The impact of lignin sulfonation on its reactivity with laccase and laccase/HBT†
Received
1st February 2019
, Accepted 1st March 2019
First published on 4th March 2019
Abstract
Lignin is a highly abundant aromatic polymer in nature, but its controlled cleavage or cross-linking is a major challenge and currently hindering industrial applicability. Laccase (L) and laccase/mediator systems (LMS) are promising tools for enzymatic lignin modification, but to date, their overall reaction outcome is hard to predict and control. This research aimed to understand the reactivity of native and sulfonated β-O-4 linked lignin structures in L and LMS treatments. Trametes versicolor laccase, and the mediator hydroxybenzotriazole (HBT) were used, and reaction products were analyzed using UHPLC-MSn and MALDI-TOF-MS. Polymerization was observed for both the native and sulfonated phenolic compounds, suggesting that sulfonation does not affect radical coupling of the phenolic lignin subunits. In contrast, sulfonation of the non-phenolic lignin structure prevented Cα oxidation and cleavage by L/HBT, which was explained by an increased Cα–H bond dissociation energy of ∼10 kcal mol−1 upon sulfonation. Overall, our results indicate that lignin sulfonation drives the overall outcome of LMS incubations towards polymerization.
Introduction
Lignin is one of the main polymeric constituents of plant cell walls and is considered a renewable precursor for chemicals and materials.1–4 Plants synthesize lignin via radical coupling of the precursors sinapyl alcohol, coniferyl alcohol and p-coumaroyl alcohol (S, G and H units, respectively). Upon polymerization, a variety of C–C and C–O inter-unit linkages is formed, of which the β-O-4 linkage is the most abundant one, accounting for 45–94% of the total inter-unit linkages.3 Polymeric lignin consists of 10–30% phenolic subunits, mainly being the endcaps of the polymer, and 70–90% non-phenolic subunits, forming the lignin backbone.5
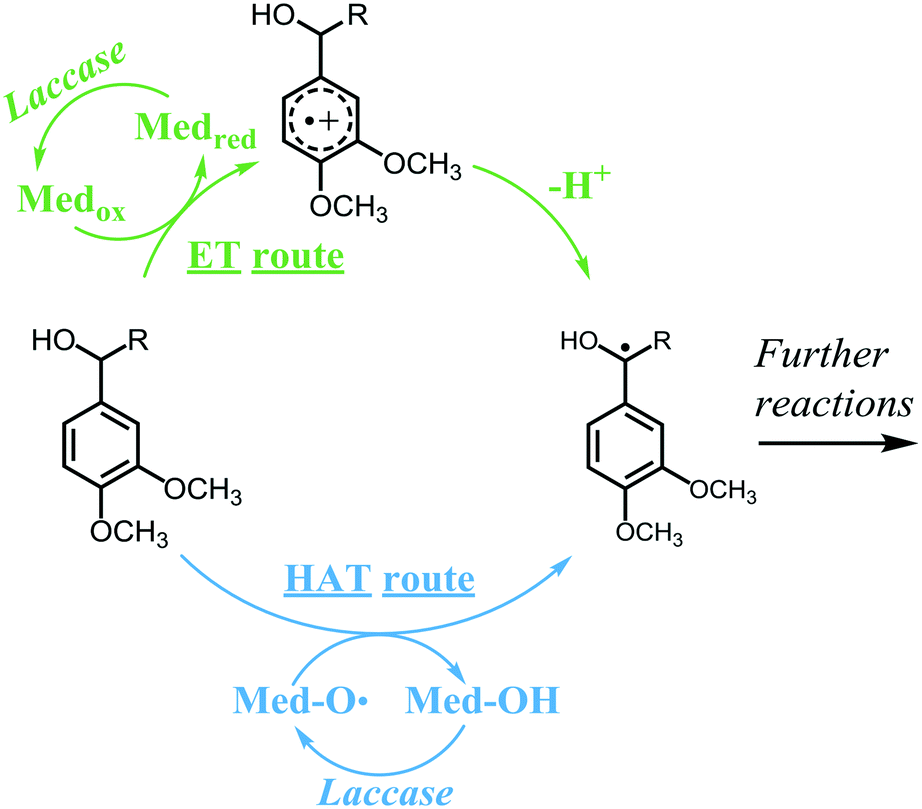
Currently, lignin is still underutilized in industrial applications. Depending on the intended application, lignin can be valorized via different routes, such as polymerization, depolymerization and grafting.6–8 Preferably, valorization should occur via sustainable approaches, such as enzymatic modifications. One of the few enzymes known to be active toward lignin is laccase (E.C. 1.10.3.2), an oxidase that couples the reduction of molecular oxygen to the one-electron oxidation of a wide variety of aromatic substrates. Laccases have relatively low redox potentials: the most powerful laccases, produced by white-rot fungi, have redox potentials up to 800 mV vs. NHE.3 In contrast, the non-phenolic subunits have redox potentials up to 1500 mV vs. NHE and are, therefore, recalcitrant to oxidation by laccase alone.9 Consequently, laccases can only oxidize the phenolic subunits of lignin. Nevertheless, when laccase is combined with a mediator, also the non-phenolic lignin structures can be oxidized.10 In such a laccase/mediator system (LMS), laccase oxidizes the mediator, which, in turn, can react with a non-phenolic lignin subunit, via different mechanisms, dependent on the mediator. Among the most widely used mediators are the synthetic compounds HBT and ABTS, but also small lignin-derived phenolic compounds, such as syringaldehyde and methyl syringate.11–18 HBT and natural phenolics have been reported to oxidize non-phenolic lignin subunits via a radical hydrogen atom transfer (HAT) mechanism, whereas ABTS is suggested to operate via electron transfer (ET) (Scheme 1).19 In the HAT mechanism, the oxidized mediator is generally an oxygen centered radical species that abstracts a radical hydrogen atom from the benzylic position of the lignin structure, forming back the non-radical mediator species and a benzylic radical. The efficiency of this reaction depends on the bond dissociation energies (BDEs) of the mediator O–H bond and the lignin Cα–H bond. In contrast, in the ET mechanism, the mediator is an oxidized species that abstracts an electron from the lignin structure, forming back the reduced mediator species and a radical cation. The radical cation then spontaneously loses a proton to form a benzylic radical. The efficiency of the ET reaction depends on the redox potentials of the mediator and the lignin structure.20 After oxidation by laccase or LMS, lignin may undergo a variety of follow-up reactions, such as radical coupling, Cα oxidation and cleavage reactions.
