
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting photobioredox catalysis by morpholine electron donors under aerobic conditions†
Leticia C. P.
Gonçalves
 *a,
Hamid R.
Mansouri
a,
Shadi
PourMehdi
a,
Mohamed
Abdellah
bc,
Bruna S.
Fadiga
bd,
Erick L.
Bastos
d,
Jacinto
Sá
be,
Marko D.
Mihovilovic
a and
Florian
Rudroff
*a
*a,
Hamid R.
Mansouri
a,
Shadi
PourMehdi
a,
Mohamed
Abdellah
bc,
Bruna S.
Fadiga
bd,
Erick L.
Bastos
d,
Jacinto
Sá
be,
Marko D.
Mihovilovic
a and
Florian
Rudroff
*a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, 1060 Vienna, Austria. E-mail: leticia.goncalves@tuwien.ac.at; florian.rudroff@tuwien.ac.at
bPhysical Chemistry Division, Department of Chemistry, Ångström Laboratory, Uppsala University, 75120 Uppsala, Sweden
cDepartment of Chemistry, Qena Faculty of Science, South Valley University, 83523 Qena, Egypt
dDepartment of Fundamental Chemistry, Institute of Chemistry, University of São Paulo, 03178-200 São Paulo, Brazil
eInstitute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Poland
First published on 23rd April 2019
Abstract
Light-driven reduction of flavins, e.g. FAD or FMN, by sacrificial electron donors emerged as a convenient method to promote biocatalytic transformations. However, flavin activation has been restricted to oxygen-free conditions to prevent enzyme deactivation caused by reactive oxygen species (ROS). Herein, we show that the photoreduction of FMN by morpholines, including 3-(N-morpholino)propanesulfonic acid (MOPS), lessens the deactivation of the enoate reductase XenB from Pseudomonas sp. during the stereoselective asymmetric enzymatic reduction of a model α,β-unsaturated diketone under aerobic conditions, leading to a 91% GC-yield and a stereoselectivity greater than 94%. The kinetic stability of the thermolabile XenB was increased by more than 20-fold in MOPS buffer compared to that in Tris-HCl buffer, and a pronounced positive effect on the transition midpoint temperature was observed. The reactive form of the FMN photocatalyst is stabilized by the formation of a 3[FMN˙−–MOPS˙+] ensemble, which reduces the formation of hydrogen peroxide and other ROS in the presence of oxygen. These results contribute to broaden the application of photobiocatalytic transformations using flavin-dependent reductases.
Introduction
Enzymes have been used as biocatalysts in myriad chemical transformations of high economic interest.1,2 Flavin-dependent oxidoreductases are a class of versatile and efficient enzymes that have emerged as biocatalysts due to their high regio- and stereoselectivity. These enzymes often require reduced nicotinamide (e.g. NADH/NADPH) and flavin cofactors (e.g. FAD/FMN) to function as catalysts. Additionally, flavin cofactors have an important structural role, preventing enzyme deactivation.3 However, the addition of these expensive cofactors and the lack of operational stability of these enzymes, make their industrial application less attractive. A cheap alternative to the in situ reduction of flavin cofactors, and a substitute for the expensive NAD(P)H cofactors, is the use of light in the presence of a sacrificial electron donor (ED).4 It is expected that the ED damages neither the substrate nor the enzyme,1,5–7 but several applied amine-based EDs, such as ethylenediaminetetraacetic acid (EDTA) and triethanolamine (TEOA),8–10 can photodecompose leading to the formation of radicals that can ultimately influence the reaction outcome.4,8,11 Furthermore, the presence of molecular oxygen in flavin-dependent photobiocatalytic systems leads to the formation of deleterious reactive oxygen species (ROS),12–14 such as superoxide anions and hydrogen peroxide, which can deactivate flavin-dependent enzymes.3,15 Hydrogen peroxide is also a side product of water splitting, which has been used as an alternative to amine-based EDs to reduce flavin cofactors.3,16,17 Attempts to reduce ROS formation by using less oxidizable deazaflavin cofactors were unfruitful.18 Therefore, most biotransformations that do not require oxygen for catalysis have been carried out under anaerobic conditions, which is technically inconvenient.3-(N-Morpholino)propanesulfonic acid (MOPS) and 3-(N-morpholino)ethanesulfonic acid (MES) have been used as alternative EDs in flavin-dependent photoredox catalysis and photobiocatalysis due to their high photostability, poor complexation properties, and buffering capacity.14,18–21 We have shown that the MOPS buffer not only acts as an ED in photobiocatalytic systems but also enhances the stability of the cyclohexanone monooxygenase from Acinetobacter calcoaceticus (CHMOAcineto).22 MOPS stabilizes the active form of the flavin cofactor FAD, reducing the production of ROS by forming a spin correlated ion pair, viz., 3[FAD˙−–MOPS˙+].22
Herein, we show that morpholine-based EDs, especially MOPS, stabilize the thermolabile enoate reductase (ERED) XenB from Pseudomonas sp.23 and the FMN cofactor, promoting photobiocatalyzed stereoselective asymmetric reduction in the presence of oxygen. Stabilization of the species formed upon the photoexcitation of FMN is related to the formation of a long-lived 3[FMN˙−–MOPS˙+] ensemble. These results are of importance for the synthesis of building blocks and fine chemicals,24 allowing the application of photobiocatalytic methods for several flavin-dependent enzyme/flavin cofactor combinations independent of the presence or absence of oxygen.
