Coupling non-thermal plasma with Ni catalysts supported on BETA zeolite for catalytic CO2 methanation†
Received
26th March 2019
, Accepted 7th July 2019
First published on 8th July 2019
Abstract
Catalytic carbon dioxide (CO2) methanation is a promising and effective process for CO2 utilisation and the production of CH4 as an alternative to using natural gas. Non-thermal plasma (NTP) activation has been proven to be highly effective in overcoming the thermodynamic limitation of reactions under mild conditions and intensifying the CO2 hydrogenation process greatly. Herein, we present an example of NTP-assisted catalytic CO2 methanation over Ni catalysts (15 wt%) supported on BETA zeolite employing lanthana (La) as the promoter. It was found that a NTP-assisted system presents remarkable catalytic performance in catalytic CO2 methanation without an external heat source. Significantly, the use of Na-form BETA zeolite and the addition of La (i.e. 15Ni–20La/Na-BETA catalyst) resulted in an improvement in CO2 conversions, surpassing the 15Ni/H-BETA catalyst, i.e. a seven-fold increase in the turnover frequency, TOF (1.45 s−1vs. 0.21 s−1), and selectivity towards CH4 (up to ca. 97%). In addition, the developed catalyst also exhibited excellent stability under NTP conditions, i.e. a stable performance over a 15 h longevity test (with a TOF of 1.44 ± 0.01 s−1). Comparative in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) characterisation of the developed catalysts revealed that the introduction of La2O3 to the Ni catalyst provides more surface hydroxyl groups, and hence enhances CO2 methanation. Additionally, by analysing the surface species over 15Ni–20La/Na-BETA comparatively under thermal and NTP conditions (by in situ DRIFTS analysis), it is proposed that both the Langmuir–Hinshelwood and Eley–Rideal mechanisms co-exist in the NTP system due to the presence of dissociated H species in the gas phase. Conversely, for the thermal system, the reaction has to go through reactions between the surface-dissociated H and carbonate-like adsorbed CO2via the Langmuir–Hinshelwood mechanism. The current mechanistic understanding of the NTP-activated system paves the way for the exploration of the reaction mechanisms/pathways of NTP-assisted catalytic CO2 methanation.
1. Introduction
Carbon dioxide (CO2) emissions from various sources (e.g. biogas and fossil fuels) are considered as the main cause of climate change.1 The amount of emitted CO2 reached 37.1 gigatonnes (Gt) in 2018 which will inevitably result in severe damage to the planet and society, foreseen by the International Energy Agency (IEA).1 To mitigate the increasingly severe global warming effects of greenhouse gases, CO2 capture and utilisation (CCU) has been proposed and has received considerable attention recently.1,2 The utilisation of CO2, as an alternative to geological storage, not only contributes to the reduction of CO2 emissions for alleviating global climate changes, but also opens up new sustainable routes for making various value-added feedstock chemicals (e.g. CO, CH3OH, and HCOOH) and fuels (e.g. CH4 and C2H4).2,3 In recent years, the hydrogenation of CO2 as a prospective technology can produce synthetic natural gas (SNG, i.e. methane), and thus helps to meet the growing energy demand and tackle the environmental issues.4 CO2 methanation (eqn (1)) employs concentrated CO2 from industrial sources and H2 from water electrolysis powered by renewable energy such as wind and solar power to produce SNG which can be easily fed into existing infrastructure of natural gas transportation networks.5| | | CO2 + 4H2 → CH4 + 2H2OΔH = −165 KJ mol−1 | (1) |
Catalytic CO2 methanation (i.e. Sabatier reaction, eqn (1)) is highly exothermic which is thermodynamically favourable at low temperatures of 200–550 °C.6,7 Therefore, under thermal activation, thermal runaway is prone to cause thermodynamic limitations and catalyst sintering.5 Therefore, alternative activation methods are proposed, such as electro- and photoreduction,8,9 and investigated to improve the performance of catalytic CO2 methanation at low temperatures. Non-thermal plasmas (NTPs) have been demonstrated to have great potential in activating and promoting heterogeneous catalysis under mild conditions which are thermodynamically beneficial to various reactions such as the water–gas shift reaction.10,11 In comparison to conventional thermal activation, a NTP has a unique feature of achieving low-temperature activation of catalytic processes due to the generation of energetic species such as ions, free radicals, electrons, etc. which can excite molecular species and break chemical bonds under ambient conditions.10
To break the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of CO2 is challenging and requires a significant amount of energy, e.g. a high temperature of >1000 °C is necessary under thermal conditions to dissociate CO2.1,12 Conversely, NTPs such as dielectric barrier discharge (DBD) plasmas can enable CO2 methanation at low temperatures of <200 °C and improve the catalytic activity and CH4 selectivity.13–16 For example, Da Costa et al.14,16,17 developed a hybrid plasma–catalyst system employing ceria–zirconia supported Ni catalysts for CO2 methanation which showed a significant enhancement in activity with high methane selectivity. Specifically, in the presence of plasma at a low temperature of 90 °C, the CO2 conversion and CH4 selectivity can reach up to ca. 80% and 100%, respectively. Conversely, in the absence of plasma, the reaction was thermodynamically limited and activated at 425 °C with a CO2 conversion of ca. 80%. In addition, Ni catalysts supported on zeolites (e.g. H-USY) have been recently applied in NTP-assisted CO2 methanation due to the strong metal–support interactions, high specific surface areas, and outstanding thermal and chemical stability of zeolites.1,18–20 However, the protonated zeolites exhibit drawbacks such as poor CO2 adsorption affinities, and thus, inhibit the catalytic activity for CO2 methanation.1 Therein, Brønsted acid sites in zeolites should be neutralised by cations (e.g. Na+) via ion exchange to improve their interaction with CO2.21 Moreover, the affinity of CO2 and the CO2 adsorption capacity of zeolites can be further improved via surface modification using rare earth metals, such as La and Ce.1 However, these strategies for improving CO2 methanation using NTPs has not yet been attempted.
O bonds of CO2 is challenging and requires a significant amount of energy, e.g. a high temperature of >1000 °C is necessary under thermal conditions to dissociate CO2.1,12 Conversely, NTPs such as dielectric barrier discharge (DBD) plasmas can enable CO2 methanation at low temperatures of <200 °C and improve the catalytic activity and CH4 selectivity.13–16 For example, Da Costa et al.14,16,17 developed a hybrid plasma–catalyst system employing ceria–zirconia supported Ni catalysts for CO2 methanation which showed a significant enhancement in activity with high methane selectivity. Specifically, in the presence of plasma at a low temperature of 90 °C, the CO2 conversion and CH4 selectivity can reach up to ca. 80% and 100%, respectively. Conversely, in the absence of plasma, the reaction was thermodynamically limited and activated at 425 °C with a CO2 conversion of ca. 80%. In addition, Ni catalysts supported on zeolites (e.g. H-USY) have been recently applied in NTP-assisted CO2 methanation due to the strong metal–support interactions, high specific surface areas, and outstanding thermal and chemical stability of zeolites.1,18–20 However, the protonated zeolites exhibit drawbacks such as poor CO2 adsorption affinities, and thus, inhibit the catalytic activity for CO2 methanation.1 Therein, Brønsted acid sites in zeolites should be neutralised by cations (e.g. Na+) via ion exchange to improve their interaction with CO2.21 Moreover, the affinity of CO2 and the CO2 adsorption capacity of zeolites can be further improved via surface modification using rare earth metals, such as La and Ce.1 However, these strategies for improving CO2 methanation using NTPs has not yet been attempted.