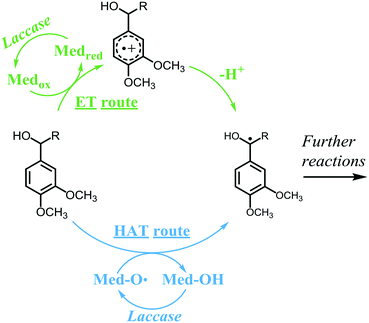 |
| | Scheme 1 Mechanisms of ET and HAT oxidation routes in laccase/mediator systems. A non-phenolic lignin model is used as an example substrate. Adapted from Baiocco et al.19 | |
In literature, LMS incubations on lignin are studied in several ways and with several purposes: (i) on biomass or pulp, in order to degrade lignin and increase the yield of saccharification of the polysaccharides present, and (ii) on technical lignins, in order to upgrade their structure for various applications. Among the technical lignins, lignosulfonate is by far the most available one,21 and is, therefore, an interesting target for lignin valorization. The main difference between native lignin and lignosulfonate is that the majority of Cα positions in lignosulfonate are substituted by a sulfonate group instead of a hydroxyl group. Lignosulfonate and native lignin also seem to differ regarding their reactions in the presence of LMS. So far, LMS incubations with lignosulfonate have only been reported to result in polymerization, whereas comparable LMS treatments mainly led to depolymerization and Cα oxidation of native lignin.3 Currently, it is unknown why these substrates react in such a different manner in LMS incubations. This is partly due to the fact that the reaction pathways of LMS-induced lignin modifications have mainly been studied using lignin model compounds that resemble the structural motifs of native lignin. The information obtained via these studies, although valuable, cannot directly be extrapolated to reactions of lignosulfonate, due to the large degree of Cα sulfonation in the latter. To date, it remains unknown whether and how Cα sulfonation influences the reactivity of lignin structures. To be able to predict or even control the reaction outcome of LMS incubations of lignin and lignosulfonate in the future, it is essential to understand how their structural features influence the reactivity with laccase and LMS.
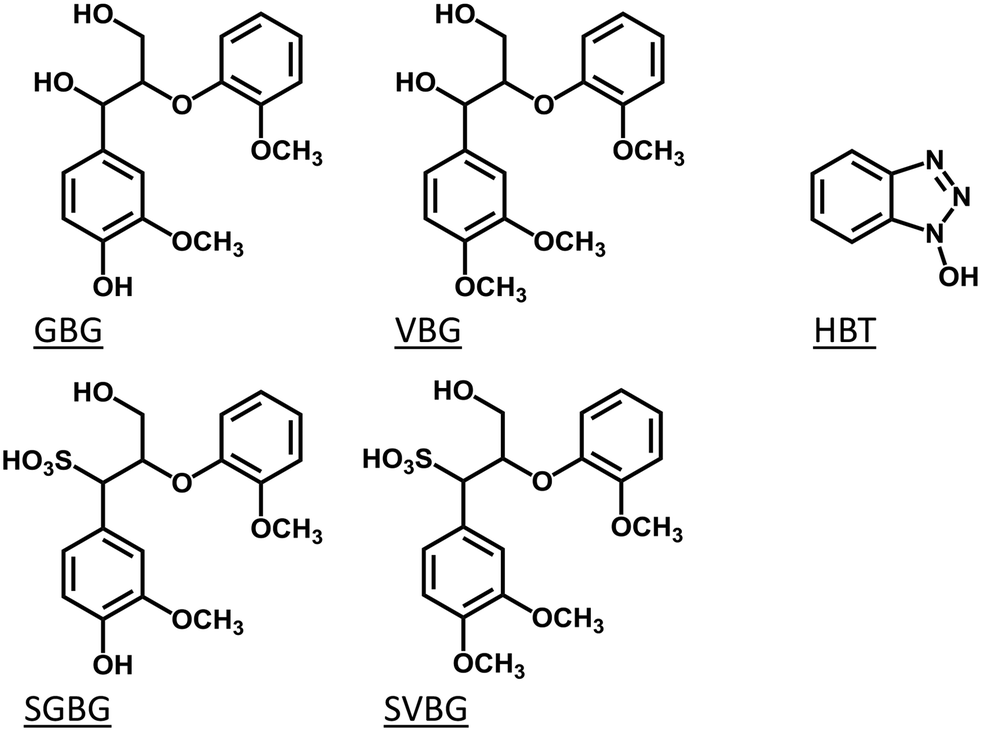
Therefore, in this study, we compared the reactivity of two types of lignin model compounds in laccase and laccase/HBT incubations: one type representing the structure of native lignin, and the other type representing the structure of lignosulfonate. To mimic native lignin, two β-O-4 linked lignin model compounds were used. Guaiacylglycerol-β-guaiacyl ether (GBG; 1-(4-hydroxy-3-methoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol) was used to mimic the phenolic lignin end caps, and veratrylglycerol-β-guaiacyl ether (VBG; 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol) was used to mimic the non-phenolic lignin backbone (Fig. 1). Their sulfonated analogues SGBG (3-hydroxy-1-(4-hydroxy-3-methoxyphenyl)-2-(2-methoxyphenoxy)propane-1-sulfonic acid) and SVBG (1-(3,4-dimethoxyphenyl)-3-hydroxy-2-(2-methoxyphenoxy)propane-1-sulfonic acid) (Fig. 1) were synthesized and used to mimic the end caps and backbone of lignosulfonate. We picked HBT as a mediator for two reasons: (i) it has been widely used in LMS incubations of both native lignin and lignosulfonate,12,18,22–24 which enables a direct comparison between our model compound study and results reported for LMS incubations of polymeric lignins, and (ii) it operates via the HAT mechanism, which makes it possible to extrapolate our results to incubations with natural phenolics as mediators.
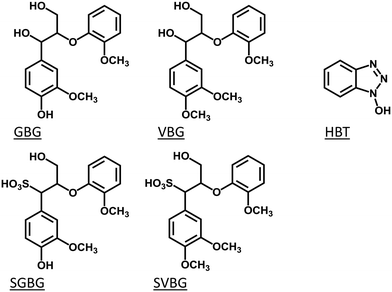 |
| | Fig. 1 Molecular structures of the lignin model compounds GBG, VBG, SGBG and SVBG, and the mediator HBT. | |
Experimental section
Materials
Guaiacylglycerol-β-guaiacyl ether (GBG; Fig. 1) was obtained from TCI chemicals (Tokyo, Japan), veratrylglycerol-β-guaiacyl ether (VBG; Fig. 1) was purchased from ABCR (Karlsruhe, Germany) and 2,5-dihydroxybenzoic acid (DHB) was bought from Bruker Daltonics (Bremen, Germany). Laccase, HBT, ABTS, SPE cartridges and all other chemicals were purchased from Sigma Aldrich (St. Louis, MO, USA). The laccase was partially purified as described earlier,25 after which the activity was determined spectrophotometrically by oxidation of ABTS (1 U = 1 μmol ABTS oxidized per minute at pH 5). Water was prepared by using a Milli-Q water purification system (Merck Millipore, Billerica, MA).