Experimental
Standard photobiocatalytic reactions
Reactions were performed in a 96-well microplate sealed with a transparent film and thermostabilized at 30 ± 3 °C. The reduction of 1 mM or 10 mM ketoisophorone was performed in the presence of 100 μM FMN and 10 μM XenB. 100 mM MOPS buffer, pH 7.5, was employed as the electron donor. Control experiments were performed in 100 mM Tris-HCl, pH 7.5, in the presence or absence of 25 mM EDTA and in 100 mM sodium phosphate buffer, pH 7.5. The reactions were irradiated using a daylight lamp (300 W, Ultra Vitalux, Osram) placed at a distance of 20 cm from the 96 well-plate. Screening samples were taken immediately after mixing and after a certain time of incubation (t). Reference reactions using XenB were performed using 5 U mL−1 GDH/12 mM glucose in the presence of 0.25 mM NADP+ to recycle the enzyme. Control experiments were performed in the dark or in the absence of the enzyme. The reported yield refers to the GC-yield based on calibrated GC-data. Enzyme expression and purification are described in the ESI.†Reactions with minimal oxygen content
Reactions with minimal oxygen content were performed in 1.5 mL vials, at 35 ± 1 °C. The system containing 50 mM MOPS, pH 7.5, and 200 μM FMN was purged with a flow of argon for 1 h before irradiation. 10 mM ketoisophorone was added to the system under an argon flow and the system was purged for 10 min. 15 μM TsOYE or 15 μM XenB was added to the reaction mixture under argon and reactions were subjected to daylight irradiation with a lamp placed at a distance of 20 cm from the vial. Sampling was performed under an argon flow. Control experiments were conducted in the dark or in the absence of the biocatalysts. Reactions were also carried out in 100 mM Tris-HCl buffer, pH 7.5, or 100 mM phosphate buffer, pH 7.5. The reported yield refers to the GC-yield based on calibrated GC-data.Enzyme activity and stability
Enzyme activity was measured by monitoring the substrate-dependent decrease in NADPH absorbance at 340 nm (ε340 = 6.22 mM−1 cm−1) in 100 mM Tris-HCl or 100 mM MOPS, at pH 7.5. The activities of XenB or TsOYE were measured using 1 μM enzyme, 100 μM NADPH and 0.5 mM cyclohexenone or ketoisophorone (0.5 mM) as substrates. The oxidation of NADPH was monitored for 120 s at 30 °C in a Shimadzu spectrophotometer (UV-1800) equipped with a thermo-controlled 6-cell positioner (CPS-240A). Enzyme activity is defined as the amount of enzyme that oxidizes 1 μmol NADPH per minute under the specified conditions. Specific activities were calculated from the observed rate constants (kobs), which were calculated by linear fitting of the initial absorbance decrease at 340 nm over time. Stability measurements were performed by incubating 10 μM enzyme at 30 °C in 100 mM Tris-HCl or 100 mM MOPS, pH 7.5, containing 100 μM FMN. Aliquots were taken at different time points and added to a cuvette containing 0.1 mM NADPH and 0.5 mM substrate to test for catalytic activity. Non-linear curve fitting was carried out using the Origin Pro 2017 software (Originlab, 2017).Transition midpoint temperature (Tm)
The effect of the concentration of MOPS and Tris-HCl, pH 7.5, on the Tm of 54 μM XenB was evaluated by employing a nanoDSF device (Prometheus NT.48, NanoTemper Technologies GmbH). Capillaries were filled directly from respective solutions (10 μL). The samples were measured in the Prometheus NT.48 in a temperature range between 25 °C and 95 °C. Data analysis was performed using NT Melting Control software (NanoTemper Technologies GmbH). The Tm was determined by fitting the tryptophan fluorescence emission ratio of 350 nm to 330 nm using a polynomial function, in which the maximum slope is indicated by the peak of its first derivative.Nanosecond laser flash spectroscopy
Nanosecond transient absorption kinetics were recorded using a frequency tripled Q-switched Nd:YAG laser coupled with an OPO (Quantel Brilliant B) to obtain the desired wavelength for the pump light (5 ns pulse at a 10 Hz repetition rate, excitation at 455 nm). A detection system from Applied Photophysics (LKS80 Spectrometer) was used, equipped with a Xe arc lamp (pulsed or continuous wave), two monochromators and an R928-type PMT, read using a 600 MHz oscilloscope (Agilent Technologies Infinium 10 GSa s−1) for kinetics. The kinetics were monitored at the maximum resolution producing 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 point traces that were logarithmically oversampled starting from 50 points from the excitation about 10 ns after the pulse. The excitation light power was controlled by means of neutral density filters. Experiments were carried out in a quartz cuvette using 100 μM FMN in 100 mM MOPS or water, pH 7.5.
