
In situ DRIFT spectroscopy insights into the reaction mechanism of CO and toluene co-oxidation over Pt-based catalysts†

Abstract
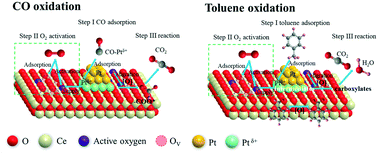
Considering that the mutual inhibition between CO and hydrocarbon (HC) co-oxidation over platinum-group metal (PGM) catalysts is a universal problem, in this study, a series of Pt-supported catalysts (Pt–Al2O3, Pt–Co3O4 and Pt–CeO2) were synthesized by combining glycol reduction and electrostatic chemical adsorption, and then were evaluated for the catalytic oxidation of CO and toluene. The Pt–CeO2 catalyst exhibited the best catalytic performance for individual CO and toluene oxidation, and effectively slowed down the competitive reaction in the presence of both CO and toluene. This superior performance over the Pt–CeO2 catalyst was associated with the formation of strong metal–support interactions (SMSIs) between the surface oxygen and adjacent Pt species. The formation of SMSIs not only enhanced the activation and migration of oxygen species and the formation of surface oxygen vacancies, but also improved the lower temperature reducibility of the catalyst. The in situ DRIFTS spectra revealed that CO was first transformed into carbon-related species (bicarbonates and carbonates), and then completely decomposed into CO2. Moreover, the reaction route for toluene oxidation may follow only one successive step: benzyl radical → benzaldehyde → benzoate → formate species, and finally completely oxidized to CO2 and H2O. The reaction pathways between CO and toluene oxidation may be independent when CO and toluene co-exist, although their reaction rates slow down due to the competitive adsorption between CO and toluene molecules at the same sites.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

