
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inductive and electrostatic effects on cobalt porphyrins for heterogeneous electrocatalytic carbon dioxide reduction†
Minghui
Zhu‡
,
Deng-Tao
Yang‡
 ,
Ruquan
Ye
,
Joy
Zeng
,
Nathan
Corbin
and
Karthish
Manthiram
*
,
Ruquan
Ye
,
Joy
Zeng
,
Nathan
Corbin
and
Karthish
Manthiram
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: karthish@mit.edu
First published on 21st January 2019
Abstract
Electrochemical carbon dioxide reduction enables conversion of carbon dioxide into fuels and chemicals with renewable energy input. Cobalt-based molecular complexes have exhibited high selectivity, activity, and stability for transforming carbon dioxide into carbon monoxide. Through evaluating immobilized cobalt porphyrins functionalized with various peripheral substituents, we demonstrated that their activity is affected not only by the electronegativity of the substituents, but importantly, also by the charge of the substituents. The performance of immobilized cobalt porphyrins can be improved by introducing electron-donating and positively charged functional groups. Through kinetic studies, we were able to understand the mechanism by which electron-donating groups enhance the observed rates of carbon dioxide reduction and how cationic functionality may contribute towards electrostatic stabilization of the intermediate formed in the rate-determining step. Our methodology provides a robust and experimentally-verified method of computationally predicting the electronic effect of peripheral substitution and hence the catalytic activity of substituted porphyrins.
1. Introduction
Electrochemical conversion of carbon dioxide (CO2) to fuel and chemicals may mitigate emissions which would otherwise contribute to global warming.1–3 Such a process is attractive because it can operate at ambient pressure and room temperature.4 Among various cathode materials that have been studied, immobilized cobalt macrocycles, including cobalt porphyrins (CoP), cobalt phthalocyanines (CoPc), and their derivatives, have drawn much attention for electroreduction of CO2 because of their high activities and selectivities for producing CO,5–8 which is a useful target that can be further reacted through existing thermochemical processes to produce diverse commodity chemicals.9–12 The structure–activity relationships of these immobilized cobalt macrocycles, however, is underexplored.Functionalization of metal macrocycles has been systematically explored in the context of homogeneous metal macrocycles, such as iron tetraphenylporphyrin (FeTPP), solubilized in non-aqueous solvents.13–17 While non-aqueous solvents have provided significant fundamental understanding, undesirable solvent oxidation at the anode and sacrificial proton donors can limit the practical use of such systems. Use of aqueous electrolytes overcomes these problems as water is oxidized in the counter reaction, leading to a sustainable and viable overall reaction. Because most metal macrocycles of interest are insoluble in aqueous electrolytes, they need to be immobilized on the electrode surface. Immobilization has the added advantage that transport limitations associated with homogenous catalysts diffusing to the electrode surface can be overcome at higher current densities. Systematic understanding of the impact of functionalization on electrochemical CO2 reduction has been achieved on neither immobilized metal macrocycles nor in aqueous electrolytes, in part due to aggregation of these complexes when deposited on substrates, which obscures their intrinsic catalytic activity.18 In fact, electron donating and withdrawing substituents have both been reported to increase catalytic activity on the same metal macrocycle, likely due to differences in catalyst loadings that make systematic understanding of structure–activity relationships difficult.19–22
In addition to the through-bond inductive effect, external electric fields are increasingly recognized to play a large role in catalysis, including thermal catalysis23 and enzyme catalysis.24 In electrochemical carbon dioxide reduction, alkali metal cations have been found to influence the distribution of products formed during the electroreduction of CO2 at a copper electrode. This was attributed to electrostatic interactions between solvated cations present at the outer Helmholtz plane and adsorbed species having large dipole moments.25 Similarly, for homogeneous iron tetraphenylporphyrin (FeTPP) based catalysts, trimethylanilinium peripheral functional groups were found to promote homogeneous CO2 electroreduction in dimethylformamide (DMF), which was attributed to the stabilization of the reactive intermediates through an electrostatic effect.17 Electrostatic effects have not yet been elucidated for heterogenized molecular complexes, likely in part due to the aggregation effects described above and due to convolution with inductive effects.