000 point traces that were logarithmically oversampled starting from 50 points from the excitation about 10 ns after the pulse. The excitation light power was controlled by means of neutral density filters. Experiments were carried out in a quartz cuvette using 100 μM FMN in 100 mM MOPS or water, pH 7.5.
Statistical analysis
All values are expressed as mean ± standard deviation (SD) of three independent replicates, unless otherwise stated. Statistical data analysis was carried out using a one-way analysis of variance (ANOVA) and Tukey's multiple comparison test (Prism 8, GraphPad 2019). In the bar graphs, *, **, ***, and **** indicate p < 0.05, p < 0.01, p < 0.001, and p < 0.0001, respectively. The level of statistical significance was taken to be p < 0.05.Results and discussion
Effect of morpholines on enzymatic photoreduction
First, we investigated the effect of MOPS on the photobiocatalytic reduction of ketoisophorone (1a) to levodione (1b) by using either TsOYE, the old yellow enzyme homologue from Thermus scotoductus SA-01 (ref. 10 and 16), or XenB, an enoate reductase from Pseudomonas sp.,23 using FMN as both an enzyme cofactor and a photocatalyst (Fig. 1). TsOYE is a thermostable enzyme, while XenB is thermolabile (half-life of 2.5 h at 30 °C in Tris-HCl buffer, Fig. S1†). Experiments were carried out under an argon atmosphere at 35 ± 1 °C, the same conditions reported by Mifsud and co-authors for the photobiocatalytic reduction of 1a in MOPS buffer using TiO2-mediated water splitting as a source of electrons and TsOYE as a biocatalyst.16 The reaction GC-yields using TsOYE and XenB in MOPS were similar to the values reported previously (63% (TsOYE) and 69% (XenB) vs. 66% (TsOYE)16) but were reached 3-times faster, i.e. 2 h of irradiation instead of 6 h (ref. 16) (Fig. 1a and b and S2†). The reaction using XenB is less stereoselective compared to that using TsOYE (ee: 74% (XenB) vs. 86% (TsOYE)), probably due to its faster enzyme deactivation (Fig. S1†), favoring the non-catalyzed photoreduction of the substrate (Fig. 1c). The relatively high reaction yield in the absence of the enzyme is related to the direct photoreduction of ketoisophorone, hence the low stereoselectivity. | ||
| Fig. 1 Photoinduced enzymatic reduction of ketoisophorone (1a) using TsOYE (a) or XenB (b), and control experiment in the absence of a biocatalyst (c). The consumption of the starting material (1a, SM cons.), product (1b) yield, and enantiomeric excess (ee) are given as percentage values; dashed lines correspond to the monoexponential fittings of the data. Reactions conditions: 1a (10 mM), enzyme (15 μM), FMN (200 μM), MOPS buffer (50 mM, pH 7.5), under an argon atmosphere and daylight irradiation (300 W) at 35 ± 1 °C. Experiments are performed at 35 °C,16 which is not the optimal operational temperature of TsOYE (65 °C); thus, its specific activity (Tables S1 and S2†) is lower than that previously reported.25,26 Rate constants are given in Table S3 and GC chromatograms are shown in Fig. S1.† The yield refers to the GC-yield and is based on calibrated GC-data. | ||
The thermolabile reductase XenB was selected as the model enzyme to further investigate the effect of MOPS and other morpholines on the photocatalytic reduction of 1a. The system XenB/FMN/1a was irradiated under aerobic conditions in the presence of MOPS, MES, 4-(2-hydroxyethyl)morpholine (HEMO), 4-methylmorpholine (MMO) or morpholine (MO). We used EDTA in Tris-HCl buffer as the positive control9,24,27 and sodium phosphate buffer (NaPi) in the absence of an ED as the negative control. After 1 h of irradiation, 1b was formed in >80% yield when the reaction was carried out in the presence of MOPS, MES, HEMO, MMO, EDTA and TEOA (Fig. 2a, Table S4†). The yields obtained in the presence of MMO and the positive control were identical, but slightly higher than those in the presence of MOPS, MES and HEMO. The stereoselectivities of the reaction in the presence of all morpholines, except MO, were as high as those in the presence of EDTA–Tris and TEOA (Fig. 2b, Table S4†). Tris-HCl improves the performance of EDTA when compared to the NaPi buffer.
