
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular water oxidation catalysis by zwitterionic carboxylate bridge-functionalized bis-NHC iridium complexes†
Raquel
Puerta-Oteo
,
M. Victoria
Jiménez
 * and
Jesús J.
Pérez-Torrente
*
* and
Jesús J.
Pérez-Torrente
*
Department of Inorganic Chemistry, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH-CSIC), University of Zaragoza-CSIC, Facultad de Ciencias, C/Pedro Cerbuna, 12, 50009 Zaragoza, Spain. E-mail: perez@unizar.es
First published on 22nd February 2019
Abstract
Zwitterionic water-soluble [Cp*IrIIICl{(MeIm)2CHCOO}] and [IrI(cod){(MeIm)2CHCOO}] complexes featuring a carboxylate bridge-functionalized bis-N-heterocyclic carbene ligand efficiently catalyzed water oxidation using ammonium cerium(IV) nitrate (CAN) or sodium periodate as sacrificial oxidants. Excellent yields with TOF50 numbers up to 1000 h−1 have been achieved using CAN as electron acceptor at [CAN]/[Ir] ratios higher than 700. The investigation of the reaction mechanism by UV-vis seems to evidence that both catalyst precursors are transformed into the same active molecular species resulting from the degradation of the hydrocarbon ligands which is partially supported by the similarity of the oxygen evolution profiles at moderate oxidant/catalyst ratios for both catalyst precursors and the same chemical oxidant. In addition, DLS studies provide evidence for the participation of homogeneous iridium molecular species as intermediates likely stabilized by the carboxylate-functionalized bis-NHC ligand.
1. Introduction
The expected energy demand in the medium term, the limited availability of fossil resources and the continuous increase of global CO2 emissions put forward the urgent need for a carbon-neutral renewable energy source. Hydrogen is an energy carrier that is regarded as the fuel of the future due to its high mass-energy density and its clean electrochemical combustion in fuel cells. In this context, water splitting to produce hydrogen and oxygen from water could be a key technological component of a hydrogen economy.1 Natural photosynthetic processes are an excellent source of inspiration for how to design a catalytic system that can use solar energy for fuel production. Green plants capture sunlight and convert it into energy-rich compounds by oxidation of water and reduction of CO2. However, an artificial photosynthetic system should follow a more practical approach using the solar energy to oxidize H2O to molecular oxygen, as a benign by-product, and then use the generated electrons to reduce protons to molecular hydrogen.2 Water oxidation is the bottle-neck of this technology since it is a thermodynamically demanding reaction of great molecular complexity from a mechanistic perspective. The oxidation of water to O2 involves the release of 4 H+ and 4 e− from two water molecules with formation of an oxygen–oxygen bond, thereby requiring both energy and an efficient catalyst.The development of active and robust water-oxidation catalysts (WOCs) is crucial for the design of artificial photosynthetic devices. A number of transition metal complexes (Mn, Fe, Co, Ni, Ru, Ir, and Cu among others)3 have been reported as efficient catalysts for water oxidation over the last few years although ruthenium and iridium are among the most active. The assessment of WOCs can be carried out under chemical, electrochemical and photochemical driven conditions.4 However, from a practical point of view, water oxidation catalysis driven by sacrificial oxidants allows a rapid screening and tuning of the catalysts.3,5 The use of an electron acceptor, such as CAN or IO4−, is key in order to re-oxidize the catalysts by removing electrons from the system. Since seminal work by Meyer on the ruthenium “blue dimer” catalyst, the first homogeneous metal-based catalytic system,6 several ruthenium(II)-based homogeneous water oxidation catalyst are known as the most active molecular catalysts for water oxidation.7 Later on, Bernhard reported in 2008 the first robust cyclometalated iridium catalyst for water oxidation using CAN as sacrificial oxidant.8 Thereafter, a series of efficient iridium catalysts featuring an electron rich Cp* ligand in combination with different N^N, N^O, N^C ligands were reported by Crabtree and others as a way to stabilize high-valent iridium intermediates.9 Further development in the field was the design of robust catalysts based on the powerful electron donating NHC ligands,10 firstly introduced by Bernhard and Albrecht in 2010,11 or even bis-NHC ligands taking advantage of the chelate effect.12 Even so, the number of M-NHC based catalytic systems for water oxidation reactions remains scarce.13
Under the harsh reaction conditions, especially when using CAN as oxidant, some hydrocarbon ligands undergo oxidative degradation resulting in situ formation of iridium oxide nanoparticles (IrOx) that also catalyse water oxidation. In this context, there has been considerable debate regarding the homogeneous or heterogeneous nature of the catalytic species.14 Thus, the characterization and identification of catalytic species under turnover conditions is of great importance. The scales of WOC activity are strongly dependent on the nature of the sacrificial oxidant due to the different electron transfer mechanism.15 In this context, the tendency of NaIO4 to form NPs is negligible compared to CAN thereby avoiding possible complications in the kinetic studies.10,14a
Our approach for the design of robust water oxidation catalysts is the use of bis-NHC ligands functionalized with a carboxylate group at the linker.16 The carboxylate function may confer hemilabile properties to the ligand while imparting water solubility to the complexes. In addition, the potential coordination of the carboxylate to the metal centre might help for the stabilization of high-valent species likely involved in water oxidation catalysis. In this respect, we have recently reported the participation of the dihydrido iridium(III) [IrIIIH2(H2O){(MeIm)2CHCOO}], stabilized by the κ3-C,C′,O coordination of the functionalized bis-NHC ligand, in the hydrogenation of CO2 to formate in water catalysed by the zwitterionic iridium(I) [Ir(cod){(MeIm)2CHCOO}] compound.17
We report herein on the catalytic activity of zwitterionic water-soluble [Cp*IrIIICl{(MeIm)2CHCOO}] and [IrI(cod){(MeIm)2CHCOO}] complexes for water oxidation driven by sacrificial oxidants. In addition, kinetic, spectroscopic, electrochemical and mass spectrometry studies have been carried to gain insight into the nature of the catalytic species.
2. Experimental
Synthesis
All the experimental procedures were performed under argon atmosphere by using Schlenk or glovebox techniques. Solvents were taken under argon atmosphere from an Innovative Technologies solvent purification system (SPS), or dried following standard procedures and distilled under argon prior to use.18 Deuterated solvents CDCl3, CD2Cl2 and DMSO-d6 (Euriso-top) were dried using activated molecular sieves and degassed by three freeze–pump–thaw cycles. D2O was purchased from Euriso-top and used as received. Standard literature procedures were used to prepare the starting materials [Cp*IrCl2]2 (Cp* = η5-C5Me5)19 and [(p-cymene)RuCl2]2.20 The zwitterionic iridium(III) and rhodium(III) compounds, [Cp*IrCl{(MeIm)2CHCOO}] (1) and [Cp*RhCl{(MeIm)2CHCOO}] (2), complex [Cp*IrCl{(MeIm)2CHCOOMe}]OTf (3),16 and zwitterionic iridium(I) compound [Ir(cod){(MeIm)2CHCOO}] (4)17 were prepared following the procedure recently reported by us.Scientific equipment
1H and 13C{1H} NMR spectra were recorded on Bruker Avance spectrometers (300, 400 or 500). Chemical shifts are reported in ppm relative to tetramethylsilane and coupling constants (J) are given in Hertz (Hz). Spectral assignments were performed, when necessary, with the help of 2D-NMR experiments. Electrospray ionization (ESI) mass spectra were recorded using a Bruker Esquire3000 plus™ ion-trap mass spectrometer equipped with a standard ESI source. High-resolution electrospray ionization mass spectra (HRMS-ESI) were recorded using a Bruker MicroToF-Q™ equipped with an API-ESI source and a Q-ToF mass analyzer. Infrared spectra were recorded on a 100 FTIR-Perkin-Elmer Spectrophotometer equipped with a universal attenuated total reflectance (UATR) accessory made by thallium bromide–iodide crystals (KRS-5). C, H and N analyses were carried out in a Perkin-Elmer 2400 Series II CHNS/O analyzer. The UV/vis spectra were recorded on a JASCO UV-vis-NIR V-670 spectrophotometer equipped with a single monochromator that covers a wavelength range from 190 to 2500 nm. Dynamic light scattering studies were carried out on a Malvern Zetasizer Nano ZS which measures the diffusion of particles moving under Brownian motion, and converts this to size and size distribution using Stokes–Einstein relationship. This device incorporates non-invasive back scatter technology (NIBS) to give the highest sensitivity simultaneously with the highest size and concentrations range in a measurement range from 0.3 nm to 10.0 microns (diameter). Electrochemical experiments were performed on a potentiostat EG&G Research Model 273. The electrochemical cell is equipped with a three electrode system: platinum disk as a working electrode, platinum thread as auxiliary electrode and a saturated calomel electrode (SCE, saturated with KCl) as a reference electrode. Electrochemical experiments were carried out under argon in about 5 × 10−4 M of the complexes and 0.1 M in tetrabutylammonium hexafluorophosphate, [N(n-Bu)4]PF6 (TBAH), or sodium tetrafluoroborate, NaBF4, in the potential range +1.8 to −1.8 (H2O) at variable scan rates. The measurements were performed at room temperature and the adjustments of pH were made via addition of HNO3. Redox potentials were referenced in acetonitrile to the Fc/Fc+ couple (0.43 V for SCE as reference electrode).Synthesis of the complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v) in the absence of light. The suspension was stirred for 6 hours and then filtered via a cannula through Celite to remove the AgCl formed. Then, the resulting solution was concentrated to ca. 1 mL under reduced pressure. Slow addition of diethyl ether afforded the compound as an orange solid which was filtered, washed with diethyl ether (3 × 3 mL) and dried in vacuo. Yield: 143.9 mg, 72%. Anal. calc. for C20H25ClN4O2Ru: C 49.03; H, 5.14; N, 11.43. Found: C, 49.25; H, 5.18; N, 11.59. 1H NMR (298 K, 300 MHz, CDCl3): δ 7.90 (d, JH–H = 1.9, 2H, CH), 6.96 (d, JH–H = 1.9, 2H, CH), 5.63 (s, 1H, CHCOO), 5.61 (d, JH–H = 6.2, 2H,
:1, v:v) in the absence of light. The suspension was stirred for 6 hours and then filtered via a cannula through Celite to remove the AgCl formed. Then, the resulting solution was concentrated to ca. 1 mL under reduced pressure. Slow addition of diethyl ether afforded the compound as an orange solid which was filtered, washed with diethyl ether (3 × 3 mL) and dried in vacuo. Yield: 143.9 mg, 72%. Anal. calc. for C20H25ClN4O2Ru: C 49.03; H, 5.14; N, 11.43. Found: C, 49.25; H, 5.18; N, 11.59. 1H NMR (298 K, 300 MHz, CDCl3): δ 7.90 (d, JH–H = 1.9, 2H, CH), 6.96 (d, JH–H = 1.9, 2H, CH), 5.63 (s, 1H, CHCOO), 5.61 (d, JH–H = 6.2, 2H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH p-cym), 5.47 (d, JH–H = 6.2, 2H, CH p-cym), 3.97 (s, 6H, NCH3), 2.38 (spt, JH–H = 6.8, 1H, CH, iPr p-cym), 2.22 (s, 3H, CH3, p-cym), 1.01 (d, JH–H = 6.8, 6H, CH3, iPr p-cym). 13C{1H} NMR (298 K, 75 MHz, CDCl3): δ 174.2 (CNCN), 163.5 (COO), 122.7, 122.3 (CH), 107.6 (C-iPr, p-cym), 104.6 (C–Me, p-cym), 92.4 (CH, p-cym), 85.5 (CH, p-cym), 75.0 (CHCOO), 38.2 (NCH3), 32.2 (CH, iPr p-cym), 22.9 (CH3, p-cym), 18.8 (CH3, iPr p-cym). MS (ESI+, CH2Cl2/MeOH, m/z, %): 491.1 ([M + H]+, 100). IR (ATR, cm−1): 1654 (COO).