In this work, we elucidated the impact of aromatic peripheral functionalization of heterogenized cobalt porphyrins for carbon dioxide reduction (Fig. 1a). We developed a strategy for quantifying the inductive effect of aromatic substituents using density functional theory (DFT), which we validated using experimental Hammett substituent constants and cyclic voltammetry of the Co(I/II) redox waves. The calculated inductive effect acts as a predictor of the experimentally measured intrinsic turnover frequency at low loadings. We are also able to deconvolute both the inductive and electrostatic effect arising from neutral and cationic substituents. These findings provide important molecular-level insights for the rational design of heterogenized cobalt-based molecular catalysts for electroreduction of CO2 to CO.
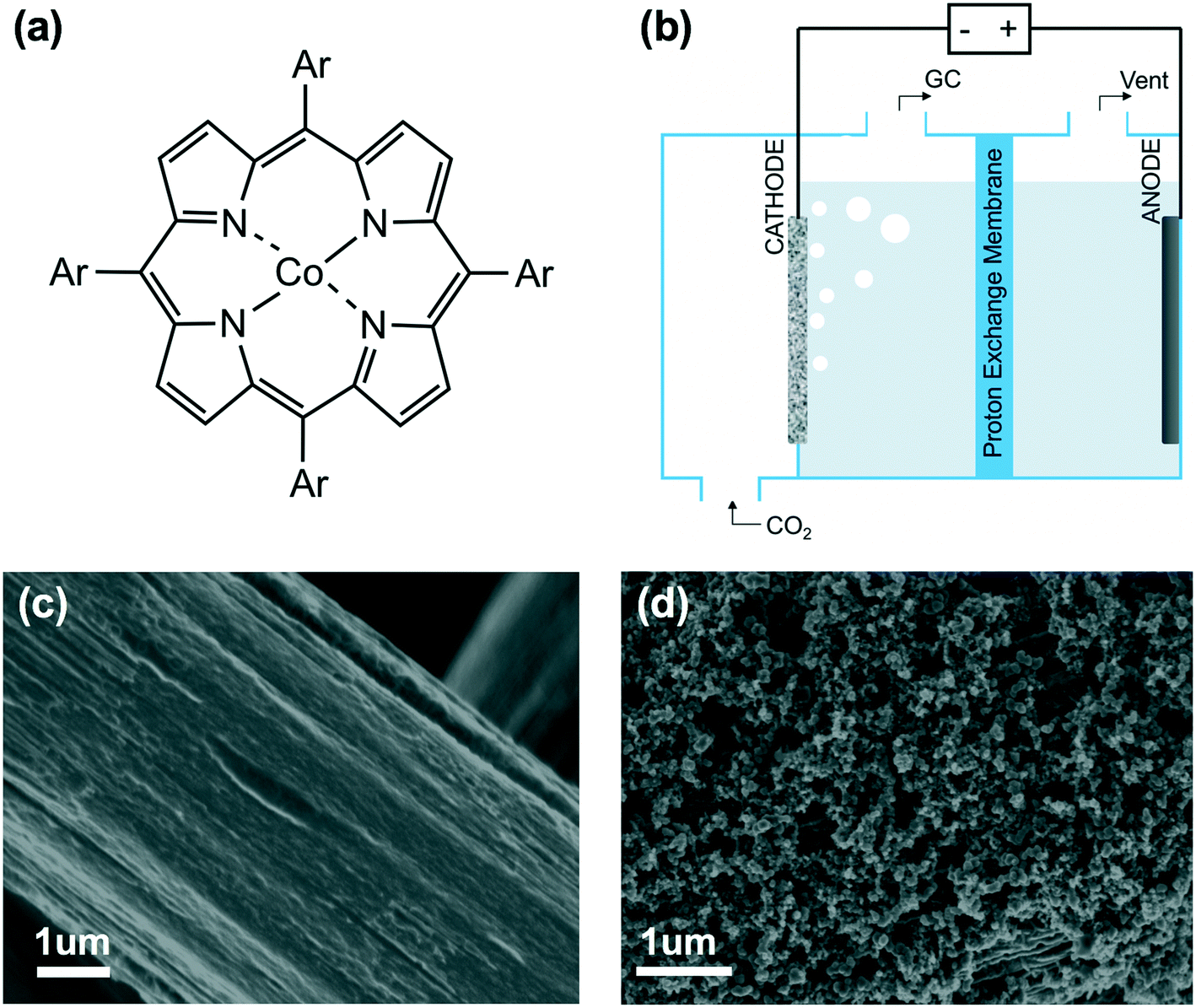 | ||
| Fig. 1 (a) Chemical structure of cobalt porphyrin with aromatic substituents (Ar). (b) Schematic of three-compartment cell configuration. (c) SEM image of pristine carbon paper. (d) SEM image of electrode with CoTPP catalyst at a loading of 1 × 10−8 mol cm−2. | ||
2. Results and discussion
2.1 Electrocatalysis at CoTPP
The catalytic behaviors of cobalt porphyrins were tested in a custom-built three-compartment cell containing 0.5 M NaHCO3 as the electrolyte (Fig. S7†).26 CO2 was introduced from a gas compartment, flowed through the working electrode into the electrolyte, and then sent to a gas chromatograph (GC) for product analysis (Fig. 1b). The working electrode was prepared by dropcasting a mixture of a particular cobalt porphyrin derivative, carbon black (conductive dispersant), Nafion (binder), and DMF onto carbon paper, which was calcined prior to dropcasting to increase hydrophilicity for better dispersion (Fig. 1c and d).27The cyclic voltammogram of cobalt tetraphenylporphyrin (CoTPP) in 0.5 M NaHCO3 under a CO2 atmosphere exhibited a prominent reductive current (Fig. 2a). Selectivity for CO was determined by electrolysis at various polarizations for 50 min; Faradaic efficiencies for CO at four tested potentials were all higher than 80% and exceeded 95% at a polarization of −0.6 V vs. RHE (Fig. 2b and Table S2†). Current densities were stable at the testing conditions of −0.6 V vs. RHE, but were less stable at more reducing potentials (Fig. S14†).