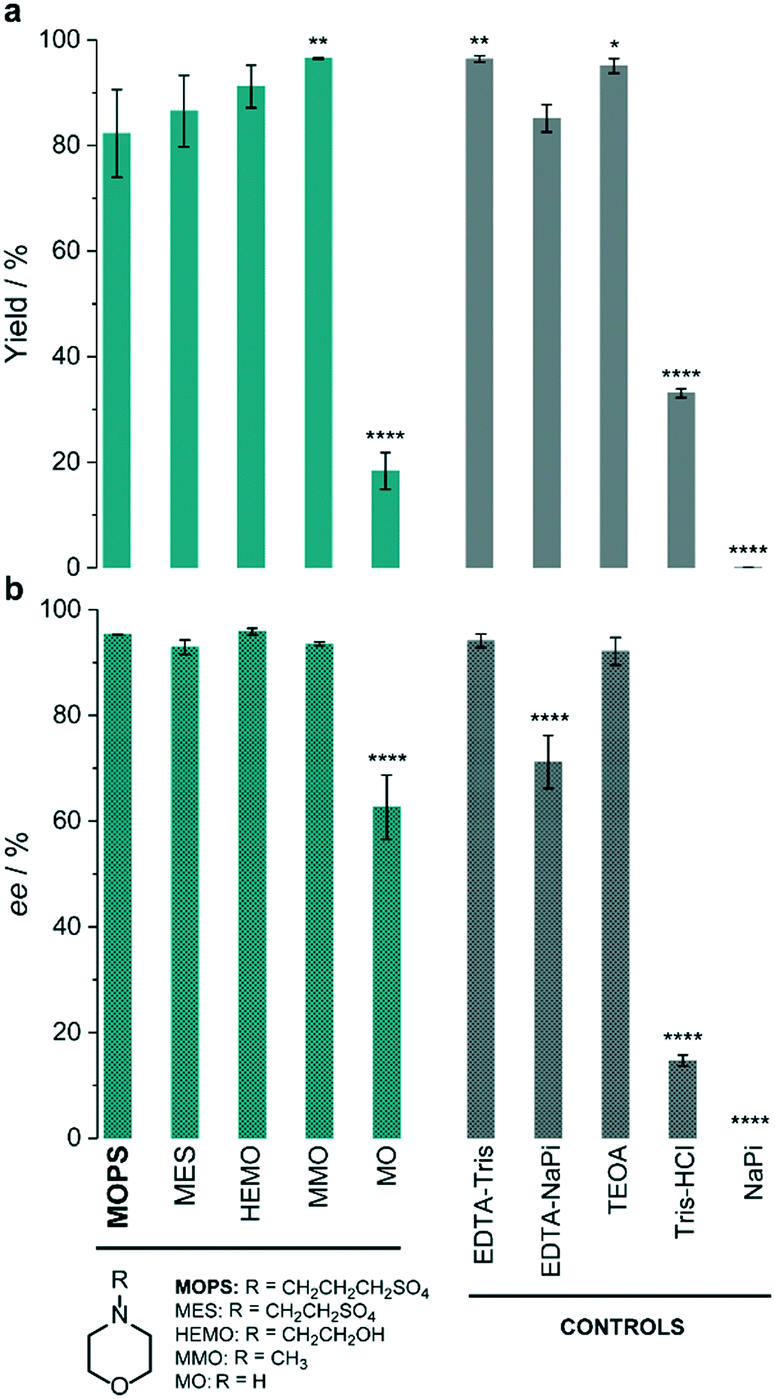 | ||
| Fig. 2 Effect of the electron donor on the photoinduced enzymatic reduction of ketoisophorone (1a) by XenB/FMN. The percentage yield (a) and enantiomeric excess (b) of levodione (1b) formation obtained in the presence of morpholines (green) are compared to those of control experiments (grey). Reaction conditions: 10 μM XenB; 100 μM FMN and 1 mM 1a; daylight irradiation for 1 h at pH 7.5. Asterisks indicate significant differences according to the analysis of variance (ANOVA) and comparison of each electron donor with MOPS using Tukey's test. The yield refers to the GC-yield and is based on calibrated GC-data. Mean values and SD are presented in Table S4.† | ||
Recently, we found that the MOPS buffer stabilized the cyclohexanone monooxygenase from Acinetobacter calcoaceticus (CHMOAcineto) and enhanced the photobiocatalytic Baeyer–Villiger oxidation of cyclohexanone to ε-caprolactone.22 The concentration of MOPS had a significant influence on the reaction yields. A similar trend, for both the reaction yield and stereoselectivity, was observed for the photoinduced reduction of 1a in the presence of XenB/FMN (Fig. 3a). The increase of the reaction yield as a function of the concentration of MOPS (on a log scale) showed a sigmoid profile, and a maximum yield of 85 ± 5% was reached when the reaction was carried out in 50–500 mM MOPS buffer. The highest stereoselectivity (approx. 93% ee) was obtained when the concentration of MOPS was within the 10–100 mM range. Therefore, 100 mM MOPS was defined as the standard buffer/ED concentration for all subsequent experiments with the XenB/FMN/1a system. The non-catalyzed photoreduction seems to gain importance at the lowest and highest MOPS concentrations, probably due to the inactivation/degradation of the enzyme.16 To test this hypothesis, we investigated the effect of the concentration of MOPS on the specific activity of XenB by keeping the enzyme solution at 30 °C for a defined period of time (hereafter incubation period, t), before performing the reduction of 1a (Fig. 3b). The enzyme activities were measured using fresh solutions of XenB in 0.5–25 mM MOPS (t = 0 min, no incubation) and were found to be identical, but dropped drastically at higher MOPS concentrations (25–500 mM). The incubation period only affects the enzyme activity when the concentration of MOPS is lower than 5 mM (Fig. 3b). Therefore, the decrease in specific enzyme activity at high MOPS concentrations is probably not related to enzyme stabilization since changes in the incubation period do not affect the specific activity of XenB.
