
An effective strategy for creating asymmetric MOFs for chirality induction: a chiral Zr-based MOF for enantioselective epoxidation†
Kayhaneh
Berijani
a,
Ali
Morsali
 *a and
Joseph T.
Hupp
b
*a and
Joseph T.
Hupp
b
aDepartment of Chemistry, Faculty of Sciences, Tarbiat Modares University, 14115-175, Tehran, Iran. E-mail: morsali_a@modares.ac.ir
bDepartment of Chemistry, Northwestern University, Technological Institute K148, 2145 Sheridan Road, Evanston, Illinois 60208, USA
First published on 16th May 2019
Abstract
Recently the construction of chiral MOFs (CMOFs) has been very challenging and complex. For the first time, we synthesized a chiral Zr-based MOF with L-tartaric acid by solvent-assisted ligand incorporation (SALI). We show that a CMOF can be postsynthetically generated by a simple method: incorporating chiral carboxylic groups on the achiral NU-1000. The post-synthesized chiral NU-1000 was used as an asymmetric support for producing a chiral catalyst with molybdenum catalytic active centers as Lewis acid sites. Enantioselective epoxidation of various prochiral alkens to epoxids by using [C-NU-1000-Mo] is comparable to that using other asymmetric homogeneous and heterogeneous catalysts, along with high enantiomeric excess and selectivity to epoxide (up to 100%). The CMOF could be reused in the styrene oxidation after five cycles without substantial deterioration in the CMOF crystallinity or catalytic performance.
Introduction
The progress of metal–organic frameworks (MOFs) as a family of porous materials with ideal designs, useful properties and different potential applications is very impressive.1a,9a The tunability of MOF components such as organic linkers, metal nodes and even functional groups not only creates new structures with different features, but can also affect the MOFs' capability, especially their catalytic activity.1b,2 Diverse MOFs were reported as heterogeneous catalysts or as supports for the heterogenization of homogeneous catalysts.2h,i Using chiral synthetic catalysts for asymmetric catalysis has advanced considerably since the past twenty-five years. Recently, CMOFs (chiral metal–organic frameworks) have been developed due to their asymmetric applications.3 The primitive investigations on homochiral MOF applications, especially catalytic abilities, have been further performed in the time range from 2002 to 2013 and they are more widely being continued. Chirality as an attractive phenomenon and a fundamental property is an important subject in asymmetric material.4 The CMOFs could be prepared using various methods; the selection of an optimal method with suitable performance is important because their preparation is possible through only a few types of reactions. There are three main procedures for synthesizing CMOFs, and these procedures may or may not involve chiral species. However, previous studies have shown that sometimes, achiral crystals have the optical activity.5,6 When there are no chiral agents, the MOF framework topology can induce poor chirality.7,8 The synthesis of CMOFs is elaborate, but the effective method is using a chiral component such as BINOL and chiral M-salen derivatives. Examples of such CMOFs are the enantiopure 2,2′-dihydroxy-1,1′-biphenyl in homochiral biphenol-based MOF, DUT-67 as an 8-connected zirconium with L-proline (exhibiting good catalytic performance) and a simple chiral catalyst UiO-66 (synthesized by using L-proline as modulator), used for the aldol reaction.9 In the present study, the construction of a chiral Zr-MOF was conducted via the incorporation of L-tartaric acid to Zr-nodes by using the SALI method. Several examples about the incorporation of different compounds, such as carboxylates, phosphonates, and dye molecules, by SALI have been reported, with different applications. Recently, Zhou et al. investigated the functionalization of PCN-700 as a flexible Zr-MOF with a wide range of linear organic dicarboxylate ligands. In addition, the functionalization of nodes in zirconium-based MOFs with various metal centers, such as Au(I), Ir(I), Cu(II), Co(II) and V(V), has been performed in solution phase by using