Sulfonation and purification of lignin model compounds
GBG was sulfonated by heating a GBG solution (20 mL, 2 mM) for 4 h at 130 °C in the presence of sodium bisulfite (3.5 M) at pH 2.5. Similarly, VBG was sulfonated by heating for 4 h at 150 °C and pH 1.5. Reactions were performed in a Monowave 400 microwave reactor (Anton Paar, Graz, Austria). Of both incubations, five batches were prepared, which were pooled afterwards. The sulfonated lignin model compounds were desalted and purified prior to use in experiments (see S2† for details). To verify that the model compounds were sulfonated, the purified sulfonated compounds were analyzed by performing 2D HSQC and HMBC NMR spectroscopy (see S3† for details). After sulfonation and purification, diastereomeric mixtures were obtained with an isomer ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 for both SGBG and SVBG, based on UHPLC-UV280 peak areas.
:3 for both SGBG and SVBG, based on UHPLC-UV280 peak areas.
Incubation of lignin model compounds with laccase and laccase/mediator systems
The phenolic model compounds, GBG and SGBG, were dissolved at 0.05 mM in sodium phosphate buffer (50 mM, pH 4) with or without an equimolar concentration of HBT. Laccase was added to reach a final substrate and mediator concentration of 0.04 mM and a laccase activity of 0.04 U mL−1 in a total volume of 1 mL. The mixtures were incubated at 40 °C and 400 rpm in a thermomixer (Eppendorf, Hamburg, Germany). After 2, 5, 10, 20, 35 and 60 min, 100 μL of the incubation mixture was collected and 10 μL of 20 mM sodium azide was added to this aliquot to stop the reaction. Incubations with the non-phenolic model compounds, VBG and SVBG, were performed similarly, with the adaptations that the HBT concentration was 0.4 mM and the reactions were stopped after 1, 3, 6 and 24 h. The samples were centrifuged (10000g, 5 min, 20 °C) and analyzed by using RP-UHPLC-PDA-MSn. Incubations for MALDI-TOF-MS were performed similarly, with substrate and mediator concentrations of 0.4 mM and a laccase activity of 0.2 U mL−1 in all incubations. Detailed descriptions of the RP-UHPLC-PDA-MSn and MALDI-TOF-MS methods can be found in S6–S9 of the ESI.† In addition, separate oxygen consumption measurements were performed using HBT, GBG and SGBG as substrates (see S11† for details).
Computational analyses
All computational calculations were performed with the B97D functional and 6-311+G(d,p) basis set, as implemented in Gaussian 16 (version B1), using a SMD solvent model for water.26
Homolytic C–H bond dissociation energies were calculated by elongating the desired C–H bond to 5 Å (in triplet state), and subtracting the single-point energy of this geometry from the (singlet) optimized geometry.
The ionization energy of all four compounds in the neutral state was determined as the difference between the single-point energies of the uncharged ground state and that of the corresponding radical cation in the geometry of the optimized neutral ground state.
Results
Reactions of phenolic lignin model compounds with laccase and laccase/HBT
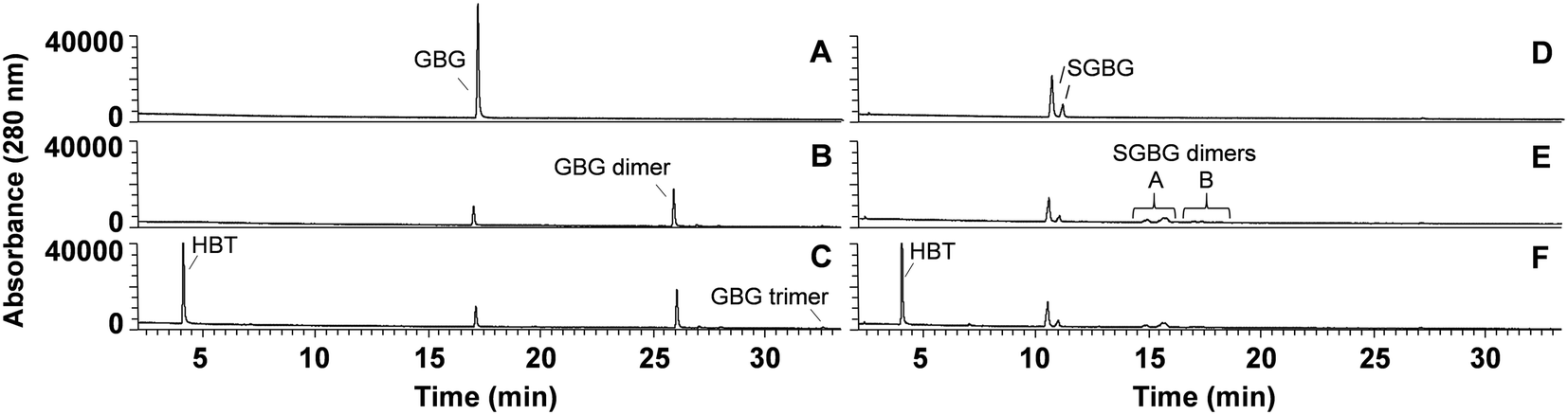
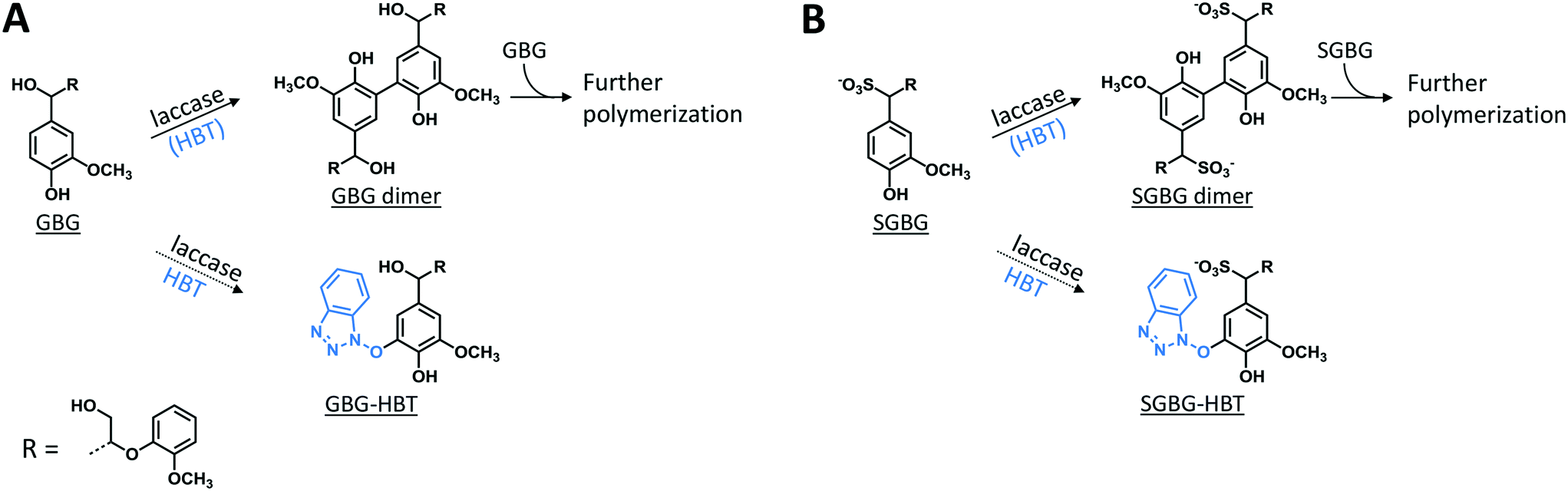
The phenolic model compounds GBG and SGBG (Fig. 1) were first incubated with laccase in the absence of HBT. After oxidation by laccase, both GBG and SGBG underwent radical coupling to form dimers (Mw = 2 × GBG–2H) (Fig. 2 and 3, Table 1, Fig. S5/S6/S9†). It should be noted that the GBG and SGBG dimers consist of four aromatic rings. From that perspective, they should be considered tetramers. Although dimerization may occur via either C–C or C–O coupling, dimerization of GBG resulted in only one clear peak. Based on previous research, this product was annotated as a C–C coupled GBG dimer.27 A second GBG dimer peak was only present in trace amounts. This dimer showed a slightly different fragmentation pattern (Fig. S10†) and was tentatively annotated as the C–O coupling product. A peak corresponding to a GBG trimer was also detected, especially after prolonged incubation times. Treatment of SGBG yielded several dimeric products, which were poorly separated by UHPLC. Due to the poor separation and relatively low intensity, the exact number of SGBG dimers formed was difficult to determine. Nevertheless, two areas of SGBG dimers could be distinguished in the chromatogram (region A and B in Fig. 2E), which showed slightly different fragmentation patterns (Fig. S11†). It was speculated that region A, containing the major SGBG dimer peaks, corresponded to C–C coupling products, and that region B, containing the minor SGBG peaks, corresponded to C–O coupling products. Within the two areas, multiple peaks were present, which can be explained by the fact that the SGBG substrate was a mixture of two diastereomers. Upon coupling, the number of diastereomers is increased, resulting in a more complex mixture of reaction products. In addition to dimers, also oligomeric reaction products were detected, by using MALDI-TOF-MS (Fig. S12/S13†). Although laccase treatment of other phenolic lignin models has been reported to result in Cα oxidized products,28 no indications for this were found in the present study. Prolonged incubation of GBG (>60 min) was also tested, but mainly resulted in precipitation of the reaction products. Upon incubation of GBG and SGBG with the laccase/HBT system, the same dimers and oligomers, as were formed in the incubations with laccase alone, were found. In addition, trace amounts of coupling products between substrates and HBT were detected (GBG–HBT and SGBG–HBT) (Table 1 and Fig. S9†). An overview of the major reaction pathways of GBG and SGBG in laccase and laccase/HBT incubations is shown in Fig. 3. In addition to product formation, the substrate conversion was monitored over time (Fig. 4). In the absence of HBT, laccase converted 68% of the GBG and 52% of the SGBG in 2 min, based on UV280 peak area. This indicated that laccase oxidizes GBG faster than SGBG. The same trends were observed for the laccase/HBT incubations: after 2 min, the laccase/HBT system converted 69% and 51% of GBG and SGBG, respectively.
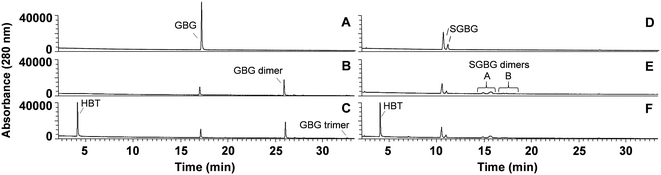 |
| | Fig. 2 RP-UHPLC-UV280 chromatograms of GBG (A–C) and SGBG (D–F) incubated for 2 min without laccase (A and D), with laccase alone (B and E) and laccase/HBT (C and F). Note that the substrate SGBG was used as a diastereomeric mixture, resulting in two chromatographic peaks. Chromatograms of other time points can be found in the ESI.† | |
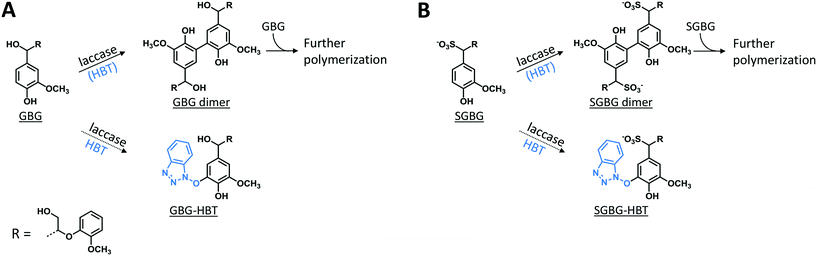 |
| | Fig. 3 Schematic representation of the outcomes of laccase and LMS incubations of GBG (A) and SGBG (B). The solid arrows show the major reaction pathway and the dotted arrows display the minor reaction pathway. Detailed information on identification of the compounds can be found in Table 1 and Fig. S9.† | |
Table 1 Compounds detected with RP-UHPLC-PDA-ESI-ITMS and RP-UHPLC-PDA-ESI-FTMS after incubation of the phenolic lignin model compounds GBG and SGBG with laccase from T. versicolor in the presence or absence of HBT. The MS2 fragments are given with their relative abundance (%) in parentheses. When no fragment with an abundance of 100% is reported, the most abundant fragment ion was equal to the parent ion. Proposed fragmentation patterns can be found in Fig. S9
| Tentative annotation |
Molecular formula |
RT (min) |
Ion |
Observed/calculated mass (Da) |
Mass error (ppm) |
MS2 fragments |
λ
max (nm) |
Incubation |
|
Although the molecular formula of this compound is known, no molecular structure could be elucidated.
|
|
Reaction products of GBG
|
| GBG |
C17H20O6 |
17.2 |
[M + Na]+ |
320.12602/320.12599 |
0.11 |
295 (44), 302 (12), 147 (8), 201 (8), 219 (8) |
277 |
Lac, lac/HBT |
| GBG dimer |
C34H38O12 |
26.0 |
[M − H]− |
638.23707/638.23633 |
1.17 |
589 (100), 483 (64), 513 (28), 435 (26), 329 (20), 541 (14), 465 (12), 359 (8) |
277 |
Lac, lac/HBT |
| Unknowna |
C24H23NO9 |
27.0 |
[M − H]− |
469.13765/469.13728 |
0.78 |
424 (100), 312 (38), 409 (36), 287 (26), 268 (16), 296 (14), 344 (12), 272 (8) |
308 |
Lac, lac/HBT |
| GBG trimer |
C51H56O18 |
32.5 |
[M − H]− |
956.34718/956.34666 |
0.54 |
907 (100), 889 (52), 919 (26), 859 (18) |
N.D. |
Lac, lac/HBT |
| GBG–HBT |
C23H23N3O7 |
19.7 |
[M − H]− |
453.15328/453.15360 |
−0.7 |
404 (100), 328 (42), 298 (38) |
330 |
Lac/HBT |
|
Reaction products of SGBG
|
| SGBG |
C17H20O8S |
10.6, 11.1 |
[M − H]− |
384.08827/384.08789 |
1.00 |
259 (100), 179 (68), 203 (38), 165 (30), 229 (22), 195 (6) |
276 |
Lac, lac/HBT |
| SGBG dimers |
C34H38O16S2 |
15.0, 15.8, 17.1 |
[M − 2H]2− |
766.16073/766.16013 |
0.79 |
561 (100), 320 (64), 123 (24), 273 (20), 449 (16), 577 (18), 437 (10) |
277 |
Lac, lac/HBT |
| SGBG–HBT |
C23H23N5O9S |
15.2 |
[M − H]− |
517.11580 517.11550 |
0.56 |
312 (100), 392 (60), 298 (34), 203 (10) |
332 |
Lac/HBT |
|
HBT and related
|
| HBT |
C6H5N3O |
3.8 |
[M − H]− |
135.04325/135.04325 |
−0.07 |
106 (100), 78 (6) |
306, 276 |
Lac/HBT |
| [M + H]+ |
91 (32), 80 (30), 107 (20), 53 (18) |
268 |
| BT |
C6H5N3 |
6.9 |
[M + H]+ |
119.04827/119.04835 |
−0.65 |
N.D. |
258, 280 |
Lac/HBT |
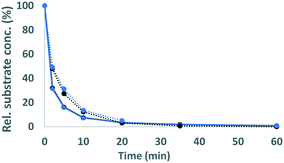 |
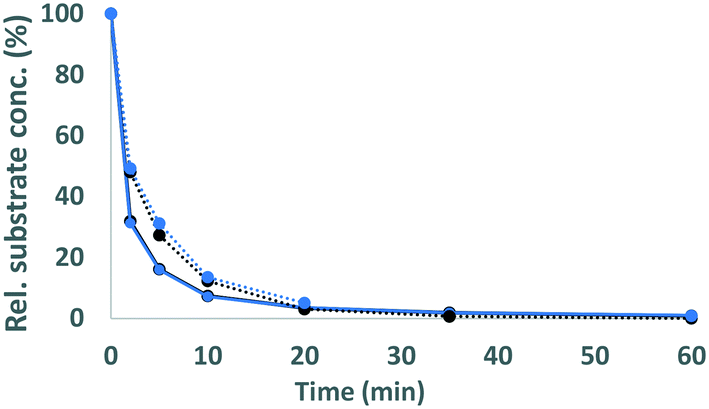
| | Fig. 4 Conversion of GBG (solid lines) and SGBG (dotted lines) in time by laccase (black) and laccase/HBT (blue). Rel. substrate conc. refers to the substrate concentration in the incubation relative to the substrate concentration at t = 0. | |
Reactions of non-phenolic lignin model compounds with laccase and laccase/HBT
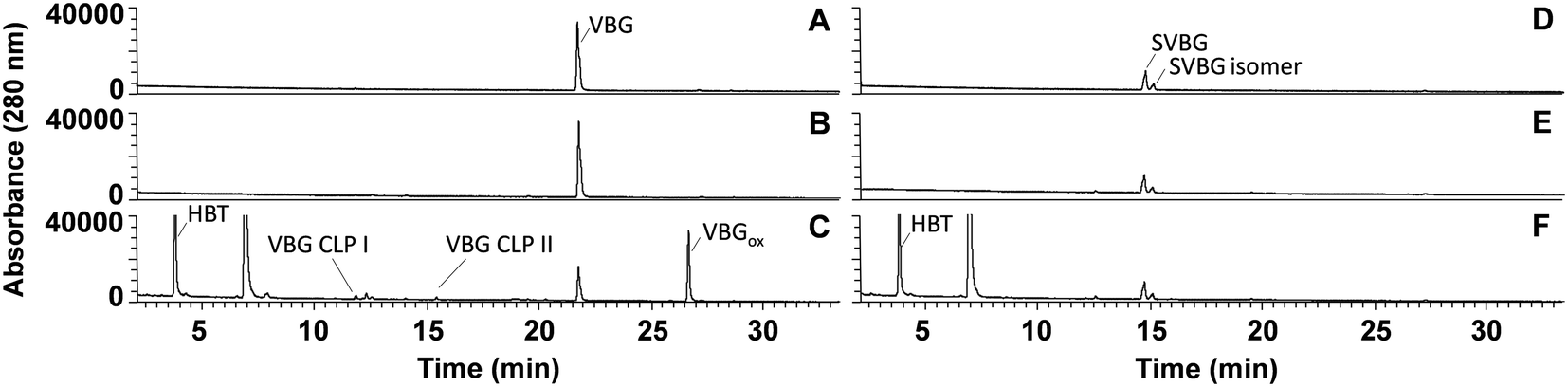
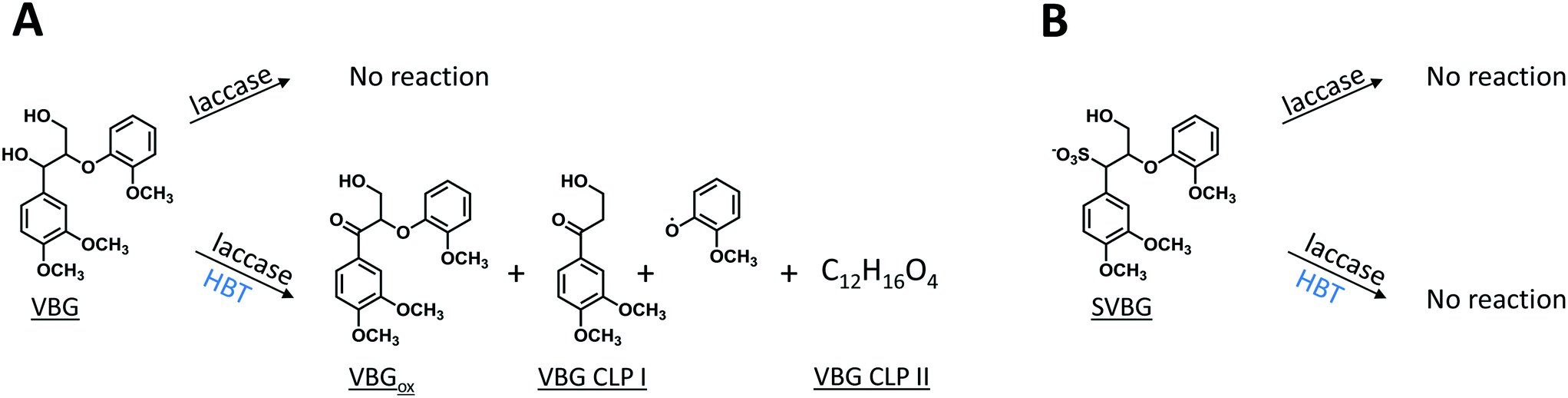
As expected, in incubations with laccase alone, no VBG and SVBG was converted (Fig. 5). In the laccase/HBT incubation, the native model compound VBG was mainly converted to its ketone analogue VBGox. In addition, small peaks corresponding to C11H14O4 and C12H16O4 were formed (Fig. 5 and 6, Table 2, Fig. S15†). These peaks were tentatively annotated as cleavage products VBG CLPI and VBG CLP II, respectively. The former cleavage product is suggested to be formed via β ether cleavage, which has been reported before.29,30 The exact structure of VBG CLP II could not be elucidated. In contrast to their phenolic analogues, VBG and SVBG did not form coupling products with HBT, suggesting that coupling to HBT occurs at the aromatic ring rather than at the benzylic position. After 24 h, 47% of the VBG was converted, based on UV280 peak area. In contrast, the laccase/HBT system was unable to convert SVBG. Even after 24 h, the areas of the SVBG peaks remained unchanged. A schematic overview of the incubation outcomes of VBG and SVBG is shown in Fig. 6.
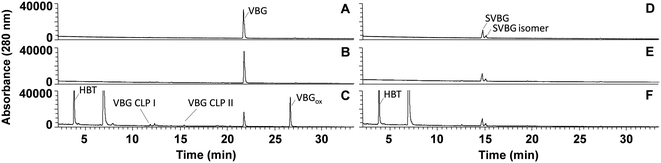 |
| | Fig. 5 RP-UHPLC-UV280 chromatograms of VBG (A–C) and SVBG (D–F) incubated for 24 h without laccase (A and D), with laccase alone (B and E) and laccase/HBT (C and F). The unlabelled peaks correspond to degradation products of the mediator. Chromatograms of other time points can be found in the ESI.† | |
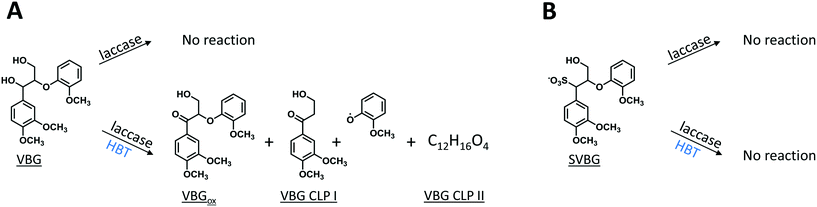 |
| | Fig. 6 Schematic representation of the outcomes of laccase and LMS incubations of VBG (A) and SVBG (B). Whereas VBG is oxidized by both LMS, SVBG is completely inert in all incubations. | |
Table 2 Compounds detected with RP-UHPLC-PDA-ESI-ITMS and RP-UHPLC-PDA-ESI-FTMS after incubation of the non-phenolic lignin model compounds VBG and SVBG with laccase from T. versicolor in the presence or absence of HBT. The MS2 fragments are given with their relative abundance (%) in parentheses. When no fragment with an abundance of 100% is reported, the most abundant fragment ion was equal to the parent ion. Proposed fragmentation patterns can be found in Fig. S15
| Tentative annotation |
Molecular formula |
RT (min) |
Ion |
Observed/calculated mass (Da) |
Mass error (ppm) |
MS2 fragments |
λ
max (nm) |
Incubation |
|
Reaction products of VBG
|
| VBG |
C18H22O6 |
21.7 |
[M + Na]+ |
334.14123/334.14164 |
1.21 |
309 (42), 215 (8), 147 (4), 233 (4) |
276 |
Lac, lac/HBT |
| VBG CLP I |
C11H14O4 |
11.9 |
[M + H]+ |
210.08920/210.08921 |
0.04 |
139 (100), 165 (20), 124 (10) |
275, 303 |
Lac/HBT |
| VBG CLP II |
C12H16O4 |
15.4 |
[M + H]+ |
224.10467/224.10486 |
−0.85 |
165 (100), 139 (18), 181 (8) |
276, 308 |
Lac/HBT |
| [M + Na]+ |
N.D. |
| VBGox |
C18H20O6 |
26.6 |
[M + H]+ |
332.12563/332.12599 |
−1.08 |
149 (100), 167 (84), 285 (84), 165 (74), 181 (72), 209 (52), 177 (30), 303 (28), 121 (20), 192 (12) |
279, 312 |
Lac/HBT |
| [M + Na]+ |
325 (100), 232 (18), 201 (10) |
|
Reaction products of SVBG
|
| SVBG |
C18H22O8S |
14.8, 15.1 |
[M − H]− |
398.10353/398.10354 |
−0.02 |
273 (100), 203 (20), 123 (18), 215 (18), 329 (8) |
277 |
Lac, lac/HBT |
|
HBT and related
|
| HBT |
C6H5N3O |
3.8 |
[M − H]− |
135.04325/135.04325 |
−0.07 |
106 (100), 78 (6) |
306, 276 |
Lac/HBT |
| [M + H]+ |
91 (32), 80 (30), 107 (20), 53 (18) |
268 |
| BT |
C6H5N3 |
6.9 |
[M + H]+ |
119.04827/119.04835 |
−0.65 |
N.D. |
258, 280 |
Lac/HBT |
Discussion
The effect of sulfonation of phenolic lignin model compounds on their conversion by laccase and laccase/HBT
The reaction pathways of GBG and SGBG were highly similar. Both model compounds underwent oligomerization in the laccase and laccase/HBT incubations. Hence, sulfonation of the phenolic end caps in lignin does not affect their proneness to undergo radical coupling reactions. Thus, both native and sulfonated lignin may undergo polymerization when oxidized by laccase. The presence of HBT had only a minor effect on the reaction pathway (Fig. 3), which can be explained by the fact that laccase oxidizes GBG and SGBG much faster than it oxidizes HBT (Fig. S14†). Consequently, GBG and SGBG are already converted before a substantial amount of HBT is oxidized.
Laccase converted SGBG more slowly than it converted GBG. The conversion rate of phenolic substrates by laccase has been reported to be dependent on the difference in redox potentials of the laccase and the substrate.31 To check whether the sulfonation may have affected the redox potential of the phenolic model compounds, we calculated their ionization energies in water using B97D/6-311+G(d,p) calculations and an SMD solvent model for water (Table 3). As the ionization energies of GBG and SGBG are very similar, it is unlikely that the lower conversion rate of SGBG as compared to GBG is due to different redox properties of the compounds. An alternative explanation might be that the sulfonation decreases the affinity of the substrate for the active site of the laccase. It has been reported that the substrate binding cavity of a laccase from T. versicolor, is dominated by negative charges.32 As the sulfonic acid group on SGBG is also negatively charged, electrostatic repulsion may occur, resulting in a lower binding affinity and, consequently, to a lower conversion rate.
Table 3 Calculated ionization energies in water (IE) and Cα–H bond dissociation energies (BDE) of the model compounds used. N.D. = not determined
|
|
Cα–H BDE (kcal mol−1) |
IE (kcal mol−1) |
| GBG |
84 |
121 |
| SGBG |
94 |
118 |
| VBG |
87 |
N.D. |
| SVBG |
94 |
N.D. |
The effect of sulfonation of non-phenolic lignin model compounds on their reactions by laccase/HBT
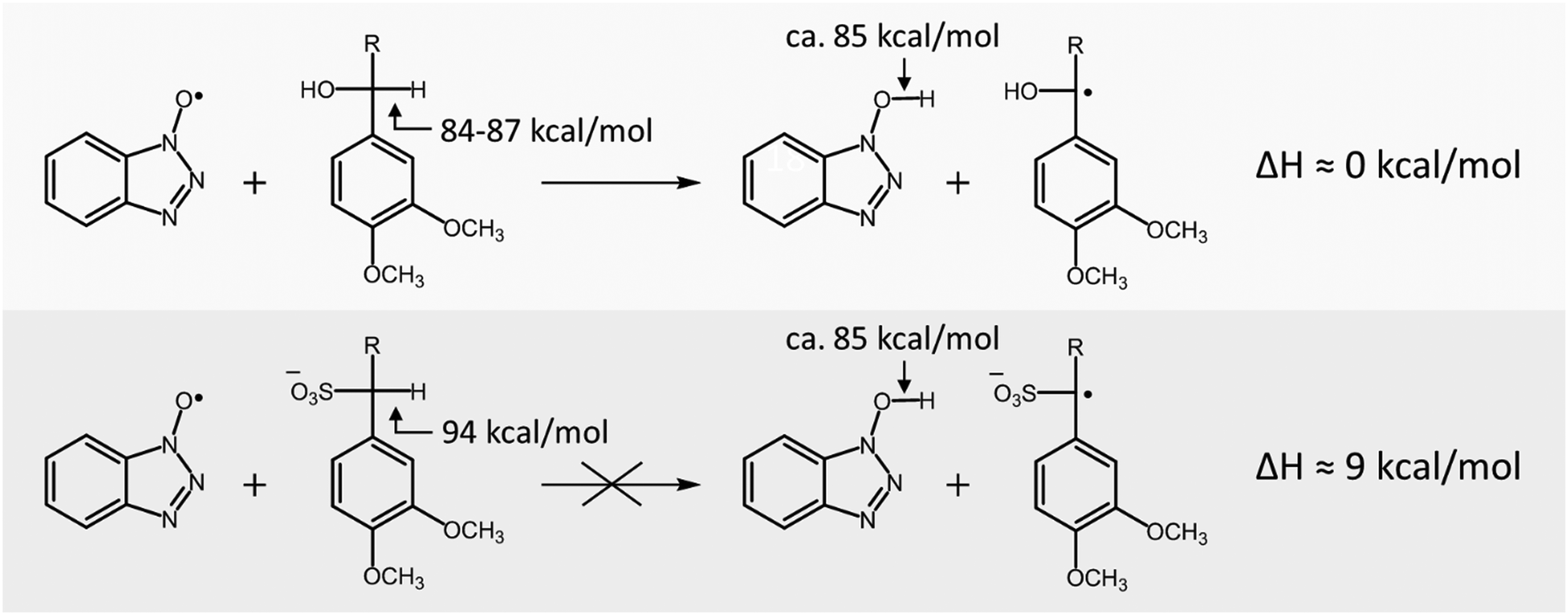
Whereas laccase/HBT slowly converted VBG to both its ketone analogue and cleavage products, it was unable to convert SVBG. Since in a laccase/HBT system oxidation of the substrate by the mediator occurs via a HAT mechanism,19 the reaction is dependent on the difference in bond dissociation energy (BDE) between the O–H bond of HBT and the Cα–H bond in the lignin model compound. In order to explain the difference in reactivity of VBG and SVBG, we calculated the Cα–H BDE of both molecules (Table 3). As can be observed, the Cα–H BDE of SVBG (94 kcal mol−1) is substantially higher than that of VBG (87 kcal mol−1), indicating that radical hydrogen abstraction is less favourable in the sulfonated structure. According to literature, the BDE of the O–H bond in HBT is approximately 85 kcal mol−1.33 As this value is in the same range as that of VBG, it seems plausible that radical hydrogen abstraction by the HBT radical can occur in the case of VBG. In contrast, the BDE of SVBG is too high for the HBT radical to abstract a radical hydrogen atom, making it inert to the laccase/HBT system, in line with our experiments (Scheme 2).
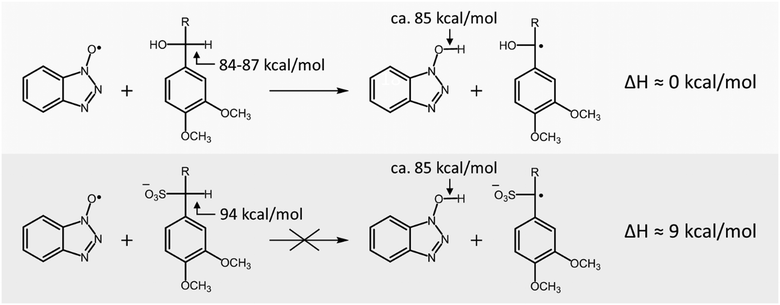 |
| | Scheme 2 Sulfonation of lignin structures increases the Cα–H BDE, and thereby makes non-phenolic lignin structures inert to conversion by the laccase/HBT system. | |
Sulfonation of lignin polymers: implications for reactivity with laccase and LMS
Several studies have investigated the effect of laccase and LMS on polymeric lignin, and both polymerization and depolymerization have been reported as outcomes.3 It has been suggested that polymerization and depolymerization are in competition with each other, and that this competition may be influenced by reaction conditions and the presence of mediators.3 Obviously, the final outcome of this competition is dependent on the reactions of both the phenolic and non-phenolic structures in lignin. It is a general trend that the activity of laccase alone leads to lignin polymerization, whereas the activity of LMS leads to depolymerization.3 This can be rationalized, as laccase alone can only oxidize the phenolic subunits, which tend to undergo radical coupling, leading to lignin polymerization. LMS also oxidizes the non-phenolic structures, which generally leads to Cα oxidation or bond cleavage.
Remarkably, when lignosulfonate is used as a substrate, both laccase and LMS incubations are shown to induce polymerization.4,6,22,23,34 To date, there was no mechanistic explanation for this observation, since the effects of laccase and LMS have only been investigated on polymeric lignosulfonate. These studies reported changes in molecular weight or overall changes in chemical structure, but did not describe detailed reaction pathways. To overcome this knowledge gap, in the present study, we used phenolic and non-phenolic sulfonated lignin model compounds. This enabled us to rationalize the observation that lignosulfonate is polymerized by both laccase and LMS: polymerization, which generally occurs upon oxidation of phenolic lignin subunits, can still occur when lignin is sulfonated. In contrast, cleavage of the β-O-4 bond, which generally occurs upon oxidation of non-phenolic lignin subunits, is impaired after sulfonation. This drives the polymerization/depolymerization competition to an overall result of lignin polymerization, even in the presence of mediators. Obviously, the extent of this effect is dependent on the degree of sulfonation of the lignosulfonate preparation and the ratio phenolic/non-phenolic subunits. In addition, it should be taken into account that the model compounds used, consist of G-units linked by a β-O-4 bond. In lignin and lignosulfonate, S and H units and other inter-unit linkages may be present, which may also play a role in the overall reaction outcome.
Conclusions
This work reports the effect of sulfonation of lignin model compounds on their reactivity in laccase and LMS incubations. Phenolic lignin model compounds, both native and sulfonated, underwent radical coupling reactions, indicating that laccase is able to induce polymerization of both native and sulfonated lignin. Sulfonation of a non-phenolic β-O-4 linked lignin model compound had a larger impact on its reactivity: whereas Cα oxidation and cleavage of the native model compound were observed in LMS incubations, the sulfonated model compound was completely unreactive in laccase and laccase/HBT incubations. Overall, since sulfonation prevents cleavage of the non-phenolic lignin backbone, while keeping radical coupling of phenolic end groups possible in LMS incubations, we conclude that sulfonation of lignin drives the overall outcome of LMS incubations toward polymerization. The insights obtained from this study can be used to rationalize and predict the effect of laccase and LMS on native lignin and lignosulfonate.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Dr. Pieter de Waard is greatly acknowledged for performing the NMR analyses.
References
- L. P. Christopher, B. Yao and Y. Ji, Front. Energy Res., 2014, 2, 12 CrossRef.
- T. D. Bugg and R. Rahmanpour, Curr. Opin. Chem. Biol., 2015, 29, 10–17 CrossRef CAS PubMed.
- L. Munk, A. K. Sitarz, D. C. Kalyani, J. D. Mikkelsen and A. S. Meyer, Biotechnol. Adv., 2015, 33, 13–24 CrossRef CAS PubMed.
- D. Areskogh, J. Li, G. R. Gellerstedt and G. Henriksson, Biomacromolecules, 2010, 11, 904–910 CrossRef CAS PubMed.
- K. Lundquist and J. Parkås, BioResources, 2011, 6, 920–926 CAS.
- A. Ortner, K. Hofer, W. Bauer, G. S. Nyanhongo and G. M. Guebitz, React. Funct. Polym., 2018, 123, 20–25 CrossRef CAS.
- Q. Chen, M. N. Marshall, S. M. Geib, M. Tien and T. L. Richard, Bioresour. Technol., 2012, 117, 186–192 CrossRef CAS.
- L. Munk, A. Punt, M. Kabel and A. S. Meyer, RSC Adv., 2017, 7, 3358–3368 RSC.
- S. R. Couto and J. L. T. Herrera, Biotechnol. Adv., 2006, 24, 500–513 CrossRef PubMed.
- R. Bourbonnais and M. G. Paice, FEBS Lett., 1990, 267, 99–102 CrossRef CAS PubMed.
- A. Rico, J. Rencoret, J. C. del Río, A. T. Martínez and A. Gutiérrez, Biotechnol. Biofuels, 2014, 7, 6 CrossRef PubMed.
- J. Rencoret, A. Pereira, C. José, A. T. Martínez and A. Gutiérrez, BioEnergy Res., 2016, 9, 917–930 CrossRef CAS.
- T. Rosado, P. Bernardo, K. Koci, A. V. Coelho, M. P. Robalo and L. O. Martins, Bioresour. Technol., 2012, 124, 371–378 CrossRef CAS PubMed.
- S. Camarero, D. Ibarra, Á. T. Martínez, J. Romero, A. Gutiérrez and C. José, Enzyme Microb. Technol., 2007, 40, 1264–1271 CrossRef CAS.
- A. I. Cañas and S. Camarero, Biotechnol. Adv., 2010, 28, 694–705 CrossRef PubMed.
- S. Shleev, P. Persson, G. Shumakovich, Y. Mazhugo, A. Yaropolov, T. Ruzgas and L. Gorton, Enzyme Microb. Technol., 2006, 39, 841–847 CrossRef CAS.
- R. Bourbonnais and M. G. Paice, Appl. Microbiol. Biotechnol., 1992, 36, 823–827 CrossRef CAS.
- A. Gutiérrez, J. Rencoret, E. M. Cadena, A. Rico, D. Barth, C. José and Á. T. Martínez, Bioresour. Technol., 2012, 119, 114–122 CrossRef PubMed.
- P. Baiocco, A. M. Barreca, M. Fabbrini, C. Galli and P. Gentili, Org. Biomol. Chem., 2003, 1, 191–197 RSC.
- C. Galli, P. Gentili and O. Lanzalunga, Angew. Chem., Int. Ed., 2008, 47, 4790–4796 CrossRef CAS.
-
R. J. A. Gosselink, Lignin as a renewable aromatic resource for the chemical industry, 2011 Search PubMed.
- D. Huber, A. Ortner, A. Daxbacher, G. S. Nyanhongo, W. Bauer and G. M. Guebitz, ACS Sustainable Chem. Eng., 2016, 4, 5303–5310 CrossRef CAS.
- E. N. Prasetyo, T. Kudanga, L. Østergaard, J. Rencoret, A. Gutiérrez, C. José, J. I. Santos, L. Nieto, J. Jiménez-Barbero and A. T. Martínez, Bioresour. Technol., 2010, 101, 5054–5062 CrossRef.
- L. Heap, A. Green, D. Brown, B. van Dongen and N. Turner, Catal. Sci. Technol., 2014, 4, 2251–2259 RSC.
- R. J. Hilgers, J.-P. Vincken, H. Gruppen and M. A. Kabel, ACS Sustainable Chem. Eng., 2018, 6, 2037–2046 CrossRef CAS.
-
M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- B. Ramalingam, B. Sana, J. Seayad, F. Ghadessy and M. Sullivan, RSC Adv., 2017, 7, 11951–11958 RSC.
- M. Lahtinen, P. Heinonen, M. Oivanen, P. Karhunen, K. Kruus and J. Sipilä, Org. Biomol. Chem., 2013, 11, 5454–5464 RSC.
- S. Kawai, M. Asukai, N. Ohya, K. Okita, T. Ito and H. Ohashi, FEMS Microbiol. Lett., 1999, 170, 51–57 CAS.
- E. Srebotnik and K. E. Hammel, J. Biotechnol., 2000, 81, 179–188 CrossRef CAS.
- F. Xu, Biochemistry, 1996, 35, 7608–7614 CrossRef CAS.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS.
- P. Astolfi, P. Brandi, C. Galli, P. Gentili, M. F. Gerini, L. Greci and O. Lanzalunga, New J. Chem., 2005, 29, 1308–1317 RSC.
- T. Gillgren, M. Hedenström and L. J. Jönsson, Int. J. Biol. Macromol., 2017, 105, 438–446 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy00249a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Megan
Twentyman-Jones
a,
Annemieke
van Dam
a,
Megan
Twentyman-Jones
a,
Annemieke
van Dam