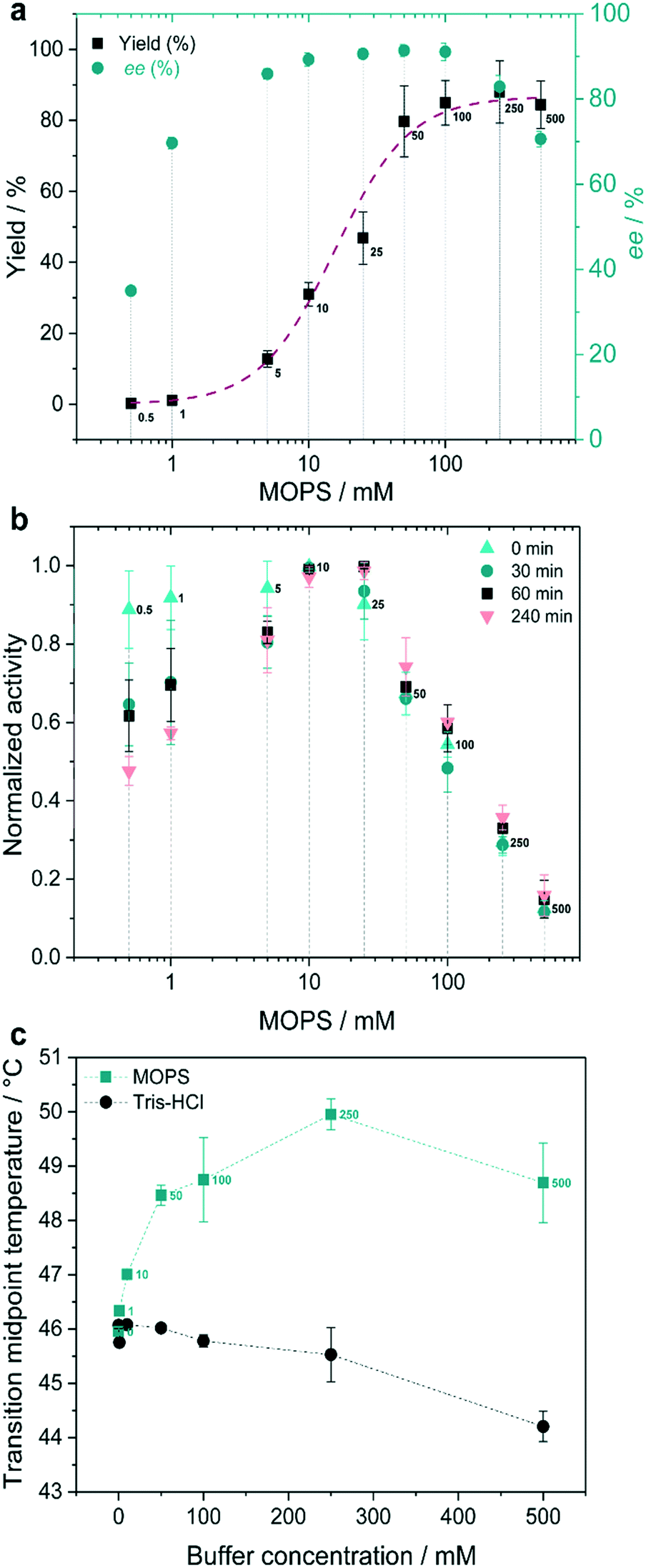 | ||
| Fig. 3 MOPS as the electron donor. (a) Effect of the concentration of MOPS on the reaction yield and enantiomeric excess (ee) of (R)-1b. (b) Effect of incubation time on the normalized specific enzyme activity. (c) Transition midpoint temperatures (Tm) of XenB at various concentrations of MOPS and Tris-HCl buffers. Reaction conditions: 1 mM 1a, 10 μM XenB and 100 μM FMN, 1 h of daylight irradiation at pH 7.5. The yield refers to the GC-yield and is based on calibrated GC-data. | ||
Finally, the effect of the concentration of XenB and FMN on the reduction of 1a (1 mM) was investigated (Fig. S3†). The reaction yield, selectivity and initial velocity depended on the enzyme concentration (Fig. S3a and S4a and b†) and the maximum yield was obtained by employing 40 μM XenB (Fig. S3 and S4†). Above 100 μM, the concentrations of FMN do not affect the reaction yield and selectivity (Fig. S3b†) and the NADP+ cofactor is not required for catalysis (Fig. S3c†). Under these conditions (1 mM 1a, 40 μM XenB, and 100 μM FMN), the desired product 1b was obtained in 91% yield and >94% ee after 40 min under daylight irradiation and in the presence of aerial oxygen (Fig. S5†).