:1, v:v) in the absence of light. The suspension was stirred for 6 hours and then filtered via a cannula through Celite to remove the AgCl formed. Then, the resulting solution was concentrated to ca. 1 mL under reduced pressure. Slow addition of diethyl ether afforded the compound as a pale yellow solid which was filtered, washed with diethyl ether (3 × 3 mL) and dried in vacuo. Yield: 82.9 mg, 83%. Anal. calc. for C26H38ClIrN4O2: C 46.87; H, 5.75; N, 8.41. Found: C, 46.74; H, 5.71; N, 8.68. 1H NMR (298 K, 300 MHz, CD2Cl2): δ 7.80 (d, JH–H = 2.2, 2H, CH), 7.05 (d, JH–H = 2.2, 2H, CH), 5.70 (s, 1H, CHCOO), 4.25 (br m, 4H, NCH2), 3.79 (br m, 4H, CH2), 1.76 (s, 15H, CH3 Cp*), 1.42 (dq, JH–H = 14.7, 7.4, 4H, CH2), 0.97 (t, JH–H = 7.4, 6H, CH3). 13C{1H} NMR (298 K, 75 MHz, CD2Cl2): δ 163.7 (COO), 151.2 (CNCN), 122.7, 120.1 (CH), 93.3 (CCH3 Cp*), 75.6 (CHCOO), 50.4 (NCH2), 20.7 (2C, CH2), 14.2 (CH3), 9.8 (CH3 Cp*). MS (ESI+, CH2Cl2/MeOH, m/z, %): 667.2 ([M + H]+, 100). IR (ATR, cm−1): 1659 (COO).
CH cod), 5.96 (m, 2H, CH cod), 4.04 (s, 6H, NCH3), 2.75 (m, 2H, >CH2 cod), 2.59 (m, 2H, >CH2 cod), 2.44 (m, 4H, >CH2 cod).
CH cod), 4.81 (m, 2H, CH cod), 3.80 (s, 6H, NCH3), 2.75 (m, 4H, >CH2 cod), 2.55 (m, 2H, >CH2 cod), 2.38 (m, 2H, >CH2 cod).
CH p-cym), 5.47 (d, JH–H = 6.2, 2H, CH p-cym), 3.97 (s, 6H, NCH3), 2.38 (spt, JH–H = 6.8, 1H, CH, iPr p-cym), 2.22 (s, 3H, CH3, p-cym), 1.01 (d, JH–H = 6.8, 6H, CH3, iPr p-cym). 13C{1H} NMR (298 K, 75 MHz, CDCl3): δ 174.2 (CNCN), 163.5 (COO), 122.7, 122.3 (CH), 107.6 (C-iPr, p-cym), 104.6 (C–Me, p-cym), 92.4 (CH, p-cym), 85.5 (CH, p-cym), 75.0 (CHCOO), 38.2 (NCH3), 32.2 (CH, iPr p-cym), 22.9 (CH3, p-cym), 18.8 (CH3, iPr p-cym). MS (ESI+, CH2Cl2/MeOH, m/z, %): 491.1 ([M + H]+, 100). IR (ATR, cm−1): 1654 (COO).
:1, v:v) in the absence of light. The suspension was stirred for 6 hours and then filtered via a cannula through Celite to remove the AgCl formed. Then, the resulting solution was concentrated to ca. 1 mL under reduced pressure. Slow addition of diethyl ether afforded the compound as a pale yellow solid which was filtered, washed with diethyl ether (3 × 3 mL) and dried in vacuo. Yield: 82.9 mg, 83%. Anal. calc. for C26H38ClIrN4O2: C 46.87; H, 5.75; N, 8.41. Found: C, 46.74; H, 5.71; N, 8.68. 1H NMR (298 K, 300 MHz, CD2Cl2): δ 7.80 (d, JH–H = 2.2, 2H, CH), 7.05 (d, JH–H = 2.2, 2H, CH), 5.70 (s, 1H, CHCOO), 4.25 (br m, 4H, NCH2), 3.79 (br m, 4H, CH2), 1.76 (s, 15H, CH3 Cp*), 1.42 (dq, JH–H = 14.7, 7.4, 4H, CH2), 0.97 (t, JH–H = 7.4, 6H, CH3). 13C{1H} NMR (298 K, 75 MHz, CD2Cl2): δ 163.7 (COO), 151.2 (CNCN), 122.7, 120.1 (CH), 93.3 (CCH3 Cp*), 75.6 (CHCOO), 50.4 (NCH2), 20.7 (2C, CH2), 14.2 (CH3), 9.8 (CH3 Cp*). MS (ESI+, CH2Cl2/MeOH, m/z, %): 667.2 ([M + H]+, 100). IR (ATR, cm−1): 1659 (COO).
CH cod), 5.96 (m, 2H, CH cod), 4.04 (s, 6H, NCH3), 2.75 (m, 2H, >CH2 cod), 2.59 (m, 2H, >CH2 cod), 2.44 (m, 4H, >CH2 cod).
CH cod), 4.81 (m, 2H, CH cod), 3.80 (s, 6H, NCH3), 2.75 (m, 4H, >CH2 cod), 2.55 (m, 2H, >CH2 cod), 2.38 (m, 2H, >CH2 cod).
NMR data for [Ir(D2O)(cod){(MeIm)2CHCOO}]2+ (d2-10). 1H NMR (298 K, 300 MHz, D2O): δ 7.66 (d, JH–H = 1.7, 2H, CH), 7.26 (d, JH–H = 1.7, 2H, CH), 6.91 (s, 1H, CHCOO), 6.84 (m, 2H, CH cod), 5.89 (m, 2H, CH cod), 4.00 (s, 6H, NCH3), 3.03 (m, 2H, >CH2 cod), 2.56 (m, 2H, >CH2 cod), 2.43 (m, 4H, >CH2 cod).
CH cod), 5.96 (m, 2H, CH cod), 4.02 (s, 6H, NCH3), 2.74 (m, 2H, >CH2 cod), 2.58 (m, 2H, >CH2 cod), 2.44 (m, 4H, >CH2 cod) (OH not observed due to H/D exchange). 13C{1H} NMR (298 K, 75 MHz, D2O): δ 168.09 (COO), 138.31 (CNCN), 125.86, 121.59 (CH), 115.88, 114.85 (CH cod), 72.60 (CHCOO), 36.87 (NCH3), 28.52, 28.40 (>CH2 cod). MS (HRESI+, CH3CN/H2O, m/z, %): 537.1491 ([M]+, 100), 515.1382 ([M-OH–H]+, 67). IR (ATR, cm−1): 3391 (OH), 1633 (COO), 787, 767, 724 (IO3−).
CH cod), 6.23 (m, 2H, CH cod), 3.89 (s, 6H, NCH3), 2.91 (m, 2H, >CH2 cod), 2.50 (m, 2H, >CH2 cod), 2.41 (m, 4H, >CH2 cod). (HRESI+, H2O, m/z, %): 599.0501 ([M + NO3]+, 57), 537.1477 ([M − H]+, 89), 519.1379 ([M-H3O]+, 79).
General procedure for catalytic water oxidation
The reactions were performed on a Man on the Moon series X102 kit micro-reactor22 with a total volume of 14.2 mL placed in a thermostatic water bath at 298 K. The reaction vessel is connected to a switchable 3-way valve via a Thorion screw through polyamide tubing. The valve can be switched between two positions, one of them connecting the reactor vessel to the exterior so that it can be used like a conventional Schlenk flask. The other position connects the flask to the pressure transducer which allows for the measurement of O2(g) evolution. A 2 mL water-solution of the sacrificial oxidant CAN (0.354 M, pH = 1) or NaIO4 (0.193 M, pH = 6–7) was placed in the glass reactor under argon atmosphere. The reactor was closed, thermally equilibrated at 298 K and then, the pressure measurement started. Once the pressure was stabilized, 0.5 mL water-solution of the catalyst was injected. Oxygen evolution was measured until constant pressure and the amount of O2(g) (mmol) produced was calculated by using the ideal gas law. Each value is averaged out form three measurements with reproducibility better than ±1%.Multi-step water oxidation reactions were performed by sequential addition of 2 mL of a solution of CAN (0.354 M, pH = 1). The first measurement was performed following the experimental procedure described above using a [CAN]/[Ir] ratio of 700. Once stable pressure of O2(g) was achieved, the pressure inside the micro-reactor was released and another 2 mL of CAN (0.354 M) were added over the remaining dark purple solution. The measurement starts as prompt as the fresh CAN solution is injected and continues until constant pressure. The space inside the micro-reactor (14.2 mL) and the total volume of solution placed inside limited the number of additions to a maximum of three.
