
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn isothermality in some commonly used plug flow reactors for X-ray based investigations of catalysts†
Mark A.
Newton
 *a,
Stefano
Checchia
b,
Amy J.
Knorpp
a,
Dragos
Stoian‡
c,
Wouter
van Beek
c,
Hermann
Emerich
c,
Alessandro
Longo
d and
Jeroen A.
van Bokhoven
*ae
*a,
Stefano
Checchia
b,
Amy J.
Knorpp
a,
Dragos
Stoian‡
c,
Wouter
van Beek
c,
Hermann
Emerich
c,
Alessandro
Longo
d and
Jeroen A.
van Bokhoven
*ae
aDepartment of Chemistry and Applied Biosciences, Institute for Chemical and Bioengineering, ETH Zürich, Switzerland. E-mail: mark.newton@chem.ethz.ch; jeroen.vanbokhoven@chem.ethz.ch
bID 15, European Synchrotron Radiation Facility, 71, avenue des Martyrs, CS 40220, 38043 Grenoble, Cedex 9, France
cSwiss Norwegian Beamlines, CS40220, 38043 Grenoble, CEDEX 9, France
dDutch-Belgian Beamline, CS40220, 38043 Grenoble, CEDEX 9, France
eLaboratory for catalysis and sustainable chemistry, Paul Scherrer Institute (PSI), Villigen, 5232 Switzerland
First published on 24th May 2019
Abstract
We compare two reactor setups commonly used to make operando measurements of catalyst behavior using X-rays in terms of the degree to which they may be considered to yield radial and axial isothermality. We use axial and radial Cu K-edge XANES mapping of copper containing zeolites after high temperature activation (773 K) in oxygen and subsequent exposure to methane (473 K), to demonstrate the ease with which it is possible to enter into substantially non isothermal situations using air blower systems of a type commonly utilized. The implications of these observations for the attainment of non-representative structure–function relations are discussed. We further show, using this chemistry and infrared imaging, how this unwanted situation can be ameliorated by adopting a different type of reactor and point out that, even in this case isothermality should not be taken for granted and should be actively verified prior to experimental investigation.
Introduction
In the last fifteen years, “operando” methods, that is to say measuring structure and performance at the same time,1–3 have gained much credence as a highly effective means to establish quantitative structure activity relationships (QSARS) that are fundamental to improved understanding of how materials work and, from that point, how to improve and optimize their function. At the same time, the use of increasingly powerful X-ray resources and techniques has become more and more important to study of a wide range of functional materials within the framework of the operando philosophy. As a result, a plethora of experimental setups have been demonstrated.In the world of catalysis, these can be broadly divided into two groups: those that present the sample in the form of a pressed and self-supporting disk, through and around which a feed flows; and those that prefer the use of packed powder beds through which a feedstock is passed. The former present the sample for study but, as the reaction feed flows around the disk, and the disk itself is of unknown porosity, they do no attain the fundamental requirements of plug flow,4,5 and cannot be regarded as being representative of real catalytic reactors. The latter, as the feed must flow through the entirety of the sample, are more representative of catalytic reactors insofar as they aim to achieve conditions of plug flow.4,5
Depending upon the preferred mode of operation of a given process, the concepts of plug flow operation, along with that of the continuously stirred tank reactor (CSTR), are fundamental to the kinetic characterization of catalysis.4,5 Plug flow provides a framework whereby systems, may be reliably quantified and compared in terms of fundamental parameters, such as contact times/space velocities, conversions, selectivity, and kinetics.
Kinetics, be they for catalytic reactions themselves, or for other aspects of importance within catalysis (for example, phase transformations in active and supporting components, changes in the domain sizes of those components, and so on), define the state of a catalyst at any given point in its lifetime and within the limits proscribed by thermodynamics. As such, proper establishment of QSARS can only achieved by experiments that can extract reliable structural and reactivity based information; fundamental aspects of characterization that are most often realised in very different manners (vide infra).
Ideally, in any measurement that aims to achieve QSARS, no gradients in flow, pressure, and temperature, should exist prior to the study of the catalysis to hand. This is not to say that the reactor system will remain gradientless during operation. Gradients in the concentration of reactants and products, and therefore in the temperature and state of the catalyst, can develop as a result of catalytic conversion, particularly in the case of highly exothermic reactions. However, it is axiomatic, that in the absence of any such chemistry, the reactor should be gradient-free in all dimensions and over the entire sample volume probed; for a tubular reactor that is to say, axially and radially.
A question that obviously arises is: to what degree these fundamentals are achieved within the various experimental solutions developed for operando study of catalysis using X-rays? In this respect some very laudable and often detailed studies have appeared6–15 regarding how appropriate some reactor designs are for reliably obtaining different types of information, and to what degree some reactors might be deemed to conform to the required starting point. In the current case we shall specifically address the notion of isothermality, how well it may be achieved in two types of commonly used tubular microreactors.
The correct establishment of a reaction temperature, and isothermality is one of the fundamental pillars upon which catalyst behavior is understood. Rates of reaction, and specifically the rate constant (k), are temperature dependent i.e.
| k = Ae−Ea/RT | (1) |
If we consider a catalyst bed having both radial and axial extension, and given constant concentration of reactants/products within these dimensions (limit of low conversion), it is intuitively easy to see that if the bed is isothermal, one can sample the bed in a variety of manners and anywhere within it and obtain results that are both consistent, and reflective of the true behavior of the material under study.
Even if the quantitative restoration of kinetic data is not the principal objective of the experiment, as kinetics pertaining all sorts of chemical transformations may be at work in a given catalyst system, it is the kinetics of (potentially) many different process that will define the actual state of the catalyst under study. Concurrently the existence of pre-existing gradients in temperature will act distort the apparent behavior across a catalyst bed according to how aspects of structure and function are being accessed.
Here we must further take note of differences that generally exist between methods used to establish the performance and structure of the sample. By and large (there are notable, if not generally available, exceptions16,17) the most commonly applied methods for establishing reactivity – for example, by measuring gas concentrations at the reactor outlet using downstream mass spectrometry or chromatography – sample the entire volume of the reactor bed. On the other hand, the probes that are used to determine structure or speciation (e.g. vibrational spectroscopies and X-ray methods) generally sample very different and technique-specific, volumes within a bed.18–20 This, in itself, is an important consideration in the design of any experiment but particularly those that seek to make combined and synchronous studies using such different methods.20
We might further note that, in respect of the use of X-rays, the evolution of synchrotron sources has resulted in a significant trend towards smaller and smaller X-ray beam sizes. Whilst this aids, and indeed permits, many new forms of study in many areas, in terms of establishing QSARS within catalytic reactors, this evolution results in an ever greater disconnect between how we measure structure and structural change, and how, in general, we measure the reactive results of this structure/structural change.
Given this increasing differential in generally available sampling methods, fundamental issues, such as obtaining isothermal operation, are even more critical to achieve if results from different probes are to be accurately and quantitatively correlated.
