
Oligomerization of ethanol-derived propene and isobutene mixtures to transportation fuels: catalyst and process considerations†
Johnny
Saavedra Lopez
 ,
Robert A.
Dagle
*,
Vanessa Lebarbier
Dagle
,
Colin
Smith
and
Karl O.
Albrecht
,
Robert A.
Dagle
*,
Vanessa Lebarbier
Dagle
,
Colin
Smith
and
Karl O.
Albrecht
Institute for Integrated Catalysis, Pacific Northwest National Laboratory, 902 Battelle Blvd., Richland, WA 99354, USA. E-mail: robert.dagle@pnnl.gov
First published on 5th February 2019
Abstract
In this paper, we report on solid acid catalysts and process conditions suitable for oligomerization of ethanol-derived mixed olefins propene and isobutene to transportation fuels, specifically targeting high-octane gasoline or jet-range hydrocarbons with high carbon efficiency. Catalytic performance was evaluated for two classes of solid acid catalysts—zeolites (H-ZSM-5, H-Y, H-beta) and polymeric resins (Amberlyst-36, Purolite-CT275)—each of which offer different catalytic properties. Two representative model olefin feeds: 1) diisobutene and 2) isobutene/propene (4/1 mol) mixture were used. Interestingly, both classes of solid acid catalysts can be tuned to produce a similar jet-distillate range hydrocarbon product with temperature determined to be the critical operating process parameter. For example, the propene conversion over H-beta zeolite was dramatically increased from 7 to 87% when the temperature was increased from 140 to 200 °C; whereas, nearly all (>99%) of the isobutene was converted over the same temperature range. However, with this relatively modest temperature increment (60 °C), formation of products in the jet-distillate range dramatically decreased from 90 to 65%, thus revealing an important activity/selectivity trade-off associated with the reaction temperature. Processing of aqueous ethanol to liquid hydrocarbon product was demonstrated in two sequential catalytic steps: 1) conversion of ethanol to an isobutene/propene-rich gas mixture over Zn1Zr2.5Oz catalyst, followed by 2) oligomerization over solid acid resin (Amberlyst-36). This processing sequence produced a highly branched olefin product primarily in the gasoline range (70% mass yield based on simulated distillation) with a high octane rating (approximate research octane number [RON] of 103). To increase the carbon yield to that of jet-range hydrocarbons, separation of the olefins from the light gases (i.e., H2, CO2, etc.) is required prior to oligomerization. Thus, in a separate set of experiments, an isobutene/propene mixture was converted over either H-beta (200 °C) or Purolite-CT275 (120 °C). This conversion step produced jet-range hydrocarbons with 80–85% mass yield. Upon subsequent distillation and hydrotreatment, both liquid products met select ASTM 7566 Annex A5 specifications for an alcohol-to-jet synthetic paraffinic kerosene (ATJ-SPK) blending component (e.g., specifications for freezing point, flash point, viscosity, and density).
1. Introduction
Currently, a need exists for alternative hydrocarbon fuel blendstocks, especially for aviation turbine and diesel fuels, from domestic resources to enhance energy security and to decrease reliance on foreign petroleum.1 An alternative technology that has received recent attention is the conversion of alcohol to jet-fuel blendstock, which often is referred to as the “alcohol-to-jet” (ATJ) process.2 The ATJ process involves alcohol dehydration, oligomerization, and hydrogenation reactions followed by a fractionation step.2 Alcohols such as ethanol and butanol have been of particular interest as feedstocks and can be produced from renewable biomass or waste sources.1,2 ASTM D7566 Annex A5 describes an approved method for creating an aviation turbine fuel containing synthesized hydrocarbons for ATJ synthetic paraffinic kerosene (ATJ-SPK). Isobutanol-derived jet fuel was the first ATJ-SPK to be specified under Annex A5 with blends originally permitted up to 30%. Recently, ethanol has been added as an approved feedstock and the blend ratio limit for ATJ-SPK fuels has been raised to 50%.3 In addition, the ethanol “blend wall” coupled with advances in production efficiency and feedstock diversification will potentially lead to excess ethanol at competitive prices available for the production of a wide range of fuels1 and commodity chemicals.4Ethanol typically is upgraded to fuel-range hydrocarbons by first undergoing dehydration to ethylene, followed by oligomerization over solid acid catalyst(s) in either one2,5 or two6 processing steps. For single-step processing, high ethanol conversion can be achieved; however, selectivity to undesirable C1–C4 light hydrocarbons as high as 40% has been reported.7–9 A competition between cracking and oligomerization reactions, extensive coke formation, and the presence of water have been reported as obstacles in producing longer chain products and high catalyst lifetimes.4,8 The control of desirable higher hydrocarbon product formation has been reported to be better facilitated with a two-step oligomerization process.6 For example, at Pacific Northwest National Laboratory, we have patented a two-step oligomerization process that produces primarily isoparaffin hydrocarbons, forms minimal aromatics, facilitates efficient conversion of high carbon fractions to distillate-range fuels, and minimizes formation of naphtha-like compounds by efficient intermediate product recycling.6 Producing isobutene directly from ethanol as intermediate olefin prior to oligomerization to fuel-range hydrocarbons represents an alternative to the ethylene route.10–13 The advantages of using isobutene as an intermediate alternative to ethylene include 1) easy conversion of isobutene into its dimer, diisobutene, which is a highly branched high-octane product that can be blended into gasoline,14 and 2) more selective conversion of isobutene to a specific targeted hydrocarbon range. Ethylene oligomerization when using zeolites typically requires activation by strong Brønsted acid sites at higher reaction temperatures, thus making selectivity control difficult to perform in one-step processing.15
ZnxZryOz mixed-oxide type catalysts with balanced acid–base sites were previously shown to be effective for converting ethanol to isobutene in a one-step process.11,12,16 In this process, ethanol undergoes a cascading sequence of reactions involving dehydrogenation, ketonization, condensation, dehydration, and decomposition to produce isobutene in a single catalytic bed.12 Recently, our group reported a greater than 50% carbon yield to isobutene-rich mixed olefins over an optimized ZnxZryOz catalyst when operating at 400 to 450 °C and atmospheric pressure.13 We also separately demonstrated oligomerization of isobutene-rich olefins, derived from an aqueous mixed oxygenate product over ZnxZryOz-type catalyst, to hydrocarbons in the jet-distillate range using the solid acid resin Amberlyst-36.10 However, oligomerization processing to the jet-range product was not optimized. In this paper, we report findings on our development of catalysts and processing conditions that enable a more selective conversion of produced olefins to targeted fuel products, specifically to hydrocarbons in the jet-fuel range or to high-octane gasoline.
Oligomerization of light alkenes to a variety of higher weight compounds is already an important and extensively studied area for the petroleum industry because it represents a route to produce motor fuels, lubricants, plasticizers, pharmaceuticals, dyes, resins, detergents, and additives17,18 Oligomerization occurs through a complex network of reactions over acid sites. The product distribution is governed by thermodynamic restrictions that favor C–C formation over cleavage at higher pressures and lower temperatures. Reverse oligomerization (olefin β-scission) and hydrogen transfer reactions (causing olefin saturation) are the main limitations for chain growth (lighter product formation).19,20 Heterogeneous solid acid catalysts typically employed for olefin oligomerization include zeolites, sulfonic acid resins (e.g. Amberlyst type),21 solid phosphoric acid (SPA),22 heteropolyacids, and sulfonated zirconia.23,24 In recent years, zeolites and sulfonic resins have received the most attention for olefin conversion to fuel-range hydrocarbons.
Zeolites allow for a higher operation temperature compared to polymeric resins, facilitating secondary reactions such as disproportionation, cracking, and isomerization in addition to oligomerization. Although these secondary reactions may enable a well distributed hydrocarbon product slate possibly desirable for fuel formulations through hydrogen transfer,20 these side reactions also can favor the formation of undesired products such as alkanes, aromatics, and products that are out of the targeted boiling-point range (naphtha, gasoline, etc., versus middle distillates).25,26 By comparison, polymeric resins have limited operating temperatures before chemical degradation occurs, making regeneration via thermal processes difficult; additionally, this lower operating temperature also limits the chemical reactions involved in the oligomerization mechanism network.27 We note that Alonso et al. has reported Amberlyst resins to have sufficient acidity—compared to ZSM-5, silica, or sulfated zirconia—to couple nonene, which is less reactive than shorter chain alkenes.28
While oligomerization is a relatively mature technology, there are still many technological challenges. Today, oligomerization of light alkenes represents an important route for the production of transportation fuels.17 However, few studies have focused describing catalysts and processes for producing blendstocks in the jet- and diesel-distillate range. Also, a big challenge facing the use of biofuels in aviation is the high quality standard requirements mandated by safety and fuel-quality specifications.29 These quality requirements are largely determined by the chemical nature of the hydrocarbon components of the fuel blend. In the preparation of finished fuels, oligomerization of intermediate olefins becomes critical to the nature of the resulting hydrocarbon products.
In this paper, we describe our study of the catalysts and processing conditions that enable selective production of high-octane gasoline or jet-fuel blendstocks from propene and isobutene olefin intermediates produced from bio-derived oxygenates (e.g., fermentation-derived ethanol). We first describe the olefins produced from aqueous ethanol feedstock over a Zn1Zr2.5Oz mixed-oxide type catalyst. We then evaluate olefin oligomerization using Amberlyst-36, a solid acid catalyst commonly reported in the literature,10,14,30 and comparatively assess different classes of zeolites and sulfonic acid polymeric resins. Diisobutene (DIB), the dimer of isobutene, was used as a model olefin feed for the oligomerization reaction. Considering its facile formation in the oligomerization of isobutene, the use of DIB as the model feed allowed desired products (dimers, trimers, tetramers) to be observed versus undesired side products (from dehydrogenation, disproportionation, cracking), likely to occur at higher temperatures (>200 °C), without influence from initial reaction steps. We then studied oligomerization of propene and isobutene intermediates. Incorporation of the less reactive propene in the product slate is a key requirement for maximizing carbon yield and increasing the carbon numbers of the product. We also describe optimization of the process conditions and the effect of recycle in the product. Finally, after distillation and hydrotreatment, we evaluate final liquid product characteristics (freezing point, flash point, viscosity, density, etc.) and compare the results to the ASTM D7566 A5 specifications for aviation turbine fuel blendstocks produced by the ATJ process.
2. Experimental
2.1. Catalyst preparation
Two types of catalysts were used in this study: 1) optimized Zn1Zr2.5Oz mixed oxide for the conversion of aqueous ethanol mixture into an olefin-rich gas stream according to methods reported in our previous publications10,13 and 2) a series of commercially available solid acid catalysts (H-form of zeolites and sulphonic acid polymeric resins) evaluated for the oligomerization of olefin feed compounds.Zn1Zr2.5Oz mixed-oxide catalyst was synthesized via wet impregnation of a Zn (NO3)26H2O solution on Zr (OH)4. The Zr (OH)4 was initially dried overnight at 105 °C to remove any excess water on the surface before impregnation. After impregnation, the catalysts were dried overnight at room temperature and then for 4 hours at 105 °C prior to calcination. The catalysts then were calcined via a 3 °C min−1 ramp to 400 °C for 2 hours, followed by a 5 °C min−1 ramp to the final calcination temperature of 550 °C for 3 hours.
Solid acid catalysts evaluated included commercially available Amberlyst-36 (Dow Chemical), CT275 (Purolite), and the hydrogen form of several classes of zeolites including H-beta, H-Y, H-ZSM-5 (all from Zeolyst), and USY (Tosoh). Before reaction, the polymeric resins were dried at 110 °C for 48 hours and then crushed and sieved to a particle size 60 to 100 mesh. The zeolite powder was pelletized using a pneumatic laboratory press (2 ton pressure force for 1 minute) before being crushed and sieved to the desired particle size (60 to 100 mesh). Before loading the reactor, the zeolites were calcined in a muffle furnace at 500 °C for 4 hours.
