gCO2/g and 0.08 gCO2/g after 10 cycles). The decrease in the CO2 uptake is significantly less pronounced in Ru/Ca3Al2O6–CaO which can be attributed to a reduction in the extent of sintering due to the presence of the stabilizer Ca3Al2O6.
Changes in the porosity of the materials with cyclic operation were monitored by N2 adsorption. The pore size distribution of the freshly calcined materials and materials that have been exposed to 10 cycles of carbonation and regeneration are plotted in Fig. 3c (values given in Table S2†). In Ru/lime and Ru/CaO the pore volume in pores with a diameter <100 nm was reduced appreciably over 10 cycles of carbonation and regeneration, viz. from 0.11 cm3 g−1 to 0.01 cm3 g−1 and from 0.19 cm3 g−1 to 0.05 cm3 g−1, respectively. On the other hand, Ru/Ca3Al2O6–CaO retains to a larger extend its pore volume (0.23 cm3 g−1 after 10 cycles of carbonation and regeneration compared to an initial value of 0.26 cm3 g−1). The reduction in pore volume (dpore < 100 nm), highlighted in gray in Fig. 3c, agrees well with the trend observed for the decay of the CO2 uptake in the kinetically controlled regime (Fig. S5†). This observation is in line with the hypothesis that the CO2 uptake in the kinetically-controlled regime is linked directly to the pore volume available in pores with dpore < 100 nm.48,49
3.4. SE-SMR performance: first cycle
In the following the SE-SMR activity of the materials synthesized was assessed at 550 °C using a feed composition of CH4:H2O:N2 = 1:4:9 (total flow rate 14 ml min−1; GHSV = 8.4 L gcat−1 h−1). Fig. 4 summarizes the results of the Ru containing materials, whereas the activity of the benchmark Ca–Ni-ex-Htlc is plotted in Fig. S6a.†Fig. 4a plots the SE-SMR performance of Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO (N2-free off-gas composition on a dry basis) in the 1st cycle. Generally, the SE-SMR reaction can be divided into three reaction stages. In the so-called pre-breakthrough stage (labelled i, Fig. 4a), high-purity H2 is produced. This stage is followed by a breakthrough stage (labelled ii) in which there is some CH4 slip. In addition, also CO2 and CO are detected in the effluent gas. In the post-breakthrough stage (labelled iii), the concentrations of CH4, CO, CO2 and H2 approach the equilibrium concentration of the conventional SMR, as the CO2 sorbent has been converted by a large extent to CaCO3. The exception from this reaction pattern is Ru/lime which shows a continuously increasing concentration of CH4, without any clear pre-breakthrough stage, indicative of a very poor SMR activity of this material. This is most likely due to the large Ru particle size and hence the low dispersion of Ru in this material (Table 1). On the other hand, Ru/CaO and Ru/Ca3Al2O6–CaO, show clearly all three reaction stages, indicative of a high catalytic activity of these two materials. For Ru/CaO, the mole fraction of CO2, CO and CH4 in the pre-breakthrough stage (t = 0–83 min for the 1st cycle) are very low (<0.03) yielding a H2 mole fraction of ≈0.97, which is close to the thermodynamic equilibrium (0.99) of the SE-SMR, and significantly higher than the thermodynamic equilibrium of the conventional SMR (0.75). This result implies that the CO2 that is produced during the simultaneous SMR and WGS reactions is immediately captured by CaO, i.e., CaO + CO2 → CaCO3. In the post-breakthrough stage, the mole fraction of CO2 in the off gases increases (0.18) while the mole fraction of H2 decreases (0.67). The three characteristic stages of the SE-SMR are also observed for Ru/Ca3Al2O6–CaO, yet the duration of the pre-breakthrough period is slightly shorter (t = 0–70 min) than for Ru/CaO. The most likely explanation for this observation is the lower content of CaO in this material due to the formation of CO2-capture-inactive Ca3Al2O6 (the CO2 uptake of Ru/CaO and Ru/Ca3Al2O6–CaO in the first cycle was 0.57 gCO2/g and 0.54 gCO2/g, respectively). The benchmark Ca–Ni-ex-Htlc has a significantly shorter pre-breakthrough period (t = 0–13 min) as this material contains 45 wt% Ni and only 21 wt% CaO (Fig. S6a†), hence possessing a lower CO2 uptake capacity.