Fig. 1 schematically illustrates two types of tubular, nominally plug flow reactors; a consideration of some of the thermal properties of some other types of reactor, and how to measure them are given as ESI.†
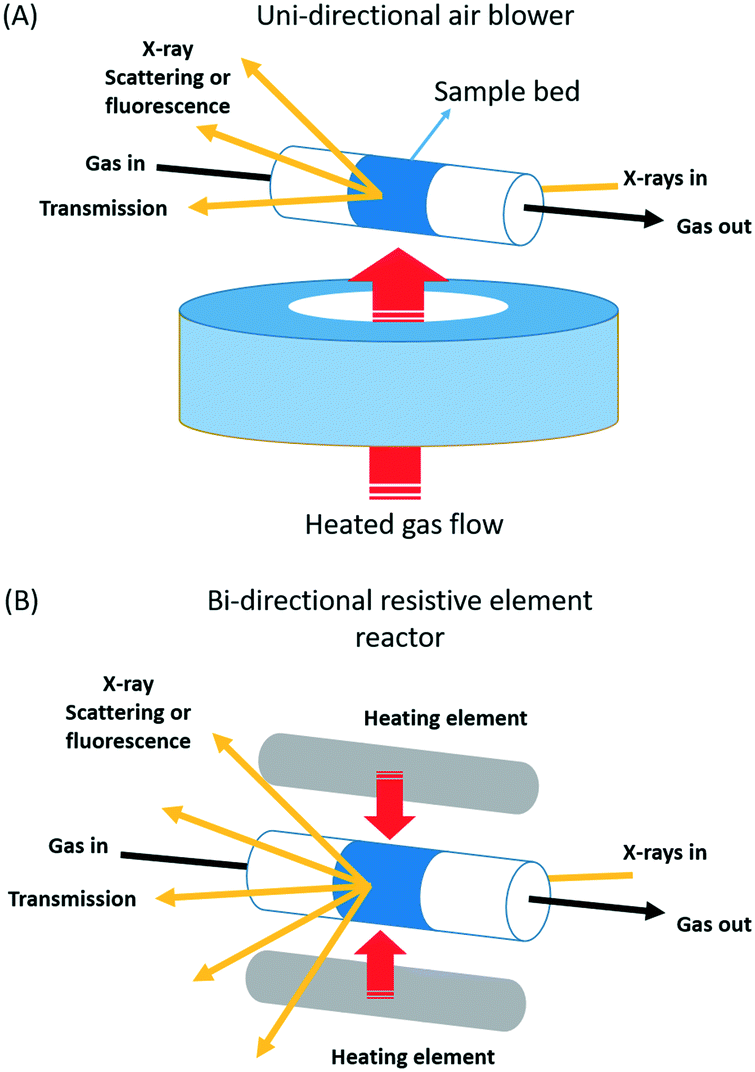 | ||
| Fig. 1 Schematic illustration of the two types of nominally plug flow reactor arrangements studied: (A): unidirectional air blower systems of the type first demonstrated by Clausen et al.6,21 and used as provided by the beamlines in this study. (B) A bi-directionally heated reactor system first developed for total X-ray scattering/PDF by Chupas and co-workers,23 a modified version of which was used in the current study. | ||
The first type the uni-directional air blower, (Fig. 1(A)) was initially implemented and demonstrated by Clausen and co-workers6,21 for the application of combined quick EXAFS and X-ray diffraction (XRD) studies of working catalysts. Since then this sort of versatile solution to the issue of sample presentation has become very widespread and popular for operando study, and many systems of this type can be found at synchrotrons around the world.
The reasons for the proliferation of air-blower systems in X-ray study can be found in a number of attractive properties that such an arrangement yields. The open nature of the sample presentation means that this sort of configuration is applicable with to a wide range of experimental circumstances and geometries. Experiments that require simple X-ray transmission (e.g. transmission XAFS), along with those that require significant solid angles of detection with varying geometry to be achieved (e.g. X-ray diffraction, total X-ray scattering, and emission spectroscopies based on fluorescence detection), or sample rotation (tomography), can all be achieved with an essential equanimity using this sort of arrangement. The range of temperatures that can be studied is also very wide, from cryogenic (90 K) to, ca. 1000 K.
In its original application and implementation6 the uni-directional air blower/capillary reactor was thoroughly assessed through direct comparison in performance for methanol synthesis with that obtained from a laboratory scale pilot reactor. However, since then, such comparisons are rarely made and the use of these sorts of reactor has been often been accompanied by significant deviations from this original and highly system specific approach (vide infra).
Further, and for the most part, detailed assessments of whether subsequently developed sample environments are indeed radially and axially isothermal are not as common as perhaps they ought to be, though examples do exist where this sort of exercise has been undertaken.8–15 There are also some elegant studies using similar systems that make use of infrared thermographic imaging to follow catalyst light off and the migration of reactive wavefronts through catalyst beds for suitably exothermic reactions.22 In these cases, as the main method used is thermal imaging, any pre-existing thermal gradients that might exist should be immediately apparent. However, outside of these examples, isothermality appears to be largely assumed rather than actively demonstrated.
The second type of reactor we shall investigate is a more recent solution developed for operando total X-ray scattering/pair distribution function measurements (PDF) by Chupas and co-workers.23 It too is extremely versatile in application and can be used in the same wide range of circumstances. The central difference between these two approaches lies in the means by which sample heating is achieved: in the former case a heated air stream is passed across the sample contained from one side; in the latter resistively heated elements are placed at either side of the sample and heating is therefore bi-directional.
To make our investigations we shall use two approaches; one based upon reactive chemistry, the other on thermal imaging using infrared. In the former case we shall make use of the formation of CuI that many XAFS based studies24–34 have shown results from the activation of methane by CuII-oxo species hosted within mordenite (MOR), that themselves are formed through calcination of the copper exchanged MOR at high temperatures (673–773 K).
This chemistry of itself is challenging in a number of ways, and the success of an experiment, in terms of having actually measured the results of the chemistry, is not easy to assess. In these systems, the products of the interaction of the activated CuII oxo-species with methane remain inside the zeolite and may only be quantified by post factum aqueous extraction/gas chromatography or through steaming using a wet gas flow.24–34
The X-ray experiment is, therefore (most often) conducted in an reactively “blind” manner. As such, to achieve reliable and meaningful results, all aspects of the experiment, including the sample environment, need to understood, controlled, and characterized to a very high level.
Indeed, previous Cu K-edge XAS studies of this process for Cu/MOR have yielded a remarkable degree of variation in results in Cu/MOR samples that are notionally very similar (e.g. similar Si/Al ratio and copper loading), and that have been treated in nominally similar fashions.26,28–31 Moreover, this variation has resulted in diametrically opposed views regarding underlying reaction mechanism and the structure of the catalytically active phase.
