
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCollective action of water molecules in zeolite dealumination†
Malte
Nielsen
a,
Anders
Hafreager
b,
Rasmus Yding
Brogaard
 *ac,
Kristof
De Wispelaere
d,
Hanne
Falsig
c,
Pablo
Beato
c,
Veronique
Van Speybroeck
*d and
Stian
Svelle
*a
*ac,
Kristof
De Wispelaere
d,
Hanne
Falsig
c,
Pablo
Beato
c,
Veronique
Van Speybroeck
*d and
Stian
Svelle
*a
aCenter for Materials Science and Nanotechnology (SMN), Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N-0315 Oslo, Norway. E-mail: stian.svelle@kjemi.uio.no
bDepartment of Physics, University of Oslo, P.O. Box 1033, Blindern, N-0315 Oslo, Norway
cHaldor Topsøe A/S, Haldor Topsøes Allé 1, 2800 Kgs. Lyngby, Denmark. E-mail: rabr@topsoe.com
dCenter for Molecular Modeling (CMM), Ghent University, Tech Lane Ghent Science Park Campus A, Technologiepark 46, 9052 Zwijnaarde, Belgium. E-mail: veronique.vanspeybroeck@ugent.be
First published on 21st June 2019
Abstract
When exposed to steam, zeolite catalysts are irreversibly deactivated by loss of acidity and framework degradation caused by dealumination. Steaming typically occurs at elevated temperatures, making it challenging to investigate the mechanism with most approaches. Herein, we follow the dynamics of zeolite dealumination in situ, in the presence of a realistic loading of water molecules by means of enhanced sampling molecular dynamics simulations. H-SSZ-13 zeolite is chosen as a target system. Monte Carlo simulations predict a loading of more than 3 water molecules per unit cell at representative steaming conditions (450 °C, 1 bar steam). Our results show that a higher water loading lowers the free energy barrier of dealumination, as water molecules cooperate to facilitate hydrolysis of Al–O bonds. We find free energies of activation for dealumination that agree well with the available experimental measurements. Clearly, the use of enhanced sampling molecular dynamics yields a major step forward in the molecular level understanding of the dealumination; insight which is very hard to derive experimentally.
Introduction
Zeolites are microporous crystalline aluminosilicates with a wide range of catalytic applications.1 The active site within Brønsted acidic zeolites arises from substitution of a Si with an Al atom and a charge-compensating proton. These active sites are deteriorated by extraction of Al from the framework when zeolites are exposed to water vapor at elevated temperatures, known as dealumination. In this work, we use enhanced sampling molecular dynamics simulations to follow this process in situ and show how multiple water molecules cooperate to hydrolyze Al–O bonds in the framework. Our work hence constitutes a significant step forward in the molecular-level understanding of an important process, which it is virtually impossible to follow in comparable detail experimentally.Dealumination by steaming occurs frequently in zeolite catalysis, be it intentional or not. In many catalytic applications of zeolites, non-reactive species, also known as ‘coke’, accumulate on and within the zeolite, reducing activity.2–5 The catalyst is regenerated by burning the coke. Because water is formed in this combustion process, the zeolite catalyst is exposed to steam at high temperature (e.g. 650–760 °C for regeneration during fluid catalytic cracking).6 This leads to irreversible dealumination of the zeolite with extraction of Al from the zeolite framework, which permanently reduces the activity of the catalyst.7 Zeolite catalysts are also subjected to steam during reaction. In the Mobil methanol to gasoline (MTG) process, the feedstock converted over the ZSM-5 zeolite catalyst has 17% initial water concentration, which will increase with conversion. The MTG reactor inlet temperature is 360 °C, the outlet temperature is 415 °C.8 Dealumination is also carried out on purpose to increase Si/Al ratio and change the mesopore structure of zeolites, such as zeolite Y, the cracking catalyst.9–11 This is typically carried out at temperatures around 500 °C and steam pressure of 2 atm during 2 hours.12
It is experimentally challenging to trace zeolite dealumination in situ because of the severe process conditions, and the majority of the reports are qualitative and descriptive rather than quantitative. Moreover, all quantitative experimental work on dealumination has been carried out for only one zeolite, i.e. ZSM-5. Sano et al.13,14 have reported an estimated rate of dealumination in the temperature range 600–800 °C. A second or third order dependency of the rate on the initial Al concentration was found. The reaction order with respect to the partial pressure of steam varied from 1.5 to 2, and the activation energy for dealumination was found to be 140 kJ mol−1. Interestingly, the rate of dealumination was seen to increase with an increasing concentration of silanol defects for partial pressures of steam below 10 kPa.13,14 Masuda et al.15 measured the dealumination rate and found the process to be 1.5 in order with respect to steam. Finally, Ong et al.16 found that the dealumination process obeyed first order kinetics with respect to Al concentration, and a rate constant for dealumination was reported.