Mechanism of catalyst stabilization by MOPS
Intrigued by these results, we wanted to gain a deeper understanding of the function of MOPS in this photobiocatalytic process. The photoexcitation of flavins in the presence of oxygen results in a series of events related to energy and electron transfers that ultimately produce detrimental ROS, including hydrogen peroxide and superoxide anions (O2˙−), which compromise the enzyme function (Fig. 4a). The concentration of H2O2 produced in Tris-HCl buffer increases over time and is roughly 10 times higher than that measured in MOPS, i.e. 144 μM vs. 15.5 μM after 4 h of irradiation (Fig. S6†). The activity of XenB remains constant for more than 50 h in MOPS, while the activity of the enzyme is halved after 2.5 ± 0.1 h in Tris-HCl and 9.4 ± 2.4 h in EDTA/Tris-HCl (Fig. 4b). | ||
Fig. 4 MOPS enhances the stability of XenB and FMN. (a) Oxidized flavin (Flox, e.g. oxidized FMN) is photoexcited producing  , which can populate the triplet state , which can populate the triplet state  via intersystem crossing (ISC). Triplet-excited flavin can oxidize the sacrificial electron donor (e.g. MOPS or EDTA) and/or react with oxygen to produce detrimental ROS. H2O2 was quantified in this work (Fig. S6†). The photoreduction of the flavin leading to a flavin anion radical (Fl˙−) is required for biocatalysis, whereas ROS production can deactivate the enzyme. (b) Residual catalytic activity of XenB in MOPS, EDTA/Tris-HCl and Tris-HCl, pH 7.5, 30 °C. (c) Effect of the buffer/ED on the remaining absorption at 446 nm after 1 h and 4 h of daylight irradiation; inset: absorption spectrum of FMN (Fig. S7†). (d) Effect of the concentration of MOPS on the remaining absorption at 446 nm after 1 h of irradiation (Fig. S9†). via intersystem crossing (ISC). Triplet-excited flavin can oxidize the sacrificial electron donor (e.g. MOPS or EDTA) and/or react with oxygen to produce detrimental ROS. H2O2 was quantified in this work (Fig. S6†). The photoreduction of the flavin leading to a flavin anion radical (Fl˙−) is required for biocatalysis, whereas ROS production can deactivate the enzyme. (b) Residual catalytic activity of XenB in MOPS, EDTA/Tris-HCl and Tris-HCl, pH 7.5, 30 °C. (c) Effect of the buffer/ED on the remaining absorption at 446 nm after 1 h and 4 h of daylight irradiation; inset: absorption spectrum of FMN (Fig. S7†). (d) Effect of the concentration of MOPS on the remaining absorption at 446 nm after 1 h of irradiation (Fig. S9†). | ||
The photodegradation of flavins is another obstacle for the broad application of flavin-photosensitized biocatalysis.28 FMN in Tris-HCl or phosphate buffers is decomposed upon irradiation, as evidenced by the decrease of the absorption at 446 nm.14 After 1 h of irradiation, the remaining FMN absorption in the presence of MOPS or EDTA/Tris-HCl is roughly 60%. However, after 4 h of continuous daylight irradiation, the remaining absorption at 446 nm in MOPS buffer does not change, although it drops by half in EDTA/Tris-HCl buffer (Fig. 4c, S7 and S8†). Furthermore, the remaining absorption at 446 nm is close to 100% when the concentration of MOPS is increased over 250 mM, indicating that the concentration of MOPS has a positive effect on the photostability of FMN (Fig. 4d, S9 and S10†).
The photoprotective action of MOPS in the XenB/FMN system could be related to the formation of a long-lived 3[FMN˙−–MOPS˙+] ensemble analogous to the spin correlated ion pair reported to prevent the deactivation of FAD-dependent CHMOAcineto.22 To verify this hypothesis, nanosecond laser flash spectroscopy experiments were carried out to determine the lifetime of the species formed upon excitation of FMN. The bleaching at 455 nm and the decays at 325 nm and 625 nm were ascribed to the ground-state recovery of FMN and to the depletion of the FMN anionic semiquinone (FMN˙−) and the triplet-excited FMN (3FMN*), respectively, as inferred from our previous results with FAD.22 The ground-state recovery of FMN is 13-times slower in MOPS than in water at the same pH (Fig. 5a) suggesting that the MOPS buffer stabilizes the species formed upon the photoexcitation of FMN. 3FMN* originates from 1FMN* by intersystem crossing29 and is reduced by the sacrificial ED producing the corresponding anionic semiquinone (FMN˙−).30 MOPS almost doubles the lifetime of 3FMN* and FMN˙− compared to the control in water (Fig. 5b and c). This result indicates that the stabilization of FMN˙− ultimately ‘freezes’ the photoinduced states preventing/retarding the oxidative pathways leading to ROS formation (Fig. 6). Consequently, enzyme stabilization by MOPS in the CHMOAcineto/FAD oxidative system22 and in the XenB/FMN reductive system likely involves the same 3[flavin˙−–MOPS˙+] ensemble.