It is therefore imperative to understand the source of these variations and whether they result from subtle differences between the materials studied or other aspects of the (numerous) approaches to sample presentation used to make these studies. It is this literature-derived observation, combined with our own experience, which has resulted in the studies that we now report. It is these studies that have also provided the foundation for the quantitative cross correlations of independently measured methanol yields and Cu K-edge XAS that we have recently presented.32,34
Results
Fig. 2 shows the results of an experiment designed investigate whether or not isothermality is achieved in a given (uni-directional air blower, Fig. 1(A)) reactor system. More precisely, a Cu/MOR sample (sieved to a 75–100 μm fraction) is presented in a larger than usual packed bed of 10 mm in length inside a large quartz tube of 2 mm diameter centered 2 mm above the heating element. This sample has then been heated (10 K min−1) to 723 K in flowing oxygen (20 ml min−1) and maintained there for about 30 minutes. The sample was then cooled to 473 K under oxygen. The system was then briefly flushed with argon (20 ml min−1) before the gas feed was switched to methane (20 ml min−1). During the switch, and subsequent to it, the development of the speciation of the copper was measured (using Cu K-edge XANES) at a single point (defined by the dimensions of the X-ray beam) within the reactor for ca. 30–60 min until the spectra acquired became invariant with time. Axial and radial mapping of the bed was then undertaken using an X-ray beam 460 × 750 μm2 in size. These measurements were made with no thermocouple present in the bed. Instead, in this case, we have relied upon a previously made “single-point” calibration of the temperature achieved inside the packed tube versus the temperature given by the control thermocouple that resides within the air-blower itself. Furthermore, the sample bed used for this exercise is, in terms of its axial dimension (10 mm) is ca. twice the size of the sorts of bed we would normally use. The reason for this is simply that, in this case, we wish to assess the reactor across the entirety of the heating zone provided and to find out where any isothermal area may exist.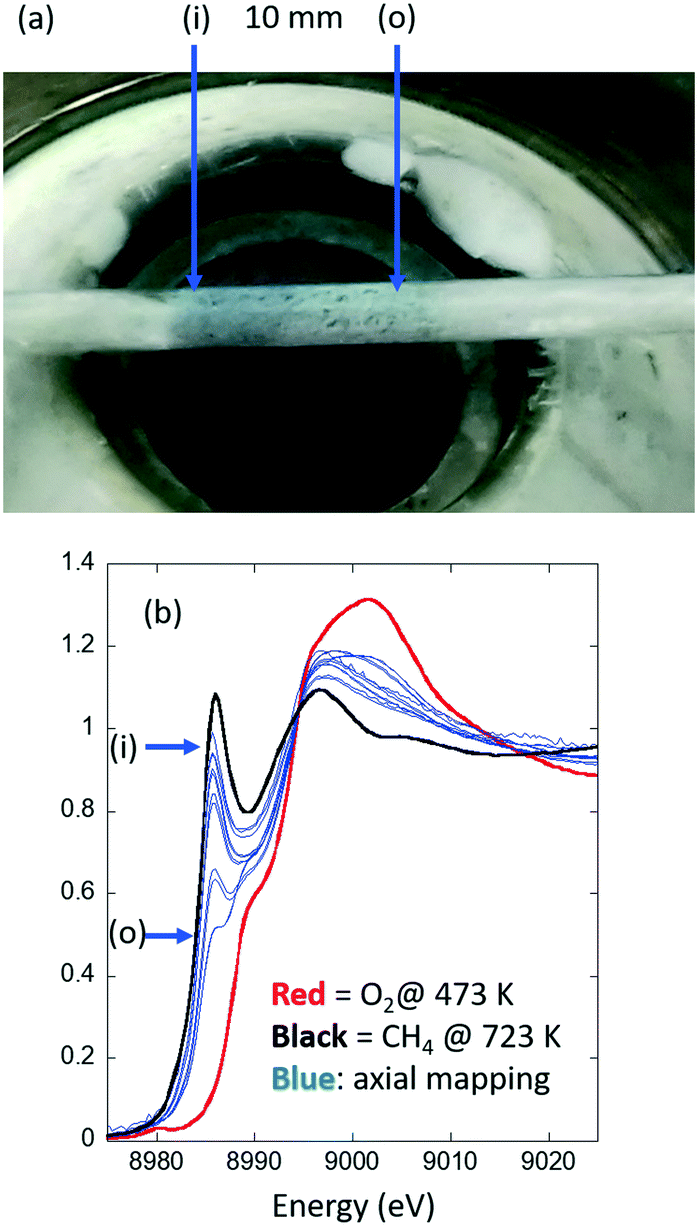 | ||
| Fig. 2 (a) The Cu/MOR sample bed in place 2 mm above the exit of the SNBL air-blower system and measured post high temperature activation in flowing oxygen to 723 K and then during subsequent exposure to methane at 473 K. An axial gradient in the colour of the sample is evident. (b) Cu K-edge XANES spectra (blue) obtained from axial mapping, 0.5 mm intervals, along the length of the sample shown in Fig. 1(A). The red spectrum is that due to the activated sample prior to exposure to methane and maintained under flowing oxygen at 473 K; the black curve that which results from the Cu/MOR sample at 723 K after heating in flowing methane. These two spectra represent, respectively, internal standards for 100% CuII and 100% CuI that are then used as a basis for linear combination analysis (LCA) to retrieve the fraction of Cu present from the Cu K-edge XANES maps. See 31 for details. The relative positions of the spectra shown in (b) are related to the picture shown in (a) with (i) = reactor inlet and (o) = reactor outlet. | ||
What has been established for the activation of methane by Cu/MOR, is that this process obeys pseudo-first order kinetics27 and results in a fraction of the starting CuII being converted into CuI. Therefore, under isothermal conditions, and at completion of the reaction (the eventual steady state), we should expect that CuI should appear at consistent levels across the entire dimension of the catalyst bed and to an extent dictated by the reaction temperature. We also note that this reaction results in a very low conversion of methane; subsequent steaming/extraction of products after such a treatment for 30 min, yields ca. 80–90 mmol g−1 CH3OH (ref. 32) for this sample at 1 bar methane pressure.
As such, this probe reaction equates to the oft-used approach to the elucidation of reaction kinetics, the limit of low conversion, wherein the possibility that any contributions from the thermicity of the reaction itself to the results obtained can be considered as negligible.
From this measurement, therefore, we also aim to understand the dimensions of the sample beds that can be used within this equipment and where they should be placed.
What is immediately evident from Fig. 2(a) is that there is an axial gradation in the colour of the Cu/MOR sample. As this result is decidedly contrary to the known kinetics of this reaction this shows that a gradient of some type exists in this system even at steady-state under the conditions applied. Fig. 2(b) makes it very clear as to the source of this gradient. The amounts of the CuI found to be present in the reacted sample vary considerably in both axial and radial directions. This strongly indicates that this experimental arrangement is not isothermal.
Moreover, the gradient observed is highly directional and there exists no substantive area of implied isothermality for the positioning of a typical (5 mm long) sample.