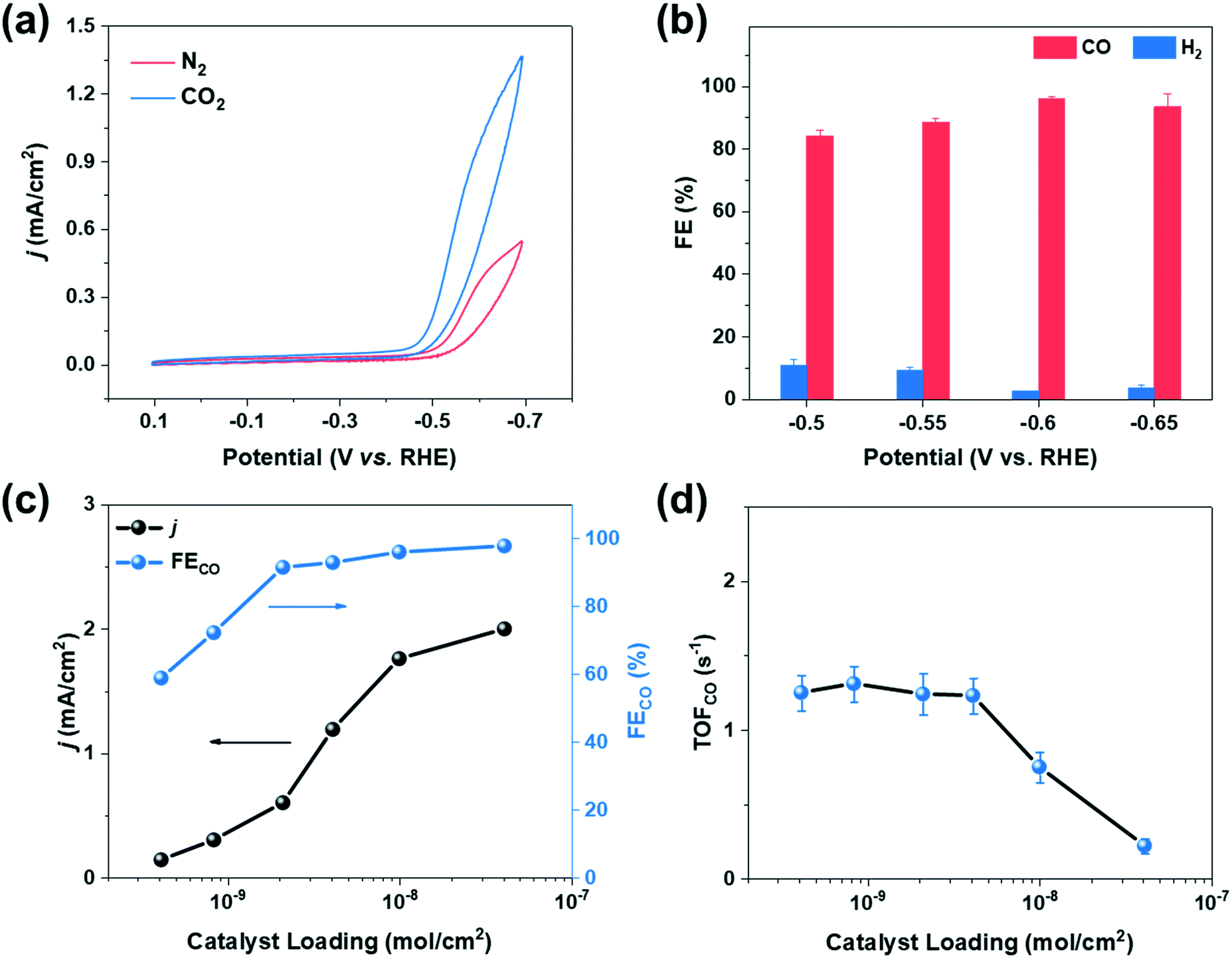 | ||
| Fig. 2 (a) Cyclic voltammograms collected at a sweep rate of 5 mV s−1 of CoTPP under N2 and CO2, respectively and (b) Faradaic efficiencies of CO2 reduction products for CoTPP at various potentials (loading: 1 × 10−8 mol cm−2). (c) Overall current densities and Faradaic efficiencies for CO of CoTPP at various loadings. (d) Turnover frequency for CO at −0.6 V vs. RHE for CoTPP at various loadings. Electrolyte solution is 0.5 M NaHCO3. | ||
We therefore conducted our study at a potential of −0.6 V vs. RHE and further evaluated the catalytic performance of CoTPP at various catalyst loadings. Specifically, the loading of CoTPP ranged from 4 × 10−10 to 4 × 10−8 mol cm−2, while the loading of carbon black was kept constant at ∼70 μg cm−2. The overall current density decreased with decreasing CoTPP loadings (Fig. 2c); at an extreme scenario where no CoTPP was loaded, the carbon black/carbon paper composite exhibited small overall current density and negligible CO partial current density (Fig. S15 and Table S3†). CoTPP loaded on carbon paper exhibited Faradaic efficiencies for CO2 to CO conversion ranging from ∼100% at high loadings to ∼60% at the lowest loading. The decrease in Faradaic efficiencies at low loadings is due to the increased contribution of the underlying carbon black/carbon fiber paper composite towards HER at low CoTPP content (Table S3†).
The turnover frequency for CO (TOFCO) was calculated to quantify the catalytic activity of cobalt porphyrins, which is defined as the number of CO molecules generated per active site per unit time. We calculated the lower bound of the TOFCO based on the total porphyrin loading from dropcasting.28,29 With decreasing catalyst