3. Results and discussion
The potential as catalysts for water oxidation of selected water-soluble iridium, rhodium and ruthenium zwitterionic complexes: [Cp*IrCl{(MeIm)2CHCOO}] (1), [Cp*RhCl{(MeIm)2CHCOO}] (2) and [(p-cymene)RuCl{(MeIm)2CHCOO}] (5), was screened in preliminary tests. A few milligrams of each zwitterionic complex was added into a vial and dissolved in 3 mL of degassed Milli-Q water. Subsequent addition of a large excess of solid CAN or NaIO4 resulted in the vigorous formation of bubbles due to oxygen evolution for the iridium precursor 1. However, no reaction was observed for the rhodium precursor 2, and just a few oxygen bubbles were observed for the ruthenium precursor 5. The colour of the solution after oxygen evolution remains purple and fades after 30 min when using CAN as chemical oxidant. However, a colour change from deep green to blue was observed with NaIO4 (see ESI†). These qualitative tests prompted us to study the catalytic activity not only of [Cp*IrCl{(MeIm)2CHCOO}] (1) but also of the related zwitterionic iridium(I) complex [Ir(cod){(MeIm)2CHCOO}] (4) featuring a 1,5-cyclooctadiene ligand (Chart 1). | ||
| Chart 1 Selected iridium, rhodium and ruthenium zwitterionic complexes as catalyst precursors for water oxidation. | ||
Catalytic activity of Ir(bis-NHC) complexes for water oxidation driven by CAN and NaIO4
Water oxidation catalytic reactions were performed under argon atmosphere on a micro-reactor equipped with a pressure transducer that allows the measurement of O2(g) evolution. The catalytic tests were performed in acidic-buffered degassed water (0.1 M HNO3, pH = 1) using a fixed amount of CAN (0.283 M) varying the concentration of catalyst precursors 1 or 4 in a range from 80 to 2820 μM. Table 1 summarizes the CAN-driven water oxidation catalytic experiments including the number of mmol of produced O2(g), TON number expressed as mmol of O2(g) per mmol of catalyst, and TOF50. The maximum amount of produced O2(g) under these experimental conditions is 0.177 mmol. Remarkably, an increase of the reaction yield was observed by reducing the catalyst concentration. Although roughly a 75% yield was achieved with a [CAN]/[Ir] ratio of 100 for catalyst 1 (entry 1), almost full conversion was attained when using ratios higher than 700 at longer reaction times (entries 6 to 11). The increase of the catalyst concentration resulted in faster reactions although the oxygen evolution stops before reaching the maximum conversion (entries 1 to 5) as was evidenced by the plateau in the O2(g) evolution plot (Fig. 1).23 However, these solutions showed catalytic activity after successive CAN additions once oxygen evolution stops which points to a likely Ir/Ce dinuclear resting state of the catalyst that becomes activated upon addition of an excess of CAN.9b,24 An induction period of 0.3–2.0 minutes was found before the O2(g) formation initiates which might be associated to the generation of active high-valent iridium species (Fig. 1, bottom). In line with these results, increasing the concentration of catalyst precursor 4 resulted in faster O2(g) evolution but the maximum yield (0.177 mmol) was not reached either. Similarly, full conversion was attained at higher [CAN]/[Ir] ratios although longer reaction times were required (entries 12–14, Table 1). The observed lag phases for 4 are roughly one minute longer than those observed for 1 at the same [CAN]/[Ir] ratio (see ESI†).| Cat. | [Ir]b | [CeIV]/[Ir] | mmol O2 | TON | TOF50c | Yield (%) |
|---|---|---|---|---|---|---|
| a Catalysts: [Cp*IrCl{(MeIm)2CHCOO}] (1) and [Ir(cod){(MeIm)2CHCOO}] (4). Reactions were carried out in 2.5 mL of acid-buffered degassed water (0.1 M HNO3, pH = 1) in a thermostatic bath at 300 K at indicated [CAN]/[Ir] ratios, [CAN]0 = 0.283 M. b [Ir] mM. c Turnover frequency (h−1) calculated at reaction time when the number of mmol of produced O2(g) reached half of the theoretically calculated. | ||||||
| 1 | 2.82 | 100 | 0.132 | 19 | 380 | 74 |
| 1 | 2.34 | 120 | 0.136 | 23 | 380 | 77 |
| 1 | 1.41 | 200 | 0.150 | 42 | 600 | 85 |
| 1 | 0.94 | 300 | 0.157 | 67 | 630 | 89 |
| 1 | 0.47 | 600 | 0.166 | 142 | 850 | 94 |
| 1 | 0.40 | 700 | 0.172 | 178 | 950 | 97 |
| 1 | 0.35 | 800 | 0.174 | 199 | 840 | 99 |
| 1 | 0.23 | 1200 | 0.177 | 308 | 1080 | >99 |
| 1 | 0.19 | 1500 | 0.177 | 375 | 1000 | >99 |
| 1 | 0.15 | 1900 | 0.177 | 471 | 1040 | >99 |
| 1 | 0.08 | 3700 | 0.175 | 891 | 1300 | 99 |
| 4 | 2.82 | 100 | 0.146 | 21 | 250 | 82 |
| 4 | 0.40 | 700 | 0.174 | 174 | 730 | 98 |
| 4 | 0.19 | 1500 | 0.170 | 357 | 900 | 96 |
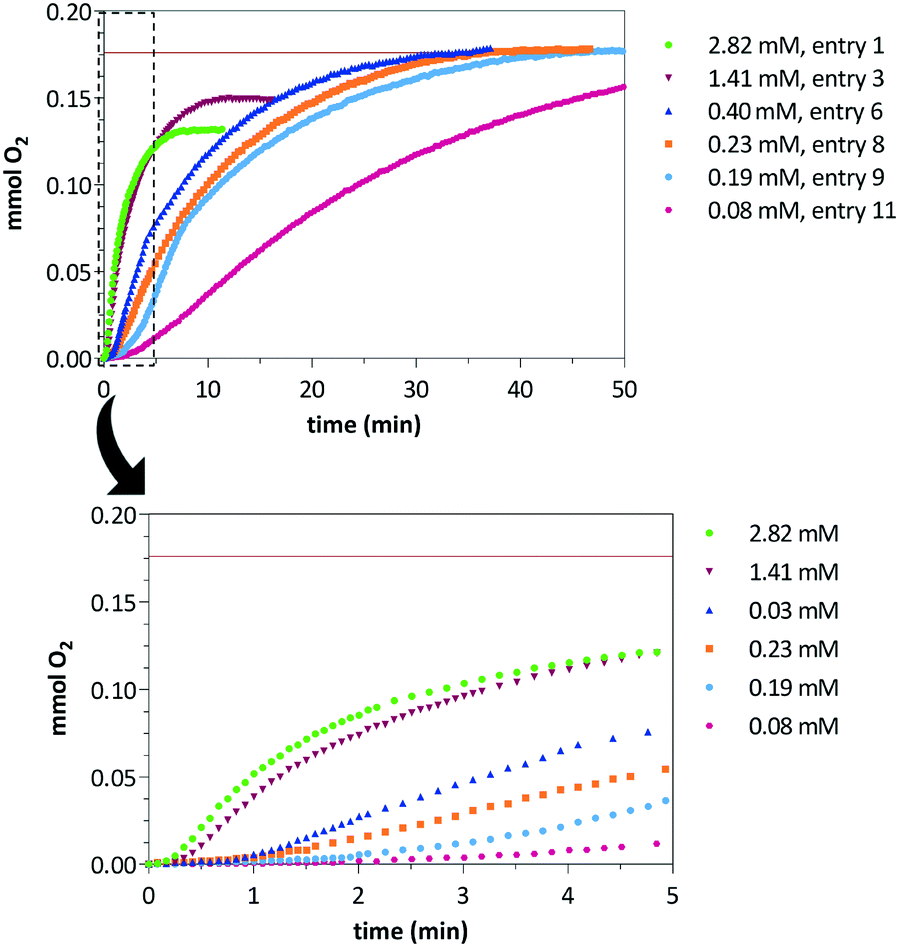 | ||
| Fig. 1 Plot of mmol of O2(g) vs. time at various concentrations of complex [Cp*IrCl{(MeIm)2CHCOO}] (1) with enlarged plots of the boxed region showing the oxygen evolution at short reaction times. The produced oxygen in all the catalytic tests is consistent with the stoichiometric limit of added CAN (horizontal line). | ||
The catalytic activity of both iridium precursors was also studied using NaIO4 as sacrificial oxidant. The reactions were carried out in degassed Milli-Q water using a fixed amount of NaIO4 (0.154 M) in a range of catalyst concentrations from 150 to 1540 μM. Under these reaction conditions the pH of the solution is approximately 7.0 and it does not change appreciably over the course of the reaction. The performed NaIO4-driven water oxidation catalytic experiments are summarized in Table 2.
| Entry | Cat. | [Ir]b | [NaIO4]/[Ir] | mmol O2 | TON | TOF50c | Yield (%) |
|---|---|---|---|---|---|---|---|
| a Catalysts: [Cp*IrCl{(MeIm)2CHCOO}] (1) and [Ir(cod){(MeIm)2CHCOO}] (4). Reactions were carried out in 2.5 mL of degassed water (pH = 7) in a thermostatic bath at 300 K at indicated [NaIO4]/[Ir] ratios, [NaIO4]0 = 0.154 M. b [Ir] mM. c Turnover frequency (h−1) calculated at reaction time when the number of mmol of produced O2(g) reached half of the theoretically calculated. | |||||||
| 1 | 1 | 1.54 | 100 | 0.178 | 46 | 160 | 92 |
| 2 | 1 | 0.51 | 300 | 0.158 | 124 | 120 | 82 |
| 3 | 1 | 0.15 | 1000 | 0.167 | 445 | 284 | 86 |
| 4 | 4 | 1.54 | 100 | 0.178 | 46 | 120 | 92 |
| 5 | 4 | 0.51 | 300 | 0.159 | 125 | 105 | 82 |
| 6 | 4 | 0.15 | 1000 | 0.163 | 435 | 285 | 84 |
Inspection of the O2(g) vs. time plots showed induction periods of 2–3 min for 1 and 3–4 min for 4 (see ESI†). Although high yields were achieved regardless of the catalyst concentration, the amount of O2(g) produced in all cases was below the theoretical maximum yield (0.192 mmol). From these results, it becomes evident that the generation of the catalytic active species is faster when CAN is used as sacrificial oxidant.25 The O2(g) evolution is also faster with CAN, as evidenced by the attained TOF50 values at similar catalyst concentration.