Theoretical calculations can potentially follow the mechanism of the dealumination process at the molecular scale, provided they account properly for the operating conditions such as realistic account of water molecules and true temperatures of the dealumination process. Based on previous theoretical calculations, the process is believed to occur through a sequence of hydrolysis reactions of Al–O–Si bonds as sketched in Scheme 1.17–21
 | ||
| Scheme 1 The reaction steps of zeolite dealumination employed in computational investigations of the process.17–21 | ||
Silaghi et al.19,20 have calculated the energy barrier for hydrolysis of the first Al–O bond for a number of zeolite framework types (MOR, FAU, MFI, CHA). A Brønsted–Evans–Polanyi-relationship linking the heat of water adsorption to the hydrolysis activation energy was established for these frameworks.20 However, these relations only strictly hold for the first hydrolysis step and are of decreasing quality for the later steps.19 This is critical, as we have shown with microkinetic modelling based on static DFT calculations that the later steps are likely rate-determining.18 This demonstrates the need for modelling beyond the BEP relations. The work in ref. 18 which was based on static DFT calculations obtained a very good correspondence with the experimental results mentioned above13–16 for the overall free energy barrier and thus rate of dealumination, as well as a reasonable agreement for the reaction order with respect to water. However, such agreement could only be obtained when using a corrected thermodynamic state of reference for water adsorbed inside the zeolite instead of the conventional gas phase reference state.18 This reference state was also used in very recent work of Stanciakova et al.,21 who did an extensive investigation of mechanisms for hydrolysis of the first Al–O bond for different Al sites in ZSM-5 zeolite including the cooperative effect of two water molecules. All these theoretical studies report that all stationary points along the dealumination reaction path are higher in free energy than the intact zeolite framework. Such a sequence of elusive intermediates sheds light on why it is difficult to investigate the reaction path with experimental methods. In such a case, theoretical calculations can provide a true added value to unravel the subsequent reaction steps. However, so far all DFT studies of zeolite dealumination have employed a static approach, in which structures are located as minima in the potential energy landscape. Free energies were subsequently derived from nuclear vibrations in the harmonic approximation, which has obvious and severe limitations,22 in particular at the high temperatures employed in dealumination. In such an approach, the configurational mobility of water and the role of surrounding water molecules are not properly accounted for.18 Having multiple water molecules in the system introduces a plethora of local minima on the potential energy surface. Hence, the static DFT approach in practice does not allow adequate sampling of all relevant states when taking the collective action of multiple water molecules into account – an action that we will demonstrate is critical in the dealumination process.