different methods.10We selected NU-1000 as a Zr-MOF due to its properties such as presence of a tetratopic organic linker, pore size and high surface area that decreased after chiralization. We achieved a novel CMOF (Chiral NU-1000) without any hardness using the SALI method that Zr–OH2 and Zr–OH collaborate in the chiralization of the Zr-nodes by chiral carboxylic acid (CFG = carboxylic-acid-containing functional group). Then, MoO2(acac)2 as a catalytic active site was immobilized on chiral NU-1000 as a chiral support. The metalation of chiral NU-1000 was performed for the enantioselective epoxidation of olefins because epoxides are intermediates or starting materials for the generation of chiral key organic compounds having industrial applications.11 Regardless of previous reports, no study has been presented similar to ours: SALI-CFG, prepared by the interaction of L-tartaric acid as an chiral organic precursor with a base species in NU-1000. The successful immobilization route of MoO2(acac)2 onto the chiral modified NU-1000 with the set conditions has been displayed in Scheme 1. In catalytic system used in this study, TBHP was chosen as a smooth and green oxidant, and the obtained results are presented as follows.12
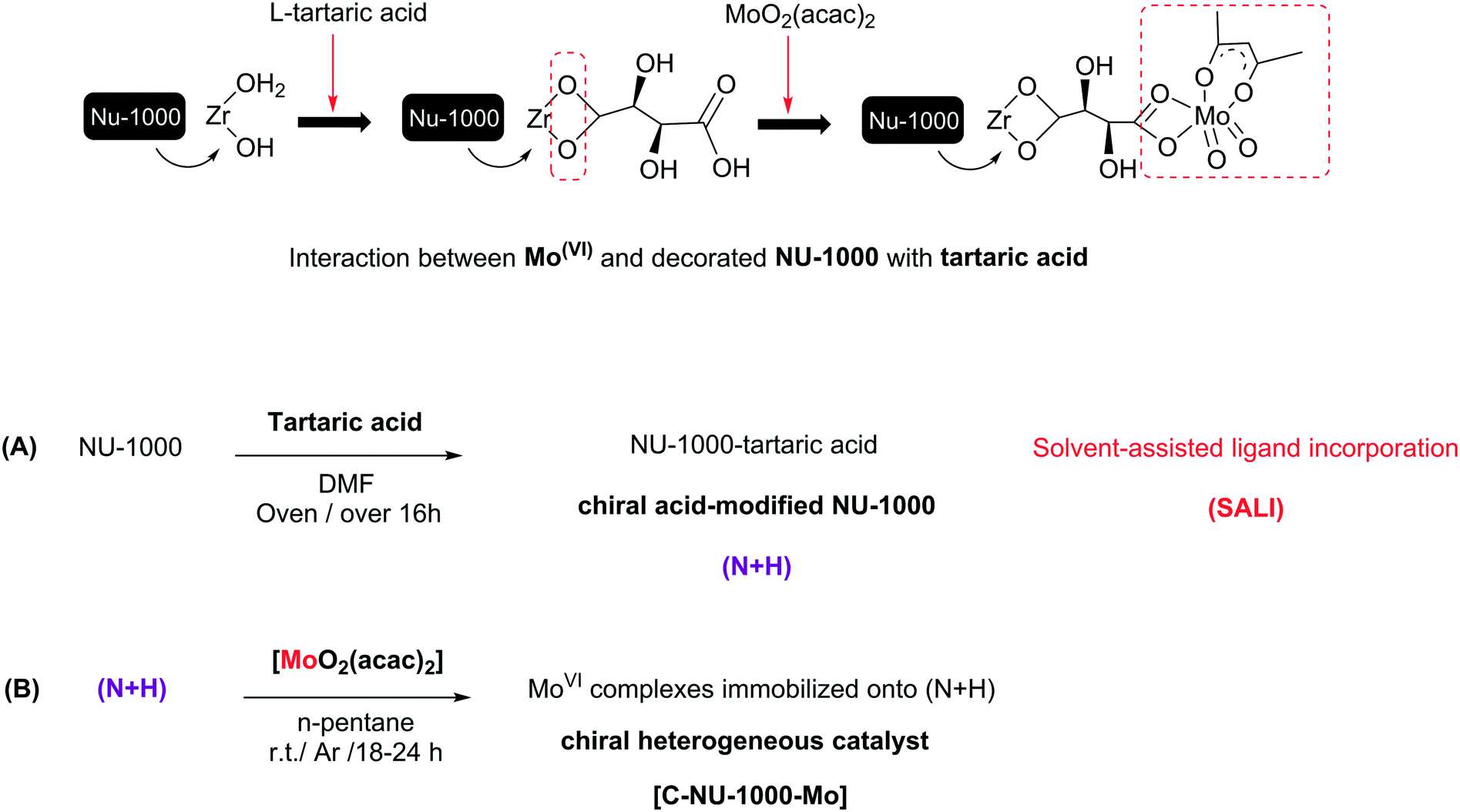 | ||
| Scheme 1 The preparation steps of (N + H) and [C-NU-1000-Mo] catalyst with the used conditions. | ||
Results and discussion
Characterization of [C-NU-1000-Mo]
In this study, molybdenyl(VI) acetylacetonate was loaded onto chiral functionalized NU-1000 with tartaric acid, and the probable interaction has been shown in Scheme 1. NU-1000 as a robust metal–organic framework showed great activity as a support for the attachment of the chiral acids. Then, the immobilization of metal active sites onto the modified NU-1000 was performed due to the accessible and ample hydroxyl and carboxyl functional groups of C-NU-1000. The asymmetric tartaric acid ligands created a chiral position close to the Mo-complexes as catalytic centers. The resultant [C-NU-1000-Mo] exhibited high activity as a chiral recyclable heterogeneous catalyst for the enantioselective epoxidation of several olefins. The simplest method for the characterization of the produced structures is FT-IR. The characteristic peaks were observed in the FT-IR spectra of NU-1000 (N), functionalized NU-1000 (N + H) and the catalyst [C-NU-1000-Mo] (Fig. 1). The appeared peaks from 2859 cm−1 to around 3500 cm−1 are characteristic of C–H stretching and O–H stretching, respectively. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond stretching peak of the benzene ring appeared in 1535 cm−1. The supported cis-MoO2 (MoO) was confirmed with the existence of the strong new doublet bands at 910 and 933 cm−1.13 The weak peak at 1718 cm−1 is related to ν(CO) vibrations of the COOH group of L-tartaric acid and the peak 1417 cm−1 is ascribed to the νs(CO2−) vibration of the present carboxylate group/O–H deformation.14 The existence of peaks at 1718 and 1417 cm−1 indicates that the chiral acid used in this study is bound to NU-1000 due to the deprotonation of carboxylate.15
C bond stretching peak of the benzene ring appeared in 1535 cm−1. The supported cis-MoO2 (MoO) was confirmed with the existence of the strong new doublet bands at 910 and 933 cm−1.13 The weak peak at 1718 cm−1 is related to ν(CO) vibrations of the COOH group of L-tartaric acid and the peak 1417 cm−1 is ascribed to the νs(CO2−) vibration of the present carboxylate group/O–H deformation.14 The existence of peaks at 1718 and 1417 cm−1 indicates that the chiral acid used in this study is bound to NU-1000 due to the deprotonation of carboxylate.15
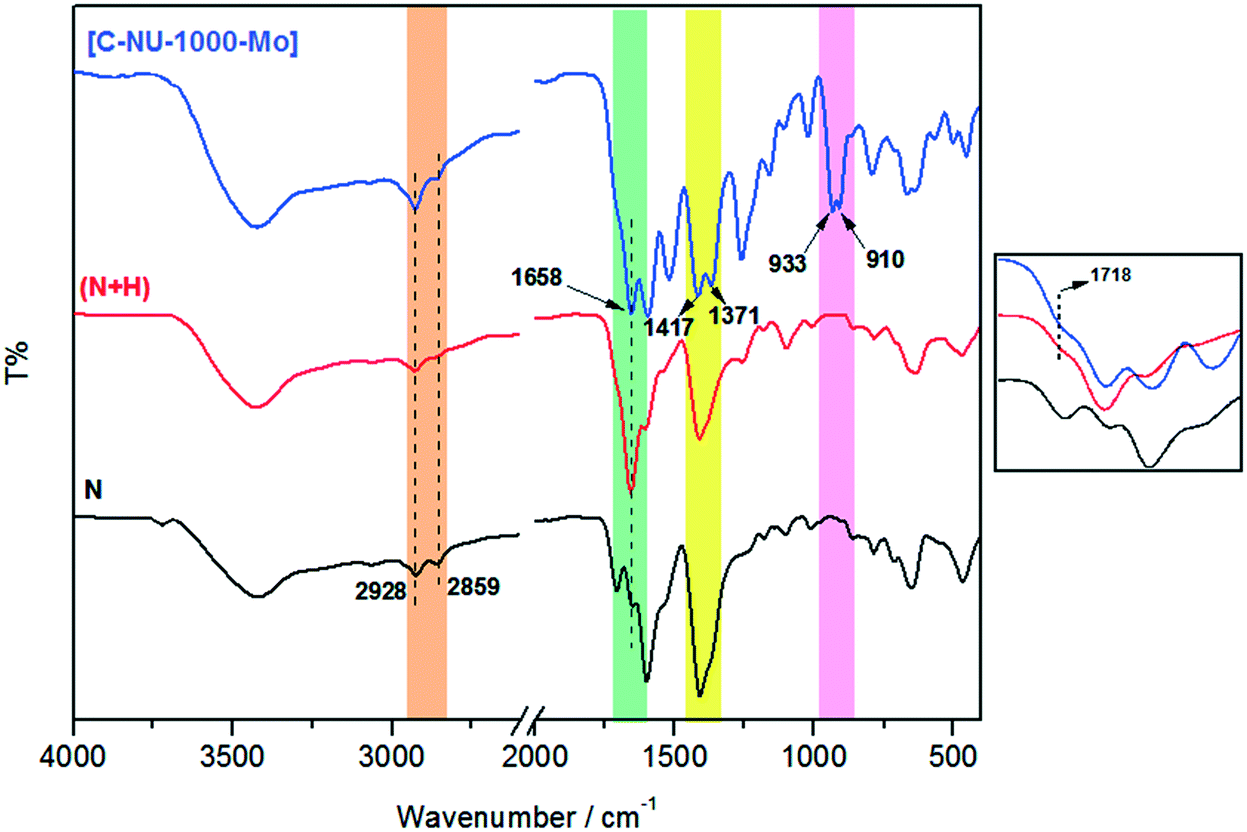 | ||
| Fig. 1 FT-IR spectra of NU-1000 (N), NU-1000 + L-tartaric acid (N + H) and [C-NU-1000-Mo]. | ||
For the molybdenum complexes using different ligands and solvents, separate UV-vis spectra were seen. Undoubtedly, electronic transitions in the catalyst depend on the type of the used ligands and nature of the metals.16 UV-vis spectra of the dispersed [C-NU-1000-Mo] (by ultrasonication) and MoO2(acac)2 were studied in DCE (Fig. S2†).