Herein, we present a catalytic study on NTP-assisted CO2 methanation using Ni catalysts supported on BETA zeolites. Specifically, (i) the performance for NTP-assisted catalytic CO2 methanation (regarding CO2 conversion and selectivities to CH4 and CO) of a series of Ni catalysts supported on H- and Na-BETA zeolites was studied systematically; (ii) the promoting effect of lanthana on the improvement of the CO2 conversion and CH4 selectivity was examined; (iii) diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed in situ to enable the mechanistic study of the catalyst surfaces of different catalysts during thermally- and NTP-activated catalytic CO2 methanation.
2. Experimental
Catalyst preparation
The catalyst support H-BETA (Si/Al = 25) was purchased from Zeolyst International. Sodium chloride (NaCl, 99.5%), nickel nitrate hexahydrate (Ni(NO3)2·6H2O, >99.0%) and lanthanum(III) nitrate hexahydrate (La(NO3)3·6H2O, 99.0%) were obtained from Sigma Aldrich and used as received. Na-BETA was prepared by ion exchange (description of a typical procedure: 5 g H-BETA or Na-BETA zeolite in 50 mL of a 0.5 M NaCl solution at room temperature (RT) for 24 h under stirring; the pH value of the solution was maintained at 7 by adding an ammonia solution during the preparation; the ion exchange procedure was repeated twice). After ion exchange, the sample was filtered, washed several times with deionised water and dried at 110 °C overnight in a convection oven, then calcined in air for 4 h at 550 °C (heating rate = 1 °C min−1). The supported Ni catalysts on BETA zeolites (with a nominal loading content of Ni of 15 wt%) were prepared via the incipient wetness impregnation method by mixing the zeolite supports in a Ni(NO3)2 aqueous solution under stirring for 12 h. After the impregnation, water was fully evaporated at 90 °C, and the sample was dried in a convection oven at 110 °C for 12 h. The resulting catalysts are denoted as 15Ni/H-BETA and 15Ni/Na-BETA. Finally, the dried catalysts were calcined in air at 550 °C for 6 h with a heating rate of 5 °C min−1. La-modified Ni catalysts supported on Na-BETA zeolite were prepared by successive impregnation of the support with Ni(NO3)2 and La(NO3)3 aqueous solutions. The nominal loading contents of La were 5, 10 and 20 wt%, respectively. The resulting catalysts are denoted as 15Ni–5La/Na-BETA, 15Ni–10La/Na-BETA and 15Ni–20La/Na-BETA, respectively. The actual metal loadings measured by inductively coupled plasma optical emission spectroscopy (ICP-OES) can be found in the ESI† (ESI, Table S1).
Characterisation of the catalysts
Nitrogen (N2) physisorption analysis of the samples was carried out at −196 °C using a Micromeritics 3Flex surface characterisation analyser. Prior to the N2 physisorption measurements, the samples (∼50 mg) were degassed at 350 °C under a vacuum overnight. The Brunauer–Emmett–Teller (BET) method was used to determine the specific surface areas of materials, while micropore and mesopore volumes were obtained by the t-plot and Barrett–Joyner–Halenda (BJH) methods (using the adsorption branch of the isotherms), respectively. The crystallinity of the powder catalysts was determined by X-ray diffraction (XRD) on a Philips X'Pert X-ray diffractometer with CuKα1 radiation (λ = 1.5406 Å) and a Ni filter, operating at 40 kV and 30 mA and diffractograms were recorded from 5 to 70°. Scanning electron microscopy (SEM) images of the catalysts were obtained using an FEI Quanta 200 ESEM at a high voltage mode of 20 kV, and energy dispersive X-ray (EDX) spectroscopy for elemental mapping was also carried out using an Oxford Ultim® Max system. All the samples were coated with gold (Au) before the SEM analysis. Transmission electron microscopy (TEM) images were collected with an FEI Tecnai G2 F20 electron microscope operated at 200 kV. The sample was prepared by dispersion of the powder catalysts in ethanol with the assistance of sonication, and a drop of the suspension was spread onto a TEM carbon grid. Hydrogen temperature programmed reduction (H2-TPR) and CO2 and CO temperature programmed desorption (CO2- and CO-TPD) experiments were carried out using a Quanta Chrome ChemBET Pulsar TPR/TPD instrument. Specifically, ∼25 mg of the samples were pre-treated at 250 °C for 30 min under a helium (He) flow to remove any adsorbed or chemisorbed H2O and CO2, and then cooled down to RT. H2-TPR was carried out under a gas mixture flow of 5% H2 in Ar at temperatures of 30–900 °C with a heating rate of 10 °C min−1. The H2 consumption was continuously monitored using a thermal conductivity detector (TCD). Before the CO2- and CO-TPD measurements, the samples were reduced at 250 °C in a 5% H2/He flow for 1 h and cooled down to RT. Subsequently, CO2 or CO (pure CO2 or 1% CO/He) was introduced to saturate the catalyst surface. The CO2 or CO saturated samples were purged using He for 60 min. CO2 or CO desorption was carried out from 30 to 900 °C with a heating rate of 10 °C min−1. X-ray photoelectron spectra (XPS) of the catalysts were obtained with a Thermo ESCALAB 250XI instrument using AlKα (hv = 1486.6 eV, at 15 kV and 10 mA). The C (1s) line at 284.6 eV was used as the binding energy reference.
NTP-assisted catalytic CO2 methanation
Fig. 1 shows the experimental rig scheme used for NTP-assisted CO2 methanation. The DBD plasma reactor consisted of a cylindrical quartz tube (6 mm O.D. × 4 mm I.D.), a ground electrode (a stainless-steel rod with a 1 mm O.D., placed along the axis of the quartz tube) and a high voltage electrode (an aluminium foil sheet wrapped around the outer surface of the tube). The discharge length and gap of the DBD reactor were 10 mm and 1.5 mm, respectively. For each experiment, 130 mg catalyst pellets (particle size of ca. 250 μm) were packed into the discharge space (between the ground electrode and the reactor body). All experiments were carried out at atmospheric pressure. Prior to the catalytic testing, the as-synthesised catalysts were reduced in situ under NTP with H2 as the discharge gas (voltage = 7.0 kV, frequency = 20.3 kHz, flow rate = 50 mL (STP) min−1). Thereafter, a H2/CO2 binary mixture (molar composition: H2/CO2 = 4) was introduced into the reactor at a weight hourly space velocity (WHSV) of 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 077 mL (STP) gcat−1 h−1via two mass flow controllers (Bronkhorst®, F-201CV-500-RAD-11-V). The DBD plasma reactor was connected to an alternating current (AC) high voltage power supply (Info Unlimited, U.S., PVM500-2500) with a peak voltage of up to 40 kV and a variable frequency of 20–70 kHz. In this work, for all the catalytic tests, the applied frequency was fixed at 20.3 kHz, while the applied peak voltage was varied from 5.5 to 7.5 kV. The applied peak voltage was measured using a digital oscilloscope (TBS1102B) with a high voltage probe (P6015A). Gas compositions (e.g. CO2, CH4, H2 and CO) in the exit of the reactor were analyzed on-line using a gas chromatograph (GC, PerkinElmer, Clarus® 590) equipped with a COL-ELITE CARBON molecular sieve packed column (N9303926), a methaniser, a TCD and a flame ionisation detector (FID). Under steady-state conditions, the samples were withdrawn using an automated six-way valve with a sample loop (1 μl) for GC analysis. At least three consecutive measurements were performed for each condition, giving a margin of error of about ±5%. Liquid products generated by the CO2 methanation reaction were continuously condensed and removed from the outlet stream using a water trap placed in an ice bath. In order to calculate the CO2 conversion (eqn (2)) and selectivities to CH4 (eqn (3)) and CO (eqn (4)), the flow rate of dry gas in the exit of the reactor was also measured using a bubble-flow meter. To benchmark the performance in NTP-activated catalysis, control experiments using a NTP-activated catalyst-free system (i.e. gas phase reaction only) and thermally activated systems were also performed.
077 mL (STP) gcat−1 h−1via two mass flow controllers (Bronkhorst®, F-201CV-500-RAD-11-V). The DBD plasma reactor was connected to an alternating current (AC) high voltage power supply (Info Unlimited, U.S., PVM500-2500) with a peak voltage of up to 40 kV and a variable frequency of 20–70 kHz. In this work, for all the catalytic tests, the applied frequency was fixed at 20.3 kHz, while the applied peak voltage was varied from 5.5 to 7.5 kV. The applied peak voltage was measured using a digital oscilloscope (TBS1102B) with a high voltage probe (P6015A). Gas compositions (e.g. CO2, CH4, H2 and CO) in the exit of the reactor were analyzed on-line using a gas chromatograph (GC, PerkinElmer, Clarus® 590) equipped with a COL-ELITE CARBON molecular sieve packed column (N9303926), a methaniser, a TCD and a flame ionisation detector (FID). Under steady-state conditions, the samples were withdrawn using an automated six-way valve with a sample loop (1 μl) for GC analysis. At least three consecutive measurements were performed for each condition, giving a margin of error of about ±5%. Liquid products generated by the CO2 methanation reaction were continuously condensed and removed from the outlet stream using a water trap placed in an ice bath. In order to calculate the CO2 conversion (eqn (2)) and selectivities to CH4 (eqn (3)) and CO (eqn (4)), the flow rate of dry gas in the exit of the reactor was also measured using a bubble-flow meter. To benchmark the performance in NTP-activated catalysis, control experiments using a NTP-activated catalyst-free system (i.e. gas phase reaction only) and thermally activated systems were also performed.
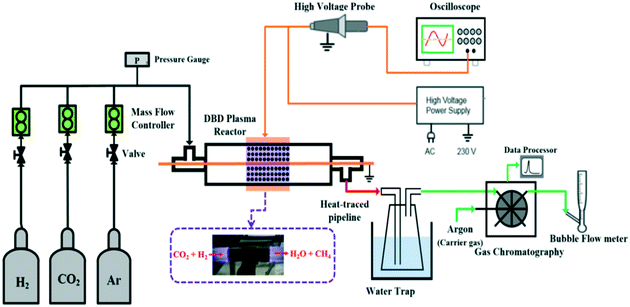 |
| | Fig. 1 Schematic diagram of the experimental rig for NTP-assisted catalytic CO2 methanation. | |
The CO2 conversion (XCO2) is calculated as follows:
| | 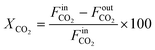 | (2) |
where
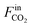
and
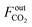
represent the molar flow rate of CO
2 in the feed and exit of the DBD plasma reactor (mol s
−1), respectively.
The selectivities to CH4 (SCH4) and CO (SCO) are calculated as follows:
| | 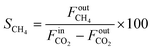 | (3) |
| | 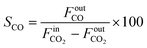 | (4) |
where
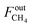
and
FoutCO represent the molar flow rate of CH
4 and CO in the outlet of the DBD plasma reactor (mol s
−1), respectively.
The specific reaction rate22 is calculated as follows:
| | 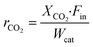 | (5) |
where
rCO2 is the conversion rate of CO
2 (mol s
−1 g
cat−1),
XCO2 is the conversion of CO
2,
Fin is the molar flow rate of CO
2 in the inlet of the DBD reactor (mol s
−1), and
Wcat is the mass of the catalyst (g).
The turnover frequencies (TOFs) of CO2 conversion, defined as moles of CO2 converted per surface nickel metal atom per second (s−1), are calculated, using the results obtained from the catalytic performance measurements and metal dispersions which are determined by using H2 pulse chemisorption (as shown in Table S2 in the ESI†), as follows:
| | 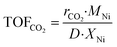 | (6) |
where
MNi (58.69 g mol
−1) is the atomic weight of Ni,
XNi is the metal content (
gNi/
gcat) of the catalyst and
D is the Ni dispersion (as shown in Tables S1 and S2,
† respectively).
In situ DRIFTS characterisation of NTP-assisted catalytic CO2 methanation
In situ DRIFTS analysis was employed to probe the surface species adsorbed on the catalysts during the thermally and NTP activated CO2 methanation aiming to understand the NTP promoted surface reactions. The hybrid NTP-DRIFTS setup was specifically described elsewhere.10 The catalysts were loaded into an infrared (IR) sample holder cup without packing or dilution, and pre-treated in a 10% H2/Ar flow under plasma (applied peak voltage: 6.0 kV, frequency: 26 kHz) for 30 min. A gas mixture (4 vol% CO2/16 vol% H2 diluted in argon, Ar) at 75 mL min−1 was then introduced into the flow cell. For the transient experiments, a constant applied peak voltage of 6.0 kV (at a frequency of 26 kHz) was applied to the power electrode in order to avoid arcing. DRIFTS spectra were recorded at every ∼56 s with a resolution of 4 cm−1 and analysed with the OPUS software.
3. Results and discussion
NTP-assisted catalytic CO2 methanation and characterisation of catalysts
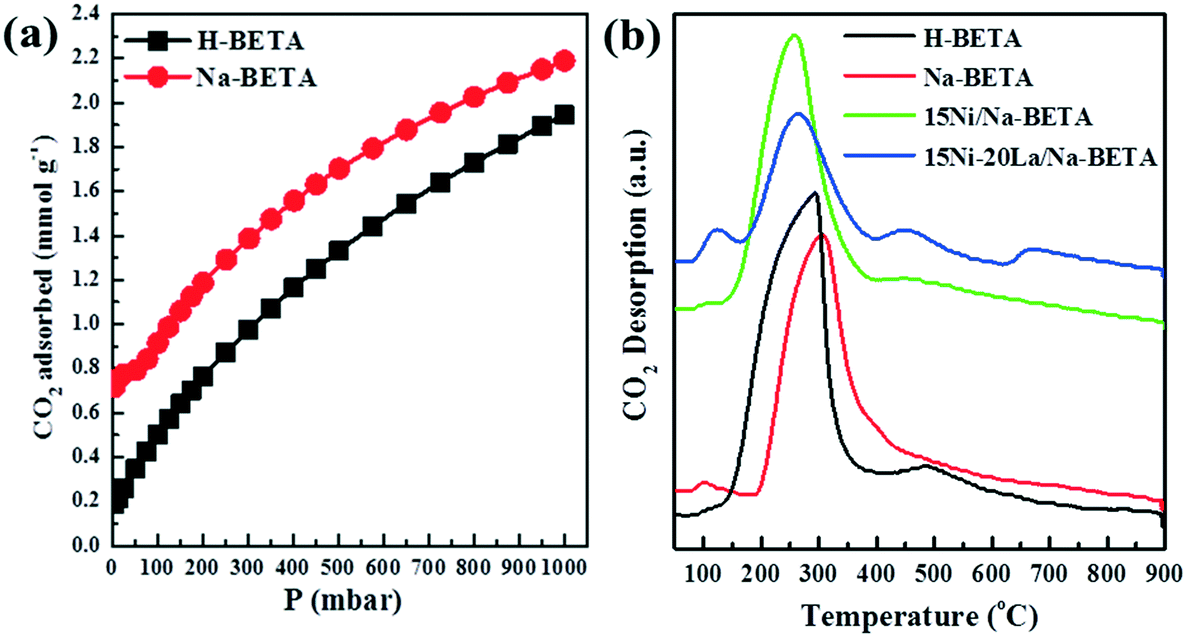
All the materials in this study were comprehensively characterised using various techniques, and the relevant information is presented in the ESI.†Fig. 2 and S1† show the relevant results of the comparative tests using catalyst-free, H-BETA, Na-BETA, 15Ni/H-BETA and 15Ni/Na-BETA systems for CO2 methanation activated by a NTP. The NTP systems without a catalyst (Fig. 2) and with an inert packing (i.e. H-BETA and Na-BETA, Fig. S1†) showed relatively low CO2 conversions at ca. 10% and no selectivity to CH4. CO was measured as the main product from the three reference experiments due to CO2 dissociation in the gas phase by plasma activation.23 Comparatively, NTP-assisted catalytic CO2 methanation over the 15Ni/H-BETA and 15Ni/Na-BETA catalysts showed significant improvements concerning both CO2 conversion and CH4 selectivity, as shown in Fig. 2 (e.g. leading to a four-fold increase in CO2 conversion at 6.5 kV). Generally, the catalytic performance of the 15Ni/H-BETA and 15Ni/Na-BETA catalysts depends on the applied peak voltage potential. However, the 15Ni/Na-BETA catalyst outperformed 15Ni/H-BETA under the conditions studied, especially at relatively low voltages of <7 kV. For example, at an applied peak voltage of 6.0 kV, CH4 selectivity was ca. 75% for 15Ni/Na-BETA versus ca. 15% for 15Ni/H-BETA. These findings support the fact that using the alkali metal exchanged zeolite supports to enhance the CO2–catalyst interactions during the catalytic reaction24 leads to an improved CO2 conversion and CH4 selectivity. Specifically, the CO2 adsorption isotherms of H-BETA and Na-BETA zeolites were measured at pressures up to 1 bar, and the results are presented in Fig. 3a. The results clearly confirm that the CO2 adsorption capacity of the Na-BETA zeolite (ca. 2.2 mmol g−1) is higher than that of the H-BETA zeolite (ca. 1.9 mmol g−1) due to the introduction of Na+ in the framework. In addition, the basicity of different samples was further examined via TPD method using CO2 as probe gas. Fig. 3b reports the CO2-TPD profiles of different samples. In general, three different basic sites can be distinguished according to the desorption temperatures, including the weak at T <150 °C, medium at T = 150–550 °C and strong one at T >550 °C.1,25–27 Both H-BETA and Na-BETA zeolites present main peaks located in the medium basicity region, whereas Na-BETA shows a small peak centred at ca. 100 °C, which could be attributed to the introduction of Na+ in the zeolite framework. After Ni impregnation, 15Ni/Na-BETA presents a similar CO2-TPD profile in comparison with Na-BETA. However, after the addition of lanthana, the 15Ni–20La/Na-BETA catalyst presents one additional TPD peak located at ca. 650 °C. The strong basicity of 15Ni–20La/Na-BETA could be beneficial to CO2 chemisorption, and hence increases its activity for catalytic CO2 methanation.28 EDX mapping of the different catalysts (as shown in Fig. 4) also indicates that Ni and La are uniformly distributed in the Na-BETA zeolites. Specifically, the amount of La element (green) increases notably as the La nominal content in the catalysts increases from 0 to 20 wt%. Additionally, in terms of the catalytic CO2 methanation performance over the developed catalysts (e.g. 15Ni/H-BETA and 15Ni/Na-BETA) under NTP conditions (as shown in Fig. 2), at a low applied voltage of <6.0 kV, the sum of CH4 and CO selectivity is lower than 100%. From the carbon balance based on only the gaseous products (i.e. CO, CH4, and CO2), it was shown that there was a ca. 15% carbon loss during the reaction. This could be mainly attributed to the formation of liquid products as exemplified in the ESI.†
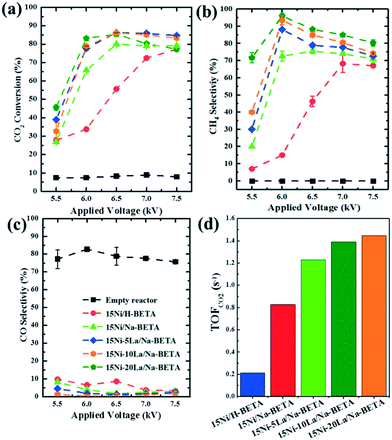 |
| | Fig. 2 Performance of the NTP-assisted catalyst-free CO2 methanation system and the NTP-activated catalytic CO2 methanation systems over the 15Ni/H-BETA, 15Ni/Na-BETA, and Ni–La/Na-BETA catalysts with different La amounts of 5, 10, and 20 wt%: (a) CO2 conversion, (b) CH4 selectivity, (c) CO selectivity, and (d) TOF of CO2 conversion at an applied voltage of 6.0 kV. | |
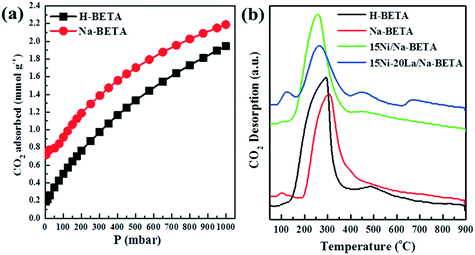 |
| | Fig. 3 (a) CO2 adsorption isotherms and (b) CO2-TPD profiles of the different samples. | |
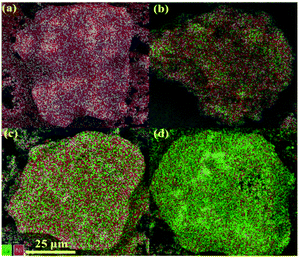 |
| | Fig. 4 EDX mapping of the different catalysts: (a) 15Ni/Na-BETA, (b) 15Ni–5La/Na-BETA, (c) 15Ni–10La/Na-BETA, and (d) 15Ni–20La/Na-BETA catalysts. | |
As also shown in Fig. 2, the addition of La to the 15Ni/Na-BETA catalyst shows a positive effect on the selective conversion of CO2 into CH4. Over the range of applied voltages, the Ni–La/Na-BETA catalysts demonstrated generally better CO2 conversions than the 15Ni/Na-BETA catalyst (Fig. 2a). The presence of La in the 15Ni/Na-BETA catalyst induced a significant effect on the selectivities towards CH4 and CO under NTP activation, particularly at low voltages, as shown in Fig. 2b and c. At the applied peak voltage of 6.0 kV, the increase of La content from 5 to 20 wt% in Ni–La/Na-BETA suppressed the CO selectivity considerably to <0.5%, while improving the CH4 selectivity up to 97%. Concerning the CH4 selectivity, both the Ni–La/Na-BETA and 15Ni/Na-BETA catalysts show optimum values in relation to the applied voltage, as shown in Fig. 2b. However, the presence of La in the catalyst shifts the optimum value to the low voltage of 6.0 kV (at 6.5 kV for 15Ni/Na-BETA). The opposite trend was also measured for the CO selectivity with the catalysts under study, indicating the promoting effect of La via a possibly different reaction pathway in the 15Ni/Na-BETA system. It should be noted that it is challenging to decouple the effects of the plasma in terms of the active site and the gas phase processes, for example, as these all contribute to the overall measured catalytic performance.10 However, in this specific system, the relatively significant CO2 conversion and CH4 selectivity were measured in the NTP activated catalyst systems, indicating the noteworthy effect due to the presence of the developed catalysts. An increase in the amount of La in the Ni–La/Na-BETA catalysts generally improves the selectivity of the systems. Additionally, as shown in Fig. 2d, the specific reaction rates (i.e. turnover frequency, TOF) for CO2 conversion over the Ni–La/Na-BETA catalysts, are especially significantly higher than that over the 15Ni/H-BETA catalysts, showing a ca. a seven-fold increase. Additionally, it was found that the La/Na-BETA catalyst (with 20 wt% La loading) is inactive for catalytic CO2 methanation under NTP conditions, showing a very low CO2 conversion of ca. 10% (shown in Fig. S1†). The latter is attributed to the uncatalysed gas phase-only reaction induced by the NTP. Therefore, in the following research, the 15Ni–20La/Na-BETA catalyst was selected for further study.
In order to highlight the critical role of NTP activation in catalytic CO2 methanation, control experiments by thermal activation (temperature range: 100–450 °C) were performed using 15Ni–20La/Na-BETA. The comparison of catalytic performances between NTP- and thermally activated catalytic CO2 methanation is presented in Fig. 5. Fig. 5a shows the theoretical thermodynamic equilibrium conversions of CO2 methanation, which were calculated using Aspen Plus 8.0 (detailed description of the calculations can be found in the ESI†), and the light-off curves of catalytic CO2 methanation by thermal activation. Under thermal conditions, the onset temperature for CO2 methanation was >250 °C and the thermodynamic equilibrium can be reached at ca. 450 °C. A high selectivity to CH4 (i.e. >95%) can be achieved by the 15Ni–20La/Na-BETA catalyst under thermal activation at temperatures higher than 250 °C, whereas the CO selectivity is only up to ca. 3%. Conversely, during NTP activation, the onset temperature for CO2 methanation is measured at ca. 110 °C (due to the Joule heating, during the catalytic tests, the NTP reactor temperatures were measured to be at a range of 100–150 °C by using an infrared (IR) thermometer, depending on the voltages applied to the system). As shown in Fig. 5a, by NTP activation of 15Ni–20La/Na-BETA, the NTP induced the highest CO2 conversion of ca. 85%, only 15% lower than the corresponding theoretical equilibrium conversion at low temperatures of <150 °C, since the catalyst was not active at these temperatures by thermal activation. Additionally, the NTP system was highly selective to CH4 with the relevant CH4 selectivity reaching up to ca. 97%. The performance of the 15Ni–20La/Na-BETA catalyst in NTP-assisted catalytic CO2 methanation was also compared with those of state-of-the-art plasma-activated catalysts (as shown in Table 1). The developed 15Ni–20La/Na-BETA catalyst demonstrates slightly better catalytic performance than the state-of-the-art catalysts (i.e. Cu/γ-Al2O3,29 Ni/CeZr,14,16 Ni/HT,17 and CeNi/Cs-USY13). However, it is worth noting that the WHSV used in this work was nearly six times higher than those in the other systems.
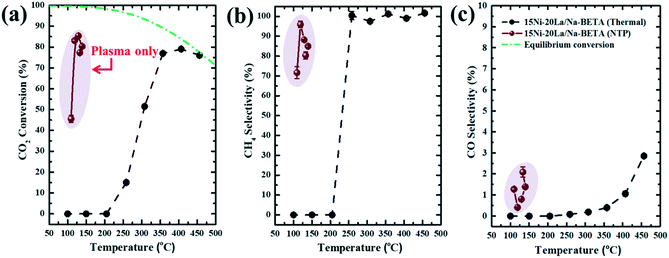 |
| | Fig. 5 Comparison of the performance of the 15Ni–20La/Na-BETA catalyst in catalytic CO2 methanation by NTP and thermal activation : (a) CO2 conversion, (b) CH4 selectivity and (c) CO selectivity (experimental conditions: feed gas composition of H2/CO2 = 4, WHSV of 23077 mL (STP) gcat−1 h−1, applied peak voltage of 5.5–7.5 kV, frequency of 20.3 kHz). Equilibrium conversions of the reaction in Fig. 5a showing the thermodynamic limitations was calculated using Aspen Plus 8.0. | |
Table 1 Comparison of NTP-assisted CO2 methanation over various catalysts at atmospheric pressure
| Catalysts |
Frequency (kHz) |
Applied peak voltage (kV) |
WHSV (mL gcat−1 h−1) |
CO2 conversion (%) |
CH4 selectivity (%) |
Ref. |
| Cu/γ-Al2O3 |
9 |
N/A |
N/A |
8 |
7 |
29
|
| Ni/CeZr |
40–41 |
13–18 |
4000 |
80 |
95 |
14, 17
|
| Ni/hydrotalcite |
40–43 |
13–18 |
4000 |
80 |
95 |
17
|
| CeNi/Cs-USY |
41–43 |
5–6 |
4000 |
70 |
95 |
13
|
| 15Ni–20La/Na-BETA |
20.3 |
6 |
23077 |
84 |
97 |
This work |
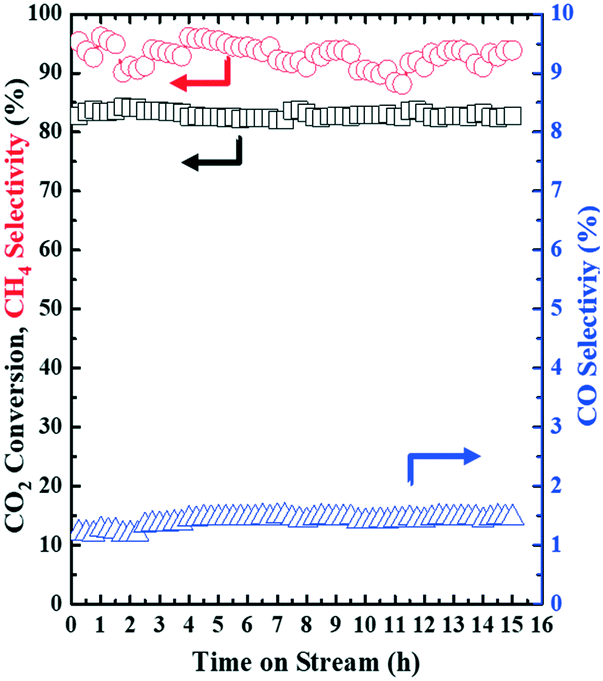
Under the NTP conditions (applied peak voltage = 6.5 kV, frequency = 20.3 kHz), the developed 15Ni–20La/Na-BETA catalyst also showed excellent long-term stability with TOF values of 1.44 ± 0.01 s−1. Over a 15 h time-on-stream evaluation, the key process performance indicators of CO2 conversion (83.0 ± 0.65%), CH4 selectivity (93.0 ± 1.9%) and CO selectivity (1.4 ± 0.1%) all remained rather stable as functions of the time-on-stream, as shown in Fig. 6.
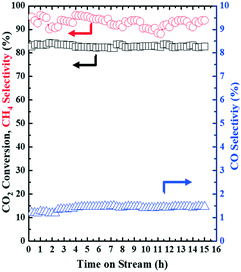 |
| | Fig. 6 Stability test of the 15Ni–20La/Na-BETA catalyst for catalytic CO2 methanation by NTP activation: CO2 conversion and selectivities to CH4 and CO (experimental conditions: feed gas composition of H2/CO2 = 4, WHSV = 23077 mL (STP) gcat−1 h−1). | |
Mechanistic study of NTP-assisted catalytic CO2 methanation
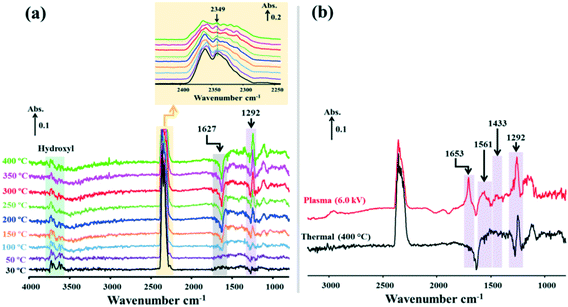
In order to gain insight into NTP-assisted catalytic CO2 methanation over the catalysts used in this work, the dynamics of surface species on the 15Ni/H-BETA, 15Ni/Na-BETA and 15Ni–20La/Na-BETA catalysts were examined using in situ DRIFTS. Fig. 7–9 present the DRIFTS spectra of the surface species adsorbed on the 15Ni/H-BETA, 15Ni/Na-BETA and 15Ni–20La/Na-BETA catalysts under plasma-off and plasma-on conditions, respectively. At ambient temperatures without plasma, the three catalysts were not active for CO2 conversion, as shown in Fig. S8† (during each experiment, mass spectrometry (MS) was employed to detect the gases such as CO2, CH4 and H2 from the outlet of the DRIFTS cell). Under the plasma-off conditions, the IR bands located at 3500–3800 cm−1 and 1500–1800 cm−1 can be attributed to OH vibrational stretching, which may originate from the hydroxyl groups adsorbed on the surface of the catalysts.30–32 Specifically, for the 15Ni–20La/Na-BETA catalyst (Fig. 9a), the intensity of the IR bands between 1500 and 1800 cm−1 are much stronger than those of the 15Ni/H-BETA (Fig. 7a) and 15Ni/Na-BETA (Fig. 8a) catalysts, indicating that the introduction of La in the catalyst leads to more hydroxyl groups formed on the surface of La2O3. Similarly, according to the previous literature reported by Ashok,33 CO2 could interact with the hydroxyl groups on a CeO2 surface, which is a rare earth metal as well. These hydroxyl groups will greatly assist in the adsorption of CO2 on the catalyst surface to inhibit gaseous CO2 dissociation in the presence of plasma.34,35 The IR band appearing at 2349 cm−1 (insets in Fig. 7–9) could be assigned to the gas phase CO2.23,35 By comparing the DRIFTS spectra of 15Ni/Na-BETA (Fig. 7a) and 15Ni–20La/Na-BETA (Fig. 8a) with that of 15Ni/H-BETA (Fig. 9a), 15Ni/H-BETA presents a much stronger IR band at 2349 cm−1, suggesting that less CO2 was adsorbed on its surface. This result confirms that the introduction of Na+ and La2O3 can positively enhance CO2 adsorption on the catalyst surface, which is in good agreement with the findings from CO2 adsorption and CO2-TPD analyses (Fig. 3).
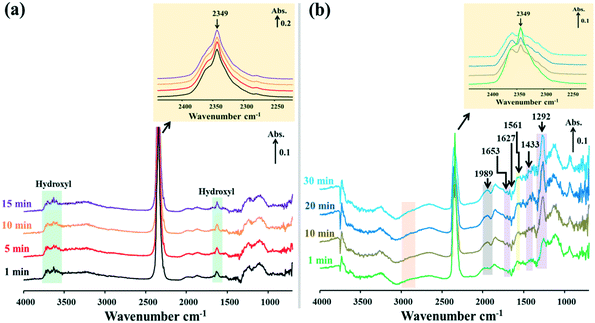 |
| | Fig. 7
In situ DRIFTS spectra of surface species on the 15Ni/H-BETA catalyst during NTP-assisted catalytic CO2 methanation: (a) plasma-off and (b) plasma-on (applied peak voltage = 6.0 kV, frequency = 26 kHz). | |
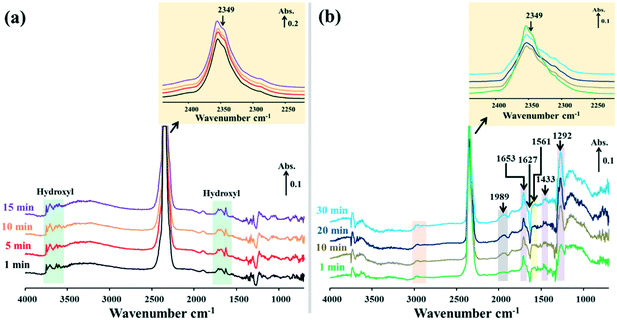 |
| | Fig. 8
In situ DRIFTS spectra of surface species on the 15Ni/Na-BETA catalyst during NTP-assisted catalytic CO2 methanation: (a) plasma-off and (b) plasma-on (applied peak voltage = 6.0 kV, frequency = 26 kHz). | |
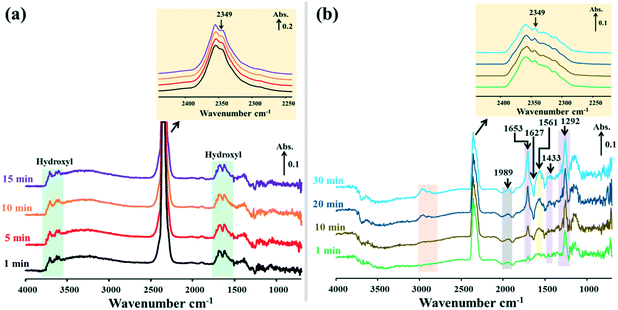 |
| | Fig. 9
In situ DRIFTS spectra of surface species on the 15Ni–20La/Na-BETA catalyst during NTP-assisted catalytic CO2 methanation: (a) plasma-off and (b) plasma-on (applied peak voltage = 6.0 kV, frequency = 26 kHz). | |
When the NTP was ignited, significant changes of the surface species were observed in the catalytic systems, as shown by the relevant DRIFTS spectra in Fig. 7b, 8b and 9b. In all cases, a gradual decrease of the IR band density at 2349 cm−1 as a function of reaction time was observed, comparatively more obvious for 15Ni–20La/Na-BETA (Fig. 9), indicating the conversion of gaseous CO2 in the presence of the catalysts and plasma. The negative band appearing at 1627 cm−1 can be attributed to the disappearance of hydroxyl groups from the surface of the catalysts in the presence of plasma.36 The IR bands at 1653, 1433, and 1292 cm−1 (shaded by the purple rectangles) can be attributed to surface carbonate species.32,33,37 Additionally, the IR band at 1561 cm−1 (shaded by the light-yellow rectangles) can be related to the surface monodentate formates.19 Specifically, as shown in Fig. 10, in comparison with the other two catalysts, the intensity of the IR band at 1561 cm−1 observed from the 15Ni–20La/Na-BETA catalyst is much stronger, indicating the favourable formation of monodentate formates on the surface of La2O3. These surface species can be further hydrogenated into CHx species leading to the formation of CH4.23,35 The C–H vibration of CHx species is characterised by the broad IR band in the 2800–3000 cm−1 region32 as shaded by the orange rectangles in Fig. 7b, 8b and 9b. It must be noted that the intensities of these IR bands on the spectra of the 15Ni–20La/Na-BETA catalyst is more intense than those of the other two catalysts (as shown in Fig. 10), suggesting more CHx species formed on the surface of the catalyst due to the existence of La2O3.
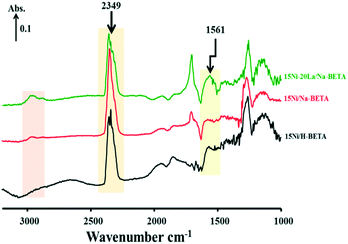 |
| | Fig. 10
In situ DRIFTS spectra of surface species on the different catalysts during NTP-assisted catalytic CO2 methanation (applied peak voltage = 6.0 kV, frequency = 26 kHz). | |
In situ DRIFTS characterisation of the catalytic system with the 15Ni–20La/Na-BETA catalyst by thermal activation was also performed, and the DRIFTS spectra are shown in Fig. 11a. Clearly, at relatively low temperatures of <150 °C, the disappearance of hydroxyl groups from the surface of the catalyst (i.e. the negative IR band at 1627 cm−1) is not significant, while the intensity of hydroxyl species bands (IR bands between 3500–4000 cm−1, shaded by a green rectangle) remains stable, proving that the catalyst was not readily active at temperatures of <150 °C, which were the measured bulk temperatures of the NTP system.
 |
| | Fig. 11 (a) In situ DRIFTS spectra of surface species on the 15Ni–20La/Na-BETA catalyst during thermally activated catalytic CO2 methanation (feed gas mixture = 10 vol% CO2/40 vol% H2 diluted in argon, total flow rate = 50 mL min−1); (b) comparative in situ DRIFTS spectra of surface species on the 15Ni–20La/Na-BETA catalyst during thermally (400 °C) and NTP (applied peak voltage = 6.0 kV, frequency = 26 kHz) activated catalytic CO2 methanation. | |
Under the thermal conditions, by continuously increasing the temperature above 150 °C, the following surface dynamics data were recorded: (i) the IR bands at 3500–3800 cm−1 (representing the hydroxyl groups, shaded by the green rectangle) started decreasing, while the intensity of the negative IR band at 1627 cm−1 increased gradually; (ii) the intensity of the IR band at 2349 cm−1, which is attributed to the gas phase CO2 (shaded by the orange rectangle), diminished gradually; (iii) the surface carbonate species (which is characterised by the sole IR band at 1292 cm−1, shaded by the purple rectangle) appeared progressively. By combining the catalytic data (Fig. 5) with the in situ DRIFTS data (Fig. 11), we can conclude that, under conventional thermal activation, CO2 methanation over the 15Ni–20La/Na-BETA catalyst requires high temperatures of >350 °C to overcome the activation barriers and enable the conversion of CO2 (about 80% at 400 °C). However, the system can be readily activated by NTP at comparatively low temperatures (i.e. <150 °C), with high CO2 conversion (of ca. 85%) and CH4 selectivity (of ca. 97%).
The comparison of the in situ DRIFTS spectra of the surface species on the 15Ni–20La/Na-BETA catalyst under thermal (400 °C, CO2 conversion of ca. 80%) and NTP activation (applied peak voltage = 6.0 kV, at ca. 125 °C, CO2 conversion of ca. 85%) conditions is shown in Fig. 11(b). NTP activation promotes the formation of various surface carbonates and formates (i.e. the IR bands at 1653, 1561, 1433 and 1292 cm−1), while thermal activation only produces a single type of surface carbonate (at 1292 cm−1), suggesting the presence of multiple reaction mechanisms for CH4 formation in the NTP system. Under the thermal conditions, the surface reaction between the surface dissociated H and carbonate-like adsorbed CO2 proceeds via the Langmuir–Hinshelwood mechanism, which is consistent with the reaction mechanisms reported in the literature.23,38 Conversely, in the NTP system, in addition to the pure surface reactions via the Langmuir–Hinshelwood mechanism, the dissociated H species in the gas phase may react with the surface-adsorbed C species directly from the gas phase via the Eley–Rideal mechanism, and hence, leading to the formation of various carbonates and formates on the catalyst surface in the NTP system under study.
4. Conclusions
Coupling non-thermal plasma (NTP) with heterogeneous catalysis is a promising technique to promote the conversion of CO2 into valuable products. In this work, Ni catalysts supported on Na-BETA zeolites employing lanthana as the promoter were developed and applied in NTP-assisted catalytic CO2 methanation without an external heat source. Experimental results revealed that neutralisation of the H-BETA zeolite by Na+via ion exchange enhances CO2 adsorption on the catalyst surface, and thus improves the catalytic performance regarding both CO2 conversion and CH4 selectivity, leading to a two-fold increase in CO2 conversion and a four-fold increase in TOF values at 6.0 kV. Moreover, the introduction of the lanthana promoter in the catalyst resulted in the highest CO2 conversion (ca. 85%) and CH4 selectivity (ca. 97%) (TOF = 1.45 s−1), while the Ni/Na-BETA catalyst gave a lower CO2 conversion (ca. 65%) and CH4 selectivity (ca. 70%) (TOF = 0.83 s−1) at 6.0 kV. In addition, a 15 h time-on-stream stability test proved that the developed catalyst is robust under the NTP conditions with regards to CO2 conversion and selectivity towards CH4. Comparative in situ DRIFTS characterisation of the 15Ni–20La/Na-BETA, 15Ni/H-BETA and 15Ni/H-BETA catalysts revealed that the addition of lanthana in the Ni catalyst enhances the formation of surface hydroxyl groups greatly, which can interact with CO2, and hence, promotes CO2 conversion. Comparative in situ DRIFTS spectra of the surface species on the 15Ni–20La/Na-BETA catalyst during thermally and NTP-activated catalytic CO2 methanation confirmed that CO2 methanation requires high temperatures (i.e. >350 °C) to overcome the activation barriers and enable the efficient conversion of CO2 (ca. 80% at 400 °C). Conversely, the catalyst can be readily activated using a NTP at comparatively low temperatures (i.e. <150 °C) with similar CO2 conversion. More importantly, based on the in situ observation of the difference in the surface species of the two systems, reactions in the NTP system presumably proceed via both Langmuir–Hinshelwood and Eley–Rideal mechanisms due to the presence of gas-phase H species. For the thermal system, on the other hand, only surface reactions between the surface-dissociated H and carbonate-like adsorbed CO2via the Langmuir–Hinshelwood mechanism are allowed. Despite the numerous studies on NTP enabled catalytic CO2 methanation systems, further research is needed to gain insight into the specific reaction mechanisms/pathways of the systems in order to fully understand the interaction of NTPs and catalysts. The findings from this work shed light upon the direction of future research, i.e. the role of NTP generated gas-phase species in the catalytic surface reactions, which may be generic in other relevant NTP-assisted catalysis processes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
HC thanks the financial support from the European Commission Marie Skłodowska-Curie Individual Fellowship (H2020-MSCA-IF-NTPleasure-748196). HX thanks the University of Manchester President's Doctoral Scholar (PDS) Award and the China Scholarship Council (CSC, file no. 201606150068) for supporting her PhD research. YS thanks the CSC for her academic visiting fellowship at the University of Manchester (file no. 201708440477) and the Foundation of Department of Education of Guangdong Province (No. 2017KZDXM085). We are also grateful to Mingyuan Cao at the University of Southern California for the thermodynamic equilibrium calculations using Aspen Plus 8.0.
References
- A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Appl. Catal., B, 2018, 238, 393–403 CrossRef CAS.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS.
- G. Centi and S. Perathoner, Greenhouse Gases: Sci. Technol., 2011, 1, 21–35 CrossRef CAS.
- C. Mebrahtu, S. Abate, S. Perathoner, S. Chen and G. Centi, Catal. Today, 2018, 304, 181–189 CrossRef CAS.
- M. Götz, J. Lefebvre, F. Mörs, A. McDaniel Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Energy, 2016, 85, 1371–1390 CrossRef.
- L. Torrente-Murciano, D. Mattia, M. D. Jones and P. K. Plucinski, J. CO2 Util., 2014, 6, 34–39 CrossRef CAS.
- R. Zhang, P. H. Winterfeld, X. Yin, Y. Xiong and Y.-S. Wu, J. Nat. Gas Sci. Eng., 2015, 27, 579–615 CrossRef CAS.
- D. Mateo, D. De Masi, J. Albero, L.-M. Lacroix, P.-F. Fazzini, B. Chaudret and H. García, Chem. – Eur. J., 2018, 24, 18436–18443 CrossRef CAS.
- R. Ahmad and A. K. Singh, J. Mater. Chem. A, 2018, 6, 21120–21130 RSC.
- S. Xu, S. Chansai, C. Stere, B. Inceesungvorn, A. Goguet, K. Wangkawong, S. F. R. Taylor, N. Al-Janabi, C. Hardacre, P. A. Martin and X. Fan, Nat. Catal., 2019, 2, 142–148 CrossRef CAS.
- C. E. Stere, J. A. Anderson, S. Chansai, J. J. Delgado, A. Goguet, W. G. Graham, C. Hardacre, S. F. R. Taylor, X. Tu, Z. Wang and H. Yang, Angew. Chem., Int. Ed., 2017, 56, 5579–5583 CrossRef CAS.
- B. Ashford and X. Tu, Curr. Opin. Green Sustain. Chem., 2017, 3, 45–49 CrossRef.
- M. C. Bacariza, M. Biset-Peiró, I. Graça, J. Guilera, J. Morante, J. M. Lopes, T. Andreu and C. Henriques, J. CO2 Util., 2018, 26, 202–211 CrossRef CAS.
- M. Nizio, A. Albarazi, S. Cavadias, J. Amouroux, M. E. Galvez and P. Da Costa, Int. J. Hydrogen Energy, 2016, 41, 11584–11592 CrossRef CAS.
-
M. Nizio, PhD thesis, Université Pierre et Marie Curie - Paris VI, 2016 Search PubMed.
- R. Benrabbah, C. Cavaniol, H. Liu, S. Ognier, S. Cavadias, M. E. Gálvez and P. Da Costa, Catal. Commun., 2017, 89, 73–76 CrossRef CAS.
- M. Nizio, R. Benrabbah, M. Krzak, R. Debek, M. Motak, S. Cavadias, M. E. Gálvez and P. Da Costa, Catal. Commun., 2016, 83, 14–17 CrossRef CAS.
- E. Jwa, S. B. Lee, H. W. Lee and Y. S. Mok, Fuel Process. Technol., 2013, 108, 89–93 CrossRef CAS.
- F. Azzolina-Jury, D. Bento, C. Henriques and F. Thibault-Starzyk, J. CO2 Util., 2017, 22, 97–109 CrossRef CAS.
- F. Goodarzi, L. Kang, F. R. Wang, F. Joensen, S. Kegnæs and J. Mielby, ChemCatChem, 2018, 10, 1566–1570 CrossRef CAS.
- S.-T. Yang, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2010, 135, 90–94 CrossRef CAS.
- R. Vakili, E. K. Gibson, S. Chansai, S. Xu, N. Al-Janabi, P. P. Wells, C. Hardacre, A. Walton and X. Fan, ChemCatChem, 2018, 10, 4238–4242 CrossRef CAS.
- F. Azzolina-Jury and F. Thibault-Starzyk, Top. Catal., 2017, 60, 1709–1721 CrossRef CAS.
- M. C. Bacariza, R. Bértolo, I. Graça, J. M. Lopes and C. Henriques, J. CO2 Util., 2017, 21, 280–291 CrossRef CAS.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS.
- Z. Boukha, L. Fitian, M. López-Haro, M. Mora, J. R. Ruiz, C. Jiménez-Sanchidrián, G. Blanco, J. J. Calvino, G. A. Cifredo, S. Trasobares and S. Bernal, J. Catal., 2010, 272, 121–130 CrossRef CAS.
- D. Wierzbicki, R. Debek, M. Motak, T. Grzybek, M. E. Gálvez and P. Da Costa, Catal. Commun., 2016, 83, 5–8 CrossRef CAS.
- T. A. Le, M. S. Kim, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293–294, 89–96 CrossRef CAS.
- Y. Zeng and X. Tu, IEEE Trans. Plasma Sci., 2016, 44, 405–411 CAS.
- S. Chilukoti, F. Gao, B. G. Anderson, J. W. H. Niemantsverdriet and M. Garland, Phys. Chem. Chem. Phys., 2008, 10, 5510–5520 RSC.
- S. Eckle, H.-G. Anfang and R. J. Behm, J. Phys. Chem. C, 2011, 115, 1361–1367 CrossRef CAS.
- J. Zheng, C. Wang, W. Chu, Y. Zhou and K. Köhler, ChemistrySelect, 2016, 1, 3197–3203 CrossRef CAS.
- J. Ashok, M. L. Ang and S. Kawi, Catal. Today, 2017, 281, 304–311 CrossRef CAS.
- C. Schild, A. Wokaun and A. Baiker, J. Mol. Catal., 1991, 69, 347–357 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, ACS Catal., 2015, 5, 6337–6349 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and S. M. Sidik, Appl. Catal., A, 2014, 486, 115–122 CrossRef CAS.
- C. Weilach, C. Spiel, K. Föttinger and G. Rupprechter, Surf. Sci., 2011, 605, 1503–1509 CrossRef CAS.
- Q. Pan, J. Peng, S. Wang and S. Wang, Catal. Sci. Technol., 2014, 4, 502–509 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy00590k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Yibing
Mu‡
a,
Yan
Shao‡
ab,
Sarayute
Chansai‡
a,
Shaojun
Xu‡
a,
Cristina E.
Stere
a,
Huan
Xiang
a,
Rongxin
Zhang
a,
Yilai
Jiao
c,
Christopher
Hardacre
*a,
Yibing
Mu‡
a,
Yan
Shao‡
ab,
Sarayute
Chansai‡
a,
Shaojun
Xu‡
a,
Cristina E.
Stere
a,
Huan
Xiang
a,
Rongxin
Zhang
a,
Yilai
Jiao
c,
Christopher
Hardacre


 and
and 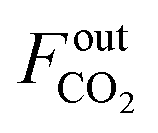 represent the molar flow rate of CO2 in the feed and exit of the DBD plasma reactor (mol s−1), respectively.
represent the molar flow rate of CO2 in the feed and exit of the DBD plasma reactor (mol s−1), respectively.


 and FoutCO represent the molar flow rate of CH4 and CO in the outlet of the DBD plasma reactor (mol s−1), respectively.
and FoutCO represent the molar flow rate of CH4 and CO in the outlet of the DBD plasma reactor (mol s−1), respectively.