Fig. 3(a–c) takes these measurements and quantifies them using step-wise temperature programmed reduction (TPR) under methane using the same, high-temperature activated, Cu/MOR sample but a second type of reactor; the bi-directionally heated cell schematically shown in Fig. 1. In this case the TPR is step-wise as at each temperature the system was followed using XAFS until such time as no further changes in the spectra were observed (to avoid any errors due to the kinetics of CuI formation under methane).
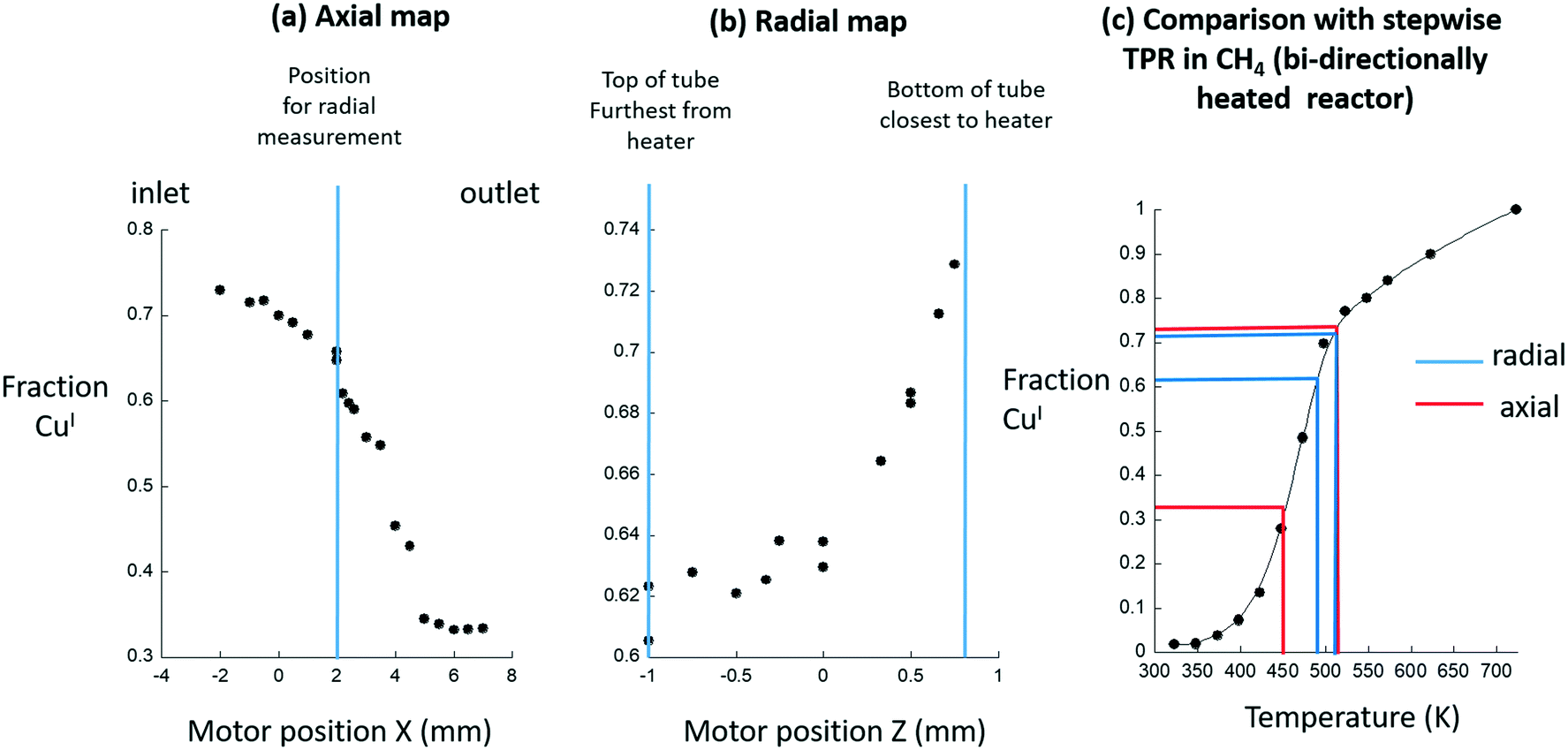 | ||
| Fig. 3 (a) LCA derived quantification of the levels of CuI returned from axial Cu K-edge XANES maps of the sample shown in Fig. 2 mounted within a uni-directional air-blower heating system. (b) The corresponding radial map obtained at the position indicated in Fig. 3(a). (c) The implied axial (red) and radial (blue) temperature ranges existing within the system are derived from comparison with step-wise TPR (●) of the Cu/MOR sample made under flowing methane using the Chupas reactor.23 | ||
Herein, we assume that this second reactor is isothermal across the dimensions of the bed used. From this assumption we may then translate the levels of CuI reported as a function of temperature in the TPR experiment (Fig. 3(c)), into temperature variations that are indicated by the variable levels of CuI formed during the methane activation reaction in the uni-directionally heated reactor (Fig. 3(a) and (b)).
This analysis shows that the levels of CuI vary from reactor inlet (>70% conversion of CuII to CuI) to the reactor outlet (ca. 33% conversion of CuII to CuI). Together, these two sets of results (axial and radial), made at steady-state under flowing methane, suggest that these variations are indeed the result of considerable differences in sample temperature across the sample bed, and that this air blower system does not provide an environment that corresponds to the basic starting point required for quantitative investigation.
Across the radial dimension (2 mm – indicated by the blue lines s in Fig. 3(c)) a 25–30 K temperature gradient is implied. That a radial gradient might exist in such a situation is, to some degree, expected simply because of the heating being applied from one side only. However, what is not, a priori, expected is the existence of the very large and directional – from the inlet to the outlet of the bed – axial temperature gradient that is implied to be of a magnitude of up to 75 K (indicated by the red lines in Fig. 3(c)).
At this point, and as stated above, these conclusions rest upon the assumption that the Chupas reactor23 is itself isothermal. As such, the same type of measurement has been performed using this latter reactor and a second air blower system. Fig. 4 compares the results obtained for the axial and radial variation of CuI obtained from within a 5 mm bed mounted within the bi-directional reactor to the radial variation obtained from a further experiment conducted using another uni-directional air blower system made available by the DUBBLE beamline at the ESRF.