The assignment of π–π*, n–π* and LMCT transitions is consistent with the transitions for the Mo-complex from 200 to 500 nm. In comparison to the neat complex spectrum (222, 267, 318 nm) for [C-NU-1000-Mo] (218, 263, 307, 423 nm), the tangible decrease in the absorption bands and a slight blue shift were seen, which could prove the immobilization Mo-complex onto NU-1000 (Fig. S2†).17
ICP analysis was performed to determine the amount of Mo loading in the present catalyst (S3). 1H NMR spectroscopy was also used to confirm the tartaric acid incorporation in the catalyst structure. [C-NU-1000-Mo] dissolved in D2SO4 was analyzed in deuterated DMSO. The obtained spectrum exhibited the signals of the protons in 1,3,6,8-tetrakis(p-benzoic-acid)pyrene (TBAPy) and L-tartaric acid that appeared at about 4.1 ppm and 7.5 to 8.3 ppm (Fig. S3†). According to the obtained data, the extent of incorporated CFG on each node was estimated (2.8 chelating L-tartrates (T)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 zirconiums:0.5 molybdenum = T:Zr:Mo ratio). The PXRD patterns of the prepared samples are shown in Fig. 2.10b The PXRD patterns showed that NU-1000 maintained crystallinity after functionalization and metalation (SALI process/immobilization of Mo-complex). The peak intensity decreased in the patterns of carboxylic acid-functionalized NU-1000 and [C-NU-1000-Mo], which is logical due to crystallinity reduction. These relative changes confirm that the functionalization of NU-1000 nodes has occurred. The XRD pattern of experimental NU-1000 matches with its simulated pattern.10bFig. 3 shows the FE-SEM images of NU-1000 and [C-NU-1000-Mo]. The images indicated that NU-1000 as a support maintained its cylindrical structure during the synthesis of [C-NU-1000-Mo], with the length of about 1.30 μm. Hence, it is concluded that tartaric acid and molybdenum-complex loading had not remarkably affected the morphology of the NU-1000 framework.
:6 zirconiums:0.5 molybdenum = T:Zr:Mo ratio). The PXRD patterns of the prepared samples are shown in Fig. 2.10b The PXRD patterns showed that NU-1000 maintained crystallinity after functionalization and metalation (SALI process/immobilization of Mo-complex). The peak intensity decreased in the patterns of carboxylic acid-functionalized NU-1000 and [C-NU-1000-Mo], which is logical due to crystallinity reduction. These relative changes confirm that the functionalization of NU-1000 nodes has occurred. The XRD pattern of experimental NU-1000 matches with its simulated pattern.10bFig. 3 shows the FE-SEM images of NU-1000 and [C-NU-1000-Mo]. The images indicated that NU-1000 as a support maintained its cylindrical structure during the synthesis of [C-NU-1000-Mo], with the length of about 1.30 μm. Hence, it is concluded that tartaric acid and molybdenum-complex loading had not remarkably affected the morphology of the NU-1000 framework.
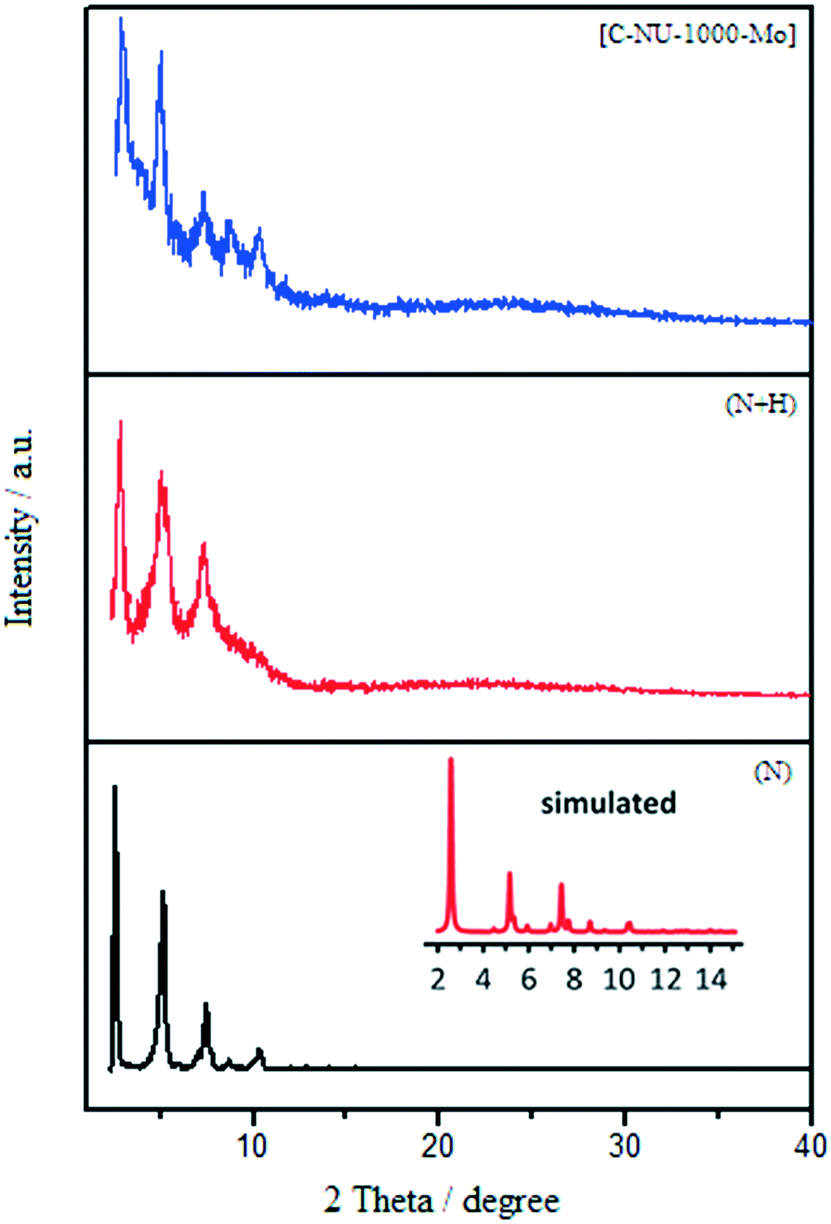 | ||
| Fig. 2 The experimental XRD patterns of NU-1000 (N) (experimental (black) and simulated (red)), NU-1000 + tartaric acid (N + H) and [C-NU-1000-Mo]. | ||
 | ||
| Fig. 3 FE-SEM images of a) NU-1000 (N) and b) [C-NU-1000-Mo]. | ||
For the porosity determination of the synthesized new porous materials, BET analysis was performed as a useful method to confirm the porosity of SALI-CFG and [C-NU-1000-Mo]. On comparing the obtained data, such as the decrease in the surface area and pore volume during the catalyst synthesis, it can be found that the incorporation of tartaric acid to NU-1000 and immobilization of MoO2(acac)2 effectively occurred, as expected.10b Further details have been summarized in Fig. S4 and Table S1.†
Furthermore, for determining the thermal stability, thermogravimetric analysis of [C-NU-1000-Mo] was conducted in the temperature range of 25–600 °C (Fig. S5†). At first, the decrease in mass was 4.05% when the temperature reached to about 110 °C, which was related to the existence of adsorbed water.17a,18 In the second and probably the third stage of mass loss, decomposition of the incorporated tartaric acids occurred.19 It seems that from the end of the third step to the last stage, the weight loss is related to the loss of the ligands of the Mo-complex.20 Hence, it can be inferred that the thermal stability of the catalyst correlates to the nature of the employed materials.10b Based on the above studies, such a chiral MOF as a Lewis-acid catalyst could act as an asymmetric catalyst with effective catalytic sites for the epoxidation of various olefins. Hence, we explored the effects of MoO2(acac)2 and NU-1000-tartaric acid in the reactions performed in the present study.