loading, TOFCO initially increased and then stabilized at ∼1.5 s−1 at a loading below 2 × 10−9 mol cm−2 (Fig. 2d), which corresponds to a TON of 4500 during the 50 min electrolysis with moderate decay in CO production (Fig. S16†). This behavior highlights the importance of comparing TOFCO of various cobalt porphyrins at low loadings to make meaningful comparisons (Table S5†).18 Such a strategy allows for structure–property relationships to be more clearly elucidated because it minimizes complicating effects that could be caused by catalyst aggregation.
2.2 Effect of peripheral functionalities
We then investigated a diverse set of cobalt porphyrins with various aromatic substituents (Ar), including the neutral porphyrins: cobalt tetraphenylporphyrin (CoTPP), cobalt tetramethoxyphenylporphyrin (CoTMPP), cobalt tetrabromophenylporphyrin (CoTBPP), and cobalt tetrachlorophenylporphyrin (CoTCPP), and the cationic porphyrins: cobalt tetra-(N-methyl-2-pyridyl)porphyrin (CoTMpyp2), cobalt tetra-(N-methyl-3-pyridyl)porphyrin (CoTMpyp3), cobalt tetra-(N-methyl-4-pyridyl)porphyrin (CoTMpyp4), and cobalt tetra-(4-N,N,N-trimethylanilinium)porphyrin (CoTMAP). Among these compounds, CoTPP, CoTMPP, CoTBPP, CoTCPP and CoTMpyp4 were commercially available. CoTMAP, CoTMpyp2 and CoTMpyp3 were synthesized using a metalation reaction and fully characterized by 1H-NMR, UV-vis spectroscopy, and high-resolution mass spectroscopy (see ESI†). The aromatic substituents in these derivatives are nearly perpendicular to the plane of the macrocycle.31 This has the ramification that the dominant through-bond effect of the substituent on the electronic structure at the cobalt center, which in turn influences adsorption of key catalytic