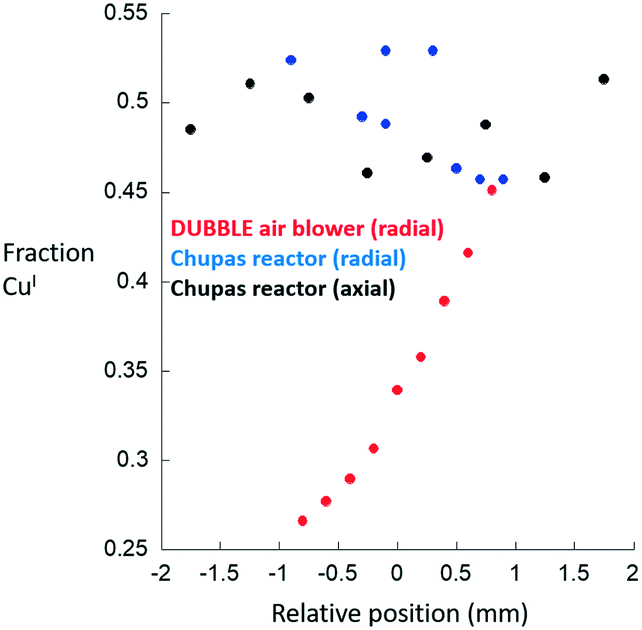 | ||
| Fig. 4 Axial (black symbols) and radial (blue symbols) variations in CuI obtained for a Cu/MOR sample (2 mm i.d. sample tube) after high temperature activation in oxygen at 723 K and subsequent exposure to methane at 473 K made using the bi-directionally heated reactor.22 The radial CuI gradient for the same sample measured under the same circumstances using the DUBBLE air blower reactor system is also given (red symbols). | ||
Only the radial gradient was measured in the latter case as, at the time of measurement, no motor existed to permit scanning of the axial extension of the sample. In both of these cases, in contrast to our first example, a thermocouple was inserted into the bed and therefore at least at one part of the bed the temperature could be reliably known.
These results show that a radial gradient in CuI is also obtained across the 2 mm diameter of the sample tube when using the uni-direction air blower system. The absolute values of the fraction of CuI present are different to the case shown in Fig. 3 as the temperature of the sample at the position at which the measurements were made was, to some degree, better established as a result of the presence within the bed of a thermocouple. Cross-referencing the magnitude of this gradient to the TPR shown in Fig. 3 once again indicates that this is representative of a radial temperature gradient of ca. 25–30 K.
Most importantly, however, are the results derived from the bi-directionally heated reactor22 in terms of the variation of CuI obtained across both radial and axial dimensions of the sample. When measured using this system we observed that, to all intents, the gradients in CuI disappear. Whilst there is some point-to-point variation in the levels of CuI reported from the analysis it is at the level of ca. ±2.5% with no accompanying signs of any sense to the scatter of the nature that might indicate the presence of a directional gradient. Moreover, and with reference to the TPR shown in Fig. 3(c), we might deduce that the scatter in the data due to the bi-directionally heated reactor would be indicative of temperature variations of only ca. 2.5–3 K over the totality of the bed sampled, and within the likely error associated with individual results and analyses.
This last observation provides a very strong indication that: firstly, the gradients we have observed with the air blower systems have nothing to do with the chemistry in question; and, secondly, that the bi-directionally heated reactor22 is, under the circumstances required for this specific measurement to be made, an acceptably isothermal environment for study across the dimensions of the sample bed.
In this respect, we can, with some degree of certitude, trust that results derived using the bi-directional reactor23 as being truly representative of the behavior of our sample at the temperature we wish to investigate it. Consequently, we may have confidence in any, structural, chemical, or kinetic derivations we might subsequently make from our experiments. The blower system, on the other hand, and under these conditions, deviates substantially from the required isothermal condition. As a result, it is clear that in these cases a range of results can be obtained from a single sample depending upon where in the bed we observe its behavior and/or on the dimensions of the X-ray beam used to probe the system.
As a final test of the bi-directionally heated reactor we have made a further assessment of using infrared imaging (Fig. 5 and 6). Fig. 5 summarises the experiment and gives an example of the raw outputs obtained from the infrared imaging camera (VarioCAM, Infratec GmbH, Dresden). Fig. 6 then goes on to quantify the axial and radial temperature variations present through extraction of data from the spatial profiles shown in Fig. 5. This imaging measurement allows us to assess any thermal gradients that may exist in this system, but also permits us to be far more precise as to over what dimensions this reactor yields an isothermal heating zone. This in turn allows us specify how large our sample beds may be, and where they must be placed within the reactor, to ensure that results derived from experiments are reliable.
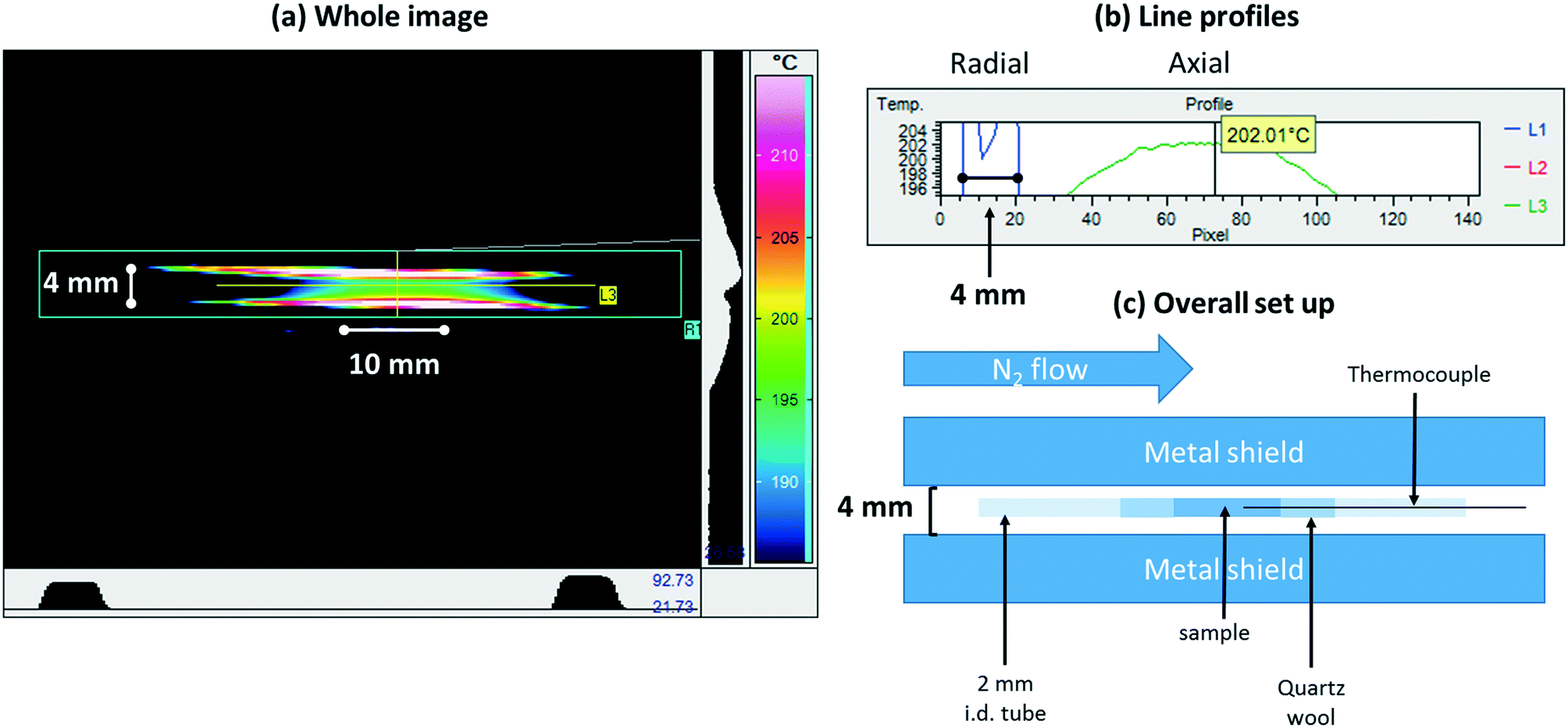 | ||
| Fig. 5 (a) Colour map infrared image derived from a sample mounted within the bi-directionally heated reactor22 (schematically represented in Fig. 1) under a flow of N2 and held at 473 K; (b) line profiles extracted from the image shown in (a); (c) a schematic description of how this measurement was made. The metal shield serves to minimize the background to the measurement and its measured spacing permits the subsequent conversion of pixels into millimeters (see Fig. 6). In making this measurement an emissivity for the sample must be assumed. In this case, the emissivity was adjusted manually until the sample temperature indicated by the camera matched acceptably that obtained from the thermocouple inserted into the sample bed. | ||
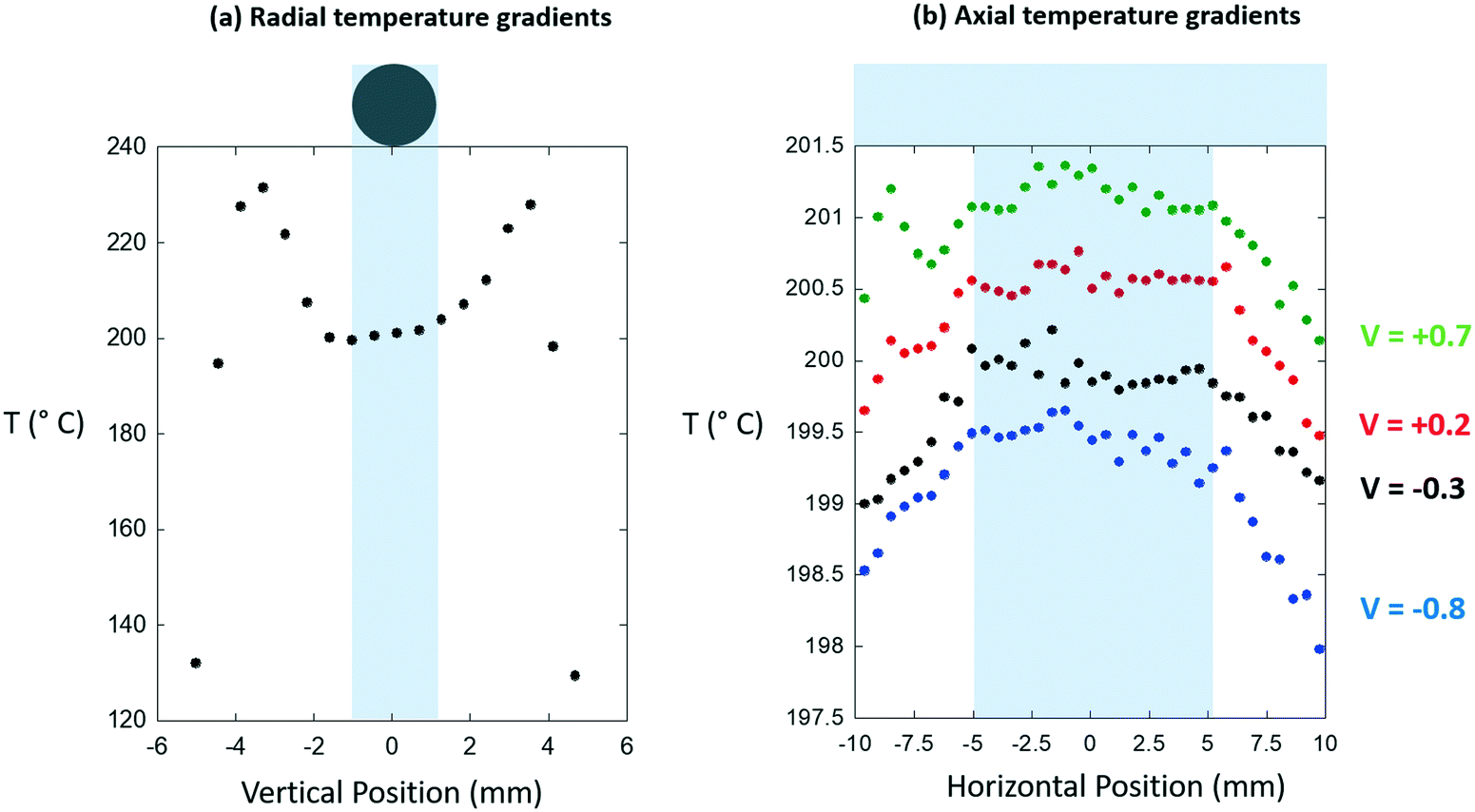 | ||
| Fig. 6 (a) Radial temperature gradients and (b) axial temperature gradients extracted from the digital infrared images shown in Fig. 5. The position of the sample bed is indicated in each case. In (a) the apparent drop of in temperature is an artifact resulting from the placement of the metal shield in front of the cell, its convolution with the sizes of the pixels, and the difference in emissivity that exists between the sample and the air gap that exists either side of it. In the case of the axial temperature gradients four vertically offset line profiles are given at the positions indicated (in mm) relative to the center of the sample tube. In (b) different V values indicate line profiles derived relative to the center of the 2 mm diameter sample tube. | ||
From these we find that the bi-directionally heated reactor, as we have implemented it, yields both axial and radial isothermality to a far more acceptable degree than was achieved using the air-blowers, and that axially we have an isothermal zone that extends over ca. 10 mm. As a result, we can conclude that we may safely use catalyst beds of 5 mm axial extension.
Discussion
The results demonstrate a number of important issues. The first is that it is all too easy to enter into a situation that is decidedly non-isothermal when using uni-directional air blower systems; and having entered into such a situation it is easy to see that highly misleading results, that have precious little to do with the intrinsic chemistry of the materials under study, can be obtained. Moreover, we might intuit that this situation will become worse the higher the temperature of operation.As noted beforehand, radial temperature gradients, must be anticipated in these systems simply as a result of the heating of the sample from only one side. The magnitude of these radial gradients will depend strongly upon the diameter of the sample bed used and we have shown that this could be as much as 30 K across a bed of only 2 mm in diameter.
The question that arises, therefore, is not whether gradients are present in this sort of reactor system, but rather whether their magnitude can be tolerated in terms of any deductions desired to be made from the experiment. In the current case, it is quite clear that they are not acceptable. Any attempt to quantify reaction kinetics, or cross-reference the X-ray data with probes of reactivity that integrate over the entirety of the bed, such as mass spectrometry or gas chromatography, will likely be subject to considerable error.