 | ||
| Fig. 5 Effect of MOPS on the transient absorption kinetics. Data were extracted at (a) 455 nm, (b) 625 nm, and (c) 325 nm, which were ascribed to FMN, 3FMN* and FMN˙−, respectively. Experimental conditions: excitation at 445 nm, kinetics monitored for 100 μs, 100 μM FMN, 100 mM MOPS, pH 7.5. | ||
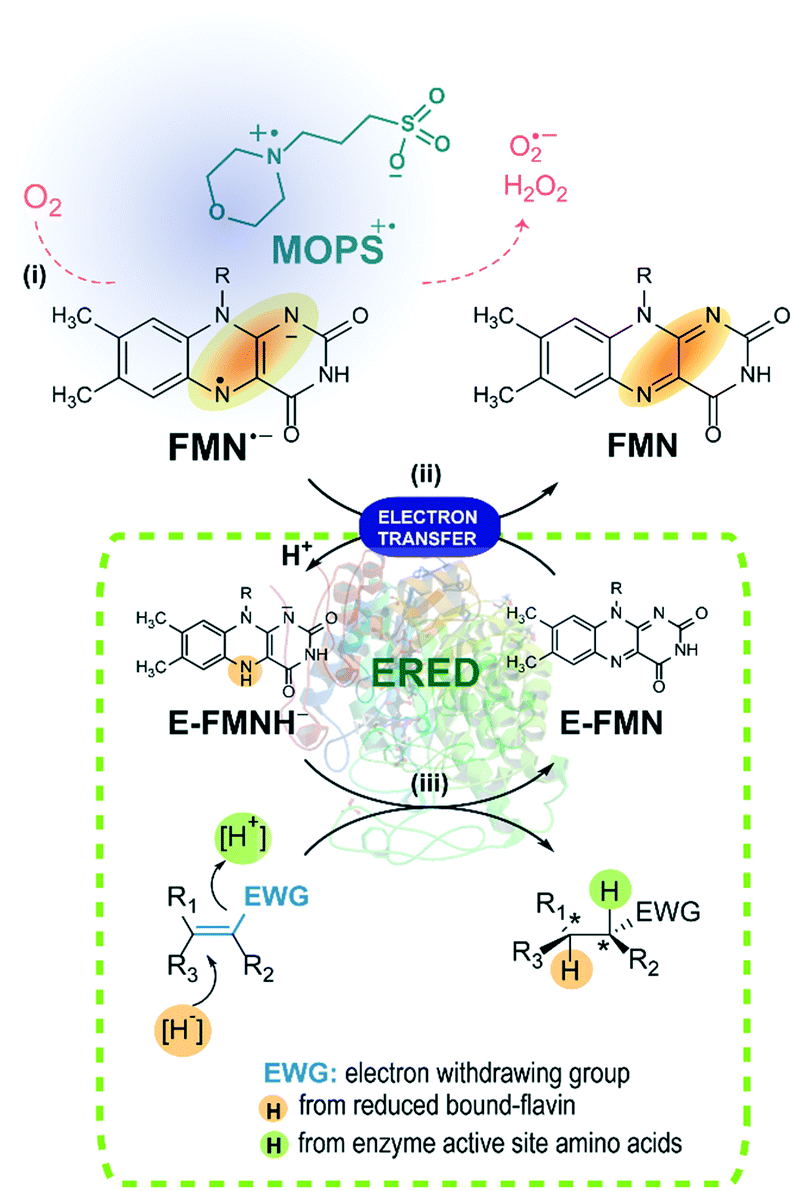 | ||
| Fig. 6 Activation of EREDs through the photoreduction of the FMN photocatalyst. (i) The FMN anionic semiquinone generated upon light irradiation of FMN in the presence of the electron donor MOPS. In the presence of MOPS, the formation of ROS is minimized. (ii) The enzyme bound FMN (E-FMN) required for the biocatalysis is reduced by FMN˙− leading to E-FMNH−. (iii) The stereoselective reduction of the substrate to the desired product is achieved in the presence of the enoate reductase (ERED). | ||
Conclusions
The morpholine buffer MOPS enhances the stability of the thermolabile XenB and the photocatalyst FMN, activating the photobiocatalyzed asymmetric reduction of ketoisophorone under aerobic conditions. Together with our previous data on the oxidation of cyclohexanone using a thermolabile monooxygenase and FAD, these results show that MOPS enhances both the stability of the biocatalyst and the flavin photocatalyst, broadening the applicability of flavin-based photobiocatalysis. In addition, the photoinduced reduction of FMN by morpholine derivatives leads to enzyme activation without requiring other electron donors making photobiocatalytic systems simpler. The ability of MOPS to form 3[flavin˙−–MOPS˙+] ensembles upon irradiation, in which flavin refers to either FAD or FMN, guarantees the use of this system under aerobic conditions without the oxidative deactivation of the enzyme, since the formation of deleterious ROS is precluded.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Austrian Science Fund FWF (grant no. M1948-N28) and by Sprint-FAPESP (2017/50130-9). We thank Dr. Thomas Bayer (University of Greifswald) for the discussions regarding the TsOYE cloning strategies, Dipl.-Ing. Sandra Orman (TU Wien) for the Ocean Optics spectrophotometer measurements and BSc. Viktor Savic (TU Wien) for the synthesis of the levodione.Notes and references
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef
.