In order to explore the influence of the alkyl-wingtip and the carboxylate functional group at the bis-NHC ligand on the catalyst performance in water oxidation, the catalytic activity of complexes [Cp*IrCl{(MeIm)2CHCOOMe}]OTf (3), [Cp*IrCl{(n-BuIm)2CHCOO}] (6) and [Cp*IrI{(MeIm)2CH2}]PF6 (7) has been evaluated (Fig. 2).
 | ||
| Fig. 2 Selected Cp*Ir(bis-NHC) compounds as catalyst for water oxidation. | ||
As can be seen in Table 3, the three selected catalysts were found to be less active than 1 at a [NaIO4]/[Ir] ratio of 100 (entries 1–4). In terms of the amount of produced O2(g), the catalytic performance of 6, featuring n-butyl wingtips, is very similar to 1 with a 93% yield, which is in agreement with their closely related structure. However, lower yields were attained with catalysts 3 (featuring a methoxycarbonyl functional group) and 7 (lacking a carboxylate group), 83 and 73%, respectively, which points to a positive effect of the carboxylate functionality in the stabilization of the active species. Similar results have been obtained using CAN as sacrificial oxidant at a [CAN]/[Ir] ratio of 100 (entries 5–8) with 1 as the most active catalyst with a TOF50 of 385 h−1. Macchioni et al. described the positive influence of a long alkyl chain wingtip (methyl vs. n-octyl) in the ancillary NHC ligand of Cp*Ir(NHC)-based catalysts in water oxidation driven by CAN.25 The presence of n-butyl substituents in 6 does not improve the catalytic activity and the observed TOF50 values were significantly lower than those attained with catalyst 1 also using a high [CAN]/[Ir] ratio of 1200 (entries 9 and 10).
| Entry | Cat. | [Ir]b | Oxid. | [oxid]/[Ir] | mmol O2 | TON | TOF50c | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| a Reactions were carried out in 2.5 mL degassed water (pH = 7.0) at 300 K, [NaIO4]0 = 0.154 M (pH = 7.0) or [CAN]0 = 0.282 M (0.1 M HNO3, pH = 1). b [Ir] mM. c Turnover frequency (h−1) calculated at reaction time of 50% of theoretically calculated O2(g). | ||||||||
| 1 | 1 | 1.54 | NaIO4 | 100 | 0.178 | 46 | 170 | 92 |
| 2 | 3 | 1.54 | NaIO4 | 100 | 0.160 | 42 | 115 | 83 |
| 3 | 6 | 1.54 | NaIO4 | 100 | 0.179 | 46 | 110 | 93 |
| 4 | 7 | 1.54 | NaIO4 | 100 | 0.140 | 36 | 140 | 73 |
| 5 | 1 | 2.82 | CAN | 100 | 0.132 | 19 | 385 | 74 |
| 6 | 3 | 2.82 | CAN | 100 | 0.149 | 21 | 265 | 84 |
| 7 | 6 | 2.82 | CAN | 100 | 0.114 | 20 | 85 | 64 |
| 8 | 7 | 2.82 | CAN | 100 | 0.143 | 20 | 175 | 81 |
| 9 | 1 | 0.23 | CAN | 1200c | 0.177 | 308 | 1080 | >99 |
| 10 | 6 | 0.23 | CAN | 1200c | 0.153 | 266 | 425 | 86 |
As can be inferred for the activity data, catalyst precursor 1 is slightly more active than 4, both showing an average activity in the range of many of the iridium complexes so far reported. Their activity is comparable to those shown by the pyridine–dicarboxylate complex, [IrCp*Cl{2,6-Py(COO)(COOH)}],26 and the solvato complex [Cp*Ir(H2O)3](NO3)2,27 with TOF50 values of 1380 h−1 and 1260 h−1, respectively; and is superior to that of [Cp*Ir(H2O)(5,5′-Me-bipy)]+ with 576 h−1.28 In addition, both complexes are more active than related bis-NHC [Cp*IrCl{(MeIm)2CH2}]PF629 and [Cp*IrCl{(MeIm)2(CH2)2}]PF630 complexes, with TOF50 of 432 h−1 and 720 h−1, respectively, and the Cp*Ir-mesoionic triazolylidene-pyridine with 960 h−1.25 On the other hand, the activities of our complexes are far away from that shown by [IrCp*(H2O)(3,3′-OH-bipy)]+ or [IrCp*(H2O)(3,3′-OH-bipym)]+, with outstanding TOF50 numbers of 13500 h−1 and 18720 h−1, respectively.31
Kinetic studies
With the aim of shed some light on the operating mechanism in our catalytic systems, we have carried out kinetic investigations on the CAN- and NaIO4-driven water oxidation catalyzed by 1 and 4. Regardless of the catalyst concentration, the highest rate of O2(g) evolution was observed at roughly 20% conversion of the sacrificial oxidant with [CAN]0 = 0.283 M and [NaIO4]0 = 0.154 M. Thus, the reaction times required to reach 20% conversion were extracted from the O2(g) evolution vs. time plots and used to determine the maximum oxygen evolution rate (μmol min−1) for each catalyst concentration. The maximum rate of oxygen evolution vs. catalyst concentration plot for the catalytic system 1/CAN is shown in Fig. 3a. The plot shows a linear relationship although a significant departure from linearity was observed at high catalyst concentrations. This linear relationship has been previously observed for related iridium-based WOCs using CAN as sacrificial oxidant.23,32 The slope of the plots for 1 and 4 with both sacrificial oxidants evidences the faster rate of oxygen evolution for CAN with both catalyst precursors. Furthermore, oxygen-evolution rate is also faster for catalyst 1 with both sacrificial oxidants (see ESI†). The activity values for the catalytic systems 1/CAN and 4/CAN (2.82–0.19 mM) are in the range of 8–19 and 2–9 TON min−1, respectively. | ||
| Fig. 3 a) Plot of maximum rate of O2(g) evolution determined at 20% conversion of CAN vs. concentration of catalyst [Cp*IrCl{(MeIm)2CHCOO}] (1). b) Log(rate)–log[1] plot and the corresponding linear fit. | ||
The log–log plot of the maximum oxygen evolving rate against catalyst concentration provides information about the reaction order in catalyst.33 Water oxidation performed with catalyst 1 driven by NaIO4 seems to proceed via apparent first-order kinetics (reaction order of 1.1) whilst complex 4 exhibits a reaction order of 0.7 under the same reaction conditions.32 A broken order was also observed for water oxidation processes driven by CAN, 0.7 for complex 1 (Fig. 3b) and 0.5 for complex 4 (see ESI†). Studies with several iridium-based catalysts also reported a fractional order dependence on the catalyst, which evidences the complexity of water oxidation reactions from the kinetic and mechanism viewpoint.23,27,32,34
The interpretation of the kinetic data with NaIO4 as oxidant entails extra difficulties since oxygen evolution could also result from decomposition of IO4− rather than water oxidation by the iridium catalyst.14f Therefore, the effect of the sacrificial oxidant concentration on water oxidation catalysis has been studied with CAN. The kinetics of CAN-driven water oxidation by catalyst 1 was studied by varying the CAN concentration from 46 to 503 mM at a fixed catalyst concentration of 182 μM. The highest rate of oxygen evolution, determined at 20% of CAN conversion, was observed at 92 mM of CAN with a value of 7.32 μmol min−1 (Fig. 4). A gradual decrease was observed at higher CAN concentrations, reaching a value as low as 4.18 μmol min−1 for a CAN concentration of 503 mM. It seems that the initial linear increase of the rate of maximum oxygen evolution was attributed to a first order dependence on CAN, and the decay of the rate of maximum oxygen evolution at high CAN concentrations has been attributed to an increase of the ionic strength of the catalytic solution or aggregation of cerium(IV) into dimeric species.35
 | ||
| Fig. 4 Variation of the maximum rate of O2(g) evolution rate (determined at 20% conversion of CAN) versus concentration of CAN at a fixed 182 μM concentration of catalyst precursor [Cp*IrCl{(MeIm)2CHCOO}] (1). | ||
In order to test whether the purple solutions obtained after CAN-driven water oxidation catalysis have the potential to restart its catalytic activity, O2(g) evolution was monitored during the step-wise addition of aqueous CAN solution (pH = 1.0) to a water solution of catalyst 1. Fig. 5 shows the plot of O2(g) evolution vs. time for three consecutive additions. Almost full conversion, consistent with the stoichiometry limit of added CAN, was attained after the first and second additions. However, TOF50 in the second addition (348 min−1) is significantly lower than that attained in the first measurement (726 min−1). However, the largest difference was observed in the third addition of sacrificial oxidant, which afforded a lower conversion value (76%) in comparison with the 98% conversion achieved in the first and second additions (see ESI†). The decrease of TOF50 values along the step-wise additions indicates partial loss of the catalytic activity that can be attributed to catalyst deactivation along time.34b It should be noted that only the first test showed an induction period of roughly 1 minute. The absence of a lag phase upon re-oxidation processes suggests that the pre-catalyst is fully transformed into the active species after the first catalytic run.
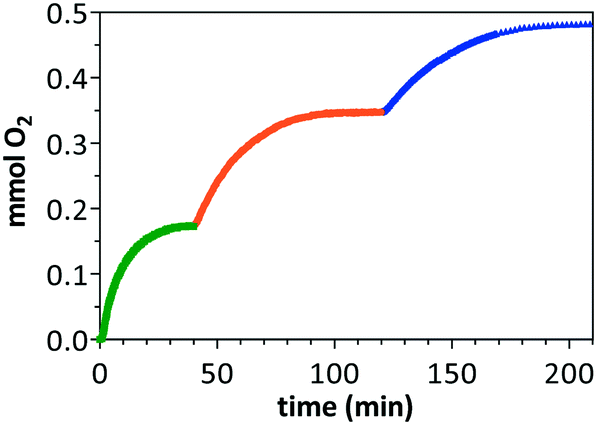 | ||
| Fig. 5 Multi-step CAN-driven water oxidation catalysis by [Cp*IrCl{(MeIm)2CHCOO}] (1). Oxygen production following the step-wise addition of 0.70 mmol of Ce(IV) to an initial 0.43 mM solution of 1 (2.5 mL, pH = 1.0) at 300 K. | ||
Water oxidation catalysis: UV-vis studies
Time-course UV-vis spectroscopic studies under catalytic conditions were conducted with the aim to identify the nature of the water oxidation catalytic active species. Variable amounts of solid CAN were added to a solution of complex 1 (0.25 mM) in acidic water (0.1 M HNO3, pH = 1) with [CAN]/[Ir] ratios in the range from 20–500. The addition of the sacrificial oxidant immediately led to an increase in the baseline spectra due to the formation of O2(g) bubbles. Regardless of the amount of added CAN, an absorption band at λ ≈ 550 nm appeared at short reaction times reaching a maximum of intensity after 13 min. From that point, the intensity of this absorption band decreased while a new band appeared at λ ≈ 630 nm as a broad shoulder (Fig. 6). Grotjahn et al. ascribed a band at 550 nm to the possible formation of IrOx nanoparticles, which show an absorption band within this spectral region.14b However, an absorption band at λ ≈ 580 nm was also attributed by Crabtree et al. to d–d transitions of octahedral iridium(IV) intermediates.36 In fact, based on Crabtree's proposal, many authors have attributed this absorption band to the presence of iridium(IV) species rather than the formation of nanoparticles.10a,32,37 | ||
| Fig. 6 Time evolution of the UV/vis spectra of solutions of catalyst precursors 1 (0.25 mM) and 4 (0.15 mM) after addition of CAN. | ||
The recorded UV/vis spectra after the addition of CAN to a buffered water solution (0.1 M HNO3, pH = 1) of catalyst precursor 4 (0.15 mM) with a [CAN]/[Ir] ratio of 500 are also shown in Fig. 6. Remarkably, under catalytic conditions both catalytic systems 1/CAN and 4/CAN exhibit similar absorption bands in the UV/vis spectra (λ ≈ 550 and 630 nm).
The catalytic systems 1/NaIO4 and 4/NaIO4 were also investigated by UV/vis spectroscopy. The recorded spectra after the addition of solid NaIO4 to degassed aqueous solution of 1 or 4 (0.25 and 0.15 mM, respectively) with [NaIO4]/[Ir] ratios in the range of 20–500 closely resemble each other. Fig. 7 shows the time evolution of the UV/vis spectra using a [NaIO4]/[Ir] ratio of 100. An absorption band at λ ≈ 585 nm, reaching a maximum after 90 minutes, was observed for the catalytic system 1/NaIO4. In the case of the catalytic system 4/NaIO4 a weak absorption band appeared at λ ≈ 595 nm together with a broad shoulder at λ ≈ 420 nm. However, a new band at λ ≈ 585 nm emerged after 10 minutes reaching a maximum absorbance after 60 minutes, which remains after 120 minutes.37a
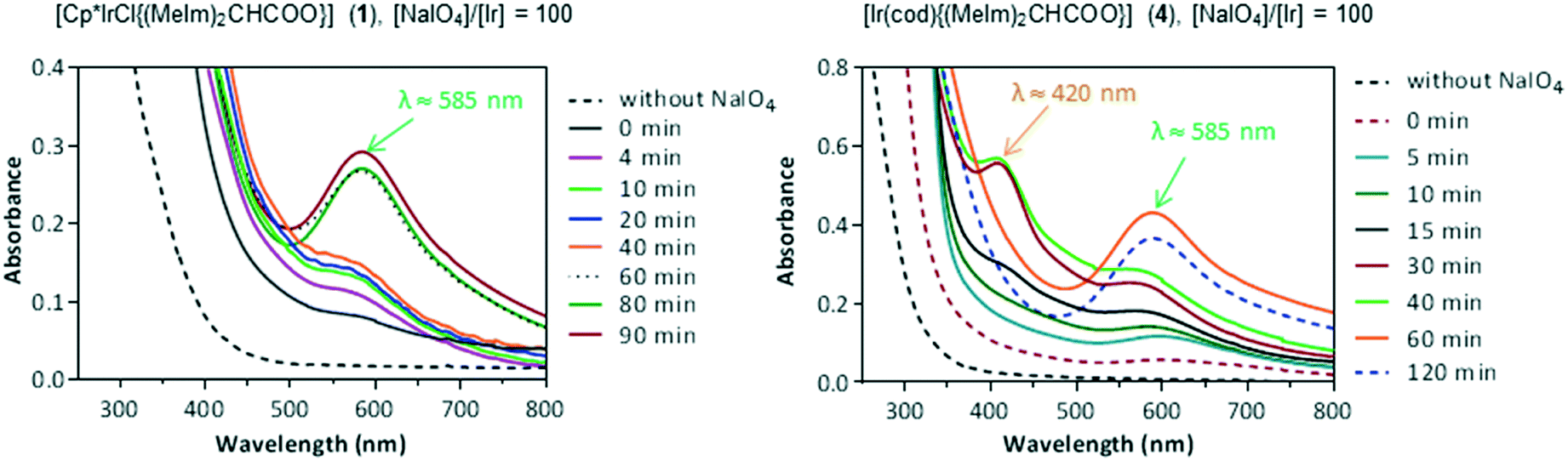 | ||
| Fig. 7 Time evolution of the UV/vis spectra of solutions of catalyst precursors 1 (0.25 mM) and 4 (0.15 mM) after addition of NaIO4. | ||
The similarity of the UV/vis spectra for the catalytic systems based on both catalyst precursors (1 and 4) with the same sacrificial oxidant (CAN or NaIO4) points to the participation of the same species in both catalytic systems. Furthermore, the active species participating in the periodate- and CAN-based catalytic systems should be closely related but different. In fact, time-course experiments revealed similar oxygen evolution profiles for both catalyst precursors. The overlay of the O2(g) evolution vs. time plots at the same concentration for both catalyst precursors showed comparable profiles for both sacrificial oxidants although a markedly shifted profile for catalyst precursor 4 using CAN as sacrificial oxidant at low catalyst concentrations (0.19 mM) is observed (Fig. 8). This shift is, in part, a consequence of the different lag phase although a lower catalytic activity for 4 under these conditions is also evident from the slope of both graphics at short reaction times.
 | ||
| Fig. 8 Plots of O2(g) evolution vs. time at various concentrations of complexes 1 and 4 using: a) CAN (0.283 M) and b) NaIO4 (0.154 M) as sacrificial oxidant. | ||
These results together with the lag phase observed at the beginning of the catalytic reactions suggest that the Cp* and cod ligands might be degraded in route to the generation of the active species, similarly to the observed by Crabtree et al. in related Cp*IrIII and IrI(cod) complexes featuring 2-(2-pyridyl)-2-propanolate ligands.37a
In fact, the GC-MS spectra of the gas phase in the micro-reactor headspace after water oxidation catalysis by the systems 1/CAN and 4/CAN showed the presence of a small peak at m/z 44 corresponding to CO2 traces which was attributed to the degradation of the hydrocarbon ligands (see ESI†). However, the degradation of the functionalized bis-NHC ligand followed by formation of IrOx (iridium oxide) nanoparticles cannot be excluded.37a
Dynamic light scattering studies
In order to investigate the possible formation of nanoparticles along water oxidation catalysis, dynamic light scattering (DLS) experiments were performed at different oxidant/catalyst ratios.10a,36,37a Interestingly, no nanoparticles were detected in solutions from the reaction of 1 and 4 neither using CAN nor NaIO4 at [oxidant]/[Ir] ratios of 100, 650 and 1200 after standing for prolonged times. These results indicate that if nanoparticles are formed, the concentration must be below the detection limit of the technique (0.1 ppm) and the sizes must be smaller than 1 nm (see ESI†). In contrast, when IrCl3 was employed as water oxidation catalyst using CAN and NaIO4 as sacrificial oxidants, with a [oxidant]/[Ir] ratio of 100, DLS analysis of the dark brown and dark green solutions obtained after 30 minutes revealed the existence of nanoparticles of 400 and 99 nm sizes, respectively (see ESI†). Literature reports on iridium nanoparticles and nanoclusters are generally characterized by their persistence along time, as it has been found in the case of IrCl3, which is in agreement with formation of IrOx particles. These results suggest that water oxidation by catalyst precursors 1 and 4 proceeds in homogeneous phase.37NMR studies
The oxidation of complexes 1 and 4 was also investigated by 1H NMR spectroscopy with the aim of identifying possible species generated in the pre-catalysts activation step. The NMR experiments were conducted in ca. 60 mM catalyst solutions in D2O (pH = 7) at room temperature. The consecutive addition of increasing amounts of sacrificial oxidant, CAN or NaIO4, led to evolution of O2(g) bubbles that made difficult to homogenize the magnetic field. Nevertheless, 1H NMR spectra were recorded under these conditions to identify possible species derived from the oxidative transformation of the catalyst precursors. PRESAT experiments were carried out in some cases in order to minimize the residual solvent signal.The 1H NMR spectrum of [Cp*IrCl{(MeIm)2CHCOO}] (1) in D2O showed the expected set of resonances for the functionalized bis-NHC ligand at δ 7.44, 7.38 (CH) and 5.94 (CH) ppm. The addition of 2 equiv. of CAN to a yellow solution of 1 produced a darkening of the solution and the incipient appearance of two singlets at δ 8.26 and 2.11 ppm which correspond to formic acid38 and acetic acid,27b,37a respectively. Increasing the amount of added CAN to 15 equiv. resulted in a dark blue solution with oxygen evolution whose 1H NMR spectrum evidenced a decrease of the amount 1, an increase of intensity of the formic and acetic acid resonances, and the appearance of a 1:1:1 signal at δ 6.68 ppm with a coupling constant JH–N = 52.4 Hz corresponding to the NH4+ coming from CAN (see ESI†). Similarly, the 1H NMR spectrum of the dark green solution obtained after addition of 10 equiv. of NaIO4 also showed the presence of formic acid and acetic acid. The intensity of both resonances increased upon addition of NaIO4 with concomitant decrease of those of 1 (see ESI†).
The 1H NMR spectrum of a dark red solution of [Ir(cod){(MeIm)2CHCOO}] (4) in D2O (pH = 7) prior the addition of NaIO4 showed the expected pattern of resonances at δ 7.43, 7.17, 6.44 (bis-NHC), and 4.83, 4.45 (CH, cod) ppm (red circles, Fig. 9a). The addition of 2 equiv. of NaIO4 produced an instantaneous colour change to green although not O2(g) bubbles were observed. However, the addition of 2 more equiv. afforded a deep green solution along with O2(g) evolution which turned into dark blue after addition of 10 equiv. of NaIO4. The 1H NMR of these solutions showed the clean formation of a new species featuring three resonances for the functionalized bis-NHC ligand at δ 7.67, 7.28 and 6.88 ppm, and two resonances for the CH olefin protons of the cod ligand at δ 6.64 and 5.96 ppm (green circles), both largely downfield shifted compared to those of 4. In particular, the shift of CHCOO resonance up to δ 6.88 ppm is a diagnostic for the κ3-C,C′,O coordination of the carboxylate-functionalized bis-NHC ligand.16 The observed chemical shifts compares well with those of the cation [IrCl(cod)κ3-C,C′,O-{(MeIm)2CHCOO}]+ which binds an electronegative ligand trans to the carboxylate moiety. Thus, the spectroscopic data are compatible with the formation of an Ir(III) octahedral species [IrX(cod){(MeIm)2CHCOO}]n+ with a O-donor ligand trans to the carboxylate fragment. This species has been prepared by reaction of 4 with one equiv. of NaIO4 in water and characterized as the hydroxo complex [Ir(OH)(cod){(MeIm)2CHCOO}][IO3] (8). Compound 8 has been isolated as a sparingly soluble white solid in 63% yield and characterized by 1H NMR and HRESI-MS (see Experimental section and ESI†). The stability of this species under the reaction conditions is remarkable. The addition of an overall of 20 equiv. resulted in the formation of only trace amounts of formic acid (δ 8.45 ppm) and free 1,5-cyclooctadiene (δ 5.50 and 2.30 ppm) after 60 min. However, an increase in the amount of formic acid with concomitant decrease of that of 8 was observed after the addition of more NaIO4 which indicates the steady transformation of 8 into the catalytic active species.
 | ||
| Fig. 9 Evolution of the 1H NMR spectra (D2O, pH = 7, 298 K) of a solution of [Ir(cod){(MeIM)2CHCOO}] (4) (60 mM) after consecutive additions of: a) solid NaIO4, and b) solid CAN (* 1,5-cyclooctadiene). | ||
This spectroscopic study was also extended to the catalytic system 4/CAN (Fig. 9b). The addition of 2 equiv. of CAN to an aqueous dark red solution of 4 in D2O (pH = 7) resulted in a colour change to yellow along with O2(g) evolution. The 1H NMR spectrum of this solution showed the presence of a single species featuring three resonances at δ 7.54, 7.14 and 6.72 ppm for the bis-NHC ligand and two resonances for the CH protons of the cod ligand at δ 6.01 and 4.76 ppm (yellow circles). The addition of 2 more equiv. of CAN afforded a green solution as a consequence of the formation of a new species exhibiting the characteristic resonances for the bis-NHC (δ 7.73, 7.37 and 7.01 ppm) and cod (δ 6.97 and 6.31 ppm) ligands (green circles). A κ3-C,C′,O coordination of the carboxylate-functionalized bis-NHC ligand is inferred for both species owing to the remarkable downfield shifted CHCOO resonance compared to 4. The yellow species is progressively transformed into the green one upon addition of CAN, which is the only observed species after addition of 16 equiv. of CAN. Subsequent addition of CAN resulted in a colour change to purple, through dark green, with steady O2(g) evolution. After addition of 24 equiv. of CAN, the species labelled in green is still observed although masked by the characteristic 1:1:1 resonance at δ 6.86 ppm with (JH–N = 52.1 Hz) corresponding to the NH4+ coming from CAN. The 1H NMR spectra also revealed the presence of formic acid (δ 8.00 ppm).
The species denoted with yellow circles has been identified as the deuterido [IrD(cod){(MeIm)2CHCOO}]+ (d-9) complex by comparison with the 1H NMR of a sample obtained by protonation of 4 with triflic acid in D2O.17 Thus, the acid media provided by the hydrolysis of Ce4+ in D2O is responsible for the protonation of 4. Although the chemical shifts of the resonances for the species labelled in green are not strictly comparable with those of 8 the spectroscopic data also point to a species also having an O-donor ligand trans to the carboxylate moiety. We hypothesize that the first oxidation product resulting from the oxidation of 4 is strongly dependent of the pH of the reaction media. Thus, the formation of the hydroxo-Ir(III) species [Ir(OD)(cod){(MeIm)2CHCOO}]+ (8) results from the oxidation of 4 in neutral medium (IO4−), whereas the aqua-Ir(III) species [Ir(D2O)(cod){(MeIm)2CHCOO}]2+ (10) might be formed in acidic medium (CAN) (Fig. 10). This species has independently prepared by protonation of 8 in water (HNO3) and isolated in low yield as colourless solid which has been characterized by 1H NMR and HRESI-MS (see Experimental section and ESI†).
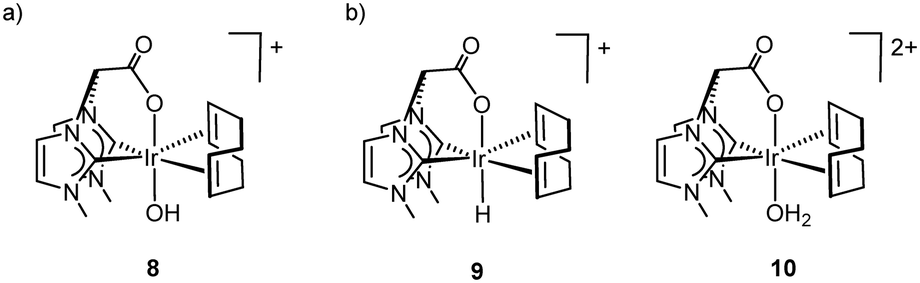 | ||
| Fig. 10 Observed species (1H NMR, D2O) in the oxidation of 4 with IO4− (a) and CAN (b) (the corresponding deuterated species are formed under the reaction conditions). | ||
The reactivity of 4 with CAN has been also studied at pH = 1 (D2O, HNO3 0.1 M) affording similar results. As expected, compound 4 is completely protonated at pH = 1 to afford the hydrido species 9. However, the step-wise addition of solid CAN showed that the conversion 9 → 10 → active species, is much faster under this conditions (see ESI†).
The transformation of the hydrido compound 9 into the aquo species 10 should follow an oxidative pathway. Then, two electron oxidation of 9 by two equiv. of CAN might afford the intermediate iridium(V)-hydride species from which the acidic hydride ligand is easily transferred to water to give 10 (Scheme 1). Although protonation of 9 with release of molecular hydrogen and concomitant coordination of H2O could be an alternative route for the formation 10, compound 9 remains unchanged at pH 1 for four hours which rule out this pathway.
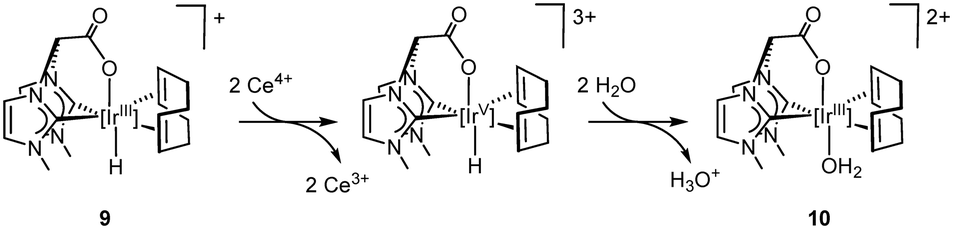 | ||
| Scheme 1 Oxidative pathway for the transformation of the hydrido-Ir(III) complex into the aquo-Ir(III) species. | ||
Mass spectrometric studies
The ESI-MS spectra of aqueous solutions of [Cp*IrCl{(MeIm)2CHCOO}] (1) and [(cod)Ir{(MeIm)2CHCOO}] (4) showed peaks at m/z 583.15 and 521.15, respectively, that correspond to the protonated molecular ions [M + H]+. In addition, the ESI-MS spectrum of 1 showed two peaks at m/z 547.17 and 503.18 for the ions [1-Cl]+ and [1-Cl–CO2]+, respectively (see ESI†). The addition of CAN to ca. 1 × 10−3 mM water solutions (pH = 7) of the catalyst precursors resulted in oxygen evolution and the immediate formation of new peaks in the MS spectra. However, at low CAN/Ir ratios the molecular ions were still detected at least 24 h after CAN addition, whereas at high CAN/Ir ratios poor MS spectra were obtained due to the blinding of the detector both in ESI+ and MaldiToF modes.The MS-ESI+ spectrum of a water solution of 1 after the addition of 50 equiv. of CAN, measured after 5 min when oxygen is evolving from the sample, showed three main species at m/z 597.11, 553.15 and 519.15, which showed the right isotopic pattern for the ions [1 + O]+, [1-CO2 + O]+ and [1-CO2–Cl + O + H]+ (Fig. 11a). These species were not observed in the MS-ESI+ spectrum recorded after 24 h that only showed the peak for [1 + H]+. The MS-MaldiToF spectrum of after the addition of 50 equiv. of CAN showed two mains peaks at m/z 463.2 and 445.0 whose isotopic distribution agreed with that of the ions [1-Cl–Cp* + OOH + H2O] and [1-Cl–Cp* + OOH], respectively (Fig. 11b).
 | ||
| Fig. 11 Mass spectra of water solutions of 1 (pH = 7) treated with 50 equiv. of CAN recorded after 5 min: a) MS-ESI+ spectrum, b) MS-MaldiToF spectrum. The insets show the simulated isotopic patterns for the identified peaks. | ||
The MS-ESI+ spectrum of a water solution of the iridium(I) complex 4 treated with 50 equiv. of CAN recorded after 10 min showed two main peaks at m/z 519.11 and 537.12, with the right isotopic pattern for the ions [4 − H]+ and [4 + H + O]+. After standing the solution for 24 h a similar MS-ESI+ spectrum was observed. The MS-MaldiToF spectrum after 24 h showed two peaks at m/z 521.1 and 599.1, that correspond to the protonated molecular ion [4 + H]+ and the ion [4 + H + O + NO3]+ (see ESI†). Unfortunately, despite the efforts made by using a range of CAN/Ir ratios, we have failed to detect any cod-free bis-NHC/Ir species in this case.
The observation of hydroperoxo species derived from [Ir{(MeIm)2CHCOO}(OOH)(H2O)x]+ in the MS spectra of the catalytic system 1/CAN is a clue for the possible participation of high-valent iridium intermediates stabilized by the carboxylate-functionalized bis-NHC ligand. On the other hand, although the oxidative degradation of Cp* and cod ligands could be anticipated in view of the spectroscopic evidences, the nature of the oxygen-containing species observed in the MS spectra cannot be determined reliably. In this regard, Macchioni et al. have recently shown that the mechanism for the degradation of the Cp* ancillary ligand in iridium-based water oxidation catalysts consists on a multistep oxidative process in which Cp*IrIII-superoxo species play a key role.38a However, the degradation pathways for the cod ligand in Ir(cod)-based water oxidation catalysts are much less understood.39,40
Electrochemical studies
The redox behaviour of complexes 1 and 4, without the chemical complications caused by chemical oxidants (CAN or NaIO4) was investigated by cyclic voltammetry (CV) using 1 mM water solutions of the complexes. The cyclic voltammogram profile for 1 shows a poor resolved anodic peak at 1.70 V, a hardly visible wave at 0.91 V and an irreversible reduction peak at 0.27 V (Fig. 12a). | ||
| Fig. 12 a) CV for 1 in water at pH = 7, b) pH-dependent CVs of complex 1 in aqueous solution (scan rate 200 mV s−1). | ||
The CVs of water solutions of complex 1 (1.0 mM) at pH = 1.0 and 7.0 in the window 0.0 to 1.3 V are shown in Fig. 12b. The observed current increase at potentials over 1.2 V is assigned to the electrocatalytic water oxidation associated to 1 which is in accordance with the lower overpotentials required for electrochemically driven water oxidation.41 The anodic response at 0.91 V at pH 7 is hardly observed at pH 1 whereas the irreversible reduction peak is shifted to 0.40 V. The oxidation peak at 0.91 V might be assigned to the IrIII/IrIV redox couple. In fact, a linear relationship between the peak current (I) for the redox couple and the square root of the scan rate (v1/2) was observed for scan rates in the range of 20–200 mV s−1 what is consistent with a rate-limiting step prior to electron transfer to the electrode.42 A linear correlation has also been observed for the reduction peaks at 0.40 V and 0.25 V at pH 1 and 7, respectively (see ESI†).
The CV for the iridium(I) compound 4 in water showed a barely discernible irreversible oxidation peak at 0.54 V which is associate to reduction peak at 0.16 V. This couple is accompanied with a quasi-reversible redox couple at ε1/2 = −0.44 V (Fig. 13). The analysis of this wave with a narrow scan window and scan rates varying from 10 to 200 mV s−1 roughly fulfils the standard reversibility criteria. In particular, the ratio of cathodic/anodic peak currents (Ipc/Ipa), that is expected to be unity for an ideal reversible process, is found to be 1.04, and the current function I(v)1/2 (v = scan rate) is approximately constant. However, the peak separation (ΔEp) of 129 mV is slightly larger than the ideal value for a reversible wave (59 mV). This electrochemical process is tentatively assigned to the redox couple IrII/IrI whereas the irreversible wave might correspond to the IrIII/IrII redox couple.43 Unfortunately, the easy protonation of compound 4 in acidic medium prevents to study the pH influence in the redox behaviour.
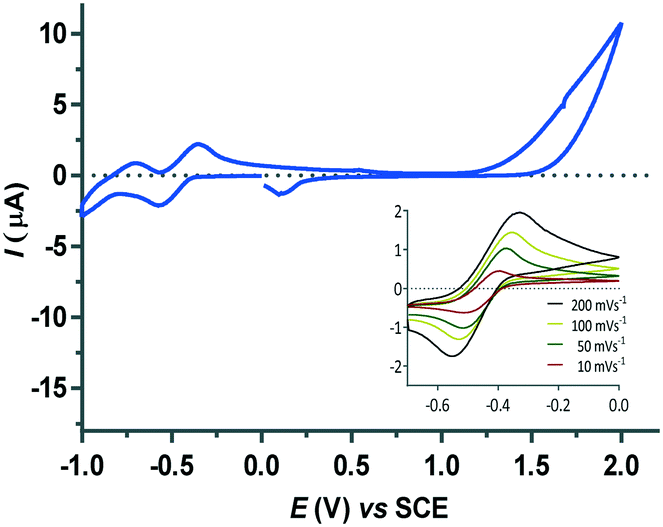 | ||
| Fig. 13 CV for compound 4 in water at pH 7. Inset: CVs of the quasi-reversible wave centred at ε1/2 = −0.44 V at different scan rates. | ||
4. Conclusions
Zwitterionic water-soluble [Cp*IrIIICl{(MeIm)2CHCOO}] and [IrI(cod){(MeIm)2CHCOO}] complexes having a carboxylate bridge-functionalized bis-NHC ligand have shown to be effective water oxidation catalysts using chemical oxidants such as CAN or NaIO4. Excellent yields and TOF50 numbers up to 1000 h−1 have been achieved when using [CAN]/[Ir] ratios higher than 700. Regardless of the oxidant employed, both catalyst precursors provided similar oxygen evolution profiles with short induction periods. This lag phase is slightly longer for the catalyst precursor [IrI(cod){(MeIm)2CHCOO}]. Changes in the wingtips of the bis-NHC ligand or in the catalyst framework did not improve the catalytic performance of the zwitterionic complex [Cp*IrIIICl{(MeIm)2CHCOO}].Kinetic, spectroscopic and DLS studies carried out with both catalysts precursors suggest the participation of common homogeneous high-valent iridium intermediate species in water oxidation catalysis. In particular, the resemblance of the UV/vis spectra and the similarity of the oxygen evolution profiles at moderate oxidant/catalyst ratios for both catalyst precursors and the same chemical oxidant, point to the likely degradation of the hydrocarbon ligands, Cp* and cod, in the pre-catalyst activation step. NMR spectroscopic studies at low oxidant/catalyst ratios have provided some hints on the pre-catalyst activation processes. In the case of [Cp*IrIIICl{(MeIm)2CHCOO}] formic and acetic acids were immediately formed regardless of the sacrificial oxidant employed. However, in the case of [IrI(cod){(MeIm)2CHCOO}], three different iridium(III) species stabilized by the tridentate coordination of the functionalized bis-NHC ligand have been identified, together with the formation of formic acid.
Taking all these results together, the molecular species responsible for water oxidation should be stabilized by the bis-NHC ligand, although the presence of the carboxylate function under the reaction conditions could be a subject of debate. Evidence for this hypothesis comes from the observation in the MS spectra of the reaction of 1 with CAN of hydroperoxo species derived from [Ir{(MeIm)2CHCOO}(OOH)(H2O)x]+. Based on these evidences, a mechanism involving high-valent IrIII/IrIV/IrV intermediate species stabilized by the carboxylate functionalized bis-NHC ligand, similar to those proposed by Crabtree13c,32 and Reek44 for Cp*Ir(III) catalysts, could be operative.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competitiveness (MINECO/FEDER, Projects CTQ2013-42532-P and CTQ2016-75884-P), and Diputación General de Aragón (DGA/FSE E42_17R) is gratefully acknowledged. R. P.-O. thanks MINECO for a predoctoral fellowship (BES-2011-045364).Notes and references
- (a) P. Sutra and A. Igau, Curr. Opin. Green Sustain Chem., 2018, 10, 60–67 CrossRef; (b) N. Armaroli and V. Balzan, Chem. – Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- (a) D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed; (b) Y. Jia, Y. Xu, R. Nie, F. Chen, Z. Zhu, J. Wang and H. Jing, J. Mater. Chem. A, 2017, 5, 5495–5501 RSC; (c) R. Razeghifard, in Natural and Artificial Photosynthesis: Solar Power as an Energy Source, Wiley, Hoboken, New Jersey, 2013 CrossRef.
- (a) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed; (b) M. D. Karkas, O. Verho, E. V. Johnston and B. Akermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed; (c) A. Llobet, in Molecular Water Oxidation Catalysis: A Key Topic for New Sustainable Energy Conversion Schemes, Wiley, United Kingdom, 2014 CrossRef; (d) I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- (a) A. Petronilho, M. Rahman, J. A. Woods, H. Al-Sayyed, H. Müller-Bunz, J. M. D. MacElroy, S. Bernhard and M. Albrecht, Dalton Trans., 2012, 41, 13074–13080 RSC; (b) R. Bofill, J. García-Antón, L. Escriche and X. Sala, J. Photochem. Photobiol., B, 2015, 152, 71–81 CrossRef CAS PubMed.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247–2252 RSC.
- (a) S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS; (b) J. A. Gilbert, D. S. Eggleston, W. R. Murphy, D. A. Geselowitz, S. W. Gersten, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 3855–3864 CrossRef CAS.
- (a) L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS PubMed; (b) J. Creus, R. Matheu, I. Peñafiel, D. Moonshiram, P. Blondeau, J. Benet-Buchholz, J. García-Antón, X. Sala, C. Godard and A. Llobet, Angew. Chem., Int. Ed., 2016, 55, 15382–15386 CrossRef CAS PubMed; (c) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210–217 CrossRef CAS PubMed.
- (a) J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730–8731 CrossRef CAS PubMed; (b) A. Bucci, G. Menendez Rodriguez, G. Bellachioma, C. Zuccaccia, A. Poater, L. Cavallo and A. Macchioni, ACS Catal., 2016, 6, 4559–4563 CrossRef CAS; (c) A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218–9219 RSC; (d) J. DePasquale, I. Nieto, L. E. Reuther, C. J. Herbst-Gervasoni, J. J. Paul, V. Mochalin, M. Zeller, C. M. Thomas, A. W. Addison and E. T. Papish, Inorg. Chem., 2013, 52, 9175–9183 CrossRef CAS PubMed; (e) M. Navarro, M. Li, H. Müller-Bunz, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2016, 22, 6740–6745 CrossRef CAS PubMed; (f) O. Diaz-Morales, T. J. P. Hersbach, D. G. H. Hetterscheid, J. N. H. Reek and M. T. M. Koper, J. Am. Chem. Soc., 2014, 136, 10432–10439 CrossRef CAS PubMed; (g) A. Lewandowska-Andralojc, D. E. Polyansky, C.-H. Wang, W.-H. Wang, Y. Himeda and E. Fujita, Phys. Chem. Chem. Phys., 2014, 16, 11976–11987 RSC.
- (a) Z. Codolà, J. M. S. Cardoso, B. Royo, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 7203–7213 CrossRef PubMed; (b) T. P. Brewster, J. D. Blakemore, N. D. Schley, C. D. Incarvito, N. Hazari, G. W. Brudvig and R. H. Crabtree, Organometallics, 2011, 30, 965–973 CrossRef CAS; (c) D. G. H. Hetterscheid and J. N. H. Reek, Chem. Commun., 2011, 47, 2712–2714 RSC.
- R. Lalrempuia, N. D. McDaniel, H. Müller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765–9768 CrossRef CAS PubMed.
- J. Graeupner, U. Hintermair, D. L. Huang, J. M. Thomsen, M. Takase, J. Campos, S. M. Hashmi, M. Elimelech, G. W. Brudvig and R. H. Crabtree, Organometallics, 2013, 32, 5384–5390 CrossRef CAS PubMed.
- (a) A. Macchioni, Eur. J. Inorg. Chem., 2019, 7–17 CrossRef CAS; (b) B. Mahanti, G. González, E. Martínez-Castro, M. Bedin, B. Martín-Matute, S. Ott and A. Thapper, ChemSusChem, 2017, 10, 4616–4623 CrossRef CAS PubMed; (c) J. M. Thomsen, D. L. Huang, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2015, 44, 12452–12472 RSC; (d) I. Corbucci, A. Macchioni and M. Albrecht, in Iridium Complexes in Water Oxidation Catalysis in Iridium Optoelectronic and Photonics Applications, ed. J E. Zysman-Colman, John Wiley & Sons Ltd., 2017, pp. 617–654 Search PubMed.
- (a) G. Menendez Rodriguez, G. Gatto, C. Zuccaccia and A. Macchioni, ChemSusChem, 2017, 10, 4503–4509 CrossRef CAS PubMed; (b) D. B. Grotjahn, D. B. Brown, J. K. Martin, D. C. Marelius, M.-C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A. Cortes-Llamas, V. Miranda-Soto, C. van Niekerk, C. E. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2011, 133, 19024–19027 CrossRef CAS PubMed; (c) S. E. Castillo-Blum, D. T. Richens and A. G. Sykes, Inorg. Chem., 1989, 28, 954–960 CrossRef CAS; (d) D. A. Pankratov, P. N. Komozin and Y. M. Kiselev, Russ. J. Inorg. Chem., 2011, 56, 1794–1799 CrossRef CAS; (e) H. Junge, N. Marquet, A. Kammer, S. Denurra, M. Bauer, S. Wohlrab, F. Gartner, M.-M. Pohl, A. Spannenberg, S. Gladiali and M. Beller, Chem. – Eur. J., 2012, 18, 12749–12758 CrossRef CAS PubMed; (f) A. R. Parent, T. P. Brewster, W. de Wolf, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2012, 51, 6147–6152 CrossRef CAS PubMed.
- R. Pokhrel, M. K. Goetz, S. E. Shaner, X. Wu and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 8384–8387 CrossRef CAS PubMed.
- R. Puerta-Oteo, M. V. Jiménez, F. J. Lahoz, F. J. Modrego, V. Passarelli and J. J. Pérez-Torrente, Inorg. Chem., 2018, 57, 5526–5543 CrossRef CAS PubMed.
- R. Puerta-Oteo, M. V. Jiménez, F. J. Lahoz, F. J. Modrego, V. Passarelli and J. J. Pérez-Torrente, Organometallics, 2018, 37, 684–696 CrossRef CAS.
- Purification of Laboratory Chemicals, ed. W. L. F. Armarego and C. L. L. Chai, Butterworth-Heinemann, Oxford, 6th edn, 2009 Search PubMed.
- C. White, A. Yates, P. M. Maitlis and D. M. Heinekey, Inorg. Synth., 1992, 29, 228–234 CAS.
- M. A. Bennett, T.-N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74–79 CAS.
- M. Vogt, V. Pons and D. M. Heinekey, Organometallics, 2005, 24, 1832–1836 CrossRef CAS.
- www.manonthemoontech.com .
- M. Li, K. Takada, J. I. Goldsmith and S. Bernhard, Inorg. Chem., 2016, 55, 518–526 CrossRef CAS PubMed.
- Z. Codolà, L. Gomez, S. T. Kleespies, L. Que, M. Costas and J. Lloret-Fillol, Nat. Commun., 2015, 6, 5865–5874 CrossRef PubMed.
- I. Corbucci, A. Petronilho, H. Müller-Bunz, L. Rocchigiani, M. Albrecht and A. Macchioni, ACS Catal., 2015, 5, 2714–2718 CrossRef CAS.
- A. Bucci, A. Savini, L. Rocchigiani, C. Zuccaccia, S. Rizzato, A. Albinati, A. Llobet and A. Macchioni, Organometallics, 2012, 31, 8071–8074 CrossRef CAS.
- (a) A. Savini, A. Bucci, G. Bellachioma, L. Rocchigiani, C. Zuccaccia, A. Llobet and A. Macchioni, Eur. J. Inorg. Chem., 2014, 690–697 CrossRef CAS; (b) A. Savini, P. Belanzoni, G. Bellachioma, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Green Chem., 2011, 13, 3360–3374 RSC.
- D. Hong, M. Murakami, Y. Yamada and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5708–5716 RSC.
- A. R. Parent, T. P. Brewster, W. De Wolf, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2012, 51, 6147–6152 CrossRef CAS PubMed.
- A. Volpe, A. Sartorel, C. Tubaro, L. Meneghini, M. Di Valentin, C. Graiff and M. Bonchio, Eur. J. Inorg. Chem., 2014, 665–675 CrossRef CAS.
- A. Lewandowska-Andralojc, D. Polyansky, C.-H. Wang, W.-H. Wang, Y. Himeda and E. Fujita, Phys. Chem. Chem. Phys., 2014, 16, 11976–11987 RSC.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- (a) J. M. Koelewijn, M. Lutz, W. I. Dzik, R. J. Detz and J. N. H. Reek, ACS Catal., 2016, 6, 3418–3427 CrossRef CAS; (b) J. A. Woods, R. Lalrempuia, A. Petronilho, N. D. McDaniel, H. Müller-Bunz, M. Albrecht and S. Bernhard, Energy Environ. Sci., 2014, 7, 2316–2328 RSC.
- A. Ikeda-Ohno, S. Tsushima, C. Hennig, T. Yaita and G. Bernhard, Dalton Trans., 2012, 41, 7190–7192 RSC.
- U. Hintermair, S. M. Hashmi, M. Elimelech and R. H. Crabtree, J. Am. Chem. Soc., 2012, 134, 9785–9795 CrossRef CAS PubMed.
- (a) U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135, 10837–10851 CrossRef CAS PubMed; (b) M. Zhou, D. Balcells, A. R. Parent, R. H. Crabtree and O. Eisenstein, ACS Catal., 2012, 2, 208–218 CrossRef CAS.
- (a) C. Zuccaccia, G. Bellachioma, O. Bortolini, A. Bucci, A. Savini and A. Macchioni, Chem. – Eur. J., 2014, 20, 3446–3456 CrossRef CAS PubMed; (b) C. Wang, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2012, 134, 19895–19908 CrossRef CAS PubMed.
- (a) M. R. Kelley and J.-U. Rohde, Dalton Trans., 2014, 43, 527–537 RSC; (b) M. P. del Río, M. A. Ciriano and C. Tejel, Angew. Chem., Int. Ed., 2008, 47, 2502–2505 CrossRef PubMed; (c) B. de Bruin, M. J. Boerakker, J. A. Brands, J. J. J. M. Donners, M. P. J. Donners, R. de Gelder, J. M. M. Smits, A. W. Gal and A. L. Spek, Chem. – Eur. J., 1999, 5, 2921–2936 CrossRef CAS.
- (a) D. G. H. Hetterscheid, M. Bens and B. de Bruin, Dalton Trans., 2005, 979–984 RSC; (b) D. G. H. Hetterscheid, M. Klop, R. J. N. A. M. Kicken, J. M. M. Smits, E. J. Reijerse and B. de Bruin, Chem. – Eur. J., 2007, 13, 3386–3405 CrossRef CAS PubMed.
- A. A. Schilt, Anal. Chem., 1963, 35, 1599–1602 CrossRef CAS.
- (a) P. Zanello, in Inorganic Electrochemistry. Theory, Practice and Applications, RSC, Cambridge, 2003 Search PubMed; (b) J. M. Savéant, in Elements of Molecular and Biomolecular Electrochemistry. An Electrochemical Approach to Electron Transfer Chemistry, Wiley, Hoboken, New Jersey, 2006 CrossRef; (c) Z. Chen, J. J. Concepcion, H. Luo, J. F. Hull, A. Paul and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 17670–17673 CrossRef CAS PubMed.
- (a) M. P. García, M. V. Jiménez, L. A. Oro, F. J. Lahoz and P. J. Alonso, Angew. Chem., Int. Ed. Engl., 1992, 31, 1527–1529 CrossRef; (b) M. P. García, M. V. Jiménez, L. A. Oro, F. J. Lahoz, J. M. Casas and P. J. Alonso, Organometallics, 1993, 12, 3257–3263 CrossRef.
- A. Venturini, A. Barbieri, J. N. Reek and D. G. Hetterscheid, Chem. – Eur. J., 2014, 20, 5358–5368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02306a |
| This journal is © The Royal Society of Chemistry 2019 |