The catalytic activity of [C-NU-1000-Mo] in the enantioselective epoxidation reactions
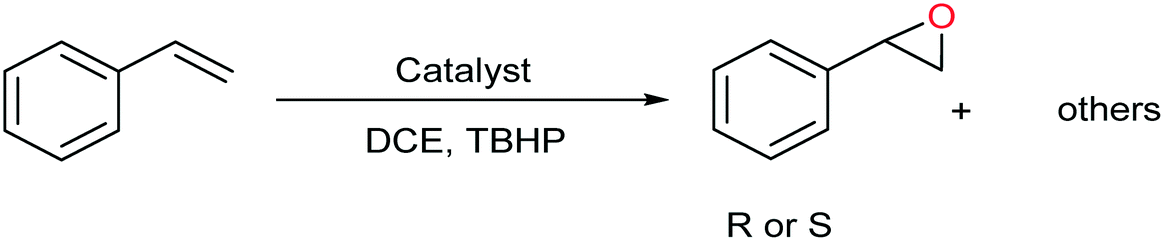
For investigation of [C-NU-1000-Mo] performance in the asymmetric epoxidation of olefins, styrene and TBHP were chosen as a model substrate and green oxidant, respectively. In the absence of [C-NU-1000-Mo], as a control experiment, no significant result was obtained, so the existence of catalyst was necessary (Table 1, entry 1). The progress of the reaction was investigated at different temperatures as one of the most important factors in the control reactions. At low temperatures, the reactions could not be completed (40 °C with conv. 54%; 60 °C with conv. 75%) and approximately 70% ee was obtained (not shown in Table 1); therefore the optimized temperature was raised until 80 °C. With regards to the influence of solvents on the reaction conversion and selectivity of product,21 the following solvents were investigated: 1,2-dichloroethane (DCE), methanol (MeOH), ethyl acetate (EtOAc) and acetonitrile (ACN) (entries 3–6). Among these solvents, DCE was found to be the favorable solvent to complete the reaction. Details for this solvent have been summarized in Table 1 (entry 3). The other solvents exhibit coordinating behavior to the metal site, which cause the reaction rate to decrease; however, DCE exhibited not significant coordinating property (Table 1, entries 4–6).22 MeOH as a polar protic solvent with high donating power was the worst solvent in our experiments because it prevented the activity of the catalyst.23 The polar solvents have a negative effect on catalyst performance because they decrease the hydroperoxo-metal species that are essential in olefin oxidation. The epoxidation proceeded slowly in the presence of NU-1000 and NU-1000 + tartaric acid, with conversion less than 15% and 10%, respectively (not shown in Table 1). Finally, in comparison to [C-NU-1000-Mo] as a heterogeneous catalyst, MoO2(acac)2 as a homogeneous catalyst was used for styrene oxidation (0.044 mmol/2 h), although no chiral agent in reaction mixture was present (Table 1, entry 2). The primary experiments for determination of the optimized conditions revealed catalyst stability, sufficient catalytic centers and chirality induction (full conversion, high degree to the wonderful enantioselectivities). To elucidate the chiral catalyst performance, enantioselective oxygenation of some of the terminal linear and aromatic olefins was performed in identical conditions (Table 2).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst/time | Solvent | Conv.b (%) | Selb (%) | Eec (%) |
|
a Reaction conditions: catalyst 50.0 mg (0.044 mmol Mo); the molar ratio of styrene:TBHP = 0.5; solvent 5 mL; 80 °C, 5 h.
b Conversions and yields were determined by GC-FID.
c ee investigated by GC on a chiral SGE-CYDEX-B capillary column. TBHP, 70 wt% in H2O. N.R. = no reaction.
|
|||||
| 1 | None/5 h | 1,2-Dichloroethane | <8 | <25 | |
| 2 | MoO2(acac)2/2 h | 1,2-Dichloroethane | 100 | <80 | |
| 3 | [C-NU-1000]/5 h | 1,2-Dichloroethane | 100 | 86 | 95 |
| 4 | [C-NU-1000]/5 h | Methanol | N.R. | ||
| 5 | [C-NU-1000]/5 h | Ethyl acetate | <40 | <70 | 52 |
| 6 | [C-NU-1000]/5 h | Acetonitrile | <50 | <60 | 68 |
| Entry | Substrate | Conv.b(%)/Sel.(%)/time (h) | Eec (%) | TONd | TOFe (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: [C-NU-1000-Mo] 50.0 mg (0.044 mmol Mo), substrates 2.0 mmol (trans-stilbene 0.5 mmol), TBHP 4 mmol, DCE 5 mL, 80 °C. b Conversion and epoxide selectivity were determined by GC-FID. c The enantiomeric excess (ee) values were determined by chiral-GC. R-(+)-limonene was used for the determining of the enantiomeric configuration of the major isomer. d TON (total turnover number) = (moles of products)/(per mole of catalyst). e Value of TOF (turnover frequency) = TON (turnover number)/(reaction time). Products were confirmed by 1H NMR. | |||||
| 1 | trans-Stilbene | 100/100/10 min | 80 (R,R) | 11.36 | 68.18 |
| 2 | Styrene | 100/86/5 h | 95 (S) | 45.45 | 9.09 |
| 3 | α-Methyl styrene | 100/83/4 h | 83 (S) | 45.45 | 11.36 |
| 4 | trans-β-Methyl styrene | 97/85/4 h | 86 (R,R) | 44.09 | 11.02 |
| 5 | 4-Chloro styrene | 88/75/7 h | 85 (S) | 40.00 | 5.71 |
| 6 | 4-Methyl styrene | 100/80/4 h | 88 (S) | 45.45 | 11.36 |
| 7 | 1-Phenyl-1-cyclohexene | 80/100/8 h | 89 (R,R) | 36.36 | 4.54 |
| 8 | 1-Octene | 72/100/8 h | 100 (R or S) | 32.72 | 4.09 |
| 9 | 1-Decene | 68/100/8 h | 97 (R) | 30.90 | 3.86 |
The enantioselective epoxidation of trans-stilbene to trans-stilbene epoxide, with 100% conversion and selectivity, generated two enantiomers with 90% yield of (R,R)-trans stilbene, 80% ee and 68.18 h−1 TOF after 10 min (Table 2, entry 1). The calculated TOF at an enantiomeric excess of 80% was a promising result.24 Different results were obtained for trans-stilbene oxidation by using various catalysts, such as like multi-wall carbon nanotubes supported molybdenyl acetylacetonate, with TOFs of 20.6 and 19.7 h−1 after 9 h.25 In the trans-stilbene epoxidation, different by-products can be obtained, for example, diol or benzaldehyde, but we did not obtain these products. This could have occurred because (1) usually a mixture of cis:trans-stilbene epoxide is obtained in a certain ratio, but in this study, stereoselectivity was 100% to trans epoxide, and (2) trans-stilbene maintained its structure without destruction and deformation.26 In the styrene epoxidation, full conversion, 86% epoxide selectivity and 95% ee were obtained after 5 h at 80 °C with TOF of 9.09 h−1. Moreover, 84% epoxide selectivity related to the styrene oxide (S) as a major product was obtained. The obtained catalytic activity of [C-NU-1000-Mo] was higher in comparison with that of the other heterogeneous catalysts such as UiO-66-NH2-SA-Mo (7.54 h−1) and MoO2(acac)-SiIm (3.8 h−1).27 Furthermore, benzaldehyde (BA) and 2-phenylacetaldehyde (PAA) were identified as minor products. The formation of BA can happen during the catalytic process via two methods: TBHP (as a nucleophile) attack on styrene epoxide or cleavage of C–C bond directly attached to CC of phenyl ring.28 2-Phenylacetaldehyde is also afforded with small percentage due to partial rearrangement of styrene epoxide.11c In addition to products, tert-butyl benzoate, benzoic acid and tBuOH can be formed as adducts.29 In the ESI,† some examples of enantioselective epoxidation of styrene with different chiral heterogeneous catalysts have been shown. These results strongly indicated that present catalytic system was very effective in contrast to the others from the point of view of enantiomeric excess. The selectivities and enantiomeric excesses rely on the substantial effects of NU-1000 and amount of L-tartaric acid. Furthermore, examples of styrene derivative oxidation and obtained conclusions are depicted in Table 2 (entry 3 to 6). In the epoxidation of α-methyl styrene (AMS) and trans-β-methyl styrene (TBMS), the conversions reached as high as 100% and 97%, separately (entry 3 and 4). Our results indicate that the oxidation of AMS showed TOF of 11.36 h−1, which was a good result compared with the other findings.27b Probably, the steric effects of the alkyl group around double bond influence the epoxide and enantioselectivity values of AMS and TBMS than styrene.30 Moreover, the difference in the reaction completion time is related to their reactivity.