In the original papers6,21 it is quite clear that isothermality was of a considerable concern and had been thought about: in these works we find detailed reference to the use of a Kapton hood to cover the sample from above in an attempt to minimize, most likely, any radial gradients present.
It is the case, however, that much larger sample holder dimensions have been used these sorts of uni-directional heating arrangements. Within our own group up to 3 diameter tubes have been used25–28 and examples of sample tubes of up to 5 mm diameter being used can be found in the literature.35
Moreover in many instances the various precautionary practices adopted by Clausen and co-workers,6,21 either abandoned or simply not indicated to have been put in place. The results of our quantification of radial temperature gradients strongly suggests that the original practices outlined by Clausen et al.6,21 should always be implemented and that heating systems of this nature should not be used with sample beds of significantly more than 1 mm in diameter.
Whilst the existence of radial gradients could be foreseen, the very considerable axial gradients revealed by our test measurements (Fig. 2 and 3) made using an air-blower system are alarming. Moreover, the source of these unacceptable gradients in temperature, are not immediately obvious. No malfunction of the heating system was evident and it had been calibrated – through a point calibration using a thermocouple – only a few weeks before the test shown in Fig. 1–3 was made.
The most likely sources of the gradients that appeared using this system, given their decided directionality, are stray air currents that arise from the air conditioning system of the hutch, and/or fans used to cool various racks of electronic equipment present in the vicinity of the sample measurement area.
On this matter, we might further add that, having demonstrated the superior behaviour of the Chupas reactor,23 in subsequent usage (also at BM31, ESRF) we have, on occasion, also derived evidence for the presence of (much smaller, but still unacceptable, ca. 15 K max) axial temperature gradients under the same reaction conditions, and that these gradients had the same directionality. We have also noted that these can appear even when the temperature – from the thermocouple inserted into one end of the catalyst bed – appears stable and as it should be. However, we have also found that these unwanted gradients can be avoided by surrounding the reactor with Kapton sheets, much as Clausen and co-workers did in their original demonstrations of the uni-directional air blower.6,21 That this is the case would support the notion that stray air currents arising from within the experimental hutch are the source of these unwanted effects. As both of the bi-directional and uni-directional air-blower systems present air gaps either side of the sample both can be seen as susceptible to such stray air currents. Clearly, however, with its dual heating of the sample, the Chupas reactor23 is a more robust (though not immune) solution in this sense, and one that retains all the versatility of the uni-directional air-blower system.
To conclude our discussion we might consider that best practice demands that isothermality, in whatever reactor system is to be used, should be verified before any experiment is entered into. We have shown that infrared imaging is a good way to achieve this end and decidedly better than relying on single point calibrations using a thermocouple. However, we must also recognize that infrared cameras are not so commonly available in X-ray beamlines or catalysis laboratories. In the absence of this solution, there are a number of other options that could be employed.
In general when at a synchrotron axial mapping of a sample bed is easily achievable using the X-ray methods available on the beamline being used. As such, it should always be possible to use X-ray spectroscopy in the manner we have done here to rapidly map the speciation of an element understudy. However, as is all too evident from the literature, axial gradients in temperature, structure, and speciation can legitimately arise as a consequence of the chemistry under study.12,13,20,36–40 As such, though easily achieved, this method may only be indicative of unwanted thermal gradients in certain cases (such as we have demonstrated for Cu/MOR in the activation of methane).
Another solution, if the beamline and sample presentation are amenable to it (in terms of the resolution that can be achieved), is to use X-ray diffraction of a known material (such as silver powder) to establish the temperature of the reactor system on the basis of its thermal expansion. Alternatively, incorporation of an internal standard material (such as boron nitride) in a packed powder bed, at levels that will not perturb the character of the system, can be used to the same effect.41 We also note, however, that incorporation of BN into pressed pellet samples can have significant and deleterious effects on the result obtained from in situ experiments.7 As ESI† we give an example of the implementation of an XRD based approach (using silver powder) to determine the actual temperature of the surface of a sample bed within a Harrick diffuse reflectance infrared cell adapted for simultaneous collection high energy X-ray scattering/infrared data.42 The result of this exercise confirms that the temperature measured using a thermocouple placed near to the sample in this specific sample environment and inside the metallic block using to heat it does indeed yield a good measure of the actual near surface temperature of the bed that is probed by both the infrared and the X-rays in this experimental arrangement.
Conclusions
Our major conclusion is that a fundamental requirement for plug flow, that of isothermal operation, should not be taken for granted; as and where possible, it should be actively verified prior to experimentation. We have shown that, for a uni-directionally heated air-blower system, it is all too easy to end up in a very much unwanted situation regarding sample isothermality; a situation that can evidently be seen to have significant potential to yield results that are simply not representative of the behavior of the sample under in numerous ways.Equally, we have shown that the bi-directionally heated reactor23 is a far more robust and reliable solution for sample presentation in X-ray based experiments and for the attainment of truly operando, and plug-flow conformant, conditions for study. Even in this case, however, proactive verification of isothermality should be entered into, as this configuration is not immune to the effects of stray air currents for which there are many sources present in typical beamline hutches.
From our measurements of radial thermal gradients in the air blower case it seems apparent that uni-directional heating systems should only be used with sample containing tubes of ≤ ca. 1 mm diameter, as was the case for their original implementation.6,21 Moreover, the sorts of remedial actions taken by Clausen et al.6,21 in these original reports to ameliorate any potential thermal issues should always be implemented; indeed, the exemplary practices detailed in these original papers should be considered as mandatory rather than as advisory. If these air blower systems are to be used outside of this original parameterisation then we suggest that they are always applied as a synchronised pair of heaters rather than from one side only, such that severe radial non-isothermality, and the enhanced likely hood of erroneous structure function relationships being derived, can be also be avoided.
Lastly, and where the objective of the experiment does not require any significant solid angle of data collection, the temperatures sought are not too extreme, and there is no requirement for very rapid heating or cooling of the sample, we might advocate that the best solution to contain the sample within an enclosed oven designed to permit X-ray transmission.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the ESRF, Swiss-Norwegian beamlines (SNBL), and DUBBLE for access to facilities. Royal-Dutch Shell is thanked for funding of the position of MAN. We are also extremely grateful for the technical support and skills of Joel Jenni and Freddy Mettler (ETH) and Geir Wiker at SNBL. Dr. Vitaly Sushkevich (Paul Scherrer Institute, Switzerland) is acknowledged for provision of the Cu/MOR sample used in this study. Dr. Dmitry Chernyshov (SNBL) is acknowledged both for the providing the Ag powder used for HXRD measurements made at ID15, and for discussions in relation of how to use XRD to determine samples temperatures. Simon Benichou (ISDD, ESRF) is thanked for provision of the infrared camera used in this study. Dr Sander van Bavel (Shell Global Solutions) is gratefully acknowledged for critical reading of the manuscript.Notes and references
- M. O. Guerrero-Perez and M. A. Banares, Chem. Commun., 2002, 1292 RSC.
- H. Topsoe, J. Catal., 2003, 216, 155 CrossRef CAS.
- B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4351 RSC.
- K. J. Laidler, Chemical kinetics, McGraw Hill, New York, 3rd edn, 1973 Search PubMed.
- C. N. Satterfield, Heterogeneous catalysis in practice, McGraw-Hill, 1980 Search PubMed.
- B. S. Clausen, G. Steffensen, B. Fabius, L. Villadsen, R. Feidenhansl and H. Topsoe, J. Catal., 1991, 132, 524 CrossRef CAS.
- J. D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037 RSC.
- Y. Yang, R. S. DisselKamp, J. Szanyi, C. H. F. Peden, C. T. Campbell and J. G. Goodwin, Rev. Sci. Instrum., 2006, 77, 094104 CrossRef.
- F. C. Meunier, D. Reid, A. Goguet, S. Shekhtman, C. Hardacre, R. Burch, W. Deng and M. Flytzani-Stephanopoulos, J. Catal., 2007, 247, 277 CrossRef CAS.
- F. C. Meunier, A. Goguet, S. Shekhtman, D. Rooney and H. Daly, Appl. Catal., A, 2008, 340, 196–202 CrossRef CAS.
- F. C. Meunier, Chem. Soc. Rev., 2010, 39, 4602 RSC.
- H. Li, M. Rivallan, F. Thibault-Starzyk, A. Travert and F. C. Meunier, Phys. Chem. Chem. Phys., 2013, 15, 7321 RSC.
- J. B. Brazier, B. N. Nguyen, L. A. Adrio, E. M. Barreiro, W. P. Leong, M. A. Newton, S. J. A. Figueroa, K. Hellgardt and K. K. Hii, Catal. Today, 2014, 229, 95 CrossRef CAS.
- B. S. Patil, P. D. Srinivasan, E. Atchison, H. Zhu and J. J. Bravo-Suárez, React. Chem. Eng., 2019, 4, 667–678 RSC.
- J. G. Moya-Cancino, A.-P. Honkanen, A. M. J. van der Eerden, H. Schaink, L. Folkertsma, M. Ghiasi, A. Longo, F. M. F. de Groot, F. Meirer, S. Huotari and B. M. Weckhuysen, ChemCatChem, 2019, 11, 1 CrossRef PubMed.
- J. Touitou, R. Burch, C. Hardare, C. McManus, K. Morgan, J. Sa and A. Goguet, Analyst, 2013, 138, 2858 RSC.
- J. Touitou, K. Morgan, R. Burch, C. Hardacre and A. Goguet, Catal. Sci. Technol., 2012, 2, 1811 RSC.
- S. Hannemen, J. D. Grunwaldt, N. van Vegten, A. Baiker, P. Boye and C. G. Schroer, Catal. Today, 2007, 126, 54 CrossRef.
- U. Hartfelder, J. Singh, J. Haase, M. Nachtegaal, D. Grolimund and J. A. van Bokhoven, Sci. Rep., 2016, 6, 37597 CrossRef CAS PubMed.
- M. A. Newton and W. van Beek, Chem. Soc. Rev., 2010, 39, 4845 RSC.
- B. S. Clausen, L. Grabaek, G. Steffensen, P. L. Hansen and H. Topsoe, Catal. Lett., 1993, 20, 23 CrossRef CAS.
- A. M. Ganzler, M. Casapu, A. Boubnov, O. Muller, S. Conrad, H. Lichtenberg, R. Frahm and J. D. Grunwaldt, J. Catal., 2015, 328, 216 CrossRef.
- P. J. Chupas, K. W. Chapman, C. Kurtz, J. C. Hanson, P. L. Lee and C. P. Grey, J. Appl. Crystallogr., 2008, 41, 822 CrossRef CAS.
- E. M. C. Alayon, M. Nachtegaal, M. Ranocchiari and J. A. Van Bokhoven, Chem. Commun., 2012, 48, 404 RSC.
- E. M. C. Alayon, M. Nachtegaal, E. I. Kleymenov and J. A. Van Bokhoven, Microporous Mesoporous Mater., 2013, 166, 131 CrossRef CAS.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16 CrossRef CAS.
- P. Vanelderen, B. E. R. Snyder, M.-L. Tsai, R. Hadt, K. Vancauwenbergh, O. Coussens, R. A. Schoonhedyt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 6383 CrossRef CAS PubMed.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi, M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2015, 17, 7681 RSC.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- S. E. Bozbag, E. M. C. Alayon, J. Pechacek, M. Nachtegaal, M. Ranocchiari and J. A. Van Bokhoven, Catal. Sci. Technol., 2016, 6, 5011 RSC.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523 CrossRef CAS PubMed.
- M. A. Newton, A. J. Knorpp, A. B. Pinar, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, J. Am. Chem. Soc., 2018, 140, 10090 CrossRef CAS PubMed.
- D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomochenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, J. Am. Chem. Soc., 2018, 140, 15270 CrossRef CAS PubMed.
- A. J. Knorpp, M. A. Newton, A. B. Pinar and J. A. van Bokhoven, Ind. Eng. Chem. Res., 2018, 57, 12036 CrossRef CAS.
- I. Lezcano-Gonzalez, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215 CrossRef CAS PubMed.
- J. D. Grunwaldt and A. Baiker, Catal. Lett., 2005, 99, 5 CrossRef CAS.
- M. A. Newton, B. Jyoti, A. J. Dent, S. Diaz-Moreno, S. G. Fiddy and J. Evans, Chem. – Eur. J., 2006, 12, 1975 CrossRef CAS PubMed.
- S. J. A. Figueroa and M. A. Newton, J. Catal., 2014, 312, 69 CrossRef CAS.
- B. M. Weckhuysen, Angew. Chem., Int. Ed., 2009, 48, 4910 CrossRef CAS PubMed.
- J. D. Grunwaldt, B. Kimmerle, A. Baiker, P. Boye, C. G. Schroer, P. Glatzel, C. N. Borca and F. Beckmann, Catal. Today, 2009, 145, 267 CrossRef CAS.
- N. E. Tsakounis, A. Voronov, M. Ronning, W. van Beek, O. Borg, E. Rytter and A. Holmen, J. Catal., 2012, 291, 138 CrossRef.
- K. A. Beyer, H. Zhao, O. J. Borkiewicz, M. A. Newton, P. J. Chupas and K. W. Chapman, J. Appl. Crystallogr., 2014, 47, 95 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials used; synchrotron based measurements and activation/reaction protocols; thermal assessment of other reaction cells. See DOI: 10.1039/c9cy00464e |
| ‡ Now at EPFL, Lausanne, Switzerland. |
| This journal is © The Royal Society of Chemistry 2019 |