intermediates, is via inductive effects as opposed to conjugation. Hence, we quantified the electronegativity of those substituents using density functional theory (DFT) via Mulliken population analysis on hydrogen charge densities (χH) in corresponding H-Ar molecules (Fig. 3a), with larger positive values representing stronger electronegativity (see ESI†).32 In this case, a probe hydrogen atom was attached to the substituent of interest and the Mulliken charge on the probe hydrogen was used as a measure of the electron withdrawing or donating nature of the substituent.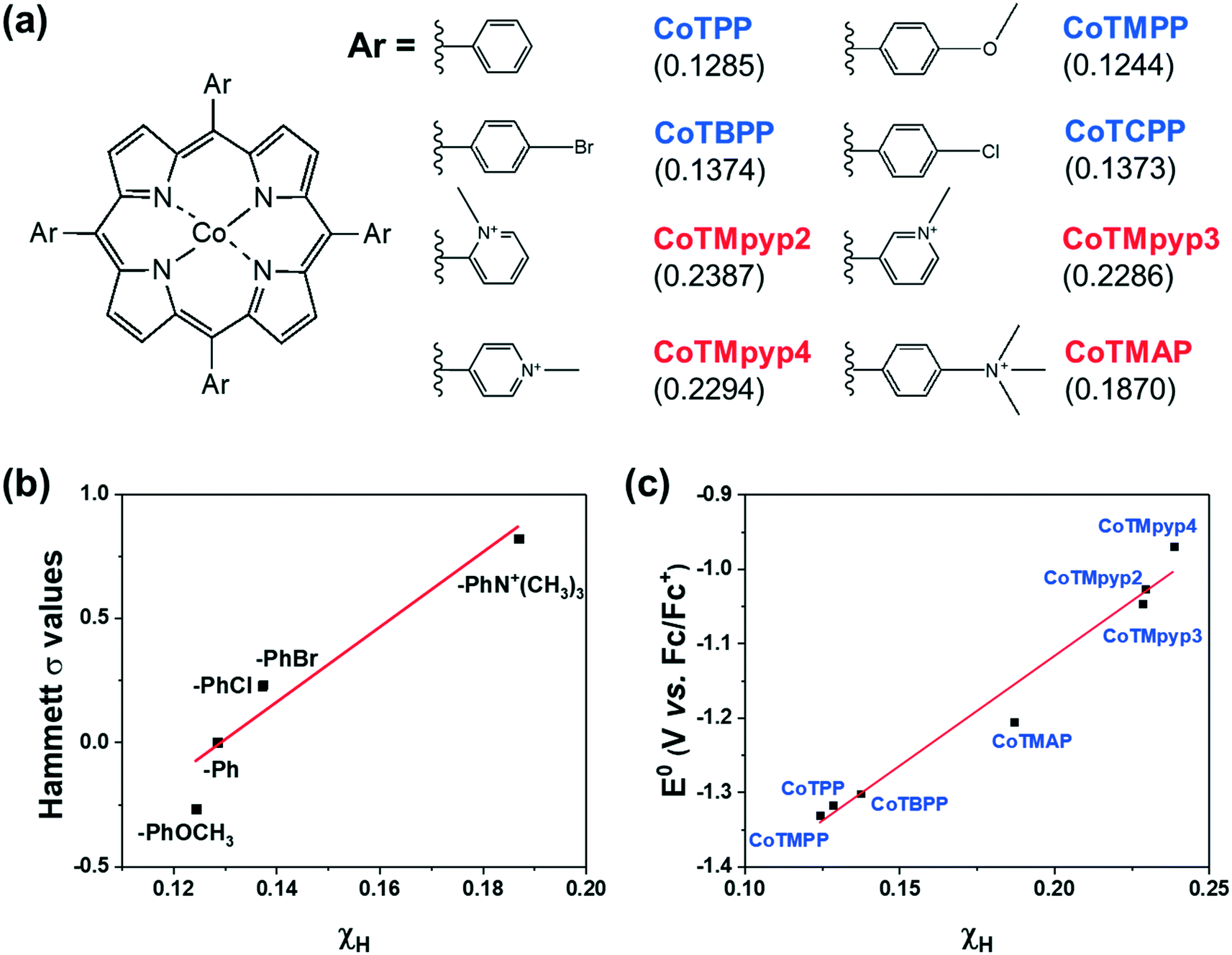 | ||
| Fig. 3 (a) The chemical structures of functionalized cobalt porphyrins and the corresponding Mulliken charge population on a probe hydrogen atom (χH) for the aromatic substituents (Ar) shown in parenthesis. (b) Correlation between calculated χH and the corresponding para position Hammett substituent constant.30 (c) Correlation between calculated χH and experimentally measured Co(I/II) redox potentials. | ||
By utilizing a calculation which involves the aromatic substituent alone instead of the entire complex, we can more easily compare the calculated χH values to experimentally tabulated Hammett substituent constants (σ). Experimentally derived Hammett values are determined from rates of protonation and equilibrium constants for deprotonation of probe carboxylic acids and are also meant to experimentally describe electron donating or withdrawing character of various functional groups. The obtained χH values correlate well with reported Hammett substituent constants (σ) in the literature (Fig. 3b),30 which confirms the fidelity of the calculated χH values in quantifying the inductive effect of the substituent.