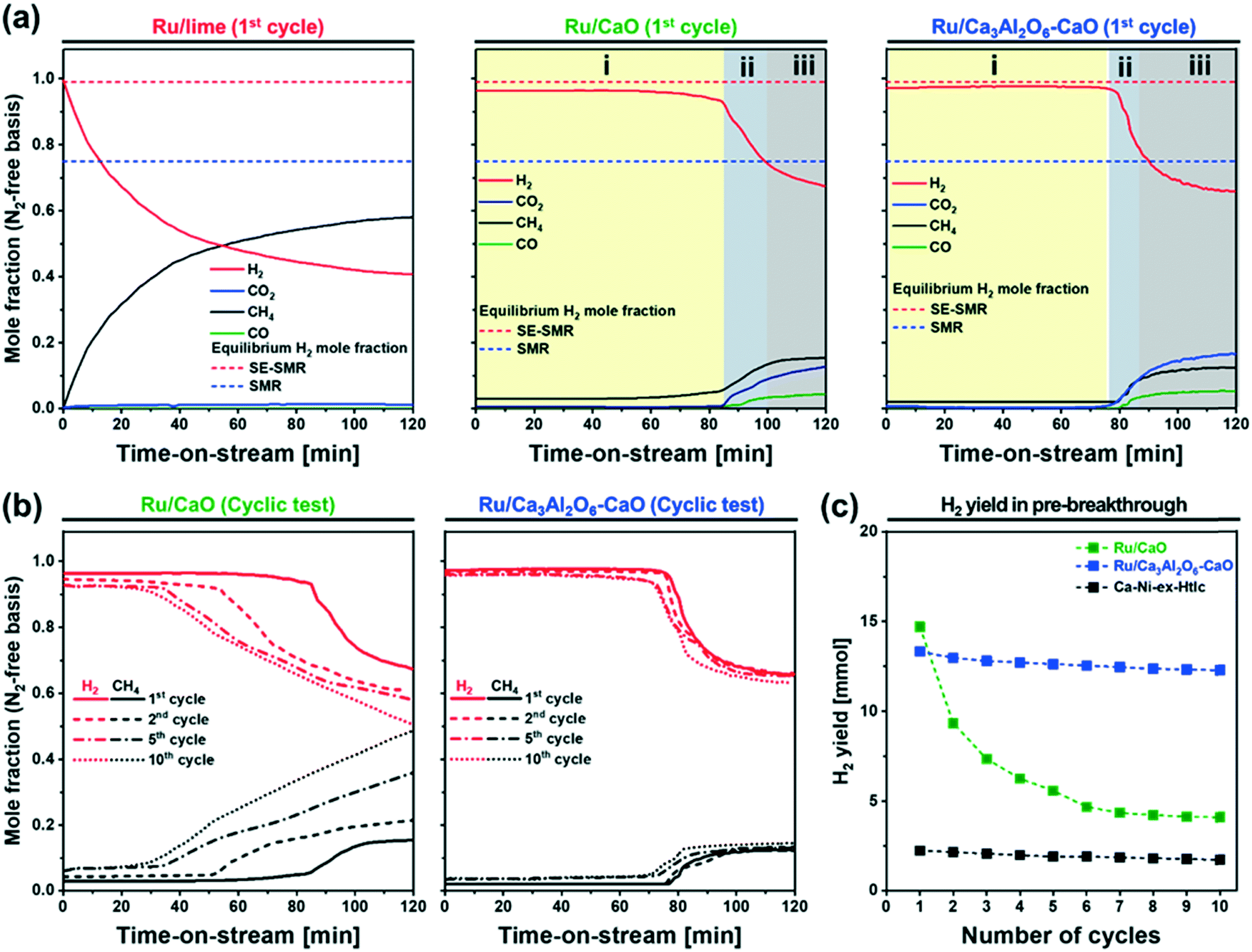 | ||
| Fig. 4 SE-SMR performance: off-gas composition for Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO in the 1st cycle. Three reaction stages can be identified in the SE-SMR step: (i) pre-breakthrough, (ii) breakthrough, and (iii) post-breakthrough stage. (b) Breakthrough curves of H2 and CH4 for Ru/CaO and Ru/Ca3Al2O6–CaO in the 1st, 5th and 10th cycles. (c) The H2 yield as a function of number of repeated SE-SMR–regeneration cycles. | ||
3.5. SE-SMR performance: cyclic performance
Fig. 4b and S6b† plot the SE-SMR performance of Ru/CaO, Ru/Ca3Al2O6–CaO and Ca–Ni-ex-Htlc as a function of cycle number (1, 2, 5 and 10 cycles). Ru/CaO shows a decreasing H2 purity in the pre-breakthrough stage over 10 cycles (decrease from 96 to 92 mole%) and a continuous increase of the concentration of CH4 at the end of post-breakthrough stage (increase from 15 to 49 mole% over 10 cycles). Moreover, for Ru/CaO the time on stream when breakthrough occurs decreases with cycle number from 83 min (1st cycle) to 53 min, 33 min and 25 min in the 2nd, 5th and 10th cycles, respectively. On the other hand, a higher cyclic stability, i.e. a reduced decline in the duration of the pre-breakthrough period with cycle number, is observed for Ru/Ca3Al2O6–CaO. After 10 cycles, Ru/Ca3Al2O6–CaO and Ca–Ni-ex-Htlc retained, respectively, 92% and 76%, of the duration of the initial pre-breakthrough period compared to 29% for Ru/CaO.Two reasons could lead to a deactivation of the materials with cycle number viz. catalyst deactivation or a reduction in the CO2 uptake capacity of the sorbent (or a combination of both effects). To obtain further insight into the cause(s) of material deactivation and to explain the improved stability of Ru/Ca3Al2O6–CaO, we investigate: 1) whether there exists a correlation between the SE-SMR performance and changes in the CO2 uptake capacity of the materials with cycle number, 2) the catalytic stability far from equilibrium conversion, 3) changes in the morphology of the materials and 4) the quantity of coke deposited.
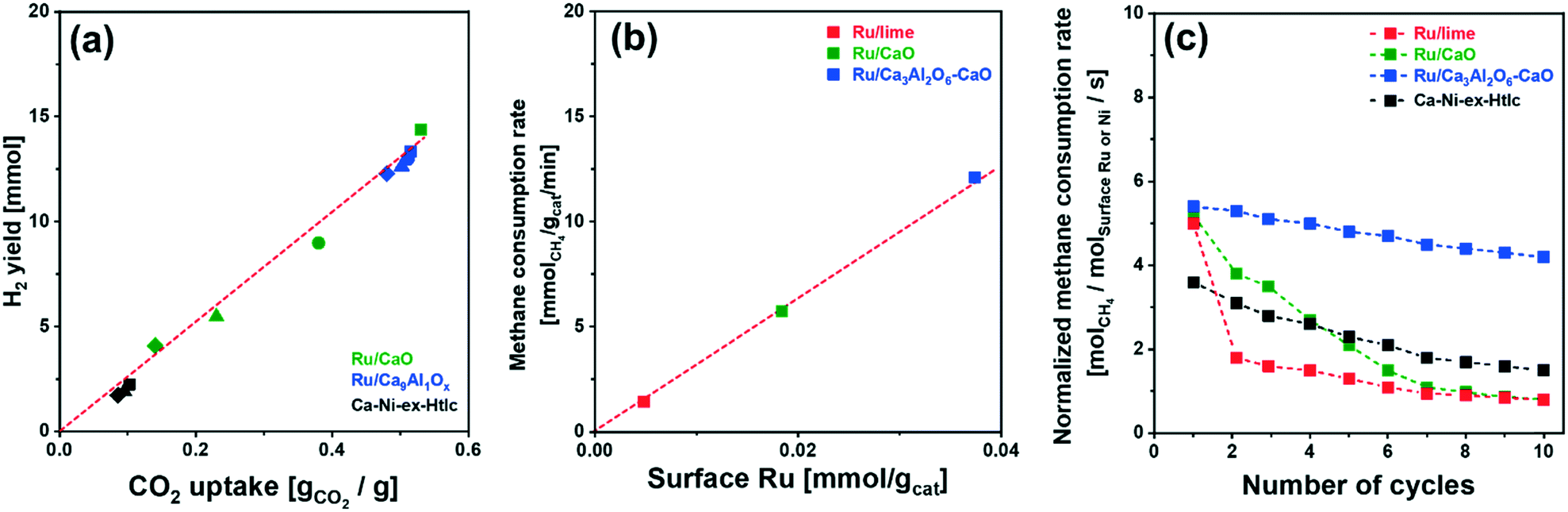 | ||
| Fig. 5 H2 yield and CH4 consumption rate: (a) H2 yield in the pre-breakthrough period as a function of the CO2 uptake for the materials studied at different cycle numbers: (■) 1st, (●) 2nd, (▲) 5th and (◆) 10th cycle (reported in Fig. S7†); (b) the rate of CH4 consumption (after 1 h of TOS) as a function of the quantity of surface Ru. (c) Rate of CH4 consumption, normalized by surface Ru or Ni, in the post-breakthrough stage of the SE-SMR reaction at 550 °C (total flow rate of 140 ml min−1 with a composition of 10 CH4/40 H2O/90 N2; GHSV = 84.0 L gcat−1 h−1). | ||
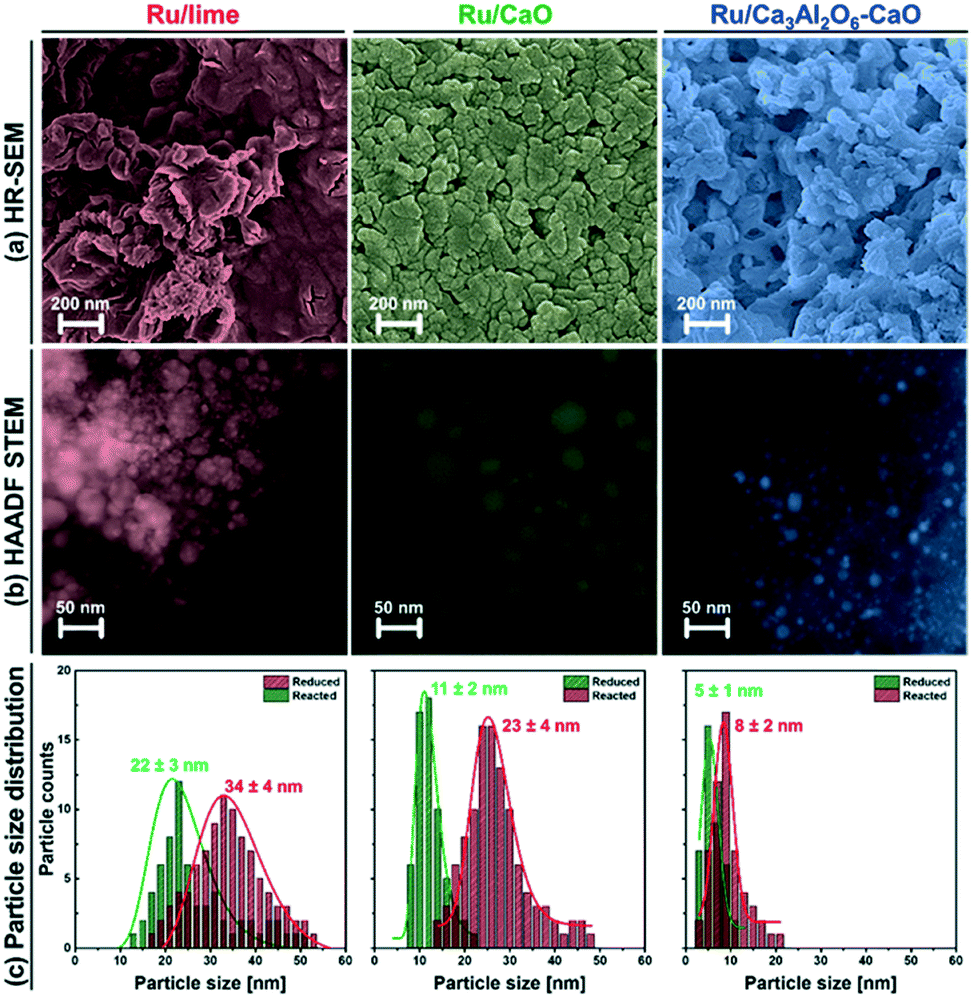 | ||
| Fig. 6 Electron microscopy-based characterization of the spent bi-functional materials: (a) HR-SEM, (b) HAADF-STEM and (c) particle size distribution of Ru/lime, Ru/CaO and Ru/Ca3Al2O6–CaO after exposure to 10 SE-SMR/regeneration cycles. | ||
Turning to the growth of the Ru nanoparticles, analysis of HAADF STEM images (freshly reduced, Fig. 2, and spent materials, Fig. 6b) reveals an increase in the size of the Ru particle size from 5.3 ± 1.3 nm to 8.4 ± 2.0 nm for Ru/Ca3Al2O6–CaO after 10 cycles (Fig. 6c). However, this particle size is still 2.7 and 4.0 times smaller than that of spent Ru/CaO (22.5 ± 4.2 nm) and Ru/lime (33.5 ± 3.6 nm), respectively. The trend in the Ru particle size as determined by HAADD STEM is in good agreement with the trend in surface Ru as quantified by H2 chemisorption (Table S3†): Ru/Ca3Al2O6–CaO (31.5 μmolRu gcat−1) > Ru/CaO (7.2 μmolRu gcat−1) > Ru/lime (2.1 μmolRu gcat−1). These measurements provide strong evidence that the decrease in the rate of methane consumption is due to the sintering of Ru particles.
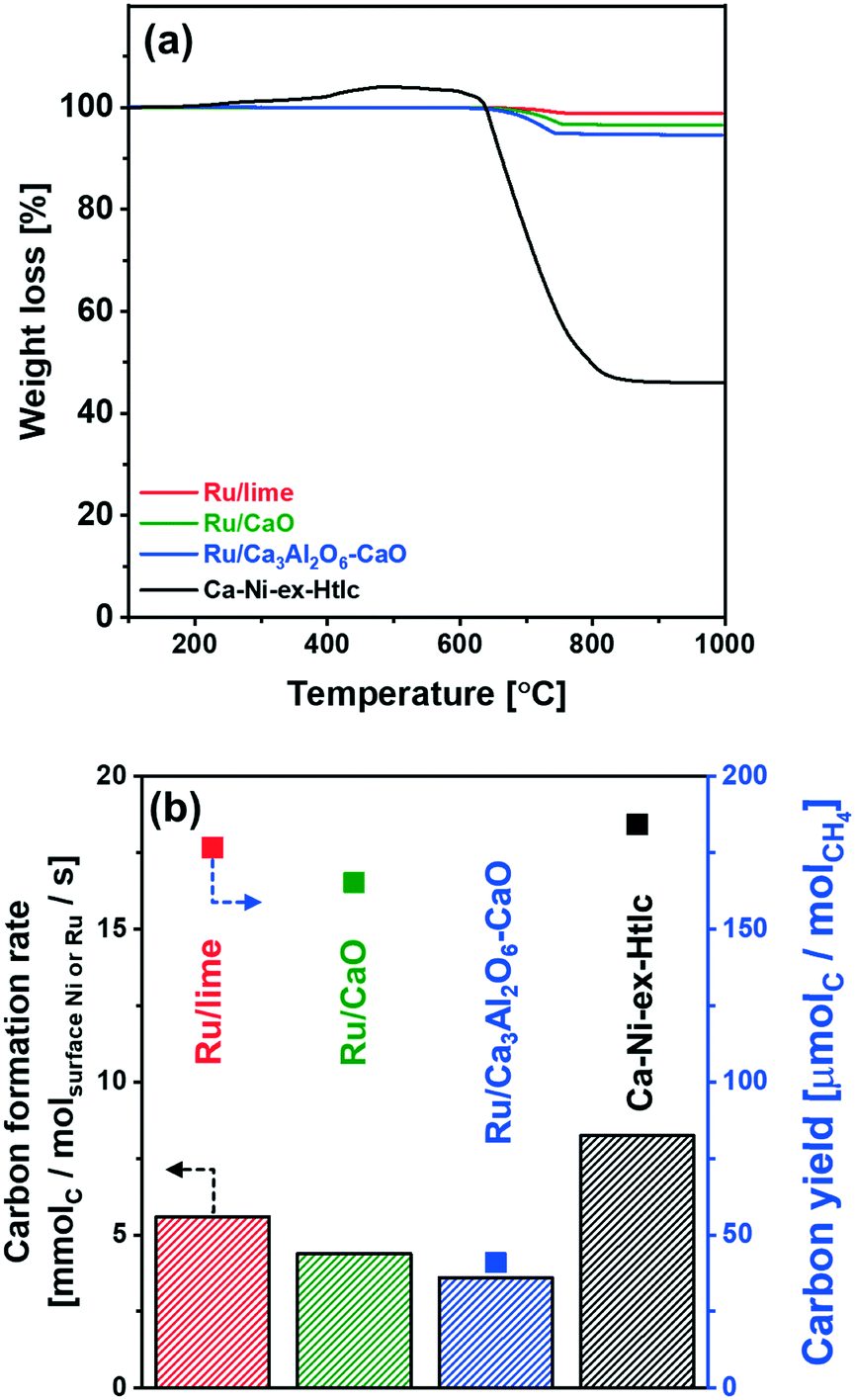 | ||
| Fig. 7 Coke formation: (a) TPO analysis of the spent materials in a TGA. Weight loss after TPO treatment is given in parenthesis. (b) Average rate of carbon deposition normalized by surface Ru or Ni, and carbon yield determined as mole of accumulated carbon per mole of reacted CH4 over 10 SE-SMR/regeneration cycles. | ||
4. Conclusions
We have developed a highly active and stable, Ru/Ca3Al2O6–CaO bi-functional SE-SMR material. XRD and XAS measurements of the as-synthesized (calcined) materials showed the formation of mixed oxides between CaO and the stabilizer, viz. Ca3Al2O6 and the catalyst, i.e. CaRuO3. In situ XAS measurements complemented by H2-TPR and XRD revealed the formation of metallic Ru upon reduction. Owing to the high activity of Ru for the SE-SMR, the fraction of CO2-capture-active CaO could be increased significantly when compared to the benchmark Ca–Ni-ex-Htlc, extending the pre-breakthrough duration and the yield of high-purity H2. The presence of Ca3Al2O6 in the material stabilized effectively the material against sintering. Furthermore, Ru/Ca3Al2O6–CaO showed very little coke formation when compared to Ca–Ni-ex-Htlc. Nonetheless, the high costs of Ru are a disadvantage and further work shall concentrate on reducing further the quantity of Ru in the material while maintaining its high activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of ETH (ETH 57 12-2) and the Swiss National Science Foundation (200020_156015). The benchmark Ca–Ni-ex-Htlc was developed by Marcin Broda. We also thank the Scientific Center for Optic and Electron Microscopy (ScopeM) at ETH Zürich for providing access to electron microscopes and the Swiss Norwegian Beamline for access and help during XAS measurements. We thank Agnieszka Kierzkowska for ICP-OES measurements and Wouter van Beek for assistance during XAS measurements.References
- J. M. Ogden, M. M. Steinbugler and T. G. Kreutz, J. Power Sources, 1999, 79, 143–168 CrossRef CAS.
- A. Midilli, M. Ay, I. Dincer and M. A. Rosen, Renewable Sustainable Energy Rev., 2005, 9, 255–271 CrossRef CAS.
- R. Ramachandran and R. K. Menon, Int. J. Hydrogen Energy, 1998, 23, 593–598 CrossRef CAS.
- K. S. Santhanam, R. J. Press, M. J. Miri, A. V. Bailey and G. A. Takacs, Introduction to hydrogen technology, John Wiley & Sons, Hoboken, NJ, 2017 Search PubMed.
- R. Soltani, M. A. Rosen and I. Dincer, Int. J. Hydrogen Energy, 2014, 39, 20266–20275 CrossRef CAS.
- M. J. Tuinier, H. P. Hamers and M. van Sint Annaland, Int. J. Greenhouse Gas Control, 2011, 5, 1559–1565 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 1993, 18, 305–324 CrossRef.
- J. C. Meerman, E. S. Hamborg, T. van Keulen, A. Ramírez, W. C. Turkenburg and A. P. C. Faaij, Int. J. Greenhouse Gas Control, 2012, 9, 160–171 CrossRef CAS.
- A. Lopez Ortiz and D. P. Harrison, Ind. Eng. Chem. Res., 2001, 40, 5102–5109 CrossRef.
- J. A. Ober, Mineral commodity summaries 2016, USGS, Reston, VA, 2016 Search PubMed.
- D. Alvarez and J. C. Abanades, Energy Fuels, 2005, 19, 270–278 CrossRef CAS.
- P. Sun, J. R. Grace, C. J. Lim and E. J. Anthony, AIChE J., 2007, 53, 2432–2442 CrossRef CAS.
- S. M. Kim, W.-C. Liao, A. M. Kierzkowska, T. Margossian, D. Hosseini, S. Yoon, M. Broda, C. Copéret and C. R. Müller, Chem. Mater., 2018, 30, 1344–1352 CrossRef CAS.
- A. Armutlulu, M. A. Naeem, H.-J. Liu, S. M. Kim, A. Kierzkowska, A. Fedorov and C. R. Müller, Adv. Mater., 2017, 29, 1702896 CrossRef PubMed.
- M. Broda, A. M. Kierzkowska and C. R. Müller, Adv. Funct. Mater., 2014, 24, 5753–5761 CrossRef CAS.
- S. M. Kim, A. M. Kierzkowska, M. Broda and C. R. Müller, Energy Procedia, 2017, 114, 220–229 CrossRef CAS.
- D. Alvarez and J. C. Abanades, Ind. Eng. Chem. Res., 2005, 44, 5608–5615 CrossRef CAS.
- A. Iulianelli, S. Liguori, J. Wilcox and A. Basile, Catal. Rev.: Sci. Eng., 2016, 58, 1–35 CrossRef CAS.
- M. S. Yancheshmeh, H. R. Radfarnia and M. C. Iliuta, Chem. Eng. J., 2016, 283, 420–444 CrossRef.
- B. Dou, C. Wang, Y. Song, H. Chen, B. Jiang, M. Yang and Y. Xu, Renewable Sustainable Energy Rev., 2016, 53, 536–546 CrossRef CAS.
- G. Vanga, D. M. Gattia, S. Stendardo and S. Scaccia, Ceram. Int., 2019, 45, 7594–7605 CrossRef CAS.
- H. Z. Feng, P. Q. Lan and S. F. Wu, Int. J. Hydrogen Energy, 2012, 37, 14161–14166 CrossRef CAS.
- G. Wu, C. Zhang, S. Li, Z. Huang, S. Yan, S. Wang, X. Ma and J. Gong, Energy Environ. Sci., 2012, 5, 8942–8949 RSC.
- M. Broda, A. M. Kierzkowska, D. Baudouin, Q. Imtiaz, C. Coperet and C. R. Müller, ACS Catal., 2012, 2, 1635–1646 CrossRef CAS.
- C. Zhao, Z. Zhou, Z. Cheng and X. Fang, Appl. Catal., B, 2016, 196, 16–26 CrossRef CAS.
- C. Dang, H. Yu, H. Wang, F. Peng and Y. Yang, Chem. Eng. J., 2016, 286, 329–338 CrossRef CAS.
- A. Di Giuliano, J. Girr, R. Massacesi, K. Gallucci and C. Courson, Int. J. Hydrogen Energy, 2017, 42, 13661–13680 CrossRef CAS.
- A. L. García-Lario, G. S. Grasa and R. Murillo, Chem. Eng. J., 2015, 264, 697–705 CrossRef.
- M. R. Cesário, B. S. Barros, C. Courson, D. M. A. Melo and A. Kiennemann, Fuel Process. Technol., 2015, 131, 247–253 CrossRef.
- H. R. Radfarnia and M. C. Iliuta, Chem. Eng. Sci., 2014, 109, 212–219 CrossRef CAS.
- P. Xu, Z. Zhou, C. Zhao and Z. Cheng, Catal. Today, 2016, 259, 347–353 CrossRef CAS.
- K. D. Dewoolkar and P. D. Vaidya, Energy Fuels, 2015, 29, 3870–3878 CrossRef CAS.
- C. Dang, Y. Li, S. M. Yusuf, Y. Cao, H. Wang, H. Yu, F. Peng and F. Li, Energy Environ. Sci., 2018, 11, 660–668 RSC.
- US Pat., US3330697A, 1967 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- R. A. Dalla Betta, J. Catal., 1974, 34, 57–60 CrossRef CAS.
- J. G. Goodwin Jr, J. Catal., 1981, 68, 227–232 CrossRef.
- H. Kubicka, React. Kinet. Catal. Lett., 1976, 5, 223–228 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- P. Scherrer, Nachr. Ges. Wiss. Goettingen, Math.-Phys. Kl., 1918, 98–100 Search PubMed.
- Q. Zhou, B. J. Kennedy, Z. Zhang, L.-Y. Jang and J. B. Aitken, Chem. Mater., 2009, 21, 4203–4209 CrossRef CAS.
- P. G. J. Koopman, A. P. G. Kieboom and H. van Bekkum, React. Kinet. Catal. Lett., 1978, 8, 389–393 CrossRef CAS.
- P. Betancourt, A. Rives, R. Hubaut, C. E. Scott and J. Goldwasser, Appl. Catal., A, 1998, 170, 307–314 CrossRef CAS.
- M. R. Goldwasser, M. E. Rivas, E. Pietri, M. J. Pérez-Zurita, M. L. Cubeiro, L. Gingembre, L. Leclercq and G. Leclercq, Appl. Catal., A, 2003, 255, 45–57 CrossRef CAS.
- J. C. Abanades and D. Alvarez, Energy Fuels, 2003, 17, 308–315 CrossRef CAS.
- R. Barker, J. Appl. Chem. Biotechnol., 1973, 23, 733–742 CrossRef CAS.
- J. S. Dennis and R. Pacciani, Chem. Eng. Sci., 2009, 64, 2147–2157 CrossRef CAS.
- R. Pacciani, C. R. Müller, J. F. Davidson, J. S. Dennis and A. N. Hayhurst, Can. J. Chem. Eng., 2008, 86, 356–366 CrossRef CAS.
- J. R. Rostrupnielsen and J. H. B. Hansen, J. Catal., 1993, 144, 38–49 CrossRef CAS.
- S. R. Challa, A. T. Delariva, T. W. Hansen, S. Helveg, J. Sehested, P. L. Hansen, F. Garzon and A. K. Datye, J. Am. Chem. Soc., 2011, 133, 20672–20675 CrossRef CAS PubMed.
- J.-H. Kim, D. J. Suh, T.-J. Park and K.-L. Kim, Appl. Catal., A, 2000, 197, 191–200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01095e |
| This journal is © The Royal Society of Chemistry 2019 |