- C. M. Clouthier and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585–1605 RSC
- L. C. P. Goncalves, D. Kracher, S. Milker, M. J. Fink, F. Rudroff, R. Ludwig, A. S. Bommarius and M. D. Mihovilovic, Adv. Synth. Catal., 2017, 359, 2121–2131 CrossRef CAS
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS PubMed
- C. T. Walsh and T. A. Wencewicz, Nat. Prod. Rep., 2013, 30, 175–200 RSC
- D. Monti, G. Ottolina, G. Carrea and S. Riva, Chem. Rev., 2011, 111, 4111–4140 CrossRef CAS PubMed
- V. Piano, B. A. Palfey and A. Mattevi, Trends Biochem. Sci., 2017, 42, 457–469 CrossRef CAS PubMed
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS
- A. Taglieber, F. Schulz, F. Hollmann, M. Rusek and M. T. Reetz, ChemBioChem, 2008, 9, 565–572 CrossRef CAS PubMed
- S. H. Lee, D. S. Choi, M. Pesic, Y. W. Lee, C. E. Paul, F. Hollmann and C. B. Park, Angew. Chem., Int. Ed., 2017, 56, 8681–8685 CrossRef CAS PubMed
- B. Höbel and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 1998, 509–514 RSC
- D. Holtmann and F. Hollmann, ChemBioChem, 2016, 17, 1391–1398 CrossRef CAS PubMed
- V. Massey, J. Biol. Chem., 1994, 269, 22459–22462 CAS
- S. Alonso-de Castro, E. Ruggiero, A. Ruiz-de-Angulo, E. Rezabal, J. C. Mareque-Rivas, X. Lopez, F. Lopez-Gallego and L. Salassa, Chem. Sci., 2017, 8, 4619–4625 RSC
- J. A. Imlay, Nat. Rev. Microbiol., 2013, 11, 443–454 CrossRef CAS PubMed
- M. Mifsud, S. Gargiulo, S. Iborra, I. W. C. E. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5, 3145 CrossRef PubMed
- W. Zhang, E. Fernandez-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55–62 CrossRef PubMed
- M. M. C. H. van Schie, S. H. H. Younes, M. C. R. Rauch, M. Pesic, C. E. Paul, I. W. C. E. Arends and F. Hollmann, Mol. Catal., 2018, 452, 277–283 CrossRef CAS
- C. J. Seel, A. Králík, M. Hacker, A. Frank, B. König and T. Gulder, ChemCatChem, 2018, 10, 3960–3963 CrossRef CAS
- W. J. Ferguson, K. I. Braunschweiger, W. R. Braunschweiger, J. R. Smith, J. J. McCormick, C. C. Wasmann, N. P. Jarvis, D. H. Bell and N. E. Good, Anal. Biochem., 1980, 104, 300–310 CrossRef CAS PubMed
- N. E. Good, G. D. Winget, W. Winter, T. N. Connolly, S. Izawa and R. M. Singh, Biochemistry, 1966, 5, 467–477 CrossRef CAS PubMed
- L. C. P. Gonçalves, H. R. Mansouri, E. L. Bastos, M. Abdellah, B. S. Fadiga, J. Sá, F. Rudroff and M. D. Mihovilovic, Catal. Sci. Technol., 2019, 9, 1365–1371 RSC
- C. Peters, R. Kölzsch, M. Kadow, L. Skalden, F. Rudroff, M. D. Mihovilovic and U. T. Bornscheuer, ChemCatChem, 2014, 6, 1021–1027 CrossRef CAS
- M. M. Grau, J. C. van der Toorn, L. G. Otten, P. Macheroux, A. Taglieber, F. E. Zilly, I. W. C. E. Arends and F. Hollmann, Adv. Synth. Catal., 2009, 351, 3279–3286 CrossRef CAS
- D. J. Opperman, L. A. Piater and E. van Heerden, J. Bacteriol., 2008, 190, 3076–3082 CrossRef CAS PubMed
- D. J. Opperman, B. T. Sewell, D. Litthauer, M. N. Isupov, J. A. Littlechild and E. van Heerden, Biochem. Biophys. Res. Commun., 2010, 393, 426–431 CrossRef CAS PubMed
- F. Hollmann, A. Taglieber, F. Schulz and M. T. Reetz, Angew. Chem., Int. Ed., 2007, 46, 2903–2906 CrossRef CAS PubMed
- M. Insińska-Rak and M. Sikorski, Chem. – Eur. J., 2014, 20, 15280–15291 CrossRef PubMed
- T. E. Swartz, S. B. Corchnoy, J. M. Christie, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, J. Biol. Chem., 2001, 276, 36493–36500 CrossRef CAS PubMed
- C. A. Dodson, P. J. Hore and M. I. Wallace, Trends Biochem. Sci., 2013, 38, 435–446 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental conditions, supplementary figures and references. See DOI: 10.1039/c9cy00496c |
| This journal is © The Royal Society of Chemistry 2019 |