The inductive effect of a substituent is important as a descriptor to the extent that it captures the electronic effect at the cobalt center. To determine this, we experimentally measured the CoI/II redox potential (Fig. S19†), which correlated well with the calculated χH values (Fig. 3c), demonstrating that the inductive effect of the substituent is associated with the electronic structure at the catalytically relevant cobalt center. The advantage of using a χH value as a descriptor of catalytic activity is that it can be computationally predicted even when the experimentally tabulated Hammett substituent constants are not available, and does not require the synthesis of a complex to experimentally measure its CoI/II redox potential.
We evaluated the catalytic activity of cobalt porphyrin derivatives (Fig. 3a) at a potential of −0.6 V vs. RHE at a low loading of 8 × 10−10 mol cm−2 to avoid aggregation (Table S4†). A clear TOFCO–χH relationship exists after peripheral charge is considered (Fig. S20†). Given that there exists a linear relationship between χH and Hammett substituent constants (Fig. 3b), we can convert the calculated χH to Hammett σ and plot TOFCOvs. these Hammett σ values (Fig. 4). For neutral porphyrins, TOFCO exhibited an inverse dependence on the σ value. In other words, more electron withdrawing substituents decreased the catalytic activity for electroreduction of CO2. Such a trend also held true for the cationic porphyrins, with CoTMAP showing the highest TOFCO (∼5 s−1 at −0.6 V vs. RHE), which is higher than that of other reported porphyrin-based complexes (Table S5†). The existence of two distinct TOFCO-σ bands corresponding to neutral and cationic porphyrins demonstrates that the activity of immobilized cobalt porphyrins is not exclusively determined by the electronegativity of substituents. Other factors, in this case, charge of peripheral functionalities, are also important for determining catalytic activity of these molecular catalysts. Specifically, we find that cationic porphyrins exhibited an upshifted trendline compared to neutral porphyrins, indicating that cationic peripheral substituents lead to increased rates of CO2 reduction while holding the inductive nature of the substituent constant.
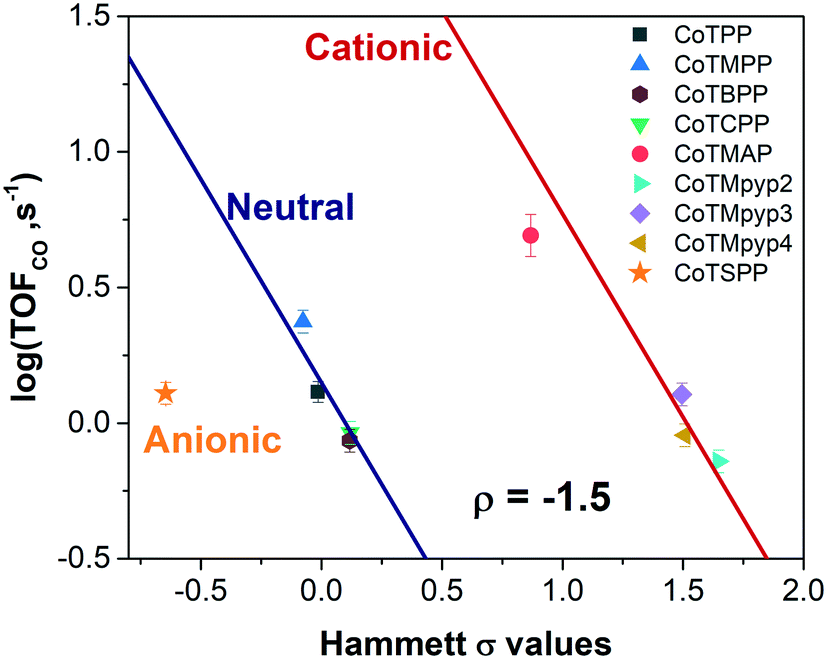 | ||
| Fig. 4 TOFCO of cobalt porphyrin derivatives with various functionalities versus calculated Hammett σ values (potential: −0.6 V vs. RHE, loading: 8 × 10−10 mol cm−2). | ||
2.3 Mechanistic interpretation
To further understand the promotion mechanism of electron-donating and cationic functionalities, we also examined the reaction mechanism of CO2 reduction on cobalt porphyrins. CoTPP at a low loading of 8 × 10−10 mol cm−2 exhibits a Tafel slope of 119 mV dec−1 (Fig. S21†), indicating that the first electron transfer is involved in the rate-determining step of CO2 reduction. The partial current density of CO (Fig. S29 and S37†) has a first-order dependence on CO2 partial pressure and zeroth-order dependence on bicarbonate concentration. The zeroth-order bicarbonate dependence also implies a zeroth-order pH dependence, because bicarbonate and proton concentrations are directly correlated under conditions of CO2-saturation.33 Together, the Tafel slope and reaction orders suggest the rate-determining step involves the binding of CO2 accompanied by an electron transfer.6,28,34 Such a conclusion also holds true for other catalysts that we tested which exhibit a wide range of TOFCO (Fig. S21–S44†). We can then rationalize that the introduction of electron-donating substituents increases the electron density of the catalytically active Co center via inductive effects, which facilitates rate limiting binding of CO2 and, therefore, results in an increase of the activity for CO2 electroreduction. Surprisingly, this is the opposite trend as observed for porphyrin-based covalent organic frameworks, in which electron withdrawing functionality is observed to promote carbon dioxide reduction.35In our case, the TOFCO for a given complex relative to the TOFCO for CoTPP is given by the following relation:
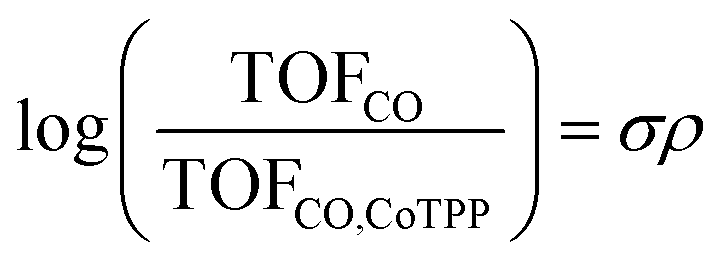
The reaction constant ρ provides an understanding of the sensitivity of the reaction to substituents and can be determined from the slope of turnover frequency as a function of σ (Fig. 4), from which we obtain ρ = −1.5. A negative reaction constant demonstrates that the rate determining step involves loss of negative charge, consistent with our mechanism indicating that charge transfer from the cobalt center to carbon dioxide is rate limiting. The magnitude of this value of ρ is small compared to ρ values for other organic reactions, many of whose magnitudes range from one to over three.36 A reason for the lower reaction constant could be that the inductive effects of the substituents must be transduced through several bonds before influencing the cobalt center, although the presence of four identical substituents does partly compensate for the larger distance.
The promotion mechanism of cationic functionalities is less understood, although similar effects have been observed in the case of homogeneous FeTPP derivatives in DMF with phenol as proton donor.17,37 In the context of metallic electrocatalysts, several studies have suggested local electric fields originating from alkali metal cations near the electrode can stabilize CO2 reduction intermediates, resulting in higher catalytic activities.38–40 Given that our kinetic data suggests that binding of carbon dioxide with concomitant electron transfer is rate limiting, the observation of two distinct bands corresponding to neutrally and positively charged porphyrins may be due to favorable, stabilizing electrostatic interactions between the cationic substituent and the transition state formed during rate limiting electron transfer to carbon dioxide. Given that cationic complexes promote electrochemical carbon dioxide reduction, we would expect that anionic complexes would lead to reduced rates of carbon dioxide reduction; we tested an anionic porphyrin, cobalt tetrasulfonatophenylporphyrin tetrasodium (CoTSPP), whose TOFCO (1.77 s−1) fell below the log(TOFCO)-Hammett σ trendline for neutral porphyrins (Fig. 4), further confirming the electrostatic hypothesis. Since electrostatic interactions drop off sharply with distance, this may explain why the TOFCO–χH relationship for the cationic porphyrins exhibit deviations from linearity that correlate with the distance between the cationic functionality and a catalytic intermediate bound at the cobalt center. This may be coupled to changes in the planarity of the porphyrin molecules. For instance, while CoTMpyp4 is expected to be nonplanar, CoTMpyp2 and CoTMpyp3 are thought to be planar, which would impact the distances associated with the electrostatic interactions.41,42 We expect that these electrostatic interactions are relatively complex since they occur within the electrochemical double layer.43
3. Conclusions
The findings of this study provide fundamental insights into the molecular engineering of immobilized cobalt porphyrins for electroreduction of CO2 in aqueous electrolyte. We demonstrated the importance of low catalyst loading for alleviating the issue of aggregation that could otherwise complicate the calculated turnover frequencies. With detailed mechanistic studies, we elucidated the rate-determining-step for CO2 reduction on cobalt porphyrins, which involves both CO2 and a single electron-transfer to carbon dioxide. Our results indicate that electron-donating substituents tend to increase the cobalt porphyrin's activity, likely by