Surely, the existence of the large channels in this mesoporous catalyst can strongly affect the substrate diffusion and oxidation rate. The examined functionalized styrenes were 4-chloro styrene and 4-methyl styrene that were oxidized with 88% and 100% conversion, respectively (Table 2, entries 5 and 6). The electronic effect of Cl- and Me-groups on the reaction rate is significant and cannot be ignored. It was found that in the epoxidation of styrene and its derivatives, apart from benzaldehyde, as an inseparable by-product, phenylacetaldehyde was rarely generated. Sometimes, acetophenone and benzoic acid can be produced depending on catalysis conditions.31 [C-NU-1000-Mo] indicated good catalytic performance in the proceeding of the 1-phenyl-1-cyclohexene epoxidation (conv%: 80%) to epoxide with great enantioselectivity (ee%: 89% (R,R)) (Table 2, entry 7), so the enantiomers were measured to be in unequal amounts. Two aliphatic hydrocarbons, 1-octene and 1-decene, were also investigated, which were oxidized to epoxides in 72% and 68% conversions, separately (Table 2, entries 8 and 9). For the epoxidation of both hydrocarbons, high epoxide selectivity was achieved (100%) after 8 h. Apparently, the conversion of linear olefins to epoxide products can be selective with a specific catalytic system.31 These findings show the presence of steric hindrance and low electron density.32 Since 1-decene is longer than 1-octene (shorter α-olefin) in term of the length, it certainly exhibits steric effect.33Table 3 presents the carbon balance in some of olefin oxidations. The percent of carbon at the initial and final time of reaction has been indicated. The interesting point was that the double bonds in used olefins underwent high chemoselectivity to chiral epoxide. Truly, a high impact of the starting tartaric acid stereochemistry was observed on the enantiomeric excess of the formed products.34 Therefore, [C-NU-1000-Mo] demonstrated an ideal chiral platform due to its physical and chemical features. Previously, the epoxidation mechanism by the Mo center as a Lewis acid was reported. Briefly, on immobilizing MoO2(acac)2 onto NU-1000-tartaric acid, acetylacetone ligand (Hacac) was lost.35 During TBHP attack on molybdenum, the proton of the terminal O atom in TBHP transfers to one oxygen atom in the MoO terminal bond and then, the alkyl peroxo-Mo intermediate was generated. Hence, the TBHP anion coordinates to Mo(VI)-catalyst with high Lewis acidity, which is a key factor in oxidation reactions. If there are free zirconium nodes, their OH2 groups can react with tBuOOH and then, node-tBuO will be created.36 With regards to the chirality induction mechanism, it can be said that the olefins with pro-S face or R-face can approach the catalytic active site. When the olefin approaches the catalytic center, chirality induction happens by H-bond interaction between the olefin (H atom on double bond) and tartrate (OH group). Herein, two generated chiral intermediates in chirality induction have been shown (Fig. 4, a and b). For example, in the enantioselective epoxidation of 1-decene, the proposed transition state is (a). Similar intermediate has been suggested in the asymmetric epoxidation of olefins in the presence of chiral amines.37
| Substrate | Carbon (%) in initial time | Carbon (%) in final time |
|---|---|---|
| Styrene/5 h | 92.26 | 0 |
| Styrene epoxide | — | 79.344 |
| Benzaldehyde | — | 9.226 |
| Phenylacetaldehyde | — | 3.690 |
| 1-Phenyl-1-cyclohexene/8 h | 91.08 | 18.216 |
| 1-Phenyl-1-cyclohexene epoxide | — | 72.864 |
| 1-Octene/8 h | 85.63 | 23.977 |
| 1-Octene epoxide | — | 61.654 |
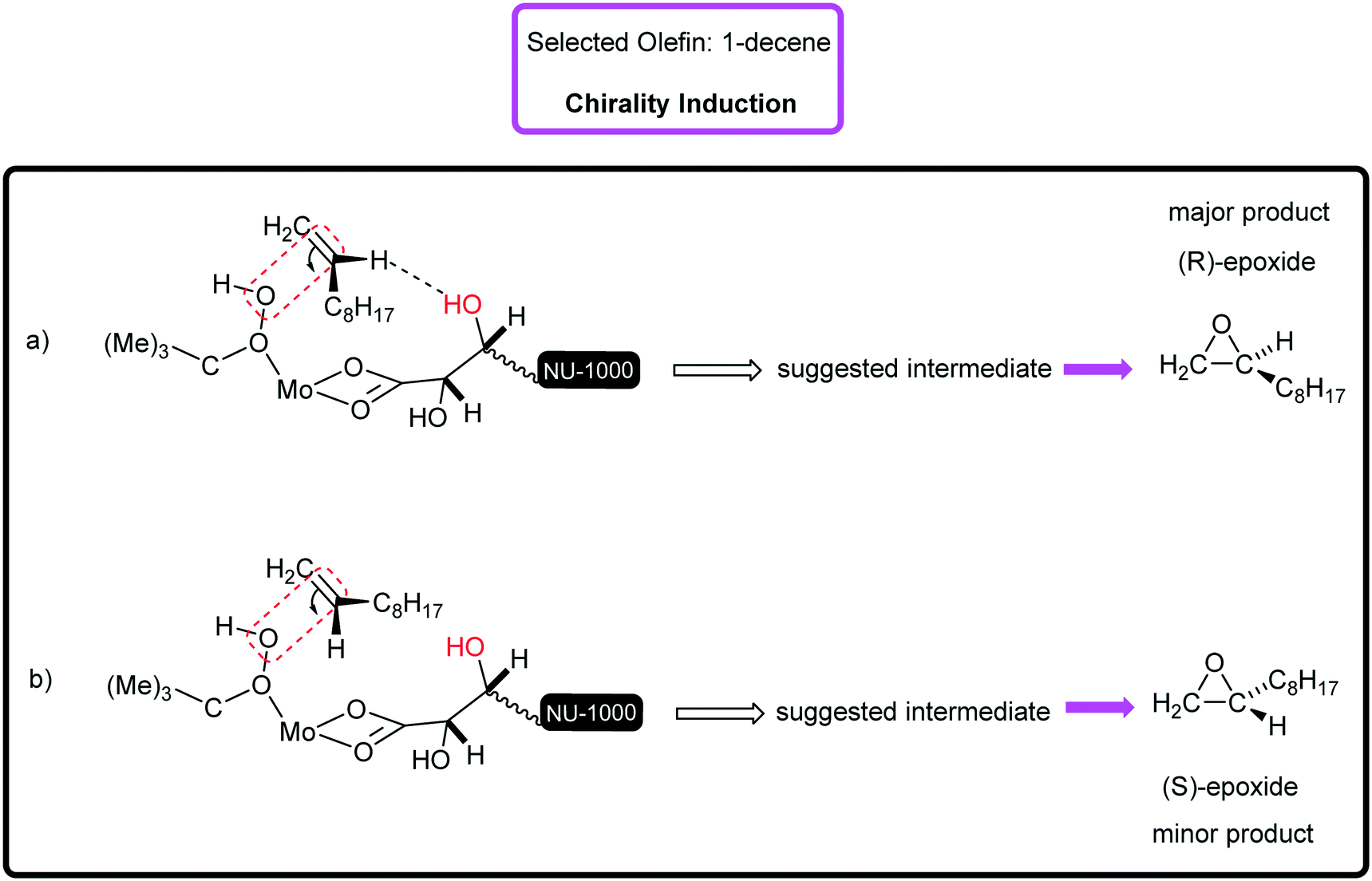 | ||
| Fig. 4 The chirality induction mechanism (a) the closing of 1-decene through pro-S face and (b) pro-R face with suggested transition states. | ||
Recycling, leaching and hot filtration tests
Our heterogeneous catalyst could be easily separated from the reaction mixture by filtration and repeatedly reused in the new styrene oxidation. To investigate the [C-NU-1000-Mo] stability, the oxidation of styrene as a model substrate was performed under the same catalytic condition for 5 runs; no significant drop was seen in conversion in each run (conv%: 100%). It is important to note that the enantiosmeric excess and epoxide selectivity from 95 and 86% in the fresh-catalyst step changed to 92% and 85% in the fifth step, respectively. Hence, considerable changes were not observed in the mentioned parameters in each of the recycled solutions (Fig. 5). FT-IR, XRD (Fig. S6†), SEM, TGA (Fig. S5†) and BET (Table S1†) analyses of the used catalyst were also performed; notably, important changes in the catalyst structure were not observed after recycling compared with the fresh catalyst. These techniques confirmed the retention of the chemical stability of the recovered [C-NU-1000-Mo]; therefore, it had an intact crystal structure. All of the solutions of the recycle reactions up to the fifth stage were investigated by ICP analysis (Table 4). The Mo content was evaluated from the fresh stage to the fifth stage, but leaching of Mo ions was observed only in the sixth step. In the Table 4, conversion, ee, epoxide selectivity and Mo concentration have been reported for each recycle.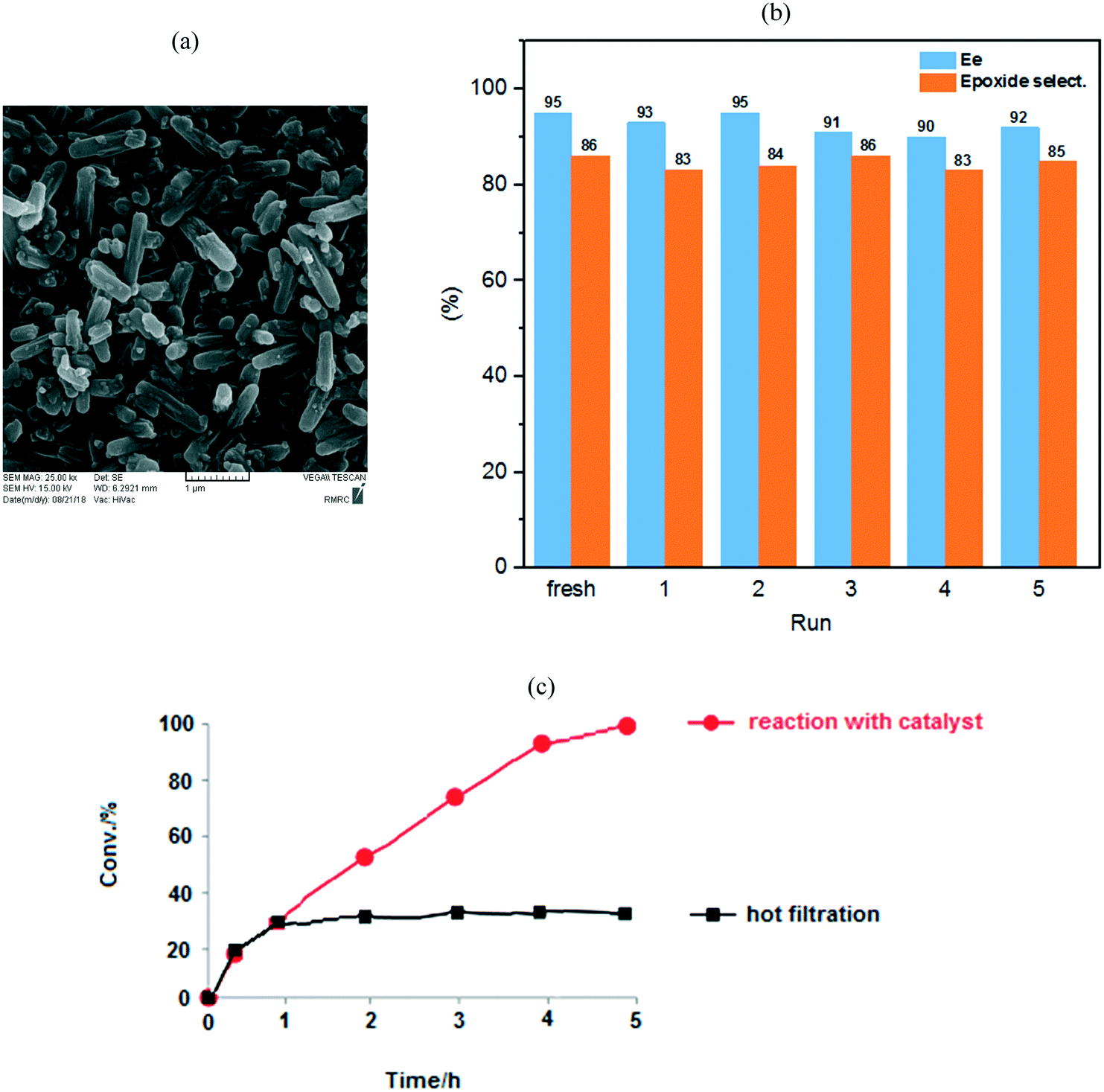 | ||
| Fig. 5 (a) The SEM image of the used catalyst in the fifth run. (b) The obtained ee and epoxide selectivity by using the reusable [C-NU-1000-Mo] in the oxidation of styrene to five recycles. (c) Hot filtration test was carried out at 80 °C. The epoxidation of styrene with catalyst (red) and after filtering the catalyst from reaction mixture after 1 h (black). | ||
| Run | [C-NU-1000-Mo] | |||
|---|---|---|---|---|
| Conv. (%) | Ee (%) | Epoxide selectivity | Mo leachedb (%) | |
| a The reaction condition is similar to Table 1 (optimized condition). b Based on Mo concentration (ppm). Determined by ICP. | ||||
| Fresh | 100 | 95 | 86 | −0.055 |
| 1 | 100 | 93 | 83 | −0.049 |
| 2 | 100 | 95 | 84 | −0.034 |
| 3 | 100 | 91 | 86 | −0.031 |
| 4 | 100 | 90 | 83 | −0.031 |
| 5 | 100 | 92 | 85 | −0.030 |
Accordingly, the recycling experiment results displayed that [C-NU-1000-Mo] is a stable Zr-MOF. Furthermore, BET analysis of the recovered catalyst was performed after 5 cycles. The SBET from 1412 m2 g−1 in fresh catalyst decreased to 1043 m2 g−1, which most probably indicated the occurrence of aggregation. In the last recycle step, the decrease in the Vtot and Dav was not significant compared with those of the fresh catalyst (0.64 cm3 g−1 and 24.67 Å, respectively). Hot filtration test was also performed to show the positive effect of the catalyst in the epoxidation process of styrene. After 1 h, the catalyst was separated from the reaction mixture and then, the present solution was stirred for 4 h. The reaction proceeded with no appreciable increase in conversion (ca. 25%, Fig. 5(c)). Hence, these observations corroborate those for the anchored Mo species on the novel chiral Zr-MOF.
Conclusions
In summary, for the first time, we present a unique CMOF based on NU-1000 and chiral tartaric acid by using the SALI route. Its preponderance was attributed to the simple design with high density of the incorporated L-tartaric acid on the Zr6 nodes and Mo-complex as the Lewis acid site. We believe that we have created a new simple CMOF with a facile chiralization strategy in comparison to the other CMOFs. The simplified design is an ideal and noteworthy aspect for this chiral catalyst. The participation of three components in this catalyst, namely, NU-1000, tartaric acid and Mo-complex plays an essential role in enantioselective epoxidation. The asymmetric catalytic activity of [C-NU-1000-Mo] was assayed in the enantioselective epoxidation of various prochiral alkenes to form enantiomers of epoxides with excellent enantioselectivities. Tartaric acid with two chiral centers and appropriate orientation has a significant effect on chirality induction to epoxides. [C-NU-1000-Mo] had the capability of the sensibly discriminating the R configuration or S configuration in epoxides; therefore, the racemic mixture (50:50) was not obtained. In addition, [C-NU-1000-Mo] as a heterogeneous catalyst can be separated from the reaction mixture and reused without any considerable degradation in activity.
Experimental methods
Materials and characterization
(2R,3R)-(+)-Tartaric acid (L-(+)-tartaric acid), MoO3, D2SO4, the various selected olefins, oxidant and the other reagents such as solvents were bought from different chemical companies. NU-1000 was successfully synthesized based on a previous report.38 In this research, different analytical techniques were employed such as GC-FID, 1H NMR, UV-vis, FT-IR, XRD, FE-SEM, BET, TGA and ICP.Echrom GC A90 gas chromatography with flame-ionization detector (China) was employed (Agilent HP-5 capillary column, 30 m × 0.320 mm × 0.25 μm, temperature limits from 60 to 325 °C) for analyzing the products obtained from the oxidative reactions. For determining the enantiomeric excess (ee), a chiral column was used (Agilent CYCLODEX-B capillary column, 30 m × 0.25 mm × 0.25 μm, temperature limits from 50 to 230 °C). 1H-NMR spectra were recorded by using a INOVA 500 MHz spectrometer. The UV-vis spectra were recorded by using a UV/VIS-Double Beam Spectrophotometer with RAYLEIGH model UV-2601. The Thermo Nicolet IR 100 FT-IR spectrometer was used for studying the structures of the generated compounds in the 4000–400 cm−1 region (mid-infrared). X-ray diffraction analysis was performed by using X'Pert Pro-MPD powder diffractometer that has been made by Philips Company, Netherlands (tube: Co, λ = 1.78897 Å, voltage: 40 kV, current: 40 mA). A scanning electron microscope (SEM, Tescan VEGA-II, voltage = 15 KV) was used for studying the morphology of the synthesized materials to main catalyst. The surfaces sorption was analyzed by using the Micrometrics TriStar II 3020 with N2 at 77 K. The NETZSCH Thermal Analyzer was used for investigating the catalyst thermal behavior with the following conditions: heating range from 25–600 °C at 10 °C min−1 under N2 atmosphere. Then, for the determination of Mo amount in the fresh catalyst and recycle solutions, an inductively coupled plasma-optical emission (ICP-OES) spectrometer (VISTA PRO-axial CCD spectrometer from Varian) was used.
Preparation of the materials
Reusable solid catalyst
For addressing the high cost and difficult conditions, especially in industrial processes, reusable heterogeneous catalysts have been considered. For checking the reusability and stability of [C-NU-1000-Mo], it was examined in the enantioselective epoxidation of styrene. After completing the reaction in each cycle, the catalyst was filtered, washed with acetonitrile and dried. Then, it was used for a new reaction with fresh styrene, TBHP and DCE. ICP-OES was employed for determining of Mo leaching in filtrates. Until the fifth recycle, no Mo leaching was detected. [C-NU-1000-Mo] was much more stable for five cycles under the initially determined conditions. The reaction conversion of styrene was 100% each time. The significant decrease in styrene oxide selectivity and ee after five cycles were not consecutively seen. The SEM, BET, TGA, FT-IR and XRD analyses for [C-NU-1000-Mo] were performed for comparing the structure of the used and fresh catalyst. Clear difference was not observed in the structure and morphology of the catalyst in two mentioned cases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Tarbiat Modares University for supporting this research.References
- (a) Q. Xia, Z. Li, C. Tan, Y. Liu, W. Gong and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8259–8266 CrossRef CAS PubMed; (b) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- (a) M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef CAS PubMed; (b) B. Chen, L. Wang, Y. Xiao, F. R. Fronczek, M. Xue, Y. Cui and G. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS PubMed; (c) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (d) K. M. L. Taylor, W. J. Rieter and W. Lin, J. Am. Chem. Soc., 2008, 130, 14358–14359 CrossRef CAS PubMed; (e) P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed; (f) L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed; (g) T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (h) K. Sabyrov, J. Jiang, O. M. Yaghi and G. A. Somorjai, J. Am. Chem. Soc., 2017, 139, 12382–12385 CrossRef CAS PubMed; (i) S. Yuan, L. Zou, H. Li, Y. P. Chen, J. Qin, Q. Zhang, W. Lu, M. B. Hall and H. C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 10776–10780 CrossRef CAS PubMed.
- (a) Y. Samoilichenko, V. Kondratenko, M. Ezernitskaya, K. Lyssenko, A. Peregudov, V. Khrustalev, V. Maleev, M. Moskalenko, M. North, A. Tsaloev, Z. T. Gugkaeva and Y. Belokon, Catal. Sci. Technol., 2017, 7, 90–101 RSC; (b) K. K. Bisht and E. Suresh, J. Am. Chem. Soc., 2013, 135, 15690–15693 CrossRef CAS PubMed; (c) Y. Peng, T. Gong, K. Zhang, X. Lin, Y. Liu, J. Jiang and Y. Cui, Nat. Commun., 2014, 5, 4406 CrossRef CAS PubMed; (d) C. Gong, H. Guo, X. Zeng, H. Xu, Q. Zeng, J. Zhang and J. Xie, Dalton Trans., 2018, 47, 6917–6923 RSC.
- J. P. Abberley, R. Killah, R. Walker, J. M. D. Storey, C. T. Imrie, M. Salamończyk, C. H. Zhu and E. G. D. Pociecha, Nat. Commun., 2018, 9, 228 CrossRef PubMed.
- (a) X. Wu, H. B. Zhang, Z. X. Xu and J. Zhang, Chem. Commun., 2015, 51, 16331–16333 RSC; (b) P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Chem. Sci., 2016, 7, 3085–3091 RSC; (c) Y. H. Han, Y. C. Liu, X. S. Xing, C. B. Tian, P. Lin and S. W. Du, Chem. Commun., 2015, 51, 14481–14484 RSC; (d) Q. Sun, A. L. Cheng, K. Wang, X. C. Yi and E. Q. Gao, CrystEngComm, 2015, 17, 1389–1397 RSC; (e) B. Q. Song, C. Qin, Y. T. Zhang, X. S. Wu, L. Yang, K. Z. Shao and Z. M. Su, Dalton Trans., 2015, 44, 18386–18394 RSC; (f) R. X. Yao, X. Cui, J. Wang and X. M. Zhang, Chem. Commun., 2015, 51, 5108–5111 RSC.
- (a) M. V. Hobden, Nature, 1967, 216, 678 CrossRef CAS; (b) J. K. O'Loane, Chem. Rev., 1980, 80, 41–61 CrossRef; (c) K. Claborn, C. Isborn, W. Kaminsky and B. Kahr, Angew. Chem., Int. Ed., 2008, 47, 5706–5717 CrossRef CAS PubMed.
- D. B. Llewellyn, D. Adamson and B. A. Arndtsen, Org. Lett., 2000, 2, 4165–4168 CrossRef CAS.
- L.-L. Xu, H.-F. Zhang, M. Li, S. W. Ng, J.-H. Feng, J.-G. Mao and D. Li, J. Am. Chem. Soc., 2018, 140, 11569–11572 CrossRef CAS PubMed.
- (a) L. Meng, Q. Cheng, C. Kim, W.-Y. Gao, L. Wojtas, Y.-S. Chen, M. J. Zaworotko, X. P. Zhang and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 10082–10085 CrossRef CAS PubMed; (b) J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334–3337 CrossRef CAS PubMed; (c) J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS; (d) Y.-Z. Chen, Y.-X. Zhou, H. Wang, J. Lu, T. Uchida, Q. Xu, S.-H. Yu and H.-L. Jiang, ACS Catal., 2015, 5, 2062–2069 CrossRef CAS; (e) K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877–882 RSC; (f) Z. Yang, C. Zhu, Z. Li, Y. Liu, G. Liu and Y. Cui, Chem. Commun., 2014, 50, 8775–8778 RSC; (g) K. Mo, Y. Yang and Y. Cui, J. Am. Chem. Soc., 2014, 136, 1746–1749 CrossRef CAS PubMed; (h) K. D. Nguyen, C. Kutzscher, F. Drache, I. Senkovska and S. Kaskel, Inorg. Chem., 2018, 57, 1483–1489 CrossRef CAS PubMed; (i) X. Feng, H. S. Jena, K. Leus, G. Wang, J. Ouwehand and P. Van Der Voort, J. Catal., 2018, 365, 36–42 CrossRef CAS.