Results and discussion
In this contribution, we take a major step forward in the mechanistic understanding of zeolite dealumination by using DFT-based molecular dynamics (DFT-MD) simulations and enhanced sampling methods to investigate the reaction steps (Sections S1 and S2†). The calculations target H-SSZ-13 zeolite (36 T-atom cell) as a representative model system at conditions of T = 450 °C and atmospheric steam pressure, similar to earlier experimental and theoretical work.16,18 We chart the entire free energy landscape for complete dealumination, starting from an initial state with a realistic loading of multiple water molecules in the zeolite pore system. This essentially corresponds to following the dealumination process in situ at realistic conditions.The water loading in the zeolite at realistic operating conditions (T = 450 °C and atmospheric steam pressure) is determined using grand canonical Monte Carlo (GCMC, Section S3†). Using the ReaxFF force field23,24 the GCMC simulation constitutes a realistic model of the equilibrium between water vapor and water adsorbed in the zeolite pores, including specific interactions with the Brønsted acid sites. We reach equilibrium at 3–3.5 water molecules per unit cell. This result demonstrates the need to include multiple water molecules in realistic simulations of the zeolite steaming process.
To simulate the Al–O hydrolysis reactions using DFT-MD, enhanced sampling techniques are necessary to explore higher free energy regions. Herein, the umbrella sampling methodology was used,25 as it has proven a robust and successful method in other studies.26–28 The technique relies on the definition of some degrees of freedom, called collective variables (CVs), along which the sampling is enhanced. It should be noted that it is not trivial to define CVs that facilitate sampling of the reaction pathway. However, as discussed in detail in Section S4,† we were able to obtain free energy profiles for dealumination steps along a single CV. The CV was chosen as the coordination number from all hydrogen atoms in water molecules (indicated in blue in Scheme 2) to the framework oxygen atoms in Al–O–Si bonds (indicated in red in Scheme 2, Section S4†). This collective variable captures hydrolysis of Al–O bonds by transfer of a proton from a water molecule to create a framework silanol group, leaving a hydroxy group bound to Al. This CV is not the only one possible. However, it is simple and does not introduce excessive bias in the system: the CV involves only coordination between water hydrogens and zeolite oxygens, but still leads to a smooth reaction pathway passing through a transition state with simultaneous breaking of the H–O bond of a water molecule and formation of the Al–O bond to the framework (see below).
 | ||
| Scheme 2 Representation of the collective variable applied in the simulation of the hydrolysis of the first Al–O bond. The variable is defined as the coordination number (CN) derived from the distances ri between the framework oxygen atoms in red and the hydrogen atoms of water in blue. | ||
In the previously suggested dealumination pathways,17–19 the reaction follows four hydrolysis steps (Scheme 1). The first three steps each hydrolyze one Al–O–Si bond, while in the fourth, a water molecule is coordinated to the Al(OH)3 species, replacing the weak Al–OH–Si bond to the framework. Using DFT-MD, we find that the aluminum species is able to move freely after hydrolyzing the three Al–O–Si bonds, and thus that the fourth step occurs spontaneously (see below). This agrees with our previous static calculations, where the fourth step was found to be nearly barrierless.18
In order to specifically investigate the influence of surrounding water on the dealumination mechanism, two sets of simulations have been performed. In the first case, only the water molecule necessary for the hydrolysis step was considered. This means that one water molecule was added in each reaction step of the simulation series. This approach of adding water molecules is identical to the static calculations performed in literature and hence facilitates comparison between this and our previous work (Section S5†).18 In the second set of simulations four water molecules were initially present in the unit cell, a loading close to the estimated realistic value determined from GCMC simulations (see above).
The second set of simulations allows us to investigate the impact of a realistic water loading on the whole reaction pathway. It further allows us to construct a free energy profile for the full dealumination path by connecting free energy profiles of subsequent elementary steps, as the number of atoms is kept constant throughout the entire simulation series. The profile is shown in Fig. 1 together with the intrinsic barriers obtained in the single-water simulations and structural snapshots of transition states (Section S6† includes a quantitative discussion of structural parameters). As a general trait, the reacting water is adsorbed on Al in the anti position to the Brønsted acid site, paralleling the mechanism proposed by Silaghi et al.19,20 Note that as water molecules are consumed along the multi-water path, the CVs for each elementary step are not mathematically identical (see definition above), although they chemically describe the same reactions (Section S4†). This means that we cannot formally guarantee that the samplings of product and reactant state of subsequent steps match exactly. This translates to an uncertainty in the relative free energies of states not directly connected by an elementary step. More research is needed to address this issue, research that is clearly outside the scope of the current work. However, the overall free energy of activation derived from the multi-water simulation series (226 kJ mol−1, Fig. 1) in fact agrees well with experiments (246 kJ mol−1 can be calculated from experiments16,18). This is remarkable, as the computational result now is obtained without the correction of the reference free energy of water inside the zeolite employed previously.18
 | ||
| Fig. 1 Free energy diagram of a complete dealumination reaction (Scheme 1) in H-SSZ-13 as obtained from DFT-MD umbrella simulations. Numbers indicate intrinsic free energy barriers of elementary steps in the single-water (grey) and multi-water (blue) pathways. The latter is constructed by connecting the free energy profiles of elementary steps, aligning the reactant energy of each step with the product energy of the preceding step. The insets are snapshots of the single- and multi-water transition states. Section S7† shows the free energy profiles of all elementary steps. | ||
The obtained intrinsic barriers from the DFT-MD simulations of the single-water pathway are all close to 100 kJ mol−1 with a variation of 15 kJ mol−1. This is in contrast to our previous static DFT results that exhibited a variation of more than 90 kJ mol−1 (Table S2†). We take this as an indication of the inherent challenges of using static DFT combined with the harmonic approach at these elevated temperatures (Section S5†).
The DFT-MD results clearly show the beneficial effect of extra water in the pores of the zeolite on the intrinsic free energy barriers of the three dealumination steps. Importantly, this effect is induced by just one additional water molecule per hydrolysis step, as will be explained in the following where we address how two water molecules cooperate to lower the barriers. Key differences are observed when considering the TSs for each hydrolysis step, as shown as insets in Fig. 1. All the TSs of the single-water pathway involve strained four-membered transition states. In the multi-water pathway one additional water stabilizes the TSs in each step. In step 1 and 2 the TSs are 6-membered, with two water molecules shuttling a proton to the framework oxygen. This proton-shuttling mechanism was also found for the first hydrolysis step in ZSM-5 zeolite using static DFT.21 The mechanism reflects a delicate balance between the entropy loss of fixating the additional water molecule and the enthalpy gain of relieving strain in the cyclic TS by expanding the ring size. The preference for 6-membered over 4-membered cyclic TSs is quite general.29 In step 3, the stabilization is of a different nature. As opposed to the other steps, the product state of step 3 is significantly stabilized in the multi-water pathway compared to the single-water pathway (25 kJ mol−1, Section S7†) by coordination of water to Al (Fig. 2). A significant part of this stabilization is already recovered in the TS (19 kJ mol−1, Fig. 1), where the additional water occupies the empty coordination site in the trigonal bipyramidal coordination of Al (compare snapshots of TS of single- and multi-water pathways in Fig. 1). The additionally coordinated water molecule makes a second type of TS without typical 4-ring strain accessible (Fig. 2). In the product state of step 3, the Al species leaves the framework (Fig. 2), showing that step 4 occurs spontaneously.
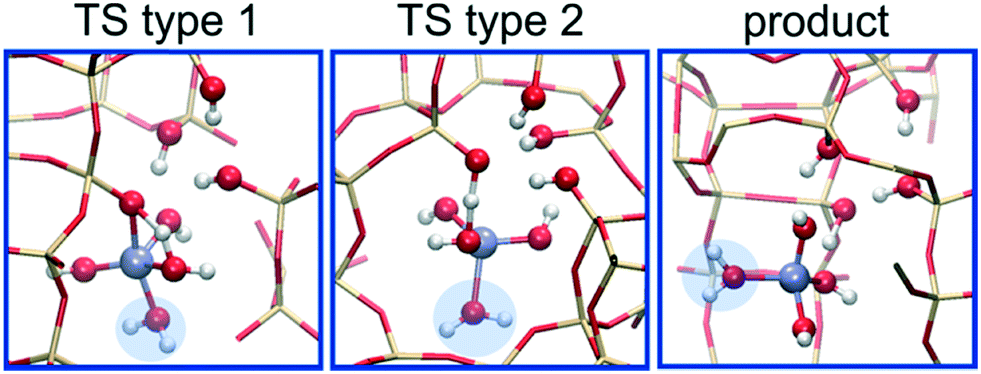 | ||
| Fig. 2 Snapshots of two configurations (TS type 1 and 2) sampled in the transition state region as well the product state (product) of step 3 of the dealumination process (Scheme 1) in the multi-water pathway (Fig. 1). An additional water molecule (highlighted in blue) stabilizes both states by coordinating to Al. | ||
It thus becomes clear that water molecules directly cooperate to facilitate hydrolysis of Al–O bonds in the entire zeolite dealumination process. Note that this cooperation effect is dynamic, as it is only present close to the transition states of the reaction pathway. Such a cooperative effect of two water molecules was also found in theoretical work on the first hydrolysis step in ZSM-5.21 Further, we have confirmed that the decrease in the free energy barriers is not due to stabilization of the TSs by unspecific solvation from the surrounding water molecules (Section S8†).
Conclusions
In summary, we were for the first time able to conduct an in situ investigation of zeolite dealumination at real steaming conditions using advanced DFT-MD simulations. The results reveal that water molecules cooperate, in part by proton shuttling, to decrease the free energy barriers of the Al–O hydrolysis steps. The work hence demonstrates that the collective action of water molecules should be considered in future experimental and theoretical investigations of the kinetics of zeolite dealumination. Finally, it must be emphasized that the present DFT-MD approach with advanced sampling allows us to efficiently sample the free energy surface and alleviates the shortcomings related to anharmonicity associated with static DFT. Thus, very good quantitative agreement with the available experimental data is achieved.Methods
DFT-based molecular dynamics (DFT-MD) calculations were performed using the CP2K 2.7 program.30 The Grimme DFT-D3 dispersion corrected31,32 revPBE exchange correlation functional33 was applied using a combination of Gaussian and plane wave basis sets34,35 with kinetic energy cutoff at 350 Ry. GTH basis sets and pseudopotentials were utilized.36H-SSZ-13 zeolite was modeled with the 36 T-site CHA unit cell with one substitution of Si with Al and addition of a proton (Si/Al = 35). The equilibrium cell parameters at steaming conditions were obtained from DFT-MD simulations in the constant temperature and pressure (NPT) ensemble. These cell parameters were used in all simulations.
Enhanced free energy sampling was made using umbrella sampling37 by coupling the CP2K MD engine to the PLUMED interface (version 2.1.3).38 Free energy profiles were derived from the results of umbrella simulations using the weighted histogram analysis method (WHAM).37
Monte Carlo simulations in the grand canonical ensemble were performed with LAMMPS. The simulations used a recent ReaxFF force field, shown to have realistic water–zeolite interactions.23,24 The simulations employed 3 × 3 × 3 unit cells (2943 atoms).
Further computational details can be found in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the European Industrial Doctorates project ZeoMorph (FP7 ITN-EID, grant agreement no. 606965) and the Research Board of Ghent University (BOF). The computational resources and services used were provided by Ghent University (Stevin Supercomputer Infrastructure), the VSC (Flemish Supercomputer Center), funded by the Research Foundation – Flanders (FWO), and the Norwegian High Performance Computing program (project no. NN4683K). K. D. W. is a fellow funded by the FWO (FWO16-PDO-047). V. V. S. acknowledges funding from the ERC, consolidator grant agreement no. 647755 – DYNPOR (2015–2020).References
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- M. Guisnet, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2009, 305, 69–83 CrossRef CAS.
- M. Guisnet, J. Mol. Catal. A: Chem., 2002, 182–183, 367–382 CrossRef CAS.
- D. Rojo-Gama, S. Etemadi, E. Kirby, K. P. Lillerud, P. Beato, S. Svelle and U. Olsbye, Faraday Discuss., 2017, 197, 421–446 RSC.
- H. Schulz, Catal. Today, 2010, 154, 183–194 CrossRef CAS.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- M. Clough, J. C. Pope, L. T. Xin Lin, V. Komvokis, S. S. Pan and B. Yilmaz, Microporous Mesoporous Mater., 2017, 254, 45–58 CrossRef CAS.
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- H. Wang, L. Su, J. Zhuang, D. Tan, Y. Xu and X. Bao, J. Phys. Chem. B, 2003, 107, 12964–12972 CrossRef CAS.
- J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- L. Karwacki, D. A. M. de Winter, L. R. Aramburo, M. N. Lebbink, J. A. Post, M. R. Drury and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2011, 50, 1294–1298 CrossRef CAS PubMed.
- R. A. Beyerlein, C. Choi-Feng, J. B. Hall, B. J. Huggins and G. J. Ray, Top. Catal., 1997, 4, 27–42 CrossRef CAS.
- T. Sano, N. Yamashita, Y. Iwami, K. Takeda and Y. Kawakami, Zeolites, 1996, 16, 258–264 CrossRef CAS.
- T. Sano, H. Ikeya, T. Kasuno, Z. B. Wang, Y. Kawakami and K. Soga, Zeolites, 1997, 19, 80–86 CrossRef CAS.
- T. Masuda, Y. Fujikata, S. R. Mukai and K. Hashimoto, Appl. Catal., A, 1998, 172, 73–83 CrossRef CAS.
- L. H. Ong, M. Dömök, R. Olindo, A. C. van Veen and J. A. Lercher, Microporous Mesoporous Mater., 2012, 164, 9–20 CrossRef CAS.
- S. Malola, S. Svelle, F. L. Bleken and O. Swang, Angew. Chem., Int. Ed., 2012, 51, 652–655 CrossRef CAS PubMed.
- M. Nielsen, R. Yding Brogaard, H. Falsig, P. Beato, O. Swang and S. Svelle, ACS Catal., 2015, 5, 7131–7139 CrossRef CAS.
- M.-C. Silaghi, C. Chizallet, J. Sauer and P. Raybaud, J. Catal., 2016, 339, 242–255 CrossRef CAS.
- M.-C. Silaghi, C. Chizallet, E. Petracovschi, T. Kerber, J. Sauer and P. Raybaud, ACS Catal., 2015, 5, 11–15 CrossRef CAS.
- K. Stanciakova, B. Ensing, F. Göltl, R. E. Bulo and B. M. Weckhuysen, ACS Catal., 2019, 9, 5119–5135 CrossRef CAS.
- G. Piccini, M. Alessio and J. Sauer, Angew. Chem., Int. Ed., 2016, 55, 5235–5237 CrossRef CAS PubMed.
- A. C. T. van Duin, C. Zou, K. Joshi, V. Bryantsev and W. A. Goddard, RSC Catal. Ser., 2014, 14, 223–243 CAS.
- K. L. Joshi, G. Psofogiannakis, A. C. T. van Duin and S. Raman, Phys. Chem. Chem. Phys., 2014, 34, 18433–18441 RSC.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- B. Ensing, A. Laio, F. L. Gervasio, M. Parrinello and M. L. Klein, J. Am. Chem. Soc., 2004, 126, 9492–9493 CrossRef CAS PubMed.
- B. Ensing and M. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6755–6759 CrossRef CAS PubMed.
- J. Hajek, C. Caratelli, R. Demuynck, K. De Wispelaere, L. Vanduyfhuys, M. Waroquier and V. Van Speybroeck, Chem. Sci., 2018, 9, 2723–2732 RSC.
- K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, WIREs Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- G. Lippert, J. Hutter and M. Parrinello, Theor. Chem. Acc., 1999, 103, 124–140 Search PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy00624a |
| This journal is © The Royal Society of Chemistry 2019 |