2.2. Catalyst characterization
Ammonia temperature programmed desorption (TPD) was used to measure the total acid sites and strength of the zeolite solid acid catalysts using a Quantachrome Autosorb II instrument equipped with a thermal conductivity detector. Sixty milligrams of catalyst sample was first pretreated at 500 °C for 2 hours under N2 flowing at 100 standard cm3 inside an adsorption quartz u-tube. After pretreatment, the sample was saturated with ammonia (10%vol in He) at 100 °C, then weakly adsorbed ammonia was flushed from the sample using He flow (100 standard cm3). The range of desorption temperature 50 to 800 °C was covered with a linear heating of 10 °C min−1.N2 adsorption was measured at 77 K with an automatic adsorptiometer (Micromeritics ASAP 2000). Samples were pretreated for 12 hours under vacuum (120 °C and 150 °C for resins and zeolites, respectively). The surface areas were determined from adsorption values for five relative pressures (P/P0 ranging from 0.05 to 0.2 using the Brunauer–Emmett–Teller (BET) surface method). The pore volumes were determined from the total amount of N2 adsorbed between P/P0 = 0.05 and P/P0 = 0.98.
2.3. Catalytic activity
Catalytic activity tests for the conversion of aqueous ethanol to primarily gasoline-range hydrocarbons were conducted in a two-step process using two reactors in series (see Fig. 1A). In the first reactor, a 12.75 mm outer diameter (10.92 mm) fixed-bed packed bed reactor was loaded with 7.0 g of Zn1Zr2.5Oz mixed-oxide catalyst used for producing isobutene-rich olefins from ethanol.13 Prior to testing, catalysts were first activated in situ at 450 °C for 8 hours under N2. Then, N2 was introduced into the system using a Brooks mass flow controller (5850E series). A 50% mixture of ethanol/water was fed into the system and converted to the gas phase using a vaporizer consisting of 6.35 mm inner-diameter steel tubing filled with quartz beads and an ISCO syringe pump. Catalytic tests were performed at 450 °C, 1.45 MPa, and two gas-hourly space-velocities [GHSV calculated as standard cm3 gas flow per volume of catalyst in an hourly basis (i.e., Vfeed Vcat−1 h−1)] as noted. A knockout pot placed directly downstream of the reaction zone was used to condense any remaining water and light oxygenates out of the system before the stream was introduced to the second reactor.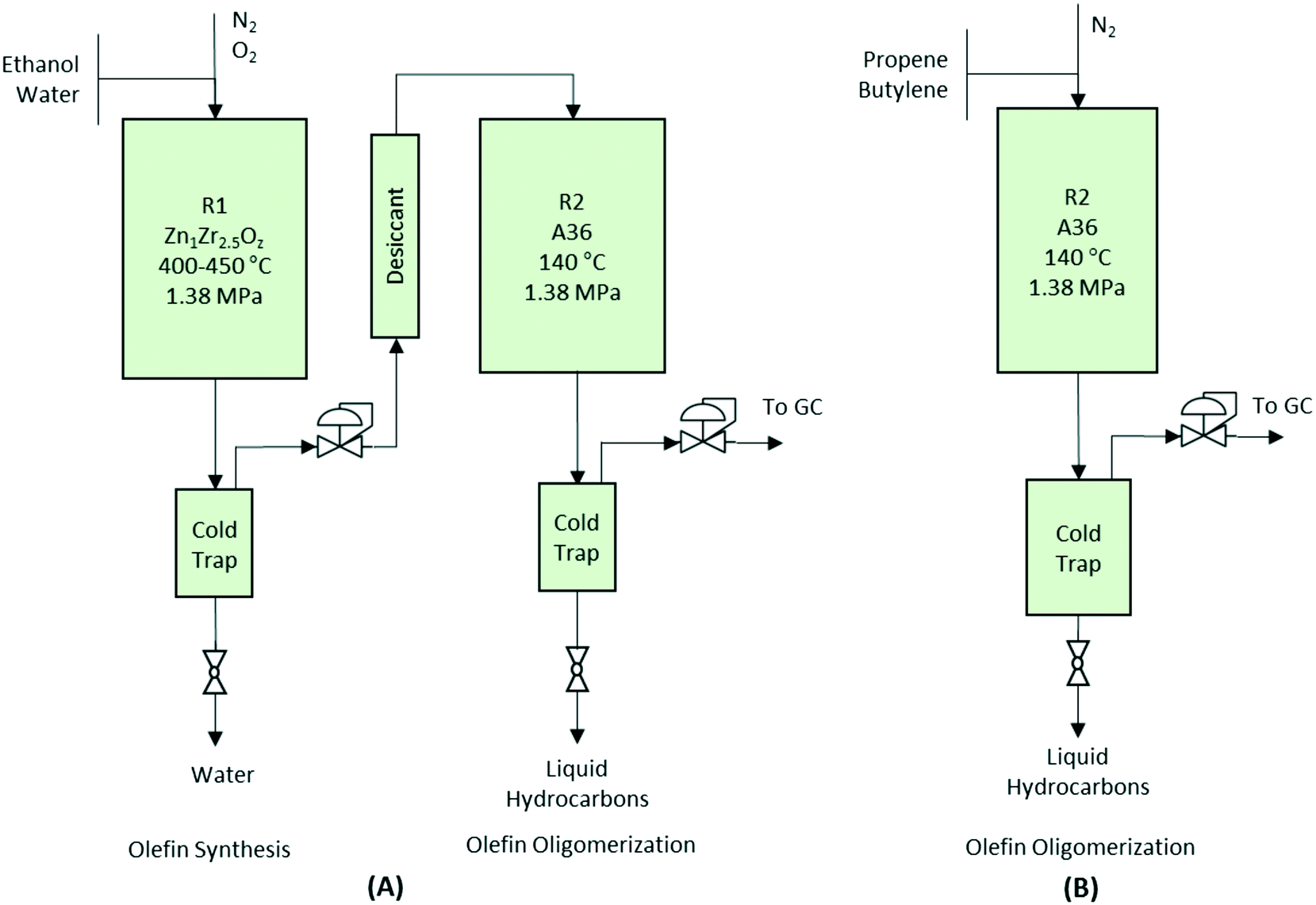 | ||
| Fig. 1 Simplified process flow diagrams depicting the bench-scale packed bed reactors used for the (A) integrated olefin production (R1) and oligomerization (R2), and (B) model olefin oligomerization (R2). | ||
Catalyst regeneration of the Zn1Zr2.5Oz mixed oxide was necessary for the 350 hour run and performed in situ at 450 °C under flowing 5% O2/He for 4 hours after isolating the reactors via a bypass. Mild oxidative treatment was performed to remove carbon/coke from the catalyst surface. During this treatment, CO2 typically was observed and considered to be indicative of carbon burn-off. Upon completion, the catalyst was cooled under N2 to the desired reactor operating temperature.
Olefins produced from the Zn1Zr2.5Oz mixed-oxide catalyst were introduced into a second reactor consisting of another fixed-bed with a 0.5 in outer diameter and 7.5 cm3 of a solid acid oligomerization catalyst sieved to a 60-to-100 mesh particle size. This reactor was operated at the pressure, range of temperatures, and GHSVs noted above. A knockout pot placed directly downstream of the reaction zone was used to condense the liquid product hydrocarbons. The reaction setup for the two-step integrated processing is depicted in Fig. 1A.
Performance evaluations for the oligomerization catalysts were performed in a single fixed-bed reactor (see Fig. 1B). A liquid feed of olefins was transported with an ISCO syringe pump and mixed with a flow of N2 prior to being fed into the reactor. WHSV (weight-hourly space velocity) was calculated based on the mass flow of olefins per weight of catalyst in an hourly basis (i.e., gfeed gcat−1 h−1). All reactors used a K-type thermocouple placed in the middle of the catalyst bed. To minimize temperature gradients within the reactor zone, an electrical resistance heating block was used for temperature control. Gaseous effluent was analyzed online using an Inficon micro GC (Model 3000A) equipped with MS-5A, Plot U, alumina, and OV-1 columns, and a thermal conductivity detector. Liquid samples collected from the knockout pot were analyzed separately ex situ using liquid chromatography (for aqueous products) and gas chromatography-mass spectrometry (GC-MS) (for hydrocarbon products).
2.4. Liquid hydrocarbon analysis and fuel property analysis
Oligomerization products consist of complex mixtures of hydrocarbons. GC-MS was used to characterize the products and estimate carbon numbers. Quantification of the mass composition of particular compounds or groups of compounds was performed using two parameters: 1) the flame ionization detector (FID) GC integrated area for a particular peak, and 2) identification based on the best match to the mass spectrometry library data.Simulated distillation profiles of the hydrocarbon products were determined following the method ASTM D2887 (ref. 31) using the GC results of a FID equipped gas chromatograph. Mass yields were based on GC-FID response and the products were separated into four boiling point ranges: naphta (<100 °C), gasoline (100–150 °C), jet range (150–300 °C), and heavies (>300 °C). RON analysis was performed using a Zeltex ZX-101XL near-infrared (NIR) portable octane analyzer using two gasolines as quality control (87 and 91 octane numbers). An additional estimation of the RON number was calculated using the automated Ignition Quality Test (IQT™) to determine a derived cetane number (DCN) based on the ignition delay of the sample. The DCN was converted to an estimated RON value by applying the inverse parabolic relationship between DCN and RON as described recently by McCormick and co-authors.32
Analysis of jet-range blendstocks included 1) flash point, determined by ASTM method D93 using a closed mini cup (7 cm3) with a Pensky-Martens apparatus; 2) freezing point, determined by reflected light scattering, which indicates the presence and disappearance of hydrocarbon crystals according to ASTM method 5972; 3) density, determined by using the change of frequency of a calibrated oscillating tube according to ASTM method D4052; and 4) viscosity, determined using ASTM method D445.
3. Results and discussion
3.1. Conversion of ethanol to isobutene and propene over Zn1Zr2.5Oz
Catalytic performance for the conversion of aqueous ethanol over Zn1Zr2.5Ox mixed-oxide catalyst is shown in Table 1. Complete ethanol conversion and greater than 50% selectivity to C3–C4 olefins was achieved. Also produced were byproducts including ethylene, CO2, CH4, H2, and trace amounts of acetone and other oxygenate compounds. As expected, a significant amount of CO2 was produced as a consequence of ketonization within the reaction mechanism. The reaction mechanism reported in our prior studies is summarized here: 1) ethanol is first dehydrogenated to form acetaldehyde (and byproduct H2); ethylene, an undesirable byproduct, also can be formed via competing ethanol dehydration; 2) acetaldehyde undergoes oxidation to acetic acid with subsequent ketonization to form acetone (and byproduct CO2); and 3) acetone undergoes a sequence of reactions that includes condensation, dehydration, and decomposition to form isobutene.10,13 These cascading sequences of reactions occur over a single bed Zn1Zr2.5Oz catalyst with tailored acid and base sites. As described in our prior report, propene is produced via hydrogenation of the acetone intermediate, and its selectivity is favored at higher operating pressures.13 We also note that undesirable methane is produced by acetone decomposition, and its formation is enhanced with decreasing space velocities.13| Compound | Gas phase effluent (%vol) | Selectivity (mol C%) |
|---|---|---|
| N2 | Balance | |
| H2 | 34.8 | |
| Isobutene | 6.2 | 44.0 |
| CO2 | 4.6 | 32.6 |
| CH4 | 1.7 | 12.1 |
| Propene | 1.2 | 8.5 |
| Ethylene | 0.3 | 2.1 |
| Acetone | 0.1 | 0.5 |
| Other oxygenates | <0.05 | 0.2 |
For the production of fuel-range hydrocarbons, the olefin-rich mixture shown in Table 1 requires oligomerization over a solid acid catalyst in a second reactor. Additional processing such as distillation and hydrotreatment after the oligomerization step is required to produce a paraffinic aviation fuel. Olefins boiling below the jet-range (less than about 150 °C) can be recuperated and reprocessed to increase the yield of a desired boiling cut (i.e., jet-range [150 to 300 °C]). The objective of this research is to develop catalytic processing for the selective conversion of this C3–C4 mixed olefin stream to targeted fuel products (i.e., high-octane gasoline or jet-range hydrocarbons).
3.2. Solid acid catalysts evaluated for olefin oligomerization
Multiple classes of solid acid catalysts have been considered for oligomerization of olefins in the literature (e.g., aqueous solutions of inorganic acids, acid modified polymeric resins, and porous aluminosilicates). However, in recent years, more attention has been dedicated to the study of two types of catalysts: 1) polymeric sulphonated resins, and 2) zeolites. Polymeric sulphonated resins have very low surface areas and low thermal stabilities. However, they have a high concentration of acid sites, and the acid strength/density can be adjusted by changing their chemical composition, crystallinity, and morphology.33 On the other hand, while zeolites generally have lower acid site concentrations, compared to resins, they exhibit higher surface areas and stabilities at elevated temperatures. In particular, zeolite H-ZSM-5 has been intensively studied since the 1970s as a catalyst for use in converting olefins to gasoline. It, however, exhibits limited selectivity towards middle distillate products.34,35 Recent studies of the oligomerization reaction indicate that factors critical for oligomerization catalytic performance include acid strength and acid density.36,37 Void space, and location of the acid sites in the microporous catalyst also affects chain length and configuration of the resulting oligomers.18,19,38 The nature of the intermediate products and reaction temperature also are critical to the reactivity of hydrocarbons over acid catalysts. For example, branched hydrocarbon intermediates may hinder mass diffusion, thus affecting product selectivity. Concurrent with oligomerization reactions, β-scission and hydrogen transfer mechanisms activated at higher temperatures (T > 200 °C) also are factors to be considered during catalyst selection and process optimization.39,40We obtained commercial solid acid catalysts from the two main groups: sulphonic acid polymeric resins (Amberlyst-36 and Purolite-CT275) and the hydrogen form of 12-member rings (large pore size – H-beta, HY, USY) and 10-member rings (medium pore size H-ZSM-5) zeolites. The total acid capacities (determined by ammonia TPD) and surface areas of the catalysts (determined by N2 adsorption) are summarized in Table 2. As seen in Table 2, the polymeric resins have significant higher acid capacities and lower surface areas relative to the zeolites (by a factor of 10). Although ammonia TPD allows a quantitative estimation of the total acid capacity to be determined, the nature of the sites cannot be quantified (e.g., Brønsted versus Lewis acid sites). Differentiation of acid strength among the different zeolites can be observed with the ammonia TPD profiles shown in Fig. 2.
| Catalyst | Amberlyst-36 (ref. 41) | CT275 (ref. 42) | H-Beta | H-Y | USY | H-ZSM-5 |
|---|---|---|---|---|---|---|
| Acid capacity (mmol g−1) | 5.4 | 5.2 | 0.86 | 0.40 | 0.46 | 0.40 |
| SiO2/Al2O3 mol ratio | n/a | n/a | 20 | 5.2 | 10 | 23 |
| BET area (m2 g−1) | 33 | 28 | 463 | 660 | 709 | 412 |
| Pore volume (cm3 g−1) | 0.2 | 0.42 | 0.18 | 0.26 | 0.15 | 0.127 |
| Pore diameter (nm) | 24 | 60 | 0.97 | 0.96 | 1.2 | 1.3 |
| Max Op. temp (°C) | 150 | 130 | n.a | n.a | n.a | n.a |
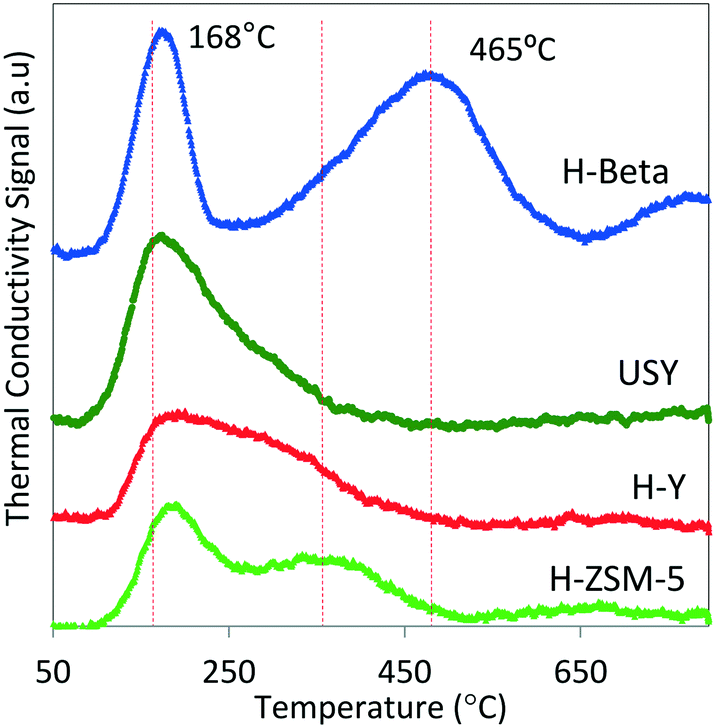 | ||
| Fig. 2 TPD profiles of ammonia chemisorbed at 100 °C on 60 mg of zeolite. The TPD profiles illustrate the integrated area (decreasing from top to bottom) were normalized to the total surface area and presented as total surface acid capacity (mmol g−1) data shown in Table 2. | ||
Table 2 shows that this particular H-beta zeolite exhibits greater acid capacity compared to H-ZSM-5 (>2 times), in spite of having similar silica/alumina ratios (20 and 23, respectively), and also compared to the other zeolites (H-Y and USY). Zeolite H-Y and H-ZSM-5 have the same total acid capacity (0.4 mmol g−1), with markedly different silica/alumina ratios (4.4 times greater for H-ZSM-5). These marked differences in acidity are related not only to the intrinsic Si/Al ratio measured for each catalyst but also to the presence of binders and additives to the zeolites formulation that affect the acidity of the resulting material as literature studies have shown.43 In terms of acid strength, ammonia TPD profiles (Fig. 2) show a higher fraction of acid sites (desorption peak centered at 465 °C) on zeolite H-beta compared to the other zeolites that exhibit weaker acid sites (lower ammonia desorption temperatures). There are no particular requirements for acid solid acid catalysts to be used in olefin oligomerization; however, the literature suggests that catalysts active for oligomerization have to satisfy at least three particular conditions: 1) provide acids sites in close proximity to allow formation of oligomers,36 2) operate at temperatures to avoid deactivation by heavy oligomers (equilibrium oligomerization versus β-scission) or coking (enhanced by hydrogen transfer mechanism),26 and 3) possess average pore sizes large enough to avoid deactivation by accumulation of heavy oligomers,24 which has shown to be rate controlling. The catalysts we chose for this study are representative of solid acid catalysts found in the literature for oligomerization of light olefins, and also span a wide range of catalytic characteristics such as acid strength/capacity, pore structure, and surface area.
3.3. Diisobutene model feed over solid acid catalysts
DIB was used as a model feed for the oligomerization reaction over the series of solid acid catalysts presented in Table 1. DIB was chosen as model feed to compare reactivity starting from the isobutene dimer and to evaluate the formation of side reaction byproducts (e.g., from isomerization, dehydrogenation, disproportionation, and cracking reactions) using three different reaction temperatures (140, 200, and 260 °C). Isobutene is reported to readily form DIB at low reaction temperatures (70 °C) over acid catalysts.23,44 Hydrogen transfer and β-scission mechanisms compete with oligomerization and are more likely to occur at higher reaction temperatures (>200 °C).20,26,39 Isomerization reactions, fast with respect to β-scission, allow for the formation of many hydrocarbon products that are useful in fuels preparation.45,46 Because of thermal stability limitations, the polymeric resins were evaluated at lower temperatures (120 °C and 140 °C for CT275 and Amberlyst36, respectively).Comparative catalyst performances are summarized in Fig. 3. Fig. 3A shows DIB conversion and a simulated distillation profile for the resulting liquid categorized into four different boiling ranges (i.e., naphtha, gasoline, jet-range hydrocarbons, and heavies fractions) using three boiling-point cuts (i.e., 100, 150, and 300 °C). Fig. 3B shows the selectivity towards gas products formed as a function of the oligomerization operating temperature. Results in Fig. 3A show that under complete DIB conversion, oligomerization products over zeolite H-beta and CT275 catalysts have improved selectivity towards jet-range products (>80 wt% in the 150 to 300 °C boiling-point range) compared to the other catalysts screened. A significantly higher acid capacity for H-beta zeolite (∼2 times) may be responsible for the increased yield towards the jet-range products when compared to the other zeolite catalysts. Fig. 3B shows that the formation of gas products (propene and butene) increases with reaction temperature. Selectivity to butene is particularly high at 260 °C over H-Y and USY catalysts, at approximately 7% and 11% (mol C%). For all the other catalysts, selectivity to gas products is low at approximately <1%. Gas olefin byproducts propene and butene can be formed through two main mechanisms: 1) disproportionation, where long chain olefins decompose to form a shorter chain olefin and a longer chain olefin, and 2) cracking of long chain hydrocarbons through the β-scission mechanism.36 Although our experimental results in this study do not differentiate the exact mechanism of gas olefin formation, the importance of catalyst selection and operating temperature is evident. The selection of oligomerization temperature is also critical in process development when less reactive olefins are incorporated to the reactive mixture.
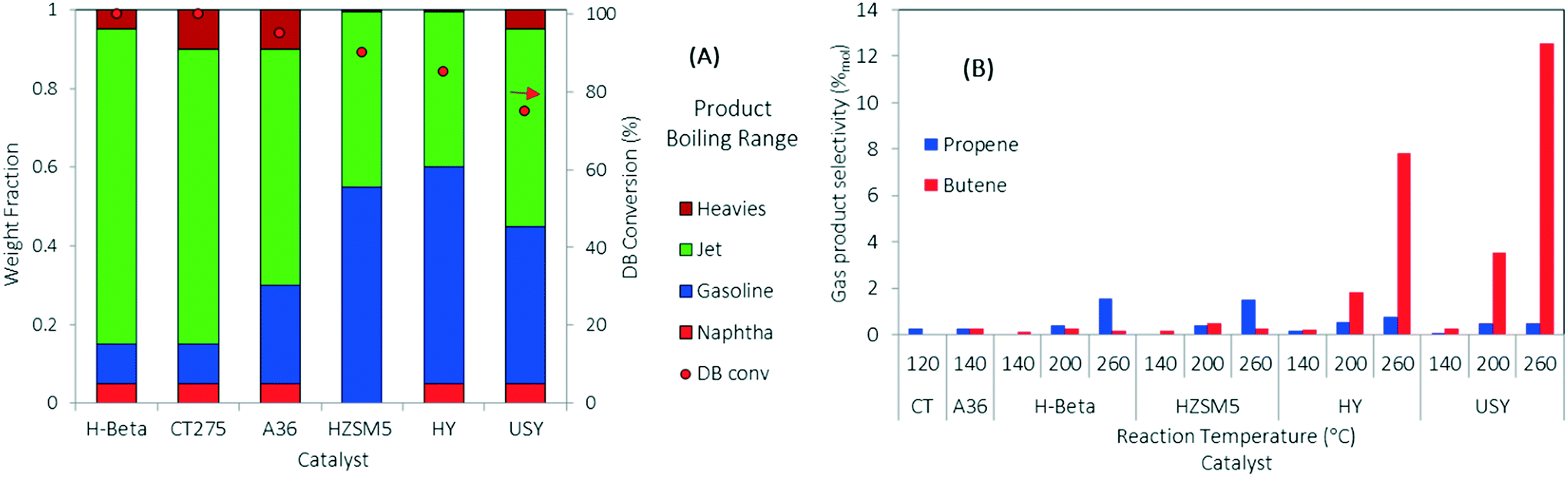 | ||
| Fig. 3 Analysis of the oligomerization products of the DIB reaction over zeolites and polymeric resins. (A) Boiling-point ranges determined by simulated distillation for the liquid product of DIB oligomerization at 140 °C (120 °C for CT275 resin). (B) Product selectivity for the DIB conversion into gas products at different reaction temperatures. Gas and liquid samples were collected under steady state after 3 hours time-on-stream (TOS) (1.38 MPa, WHSV = 1.1 h−1). | ||
Fig. 4 further illustrates the effect of varying operating temperature for the oligomerization of DIB over H-beta zeolite. The figure also shows the simulated distillation profiles of the liquid product (Fig. 4A), categorization by boiling-point range (Fig. 4B), and chemical functionality (Fig. 4C). DIB (b.p. 101 °C) is fully converted to a mixture with a wide boiling-point distribution, from naphtha to heavy products, a dramatic decrease in the yield to jet-range hydrocarbons occurs with increasing operating temperature. For example, as illustrated in Fig. 4B, as the reaction temperature increases from 140 to 260 °C, the yield to jet-range hydrocarbons dramatically decreases from 80% to 40%. This decrease in jet-range product yield is compensated by increased formation of both, naphtha and heavies fractions. Fig. 4C also illustrates how the product chemical functionality also is affected by temperature. For example, when increasing the temperature from 200 to 260 °C, the olefin selectivity was reduced from ∼65% to ∼40%, while a corresponding increase in paraffins was observed. Aromatics, a product from cyclization and dehydrogenation of reaction intermediates, also are formed at higher temperatures (i.e., >260 °C). We attribute this diversity in chemical functionalities occurring at low oligomerization temperatures (branched olefins and naphtenics) to kinetically favored skeletal and double-bond isomerizations caused by catalyst acidity.45 On the other hand, changes in carbon number distribution, accompanied by the selective formation of paraffins and aromatics, can be explained by chain breaking and hydrogen transfer mechanisms both occurring over acid sites at high reaction temperatures.20,26 Thus, reaction temperatures >200 °C lead to preferential formation of products in the naphtha and heavies range while also producing more saturated products. From these results, it can be seen that lower oligomerization temperatures are necessary to increase the yield to jet-range hydrocarbons. We note that the production of paraffins limits the potential for recycle for any oligomerization processing scheme.
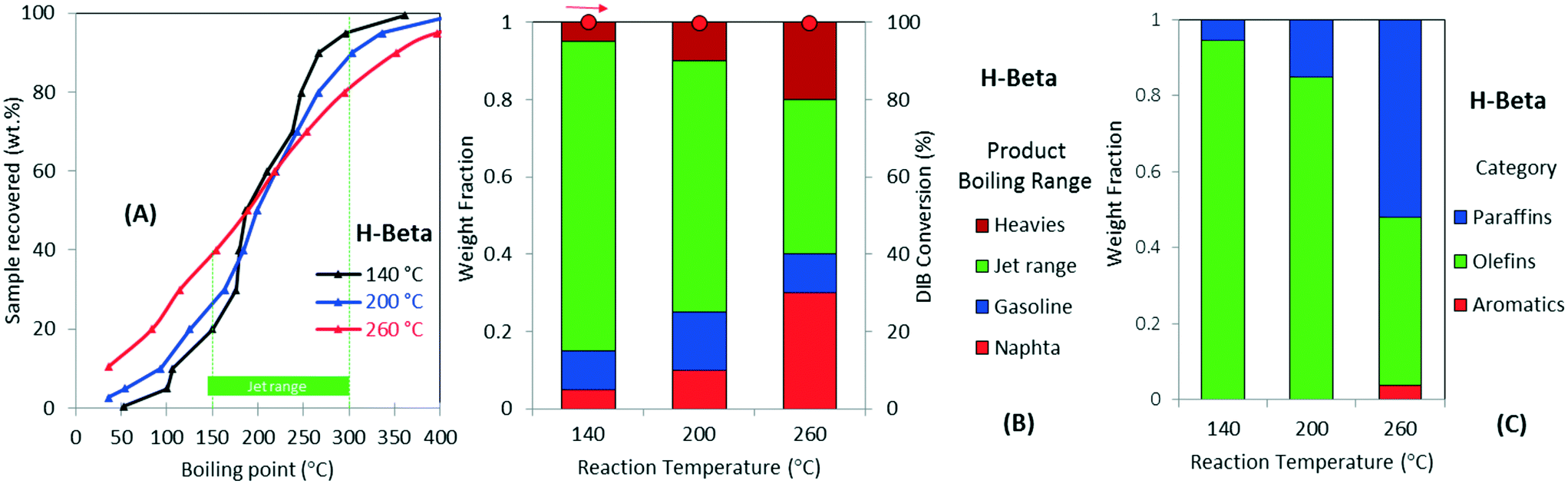 | ||
| Fig. 4 (A) Simulated distillation and GC-MS analysis for the liquids products derived from DIB feed reaction over H-beta zeolite as classified by (B) by boiling-point range and (C) chemical functionality. DIB (b.p. 101 °C) is fully converted to a wide boiling mixture of hydrocarbons. | ||
3.4. Propene and isobutene model feed over solid acid catalysts
The previous section showed that when using the DIB model feed, the yield to jet-range hydrocarbons can be improved with the proper selection of catalyst and operating temperature. To correlate these results with actual ethanol-derived olefin product, an isobutene/propene mixture (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) was used as a model feedstock. This mixture represents a typical composition derived from ethanol over Zn1Zr2.5Oz-type catalyst when operated at 450 °C (Table 2).13 Oligomerization results are shown in Table 3 and Fig. 4 using, H-beta, the most promising zeolite determined from above, and compared to the polymeric resins CT275 and Amberlyst-36. Table 3 shows that isobutene conversion is nearly complete for all the catalysts in the temperature range studied. However, propene conversion is hampered at low operating temperatures. For example, over H-beta catalyst, propene conversion drops to 7.2% when the reaction temperature is lowered from to 200 °C to 140 °C. A similar dramatic drop in propene conversion occurs over CT275 with just a 20 °C decrease in reaction temperature. However, isobutene conversion remains high in all cases.
:1 molar) mixture over H-beta zeolite and CT275 and Amberlyst-36 (A36) resins (3 hours TOS, weight hour space velocity [WHSV] = 1.1 h−1)
:1 molar ratio) was used as a model feedstock. This mixture represents a typical composition derived from ethanol over Zn1Zr2.5Oz-type catalyst when operated at 450 °C (Table 2).13 Oligomerization results are shown in Table 3 and Fig. 4 using, H-beta, the most promising zeolite determined from above, and compared to the polymeric resins CT275 and Amberlyst-36. Table 3 shows that isobutene conversion is nearly complete for all the catalysts in the temperature range studied. However, propene conversion is hampered at low operating temperatures. For example, over H-beta catalyst, propene conversion drops to 7.2% when the reaction temperature is lowered from to 200 °C to 140 °C. A similar dramatic drop in propene conversion occurs over CT275 with just a 20 °C decrease in reaction temperature. However, isobutene conversion remains high in all cases.
:1 molar) mixture over H-beta zeolite and CT275 and Amberlyst-36 (A36) resins (3 hours TOS, weight hour space velocity [WHSV] = 1.1 h−1)
| Catalyst | T (°C) | Propene conversion (%) | Isobutene conversion (%) |
|---|---|---|---|
| H-Beta | 140 | 7.2 | 99.9 |
| H-Beta | 200 | 87.1 | 99.7 |
| H-Beta | 260 | 83.0 | 99.8 |
| CT275 | 100 | 36.6 | 99.9 |
| CT275 | 120 | 98.2 | 99.9 |
| A36 | 140 | 89.2 | 99.9 |
Oligomerization temperature not only effects overall olefin conversion but also the product slate. Similar to the results obtained using DIB, yield to the jet-distillate range also is severely affected by changes in reaction temperature with the propene/isobutene mixture feedstock. Fig. 5 shows how the jet-distillate yield decreases to about 30% over the H-beta zeolite when the temperature is increased to 260 °C. With the CT275 resin, only a 20 °C change drastically affects the simulated distillation profile as shown in Fig. 6. At 100 °C, the resulting simulated distillation profile indicates the presence of products primarily in two main boiling ranges of ∼100 °C and 170 to 180 °C. These two groups are consistent with the presence of C8 and C12 branched olefins, respectively. However, when operating at 120 °C, more C8 intermediate olefins are incorporated into the jet-range fraction of the final product. Additionally, the profile is smoother, which indicates the formation of an extensively isomerized product with similar carbon numbers. The formation and control of a wide boiling-point range, intimately related to the formation of isomerization products from relatively few oligomerization products, is a particularly useful catalyst performance characteristic that is desirable within the scope of fuels manufacturing.
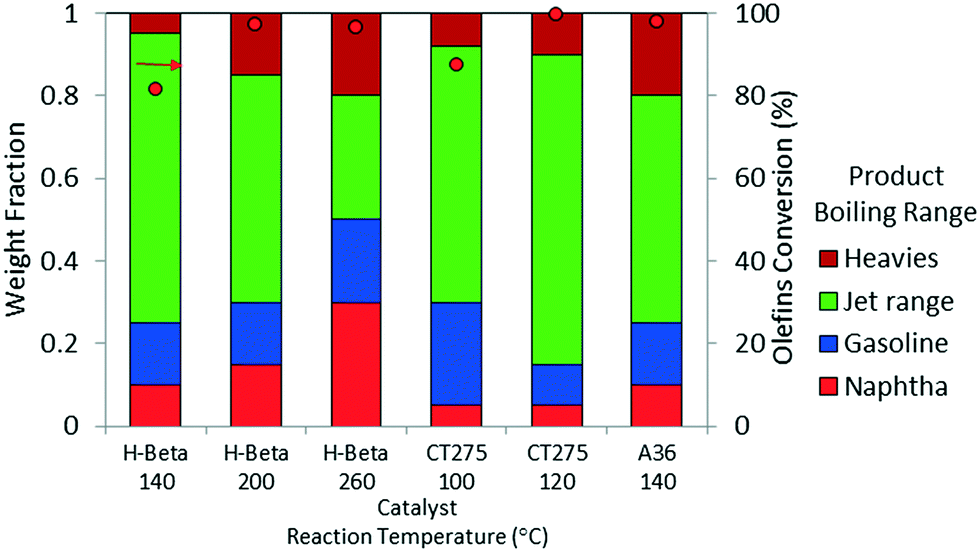 | ||
| Fig. 5 Product yields based on simulated distillation for the oligomerization liquid product of model isobutene/propene (4:1 molar ratio; 3 hours TOS, p = 1.38 MPa, WHSV = 1.1 h−1). | ||
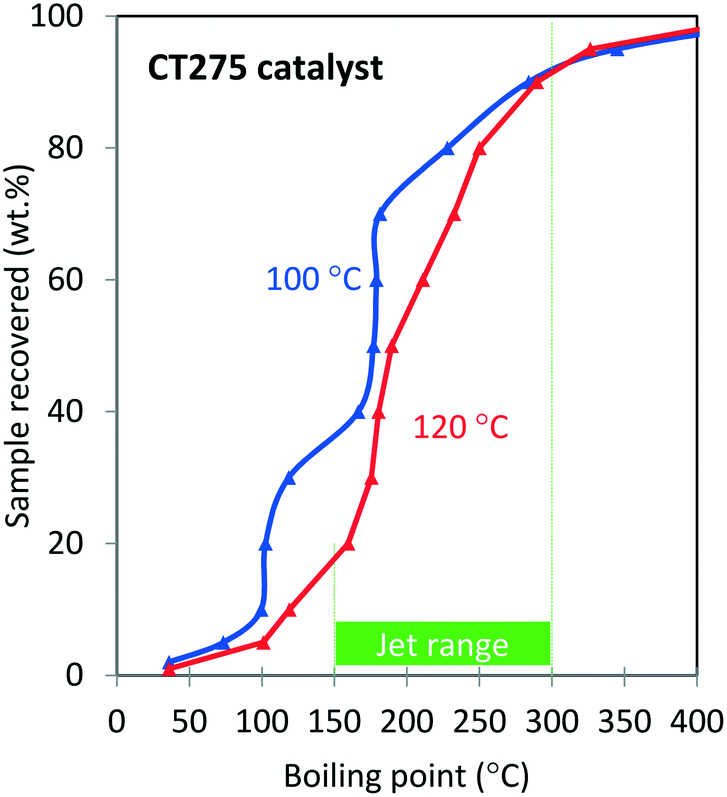 | ||
| Fig. 6 Effect of oligomerization temperature on simulated distillation profiles for the oligomerization product of a model isobutene/propene model feed over CT275 catalyst (4:1 molar ratio; 3 hours TOS, p = 1.38 MPa, WHSV = 1.1 h−1). | ||
Results from these model studies emphasize the importance of reaction temperature on the two primary metrics for catalytic performance: 1) total olefin conversion and 2) yield to desired distillate range product. Another important conclusion derived from these results is that underlying mechanisms such as isomerization, hydrogen transfer and dehydrogenation (aromatization) activity can be controlled by carefully selecting the catalyst and operating temperature. Analysis of GCMS data provides qualitative information pertaining to chemical changes in the oligomerization products. ESI† (Table S1) presents the library match for the most abundant compounds obtained for the oligomerization products at three reaction temperatures (140, 200, and 260 °C) of the model mixture isobutene/propene over zeolite H-beta. GCMS results infer an important decrease in branching occurring in parallel to the formation of paraffins and β-scission products at higher reaction temperatures.
Catalyst stability over extended operating durations is also a critical factor. For the conditions chosen here, catalytic performance was steady for a 20 hour period. However, we note that catalyst stability cannot accurately be determined when operating at high conversion. A more rigorous evaluation of catalyst stability is required in order to understand the true extent of deactivation. Here, the conversion was too high and the duration relatively limited to thoroughly assess catalyst deactivation. As with other zeolite catalyzed processes it is anticipated that longer term deactivation would be observed and that periodic regeneration would be required. Nonetheless, from Fig. 7, it can be seen that high olefin conversion can be achieved for two different catalyst systems (H-beta zeolite and CT275 resin) and operating at different temperatures (200 °C and 120 °C, respectively) while also producing a similar liquid product distribution as evidenced by simulated distillation profile (Fig. 6).
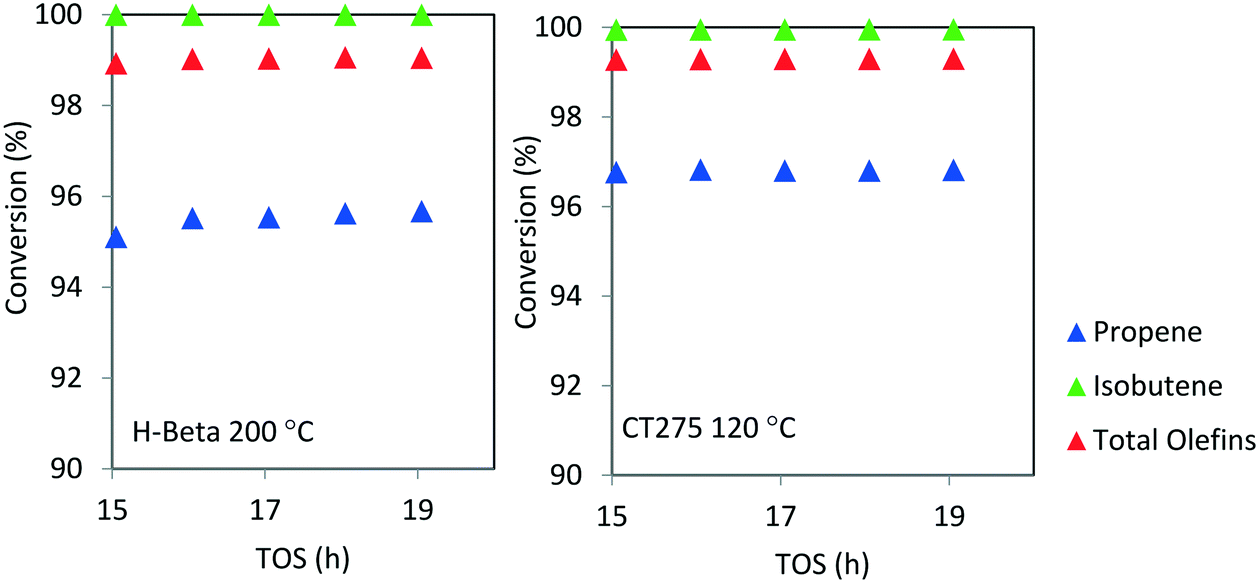 | ||
| Fig. 7 Isobutene/propene conversion over H-beta (200 °C) and CT275 (120 °C) catalysts (4:1 molar ratio, p = 1.38 MPa, WHSV = 1.1 h−1). | ||
3.5. Integrated process with oxygenate separation prior to oligomerization
Next, the production of liquid hydrocarbons was investigated via oligomerization of the isobutene and propene olefins derived from aqueous ethanol over Zn1Zr2.5Oz-type catalyst. We previously reported on oligomerization of propene and isobutene olefin intermediates using unoptimized conditions.10 Here, a 50 wt% ethanol in water mixture, representative of a fermentation-derived product, was fed over Zn1Zr2.5Oz catalyst at 450 °C. An olefin mixture rich in isobutene and propene was produced and reacted over a period of 350 hours. The results of this experiment are shown in Fig. 8. As previously reported, this reaction does facilitate coke build-up on the catalyst so periodic catalyst regeneration is required to avoid a drop in oxygenate intermediate formation (e.g., acetone formed as an intermediate).13 Therefore, we introduced periodic regenerative oxidative steps to demonstrate semi-continuous operation under full ethanol conversion without any evident loss in performance.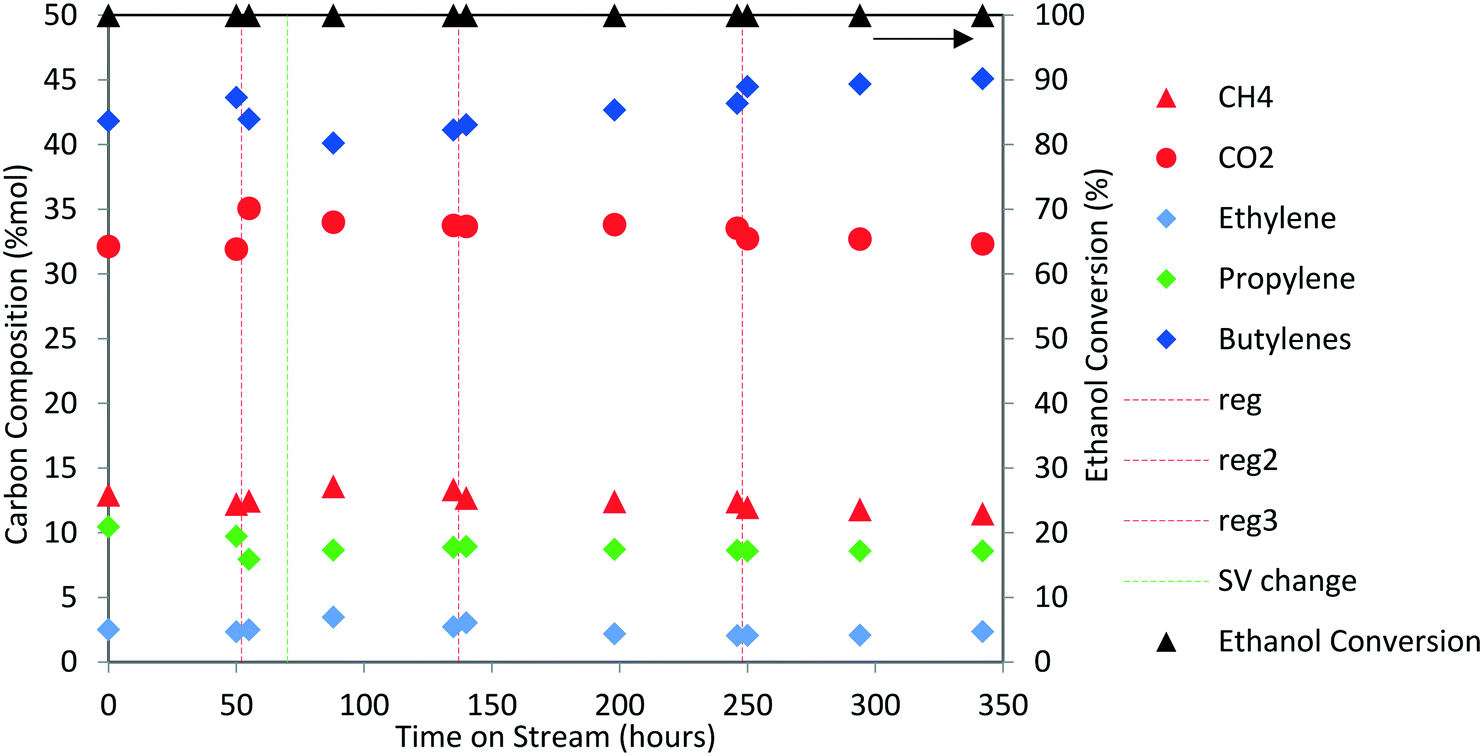 | ||
| Fig. 8 Ethanol conversion (mol%) and product selectivity (mol C%) over Zn1Zr2.5Oz (50 wt% ethanol in water feed; 450 °C, 1.38 MPa, GHSV = 320 to 660 h−1). Vertical red dotted lines represent regeneration steps and green dotted line marks a change in contact time. | ||
During the 350 hour run, the olefin-rich gas produced in the Zn1Zr2.5Oz reactor was fed to a second reactor containing Amberlyst-36 catalyst (see Fig. 1A). Water and trace oxygenates in this gas stream were removed using a cold trap and a drierite adsorption bed. Olefin conversion and characterization of the resulting oligomerization product is shown in Table 4. Isobutene and propene conversions were 89 and 37%, respectively. A neglible amount of ethene was reacted under these conditions. The liquid olefin product contained highly branched C8 and C12 hydrocarbons, and the simulated distillation profile of the hydrocarbon product is shown in Fig. 9.
| Compound | Compositionb (%w) |
|---|---|
| C5 olefins | 1.5 |
| C6 olefins | 3.3 |
| C7 olefins | 7.47 |
| C8 olefins | 37.1 |
| C8 cyclic | 4.1 |
| C11 olefins | 1.3 |
| C12 olefins | 25.6 |
| C10 cyclic | 6.9 |
| C12 cyclic | 12.6 |
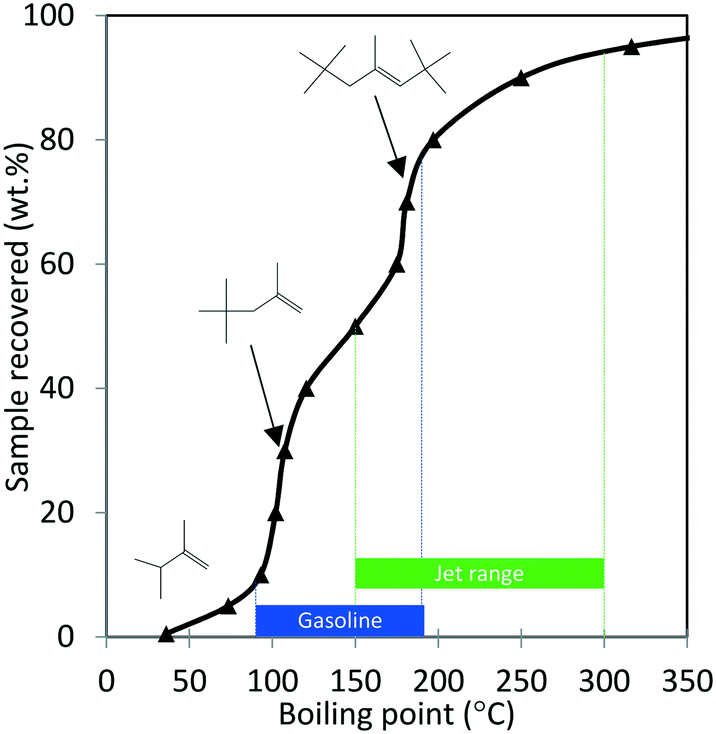 | ||
| Fig. 9 Simulated distillation profile for the oligomerization product shown in Table 4 collected after 350 hours TOS (Amberlyst-36 catalyst; p = 1.38 MPa, 140 °C, WHSV = 1.1 h−1). Proposed structures of major products with corresponding boiling points were obtained from GC-MS analysis. | ||
Results shown in Fig. 9 and Table 4 show that major components of the oligomerization product from reaction over the Amberlyst-36 catalyst were predominantly C8 and C12 olefins with boiling points corresponding to the gasoline range with a yield close to 70%. The yield in the jet-distillate range was lower at around 40%. The simulated distillation profile and GC-MS characterization of the product indicate the existence of two main reaction products: C8 (b.p. 100 to 105 °C) and C12 (b.p. 180 to 190 °C) branched olefins, which are the primary products of isobutene dimerization and trimerization, respectively. The presence of naphthenes (e.g., dimethylcyclohexane, and isobutyl-dimethylcyclohexane (C12H24), b.p. 200 to 220 °C) is an indication that isomerization (cyclization) is occurring concurrently with oligomerization. This liquid product was distilled into a gasoline range (90 to 190 °C) and found to contain a high RON (103) (Table 4), which is consistent with the large amount of branched C8 and C12 olefins present. A summary of the conversion and selectivity for each step within the integrated process for ethanol conversion to isobutene and propene olefins followed by oxygenate separation and olefin oligomerization is shown in Table 5. It should be noted that the ethanol conversion step is highly active (100% ethanol conversion). However, selectivity to isobutene (42.0 mol C%) and propene (8.5 mol C%) is limited due to the stoichiometric requirement to produce CO2via ketaonization within the reaction mechanism over Zn1Zr2.5Oz catalyst. Improved selectivity to olefins would likely be required for commercial application. Nonetheless, subsequent conversion of the produced olefins to fuel-range hydrocarbons is relatively selective.
| Property | Olefin Productiona (reactor 1) | Olefins oligomerizationb (reactor 2) | Overall process |
|---|---|---|---|
| a Ethanol over Zn1Zr2.5Oz (50 wt% ethanol in water feed; 450 °C, 1.38 MPa, GHSV = 320 to 660 h−1). b Olefin oligomerization over Amberlyst-36 catalyst (140 °C, WHSV = 1.1 h−1). | |||
| Carbon conversion (wt%) | 100% ethanol conversion | 79.1% propene + isobutene conversion | 39.9% yield to liquid hydrocarbons |
| Selectivity (%) | 42.0 mol C% isobutene | 9.0 wt% lights (b.p. < 90 °C) | 4.5 wt% lights (b.p. < 90 °C) |
| 8.5 mol C% propene | 68.0 wt% gasoline (b.p. 90–190 °C) | 35.3 wt% gasoline (b.p. 90–190 °C) | |
| 49.5 mol C% balance (CO2, CH4, ethylene) | 44.0 wt% jet-range (b.p. 150–300 °C) | 22.7 wt% jet-range (b.p. 150–300 °C) | |
| 5.0 wt% heavies (b.p. > 300 °C) | 2.5 wt% heavies (b.p. > 300 °C) | ||
| Carbon balance (wt%) | 96% | 97% | 93% |
A summary of the conversion and selectivity for each step within the integrated process for ethanol conversion to isobutene and propene olefins followed by oxygenate separation and olefin oligomerization is shown in Table 5. It should be noted that the ethanol conversion step is highly active (100% ethanol conversion). However, selectivity to isobutene (42.0 mol C%) and propene (8.5 mol C%) is limited primarily due to the stoichiometric requirement to produce CO2via ketonization within the reaction mechanism over Zn1Zr2.5Oz catalyst. Improved selectivity to olefins would likely be required for commercial application. Nonetheless, subsequent conversion of the produced olefins to fuel-range hydrocarbons is relatively selective and carbon efficiency can be greatly improved by recirculation of unreacted olefins.
3.6. Integrated process with inter-stage olefin separation and oligomerization
As shown in Table 1, the mixed olefin product from the ethanol conversion process contained a significant amount of light gases, primarily CO2 and H2. If these unreactive gases are included in the feed to the oligomerization reactor, they dilute the olefins and hinder chain growth. To increase the partial pressure of reactants in the oligomerization reactor, the olefins must be separated before the oligomerization process. This is described in our technoeconomic analysis reported for a similar process47 using unoptimized oligomerization results.10 Here we consider two updated process schemes both of which rely on inter-stage olefin separation (e.g., using olefin gas absorption as previously proposed).47 In the first process scheme, after oligomerization, the unreacted olefins and light recovered fraction are separated by distillation and then recycled to the oligomerization reactor (scheme shown in Fig. 10). Demonstration of this process scheme was performed for both H-beta zeolite and CT275 polymeric resin, and results are shown in Fig. 10. In both cases no significant improvement in the distillation profile to the jet-range distillate was observed after incorporating the light recovered fraction. With recycle, the yield to the jet-range distillate fraction improved by approximately 15% and 10% for H-beta and CT275 catalysts, respectively.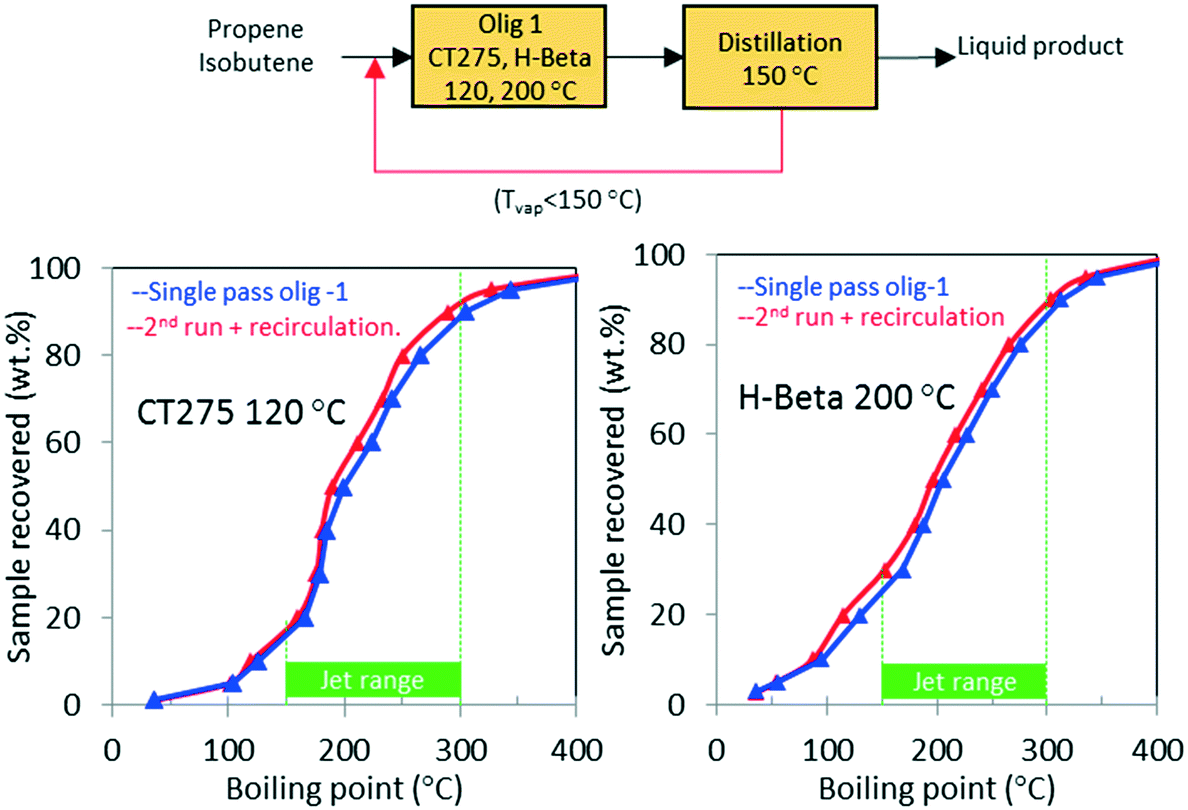 | ||
| Fig. 10 Simulated distillation results for the reaction product of model isobutene/propene (4/1 molar) feed mixture over H-beta (left side) and CT275 (right side) after single-pass conversion (blue line) and for a second run (red line) incorporating the light olefins of the first run (b.p. < 150 °C). | ||
In a second process scheme, we explored using a second oligomerization bed for converting the light fraction recovered after distillation (Fig. 11). Results reported in Fig. 11 show that with H-beta zeolite, yield to jet-range distillate was significantly improved with the use of a sequential oligomerization bed. Yield to products into the jet-distillate range was boosted by 24% and 6% over H-beta zeolite and CT275, respectively. Thus, a significant enhancement to the jet range yield was facilitated using sequential oligomerization versus using recycle alone, particularly with the H-beta zeolite case. With recycle, preferential adsorption of isobutene over intermediate oligomers (e.g., C8 branched olefins) may be responsible for limited chain growth. In contrast, when a sequential bed is used, the relative absence of isobutene in the feed allowed for activation and further reaction of C8 olefins.
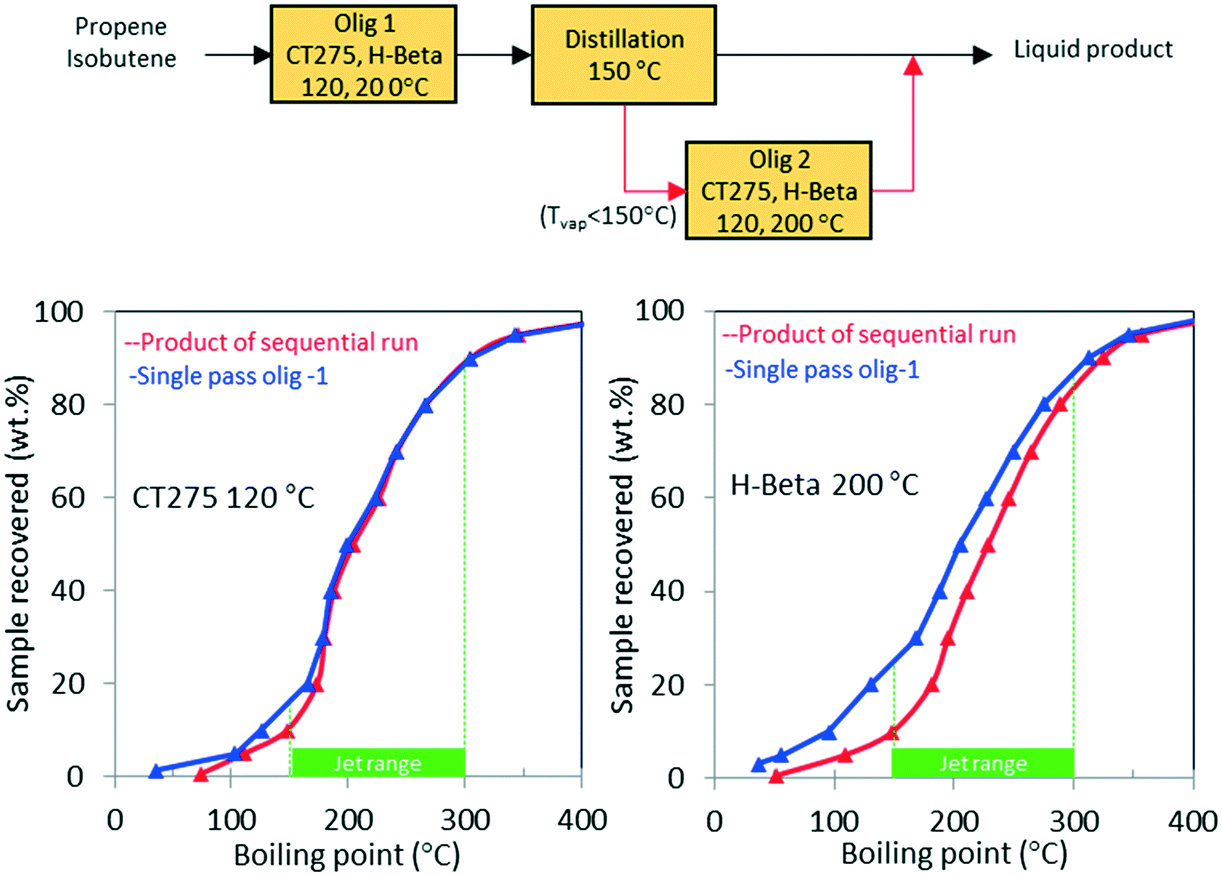 | ||
| Fig. 11 Simulated distillation results for the reaction product of model isobutene/propene (4/1 molar) feed mixture over H-beta (left figure) and CT275 (right figure) after single-pass conversion (blue line) and after a second oligomerization (red line) of the light olefins obtained in the first run (b.p. < 150 °C). | ||
The potential use of these oligomerization products as a blendstock for jet fuel would require catalytic hydrotreatment in which the olefins are converted into paraffins (ASTM has specifications for the content of olefins and other compounds). The concentration of olefins is limited in jet blendstocks and final fuel mixtures because they lead to the formation of deposits (stemming from rapid auto-oxidation compared to other hydrocarbons) and are detrimental to product stability as pertaining to long-term storage.29 In addition to boiling range requirements, energy density and strict chemical composition requirements (e.g., sulfur, aromatics, and water concentrations) also must be met.2 Thus, physicochemical characterization of the liquid products was performed (see Table 6). Oligomerization products resulting from reaction over zeolite H-beta and CT275 (using the sequential bed reaction scheme described above) were hydrotreated in a batch Parr reactor at 200 °C (5.17 MPa under H2 for 7.5 hours over 3% Pt/Al2O3 catalyst). After hydrotreatment distillation was performed and a liquid fraction representative of the jet-distillate range (b.p. 150 to 300 °C) was obtained. Fig. 12 shows GC-MS characterization results for the jet-distillate fractions produced reaction over H-beta and CT275. The hydrocarbon range consisted of compounds with between 9 and 18 carbons, with a relatively high concentration of 12 carbons (b.p. 190 to 230 °C). Before hydrotreatment, the liquid product had a high olefin content (estimated by GC-MS at 87% and 94% for H-beta and CT275, respectively). After hydrotreatment, both products were converted into an almost exclusive mixture of paraffinic hydrocarbons (>99.7%). We note that although the distillation profiles were similar for both products, the chemical composition was significantly different. GC-MS characterization details are provided in the Table S1 of the ESI.† Here, it can be seen that the liquid products produced with CT275 resin predominantly consisted of highly branched olefins with 12 and 16 carbons (estimated 64 area% by GC-MS TIC). However, over H-beta zeolite, the liquid product consisted of fewer branched olefins and a wider distribution of carbon numbers. We speculate that increased acidity of the CT275 polymeric resin readily facilitated isomerization reactions, although we note that both catalysts facilitated significant branching. Also, the higher temperature used with H-beta more readily facilitated other side reactions (e.g., cracking) that resulted in a wider distribution of carbon numbers.
| Property | CT275 | H-Beta | Blendstock requirements (ASTM D7566) | |
|---|---|---|---|---|
| Olefin conversion (wt%) | 99.3 | 98.9 | ||
| Lights yield (b.p. RT to 150 °C, wt%) | 1.6 | 2.8 | ||
| Jet-range yield (b.p. 150 to 300 °C, wt%) | 86.9 (75 single pass) | 83.5 (66 single pass) | ||
| Heavies yield (b.p. > 300 °C, wt%) | 11.5 | 13.7 | ||
| Aviation fuel properties | Freezing point (°C) | −74 | −74 | −40 max (D5972) |
| Flash point (°C) | 51.5 | 58.5 | 38 min (D445) | |
| Viscosity (mm2 s−1) | 2.0 | 2.2 | 8 max (D93) | |
| Density (kg m−3) | 780 | 780 | 775 to 840 (D4052) | |
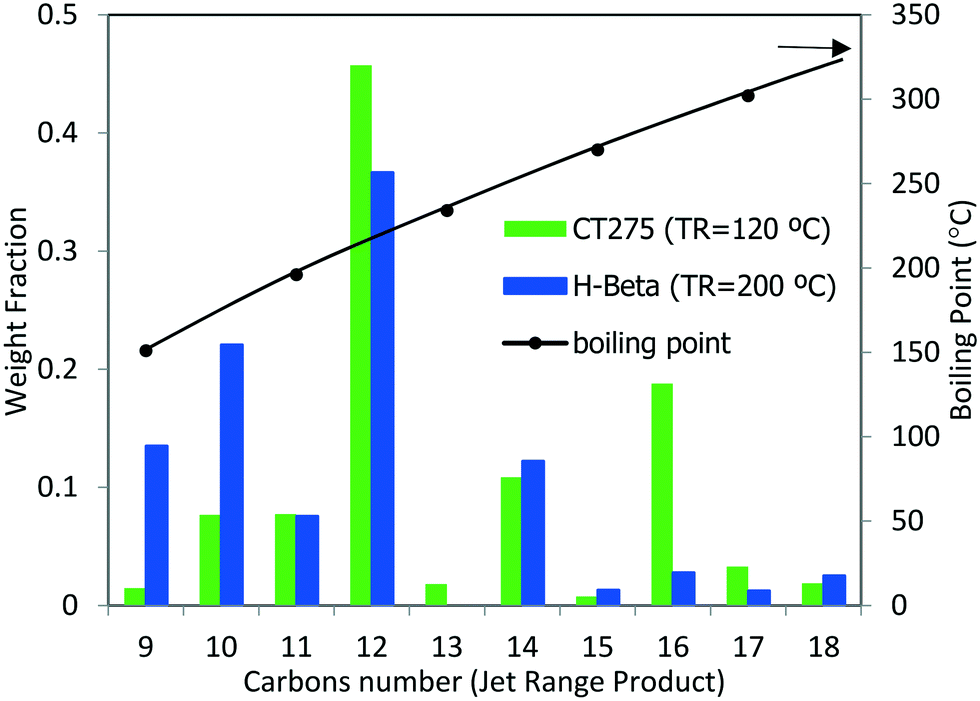 | ||
| Fig. 12 Carbon number distribution as determined by GC-MS for the reaction product via sequential oligomerization shown in Fig. 11 followed by hydrotreatment. Right axis (black line) indicated the average boiling point. Hydrotreated products comprised primarily paraffins with less than 0.3 area% of other compounds. No aromatic compounds were detected. | ||
4. Conclusions
Several zeolites and polymeric resins were evaluated for oligomerization of isobutene/propene or DIB model feeds. Of those investigated, H-beta (zeolite) and CT275 (polymer resin) were both found to be active, selective, and stable (for 20 hours) for producing fuel-range hydrocarbons. A higher oligomerization temperature was needed to achieve high conversion of propene, which was less reactive relative to isobutene probably because of the oligomerization mechanism in which propene must form a less stable secondary carbocation versus the tertiary carbocation mechanism of isobutene. However higher temperatures facilitated depletion of the desired jet-range blendstocks as a result of secondary cyclization/isomerization, hydrogenation, and cracking reactions. These secondary reaction mechanisms, activated at higher reaction temperatures, facilitate changes in carbon number distribution and chemical functionalities to the resulting finished fuel.We demonstrated a two-step process for producing a high-octane gasoline. Wet ethanol feedstock (50 wt% ethanol) was converted over Zn1Zr2.5Oz mixed-oxide catalyst into an olefin mixture with 50% yield to isobutene and propene. The catalyst was operated for 350 hours TOS with periodic regeneration of the catalyst to remove coke. After oxygenate separation, a second bed packed with polymeric resin produced a 70% olefin yield (after a single pass) of a gasoline-range hydrocarbon (b.p. 90 to 190 °C) with a high octane (RON 103).
For conversion to a jet-range hydrocarbon products (b.p. 150 to 300 °C), light gases produced from the Zn1Zr2.5Oz-derived catalyst system (i.e., H2 and CO2) must be removed to facilitate chain growth. Using model isobutene and propene feed oligomerization over both H-beta zeolite and CT275 polymer resin produced jet-range hydrocarbon products. With separation and recycle of the light olefin product, the yield to jet-distillate fraction improved by approximately 15% and 10% for H-beta and CT275 catalysts, respectively. However, when the light fraction was oligomerized over a second bed, the yield to jet-range products increased by 24% and 6% over H-beta zeolite and CT275, respectively. Particularly highlighted in the case for the H-beta zeolite, sequential bed operation facilitated the activation and oligomerization of light olefins without competition from olefin feedstock, as was the case with recycle. With both zeolite and polymeric resin catalysts, high olefin conversion was achieved, resulting in liquid hydrocarbons with similar simulated distillation profiles. However, GC-MS analysis of the liquid products suggests subtle variations in chemical compositions. The H-beta zeolite produced longer carbon chains with less branching compared to the CT275 solid acid resin. Regardless, after hydrotreatment, further characterization revealed that both liquid products met selected ASTM D7566 A5 specifications for ATJ blendstock (e.g., freezing point, flash point, viscosity, density).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Chemical Catalysis for Bioenergy Consortium (ChemCatBio) sponsored by the U.S. Department of Energy (DOE) Bioenergy Technologies Office (BETO) and performed at Pacific Northwest National Laboratory (PNNL) under contract DE-AC05-76RL0183. ChemCatBio leverages unique national laboratory capabilities to address technical risks associated with accelerating the development of catalysts and related technologies for commercializing biomass-derived fuels and chemicals. Some of this research was conducted as part of the Co-Optimization of Fuels & Engines (Co-Optima) project sponsored by the DOE's Office of Energy Efficiency and Renewable Energy, Bioenergy Technologies and Vehicle Technologies Offices. Co-Optima is a collaborative project of multiple national laboratories initiated to simultaneously accelerate the introduction of affordable, scalable, and sustainable biofuels and high-efficiency, low-emission vehicle engines. PNNL is a multi-program national laboratory operated for DOE by Battelle Memorial Institute. Advanced catalyst characterization use was granted by a user proposal at the William R. Wiley Environmental Molecular Sciences Laboratory (EMSL). EMSL is a national scientific user facility sponsored by DOE's Office of Biological and Environmental Research and located at PNNL. The authors would like to thank Teresa Lemmon, Marie Swita, Samuel Fox, and Rich Lucke for analytical support, and Rich Hallen for reviewing the manuscript and providing helpful insights. The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights.References
- G. W. Huber, S. Iborra and A. Corma, Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- W.-C. Wang and L. Tao, Bio-jet fuel conversion technologies, Renewable Sustainable Energy Rev., 2016, 53, 801–822 CrossRef CAS.
- http://www.greenaironline.com/news.php?viewStory=2469, Accessed May 18, 2018.
- J. Sun and Y. Wang, Recent Advances in Catalytic Conversion of Ethanol to Chemicals, ACS Catal., 2014, 4, 1078–1090 CrossRef CAS.
- R. Andrei, I. M. Popa, F. Fajula and V. Hulea, Heterogeneous oligomerization of ethylene over highly active and stable Ni-AlSBA-15 mesoporous catalysts, 2015 Search PubMed.
- M. A. Lilga, R. T. Hallen, K. O. Albrecht, A. R. Cooper, J. G. Frye and K. K. Ramasamy, Systems and processes for conversion of ethylene feedstocks to hydrocarbon fuels, in: USPTO, US Pat., US 20170218283 A1, 2017 Search PubMed.
- A. Galadima and O. Muraza, Zeolite catalysts in upgrading of bioethanol to fuels range hydrocarbons: A review, J. Ind. Eng. Chem., 2015, 31, 1–14 CrossRef CAS.
- F. Ferreira Madeira, K. Ben Tayeb, L. Pinard, H. Vezin, S. Maury and N. Cadran, Ethanol transformation into hydrocarbons on ZSM-5 zeolites: Influence of Si/Al ratio on catalytic performances and deactivation rate. Study of the radical species role, Appl. Catal., A, 2012, 443–444, 171–180 CrossRef CAS.
- X. Wang, X. Hu, C. Song, K. W. Lux, M. Namazian and T. Imam, Oligomerization of Biomass-Derived Light Olefins to Liquid Fuel: Effect of Alkali Treatment on the HZSM-5 Catalyst, Ind. Eng. Chem. Res., 2017, 56, 12046–12055 CrossRef CAS.
- V. L. Dagle, C. Smith, M. Flake, K. O. Albrecht, M. J. Gray, K. K. Ramasamy and R. A. Dagle, Integrated process for the catalytic conversion of biomass-derived syngas into transportation fuels, Green Chem., 2016, 18, 1880–1891 RSC.
- C. Liu, J. Sun, C. Smith and Y. Wang, A study of ZnxZryOz mixed oxides for direct conversion of ethanol to isobutene, Appl. Catal., A, 2013, 467, 91–97 CrossRef CAS.
- J. Sun, K. Zhu, F. Gao, C. Wang, J. Liu, C. H. F. Peden and Y. Wang, Direct Conversion of Bio-ethanol to Isobutene on Nanosized ZnxZryOz Mixed Oxides with Balanced Acid–Base Sites, J. Am. Chem. Soc., 2011, 133, 11096–11099 CrossRef CAS PubMed.
- C. Smith, V. L. Dagle, M. Flake, K. K. Ramasamy, L. Kovarik, M. Bowden, T. Onfroy and R. A. Dagle, Conversion of syngas-derived C2+ mixed oxygenates to C3-C5 olefins over ZnxZryOz mixed oxide catalysts, Catal. Sci. Technol., 2016, 6, 2325–2336 RSC.
- H. I. Mahdi and O. Muraza, Conversion of Isobutylene to Octane-Booster Compounds after Methyl tert-Butyl Ether Phaseout: The Role of Heterogeneous Catalysis, Ind. Eng. Chem. Res., 2016, 55, 11193–11210 CrossRef CAS.
- J. P. van den Berg, J. P. Wolthuizen and J. H. C. van Hooff, Reaction of small olefins on zeolite H-ZSM-5. A thermogravimetric study at low and intermediate temperatures, J. Catal., 1983, 80, 139–144 CrossRef CAS.
- J. Sun, R. A. L. Baylon, C. Liu, D. Mei, K. J. Martin, P. Venkitasubramanian and Y. Wang, Key Roles of Lewis Acid–Base Pairs on ZnxZryOz in Direct Ethanol/Acetone to Isobutene Conversion, J. Am. Chem. Soc., 2016, 138(2), 507–517 CrossRef CAS PubMed.
- A. Corma and S. Iborra, Oligomerization of Alkenes, Catalysts for Fine Chemical Synthesis, John Wiley & Sons, Ltd, 2006, pp. 125–140 Search PubMed.
- J. A. Martens, W. H. Verrelst, G. M. Mathys, S. H. Brown and P. A. Jacobs, Tailored Catalytic Propene Trimerization over Acidic Zeolites with Tubular Pores, Angew. Chem., Int. Ed., 2005, 44, 5687–5690 CrossRef CAS PubMed.
- M. L. Sarazen, E. Doskocil and E. Iglesia, Effects of Void Environment and Acid Strength on Alkene Oligomerization Selectivity, ACS Catal., 2016, 6, 7059–7070 CrossRef CAS.
- J. A. Martens, R. Ravishankar, I. E. Mishin and P. A. Jacobs, Tailored Alkene Oligomerization with H-ZSM-57 Zeolite, Angew. Chem., Int. Ed., 2000, 39, 4376–4379 CrossRef CAS PubMed.
- B. H. Babu, M. Lee, D. W. Hwang, Y. Kim and H.-J. Chae, An integrated process for production of jet-fuel range olefins from ethylene using Ni-AlSBA-15 and Amberlyst-35 catalysts, Appl. Catal., A, 2017, 530, 48–55 CrossRef CAS.
- C. P. Nicholas, L. Laipert and S. Prabhakar, Oligomerization of Light Olefins to Gasoline: An Advanced NMR Characterization of Liquid Products, Ind. Eng. Chem. Res., 2016, 55, 9140–9146 CrossRef CAS.
- J. W. Yoon, J.-S. Chang, H.-D. Lee, T.-J. Kim and S. H. Jhung, Trimerization of isobutene over a zeolite beta catalyst, J. Catal., 2007, 245, 253–256 CrossRef CAS.
- A. de Klerk, Oligomerization of 1-Hexene and 1-Octene over Solid Acid Catalysts, Ind. Eng. Chem. Res., 2005, 44, 3887–3893 CrossRef CAS.
- S. Bessell and D. Seddon, The conversion of ethene and propene to higher hydrocarbons over ZSM-5, J. Catal., 1987, 105, 270–275 CrossRef CAS.
- R. J. Quann, L. A. Green, S. A. Tabak and F. J. Krambeck, Chemistry of olefin oligomerization over ZSM-5 catalyst, Ind. Eng. Chem. Res., 1988, 27, 565–570 CrossRef CAS.
- K. Hauge, E. Bergene, D. Chen, G. R. Fredriksen and A. Holmen, Oligomerization of isobutene over solid acid catalysts, Catal. Today, 2005, 100, 463–466 CrossRef CAS.
- D. M. Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Production of liquid hydrocarbon transportation fuels by oligomerization of biomass-derived C9 alkenes, Green Chem., 2010, 12, 992–999 RSC.
- A. Zschocke, D. L. AG, S. Scheuermann and J. Ortner, High Biofuel Blends in Aviation (HBBA), Interim Report, ENER C, vol. 2pp. 420–421 Search PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Integrated Catalytic Conversion of γ-Valerolactone to Liquid Alkenes for Transportation Fuels, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- ASTM D86-17, Standard Test Method for Distillation of Petroleum Products and Liquid Fuels at Atmospheric Pressure, ASTM International, West Conshohocken, PA, 2017 Search PubMed.
- R. L. McCormick, G. Fioroni, L. Fouts, E. Christensen, J. Yanowitz, E. Polikarpov, K. Albrecht, D. J. Gaspar, J. Gladden and A. George, Selection Criteria and Screening of Potential Biomass-Derived Streams as Fuel Blendstocks for Advanced Spark-Ignition Engines, SAE Int. J. Fuels Lubr., 2017, 19 Search PubMed , Medium: ED; Size.
- X. Zhang, Y. Zhao, S. Xu, Y. Yang, J. Liu, Y. Wei and Q. Yang, Polystyrene sulphonic acid resins with enhanced acid strength via macromolecular self-assembly within confined nanospace, Nat. Commun., 2014, 5, 3170 CrossRef PubMed.
- C. J. Plank, E. J. Rosinski and E. N. Givens, Converting low molecular weight olefins over zeolites, US Pat., US 3,960,978, 1977 Search PubMed.
- A. de Klerk, Properties of Synthetic Fuels from H-ZSM-5 Oligomerization of Fischer–Tropsch Type Feed Materials, Energy Fuels, 2007, 21, 3084–3089 CrossRef CAS.
- M. L. Sarazen, E. Doskocil and E. Iglesia, Catalysis on solid acids: Mechanism and catalyst descriptors in oligomerization reactions of light alkenes, J. Catal., 2016, 344, 553–569 CrossRef CAS.
- A. N. Mlinar, P. M. Zimmerman, F. E. Celik, M. Head-Gordon and A. T. Bell, Effects of Brønsted-acid site proximity on the oligomerization of propene in H-MFI, J. Catal., 2012, 288, 65–73 CrossRef CAS.
- R. Gounder and E. Iglesia, Catalytic Consequences of Spatial Constraints and Acid Site Location for Monomolecular Alkane Activation on Zeolites, J. Am. Chem. Soc., 2009, 131, 1958–1971 CrossRef CAS PubMed.
- Y. T. Kim, J. P. Chada, Z. Xu, Y. J. Pagan-Torres, D. C. Rosenfeld, W. L. Winniford, E. Schmidt and G. W. Huber, Low-temperature oligomerization of 1-butene with H-ferrierite, J. Catal., 2015, 323, 33–44 CrossRef CAS.
- A. Coelho, G. Caeiro, M. A. N. D. A. Lemos, F. Lemos and F. R. Ribeiro, 1-Butene oligomerization over ZSM-5 zeolite: Part 1 – Effect of reaction conditions, Fuel, 2013, 111, 449–460 CrossRef CAS.
- MSDS specs obtained from www.Dow.com website.
- R. Bringué, M. Cadenas, C. Fité, M. Iborra and F. Cunill, Study of the oligomerization of 1-octene catalyzed by macroreticular ion-exchange resins, Chem. Eng. J., 2012, 207–208, 226–234 CrossRef.
- P. Pérez-Uriarte, M. Gamero, A. Ateka, M. Díaz, A. T. Aguayo and J. Bilbao, Effect of the Acidity of HZSM-5 Zeolite and the Binder in the DME Transformation to Olefins, Ind. Eng. Chem. Res., 2016, 55, 1513–1521 CrossRef.
- J. W. Yoon, S. H. Jhung, D. H. Choo, S. J. Lee, K.-Y. Lee and J.-S. Chang, Oligomerization of isobutene over dealuminated Y zeolite catalysts, Appl. Catal., A, 2008, 337, 73–77 CrossRef CAS.
- M. J. Wulfers and R. F. Lobo, Assessment of mass transfer limitations in oligomerization of butene at high pressure on H-beta, Appl. Catal., A, 2015, 505, 394–401 CrossRef CAS.
- M. N. Mazar, S. Al-Hashimi, M. Cococcioni and A. Bhan, β-Scission of Olefins on Acidic Zeolites: A Periodic PBE-D Study in H-ZSM-5, J. Phys. Chem. C, 2013, 117, 23609–23620 CrossRef CAS.
- E. C. D. Tan, L. J. Snowden-Swan, M. Talmadge, A. Dutta, S. Jones, K. K. Ramasamy, M. Gray, R. Dagle, A. Padmaperuma, M. Gerber, A. H. Sahir, L. Tao and Y. Zhang, Comparative techno-economic analysis and process design for indirect liquefaction pathways to distillate-range fuels via biomass-derived oxygenated intermediates upgrading, Biofuels, Bioprod. Biorefin., 2017, 11, 41–66 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02297f |
| This journal is © The Royal Society of Chemistry 2019 |