- (a) P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC; (b) P. Deria, W. Bury, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 1965–1968 RSC; (c) J.-S. Qin, S. Yuan, A. Alsalme and H.-C. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 33408–33412 CrossRef CAS PubMed; (d) C. Larabi and E. A. Quadrelli, Eur. J. Inorg. Chem., 2012, 2012, 3014–3022 CrossRef CAS; (e) D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS PubMed; (f) A. E. Platero-Prats, Z. Li, L. C. Gallington, A. W. Peters, J. T. Hupp, O. K. Farha and K. W. Chapman, Faraday Discuss., 2017, 201, 337–350 RSC; (g) P. Ji, K. Manna, Z. Lin, X. Feng, A. Urban, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 7004–7011 CrossRef CAS PubMed; (h) H. G. T. Nguyen, N. M. Schweitzer, C.-Y. Chang, T. L. Drake, M. C. So, P. C. Stair, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2014, 4, 2496–2500 CrossRef CAS.
- (a) W. J. Choi and C. Y. Choi, Biotechnol. Bioprocess Eng., 2005, 10, 167–179 CrossRef CAS; (b) B. M. Trost, Comprehensive Organic Synthesis; Pergamon Press, NewYork, 1st edn, 1991 Search PubMed; (c) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 836–840 CrossRef CAS.
- (a) P. M. Reis, C. A. Gamelas, J. A. Brito, N. Saffon, M. Gómez and B. Royo, Eur. J. Inorg. Chem., 2011, 2011, 666–673 CrossRef; (b) C. Dinoi, M. Ciclosi, E. Manoury, L. Maron, L. Perrin and R. Poli, Chem. – Eur. J., 2010, 16, 9572–9584 CrossRef CAS PubMed.
- F. J. Arnaiz, J. Chem. Educ., 1995, 72, A7 CrossRef CAS.
- M. N. Rode, S. S. Hussaini, G. Muley, B. H. Pawar and M. D. Shirsat, J. Optoelectron. Adv. Mater. Rapid Commun., 2008, 2, 855–858 CAS.
- L. Hadian-Dehkordi and H. Hosseini-Monfared, Green Chem., 2016, 18, 497–507 RSC.
- M. D. Hopkins, W. P. Schaefer, M. J. Bronikowski, W. H. Woodruff, V. M. Miskowski, R. F. Dallinger and H. B. Gray, J. Am. Chem. Soc., 1987, 109, 408–416 CrossRef CAS.
- (a) M. Farias, M. Martinelli and G. Koszeniewski Rolim, Appl. Catal., A, 2011, 403, 119–127 CrossRef CAS; (b) M. Bagherzadeh, M. Zare, M. Amini, T. Salemnoush, S. Akbayrak and S. Özkar, J. Mol. Catal. A: Chem., 2014, 395, 470–480 CrossRef CAS.
- J. Huang, L. Yuan, J. Cai and Z. Liu, Microporous Mesoporous Mater., 2015, 214, 121–126 CrossRef CAS.
- K. Berijani, A. Farokhi, H. Hosseini-Monfared and C. Janiak, Tetrahedron, 2018, 74, 2202–2210 CrossRef CAS.
- Y. Li, X. Fu, B. Gong, X. Zou, X. Tu and J. Chen, J. Mol. Catal. A: Chem., 2010, 322, 55–62 CrossRef CAS.
- H. Alper and M. Harustiak, J. Mol. Catal., 1993, 84, 87–92 CrossRef CAS.
- (a) F. E. Kühn, M. Groarke, É. Bencze, E. Herdtweck, A. Prazeres, A. M. Santos, M. J. Calhorda, C. C. Romao, I. S. Goncalves, A. D. Lopes and M. Pillinger, Chem. – Eur. J., 2002, 8, 2370–2383 CrossRef; (b) M. Minelli, J. H. Enemark, R. T. Brownlee, M. J. O'Connor and A. G. Wedd, Coord. Chem. Rev., 1985, 68, 169–278 CrossRef CAS.
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS.
- (a) N. Hosoya, R. Irie, Y. Ito and T. Katsuki, Synlett, 1991, 691–692 CrossRef CAS; (b) Y. Sun, N. Tang, X. W. Liu and W. S. Liu, Chin. Chem. Lett., 2004, 15, 973–976 CAS.
- F. Esnaashari, M. Moghadam, V. Mirkhani, S. Tangestaninejad, I. Mohammadpoor-Baltork, A. R. Khosropour and M. Zakeri, Mater. Chem. Phys., 2012, 137, 69–75 CrossRef CAS.
- N. Grover and F. E. Kuhn, Curr. Org. Chem., 2012, 16, 16–32 CrossRef CAS.
- (a) R. Kardanpour, S. Tangestaninejad, V. Mirkhani, M. Moghadamn, I. Mohammadpoor-Baltork and F. Zadehahmadi, J. Solid State Chem., 2015, 226, 262–272 CrossRef CAS; (b) S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork and K. Ghani, Inorg. Chem. Commun., 2008, 11, 270–274 CrossRef CAS.
- S. J. J. Titinchi, G. V. Willingh, H. S. Abbo and R. Prasad, Catal. Sci. Technol., 2015, 5, 325–338 RSC.
- W. R. Thiel, M. Angstl and N. Hansen, J. Mol. Catal. A: Chem., 1995, 103, 5–10 CrossRef CAS.
- S. Rayati, N. Rafiee and A. Wojtczak, Inorg. Chim. Acta, 2012, 386, 27–35 CrossRef CAS.
- M. Bagherzadeh, M. Zare, T. Salemnoush, S. Özkarb and S. Akbayrak, Appl. Catal., A, 2014, 475, 55–62 CrossRef CAS.
- M. Masteri-Farahani, F. Farzaneh and M. Ghandi, Catal. Commun., 2007, 8, 6–10 CrossRef CAS.
- (a) M. A. Villar and M. L. Ferreira, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1136–1148 CrossRef CAS; (b) M. Salavati-Niasaria and M. Bazarganipour, J. Mol. Catal. A: Chem., 2007, 278, 173–180 CrossRef.
- S. V. Gonzalez and P. Carlsen, General papers, ARKIVOC, 2011, 325–336 Search PubMed.
- C. Pereira, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mol. Catal. A: Chem., 2008, 283, 5–14 CrossRef CAS.
- (a) H. Noh, Y. Cui, A. W. Peters, D. R. Pahls, M. A. Ortuño, N. A. Vermeulen, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14720–14726 CrossRef CAS PubMed; (b) J. A. Gnecco, G. Borda and P. Reyes, J. Chil. Chem. Soc., 2004, 49, 179–184 CAS; (c) J. M. Sobczak and J. J. Ziółkowski, Appl. Catal., A, 2003, 248, 261–268 CrossRef CAS.
- (a) M. Tokles and J. K. Snyder, Tetrahedron Lett., 1986, 27, 3951–3954 CrossRef CAS; (b) Y. Tohru and N. Koichi, Chem. Lett., 1986, 15, 131–134 CrossRef.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. F. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Nat. Protoc., 2016, 11, 149–162 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR of cis-MoO2(acac)2 and L-tartaric acid, UV-vis spectra, 1H NMR, ICP analysis, N2 adsorption and BJH pore size distribution data, BET plots, structural properties of NU-1000 (N) and chiral catalyst [C-NU-1000-Mo], TGA, PXRD and FT-IR of the used catalyst, chiral GC chromatograms, table of enantioselective epoxidation examples of the styrene by various asymmetric heterogeneous catalysts. See DOI: 10.1039/c9cy00565j |
| This journal is © The Royal Society of Chemistry 2019 |