increasing electron density at the cobalt center and facilitating electrosorption of CO2. In addition, cationic functionalities led to an increase in CO2 reduction activity at the immobilized complex, likely due to electrostatic stabilization of the transition state formed during rate determining electron transfer to carbon dioxide. These results provide an approach for the rational design of highly active immobilized metal macrocycles for the electroreduction of CO2 to CO based on deconvolution of inductive and electrostatic effects. Our approach involving density functional theory calculations of the inductive effect of substituents, which are experimentally validated against Hammett substituent constants and redox potentials of complexes, provides a robust framework for computational screening of candidate catalyst structures prior to experimental testing, which may be more broadly applied.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Subodh Gupta for insightful discussions. We gratefully acknowledge financial support from Cenovus Energy and the MIT Energy Initiative Low Carbon Energy Center on Carbon Capture, Utilization, and Storage. This work made use of the NSF-funded MIT CMSE shared facilities.References
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- T. R. Karl, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- P. M. Cox, R. A. Betts, C. D. Jones, S. A. Spall and I. J. Totterdell, Nature, 2000, 408, 184–187 CrossRef CAS.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–14 CrossRef.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158 RSC.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed.
- K. Fang, D. Li, M. Lin, M. Xiang, W. Wei and Y. Sun, Catal. Today, 2009, 147, 133–138 CrossRef CAS.
- S. K. Gangwal and V. Subramani, Energy Fuels, 2008, 22, 814–839 CrossRef.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2015, 48, 2996–3006 CrossRef CAS PubMed.
- R. B. Ambre, Q. Daniel, T. Fan, H. Chen, B. Zhang, L. Wang, M. S. G. Ahlquist, L. Duan and L. Sun, Chem. Commun., 2016, 52, 14478–14481 RSC.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- M. Zhu, R. Ye, K. Jin, N. Lazouski and K. Manthiram, ACS Energy Lett., 2018, 3, 1381–1386 CrossRef CAS.
- M. Yuasa, B. Steiger and F. C. Anson, J. Porphyrins Phthalocyanines, 1997, 01, 181–188 CrossRef CAS.
- A. Bettelheim, B. A. White, S. A. Raybuck and R. W. Murray, Inorg. Chem., 1987, 26, 1009–1017 CrossRef CAS.
- E. Song, C. Shi and F. C. Anson, Langmuir, 1998, 14, 4315–4321 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS PubMed.
- F. Che, J. T. Gray, S. Ha, N. Kruse, S. L. Scott and J.-S. McEwen, ACS Catal., 2018, 8, 5153–5174 CrossRef CAS.
- S. D. Fried and S. G. Boxer, Annu. Rev. Biochem., 2017, 86, 387–415 CrossRef CAS PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC.
- D. Kong, H. Wang, Z. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed.
- X.-M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- H. M. Marques and I. Cukrowski, Phys. Chem. Chem. Phys., 2002, 4, 5878–5887 RSC.
- S. Marriott, W. F. Reynolds, R. W. Taft and R. D. Topsom, J. Org. Chem., 1984, 49, 959–965 CrossRef CAS.
- A. Wuttig, Y. Yoon, J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2017, 139, 17109–17113 CrossRef CAS PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- C. Costentin, M. Robert, J.-M. Savéant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Norskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- V. H. Le, N. Nagesh and E. A. Lewis, PLoS One, 2013, 8, 1–9 Search PubMed.
- F. X. Han, R. T. Wheelhouse and L. H. Hurley, J. Am. Chem. Soc., 1999, 121, 3561–3570 CrossRef CAS.
- S. Yoshimoto and T. Sawaguchi, J. Am. Chem. Soc., 2008, 130, 15944–15949 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy00102f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |