
Polysaccharide-based superporous hydrogel embedded with copper nanoparticles: a green and versatile catalyst for the synthesis of 1,2,3-triazoles†
Jaqueline F.
Souza
a,
Gabriel P.
Costa
a,
Rafael
Luque
 bc,
Diego
Alves
*a and
André R.
Fajardo
*a
bc,
Diego
Alves
*a and
André R.
Fajardo
*a
aUniversidade Federal de Pelotas, Brazil. E-mail: diego.alves@ufpel.edu.br; andre.fajardo@pq.cnpq.br; Fax: +55 5332757533; Tel: +55 5332757533
bDepartamento de Química Orgánica, Grupo FQM-383, Universidad de Córdoba, Campus de Rabanales, Edificio Marie Curie (C-3), Ctra Nnal IV-A, Km 396, Córdoba, Spain
cPeoples Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya str., 117198, Moscow, Russia
First published on 19th November 2018
Abstract
In this contribution, we a report a facile synthesis and stabilization of copper nanoparticles (CuNPs) in a superporous hydrogel based on chitosan (Cs) and poly(vinyl alcohol) (PVA). The functional groups of Cs and PVA stabilized the CuNPs and, as a result, several properties (e.g. thermal, morphological and liquid uptake) of the final materials exhibited remarkable improvements in terms of stability and catalytic activity. The metallic nature of CuNPs was confirmed by XRD and XPS analyses. From the XRD data, the Cu nanoparticle size was estimated to range from 3 to 7 nm. Moreover, EDS mapping analysis revealed that the CuNPs are homogeneously distributed throughout the hydrogel surface. The Cs/PVA-CuNP hydrogel was found to be an excellent catalyst for the synthesis of 1,2,3-triazoles using phenyl azide and phenylacetylene as model substrates under mild optimized conditions (>90% yield). Reuse studies revealed that this original CuNP-containing catalyst can be employed in at least 5 consecutive cycloaddition reactions without demonstrating a clear loss of efficiency. Moreover, the copper leaching from the hydrogel matrix was negligible during these consecutive reactions under the investigated conditions. The Cs/PVA-CuNP hydrogel could also efficiently catalyze cycloaddition reactions involving different substituted azide and alkyne precursors.
Introduction
Considerable efforts from the scientific community towards environmentally friendly processes and technologies in recent years have been directed to seeking alternative processes and syntheses with improved green credentials.1,2 Overall, green chemistry seeks ways to produce and process substances and materials in a more benign way considering human health and the environment.3 Catalysis is probably the most prominent field of green chemistry due to its direct relationship with the design of chemical substances and processes that reduce or eliminate the use and generation of hazardous materials.4 This aspect is very attractive from the environmental protection viewpoint. Moreover, the use of catalysts and catalytic systems allows saving time and energy, desirable from an economic viewpoint.Taking into account the different catalysts described in the literature, metal nanoparticles (MNPs) have gained great attention due to their interesting properties (high reactivity, selectivity, controllable facets, size, and shape, among others).5,6 In this context, chemical reactions catalyzed by MNPs offer great advantages in terms of green credentials as compared to other conventional catalysts such as high yield, fast reactions and reusability.7 Among the transition metals used to synthesize nanoparticles, copper (Cu) is of particular interest due to its interesting properties, low-cost preparation and many potential applications in catalysis. Copper nanoparticles (CuNPs) have been employed in numerous areas such as nanofluids, biological and optical applications, and nanodevices.8,9 Considering organic synthesis, CuNPs have been successfully utilized as heterogeneous catalysts in oxidative cross-coupling reactions and azide–alkyne cycloaddition reactions (“click reactions”).10–12 Triazoles resulting from this type of “click reaction” are of particular interest since they are used in several applications (e.g. dyes, agrochemicals, medicinal chemistry, and so on).12,13
Despite the remarkable properties of MNPs in catalysis, their use still has some limitations due to issues related to aggregation and coalescence. In nano-sized dimensions, the surface energy of such particles increases and strong attractive forces cause inherent instability in such systems.14 MNPs undergo agglomeration in order to reduce the energy associated with their large surface areas. This undesirable phenomenon impairs drastically the catalytic activity of MNPs. Numerous studies report elegant strategies to overcome this drawback and, as noticed, the immobilization of MNPs on solid supports is one of the most reliable.15,16 Solid supports utilized for this aim are generally engineered from inorganic, organic or hybrid materials.17,18 More recently, polysaccharide-based materials have been used as supports for MNPs without any undesirable consequences on their practical use and the environment.19,20 Attractive properties such as renewable origin, biodegradability, low cost, high molecular weight and high functionality encourage the use of polysaccharides for this aim. In this sense, materials designed from chitosan (Cs), a well-known polysaccharide synthesized from the total or partial deacetylation of chitin, are good candidates to support MNPs.21,22 Cs has free amine groups and hydroxyl groups on its backbone, increasing its ability to interact with metallic ions (Mn+) and coordinate with MNPs, preventing their agglomeration and leaching.23 Despite these attractive properties, the low mechanical strength and low hydrophilicity of chitosan-based materials additionally require the formation of composites with synthetic polymers. Herein, Cs was combined with poly(vinyl alcohol) (PVA), a synthetic hydrophilic and non-toxic polymer, in order to obtain a superporous hydrogel employed as a solid support for CuNPs. The synthesis of CuNPs was performed in situ and their catalytic activity was investigated towards the synthesis of 1,4-disubstituted-1,2,3-triazoles under eco-friendly conditions.
Experimental
Materials
Chitosan (Cs) (Mv 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 85% deacetylated) was purchased from Golden-Shell Biochemical (China). Poly(vinyl alcohol) (PVA) (Mw 124000 g mol−1 and 99% hydrolyzed) and glutaraldehyde (50 wt/v% in H2O) were purchased from Sigma-Aldrich (USA). Copper(II) chloride dihydrate (CuCl2·2H2O, P.A.), sodium borohydride (NaBH4, P.A.) and sodium bicarbonate (NaHCO3, 99%) were purchased from Vetec (Brazil). Acetic acid (99%) and ethanol (P.A.) were purchased from Synth (Brazil). Terminal alkynes were purchased from Sigma Aldrich and used without any purification. Organic azides were synthesized according to reported procedures.24–26 All chemicals of analytical grade were used as received without further purification.
000 g mol−1 and 85% deacetylated) was purchased from Golden-Shell Biochemical (China). Poly(vinyl alcohol) (PVA) (Mw 124000 g mol−1 and 99% hydrolyzed) and glutaraldehyde (50 wt/v% in H2O) were purchased from Sigma-Aldrich (USA). Copper(II) chloride dihydrate (CuCl2·2H2O, P.A.), sodium borohydride (NaBH4, P.A.) and sodium bicarbonate (NaHCO3, 99%) were purchased from Vetec (Brazil). Acetic acid (99%) and ethanol (P.A.) were purchased from Synth (Brazil). Terminal alkynes were purchased from Sigma Aldrich and used without any purification. Organic azides were synthesized according to reported procedures.24–26 All chemicals of analytical grade were used as received without further purification.
Synthesis of the superporous hydrogel
A Cs-solution was prepared by dissolving 250 mg of the polysaccharide in acetic acid solution (1 v/v%) under magnetic stirring for 12 h. In parallel, a second solution was prepared by dissolving 100 mg of PVA in distilled water under stirring at 80 °C for 12 h. The PVA-solution was subsequently cooled down to room temperature and mixed with the Cs-solution. The resulting solution was homogenized under magnetic stirring for 1 h. Glutaraldehyde (50 μL), the crosslinking agent, was added to the polymeric solution followed by addition of 50 mg of NaHCO3 as the gas blowing agent. The system was gently stirred for 30 min and immediately deep-frozen (−20 °C) for 24 h. The frozen hydrogel was collected and immersed in ethanol (200 mL) for 24 h. Finally, cubic samples (1 cm3) were cut from the as-prepared hydrogel and then dried under vacuum (37 °C) for 48 h. These samples were labeled as Cs-gel.In situ synthesis of CuNPs
The synthesis of the CuNPs into the Cs/PVA hydrogel matrix was performed by a two-step methodology: (i) adsorption and (ii) reduction of Cu2+ ions. Briefly, hydrogel samples (cubic shape – 1 cm3) were immersed in 50 mL of CuCl2·2H2O solution (0.05 mol L−1) under mild stirring at room temperature for 24 h. Next, Cu2+-containing samples (labeled as Cs/PVA-Cu2+) were thoroughly washed with distilled water in order to remove weakly physisorbed ions and subsequently immersed in a NaBH4 solution (0.5 mol L−1) at room temperature for 4 h. After that, the samples embedded with CuNPs (labeled as Cs/PVA-CuNP) were collected, washed with distilled water and dried under vacuum (37 °C) for 48 h. The dried samples were kept in a desiccator prior to characterization and use.Hydrogel characterization
The amount of Cu2+ ions adsorbed per hydrogel sample film was quantified using the flame atomic absorption spectroscopy (FAAS) technique. For this, aliquots of the CuCl2·2H2O solution (after the adsorption step) and washing medium were collected and analyzed in a Shimadzu spectrometer in order to determine the residual concentration of copper. The instrument was equipped with a copper hollow cathode lamp and a deuterium lamp for background correction. The operating parameters were adjusted according to the standard guidelines of the manufacturers and the atomic absorption signal was measured as a peak height mode as a function of an analytical curve (R2 ≈ 0.999). The amount of Cu2+ in each sample was calculated as per eqn (1):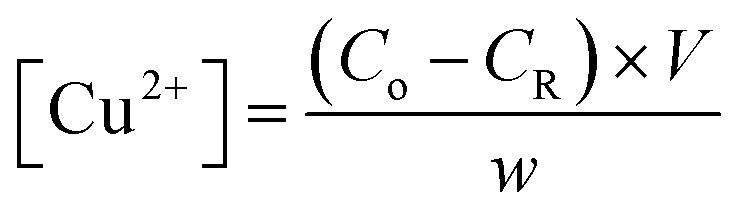 | (1) |
Fourier transform infrared spectroscopy (FTIR) analyses were performed in Shimadzu IR-Affinity-1 (Japan) equipment operating in the spectral region of 4000–400 cm−1 with a resolution of 4 cm−1. Before spectra acquisition, the samples were ground with spectroscopic grade KBr and pressed into disks. Thermogravimetric analyses (TGA) were performed in a Shimadzu DTG60 analyzer (Japan) operating in a temperature range of 25–500 °C with a scanning rate of 10 °C min−1 under a N2(g) atmosphere (flow of 20 mL min−1). X-ray diffraction (XRD) patterns were recorded from powder samples using a Siemens D500 diffractometer (Germany) equipped with a Cu-Kα radiation source at 30 kV and 20 mA. The XRD patterns were obtained in a scanning range of 5–50° with a scanning rate of 1° min−1. The surface elemental information of Cs/PVA-CuNP (binding energy and oxidation states) was determined using a Thermo Scientific Escalab 250Xi (USA) X-ray photoelectron spectrometer (XPS) at an excitation energy of Al-Kα (1486.6 eV). The XPS spectrum was obtained in the preparation chamber of the equipment with a base pressure below 5 × 10−9 mbar at 293 K for 60 min. The energy scale was calibrated by assigning 284.6 eV to the adventitious C 1s peak and 530.8 and 532 eV to O 1s. Scanning electron microscopy (SEM) images were recorded at 15 kV using a Jeol JSM-6610LV microscope (USA) equipped with an energy-dispersive X-ray (EDX) analyzer. Before SEM visualization, the samples were gold-coated by sputtering. N2(g) adsorption–desorption isotherms were obtained using a Micromeritics Tristar II Kr-3020 (USA) gas adsorption analyzer using samples previously heated to 120 °C under vacuum for 20 h. The Brunauer–Emmett–Teller (BET) specific surface area was examined using the adsorption data.
The total porosity of each hydrogel sample was examined using the liquid displacement method.27 Dry samples were immersed in a known initial volume of acetone (V1) at room temperature for 24 h. Following acetone impregnation, the volume of the set sample-acetone was determined (V2). Then, the acetone-impregnated hydrogel sample was withdrawn and the remaining liquid volume was determined (V3). The total porosity (in %) was calculated as per eqn (2). This experiment was performed in triplicate for each sample.
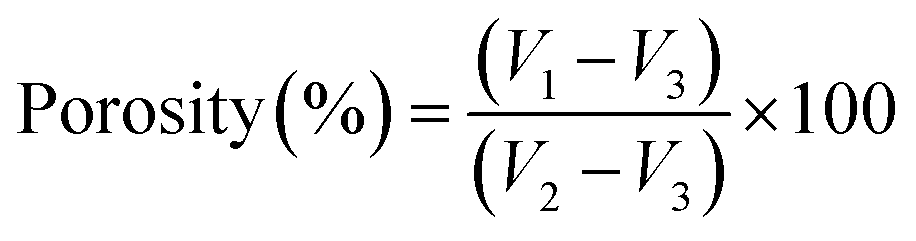 | (2) |
Swelling experiments
The water uptake capacity of the as-prepared hydrogels was investigated by swelling experiments conducted in distilled water at room temperature and mild stirring (100 rpm). Briefly, dry samples (50 mg) were weighed (wo) and then immersed in the swelling medium. At pre-determined time intervals, the swollen samples were withdrawn and weighed again (wt). Before this step, the excess liquid on the sample surface was carefully drained. The swelling degrees (in %) at different immersion times (t) were calculated as per eqn (3). This experiment was performed in triplicate for each sample.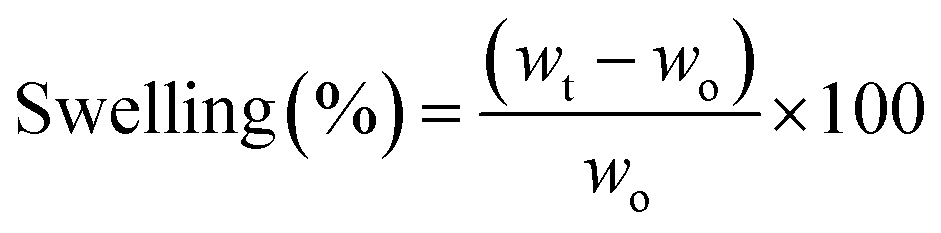 | (3) |
The effect of the pH and temperature of the swelling medium on the maximum water uptake was also investigated in separate experiments using similar procedures. Considering the pH effect, the pH of the swelling medium was adjusted to different values (2–12) using HCl (1.0 mol L−1) or NaOH (1.0 mol L−1) solutions. Samples were kept in these swelling media at room temperature for 24 h. The effect of the temperature was investigated using distilled water as the swelling medium thermostated at different temperatures (25–75 °C). Again, the samples were kept immersed under these conditions for 24 h. The maximum swelling at equilibrium for each experiment was calculated using eqn (3). Each experiment was performed in triplicate.
General experimental procedure for the synthesis of 1,4-disubstituted 1,2,3-triazoles
Aryl azides 1 (0.157 mmol), terminal alkynes 2 (0.157 mmol), the Cs/PVA-CuNP hydrogel (10 mol%, 1 mg of Cu per hydrogel sample) and a mixture of EtOH/H2O (1:1) (1.5 mL) were added to a glass vial. Then, the heterogeneous reaction mixture was sonicated for 6 h at room temperature in an ultrasonic bath. After the total disappearance of the starting materials, ethyl acetate (3 mL) was added and the reaction mixture was sonicated for additional 5 min. The reaction mixture was then separated from Cs/PVA-CuNP using a Pasteur pipette. This procedure was then repeated four times and the combined extracts were dried over MgSO4 and concentrated under vacuum. The crude products obtained were subsequently purified by column chromatography on silica gel using a mixture of hexane/ethyl acetate as the eluent to afford the desired products 3a–h. All the products were characterized by nuclear magnetic resonance (NMR) spectroscopy in a Bruker Avance DPX 400 spectrometer at 400 MHz (1H) and at 100 MHz (13C). All the NMR spectra were acquired using CDCl3 or DMSO-d6 as the deuterated solvent and tetramethylsilane (TMS) was used as the internal standard. The recovered Cs/PVA-CuNP sample was dried under vacuum and could be reused directly in subsequent reactions.
Results and discussion
Hydrogel characterization
The chemical structural properties of the as-prepared hydrogels (with and without Cu specimens) were examined by FTIR (Fig. 1a). As noticed, the Cs/PVA spectrum shows the characteristic bands of the starting materials (Cs and PVA) with some discrepancies.21,28,29 The broad band centered at 3423 cm−1 is associated with the O–H stretching of the hydroxyl groups of Cs and PVA. Moreover, this band overlaps with the band of the N–H stretching of the amino groups of Cs. The band at 1655 cm−1 is assigned to the imine bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) formed due to the crosslinking of Cs with glutaraldehyde.30 The band at 1559 cm−1 is assigned to N–H deformation (amide II band) while the band at 1412 cm−1 is attributed to C–N stretching.30 The bands in the 1150–1000 cm−1 region are ascribed to the C–O–C bending and C–OH stretching of Cs and the C–O stretching of PVA.31 In parallel, the Cu2+-containing hydrogel (Cs/PVA-Cu2+) spectrum shows an obvious modification as compared to the spectrum of the pristine hydrogel. In general, the different vibrational modes of the amino and hydroxyl groups in the Cs/PVA-Cu2+ spectrum were shifted to distinct wavenumber regions or they showed different absorbance intensities as a result of the interaction of these groups with Cu2+ ions. For instance, the band at 3423 cm−1 (O–H and N–H stretching modes) was shifted to 3445 cm−1 due to a reduction of intra and intermolecular H-bonds. The bands associated with N–H and C–N deformation modes and C–OH stretching decreased in intensity likely due to the interaction between the amino and hydroxyl groups with Cu2+ ions.21,32 These results were in good agreement with FAAS analysis, which demonstrated that the Cs/PVA hydrogel sample is able to adsorb and retain 1.0 ± 0.2 mg of Cu2+ (n ≈ 6). After the reduction of Cu2+ ions, band changes associated with the amino and hydroxyl groups of Cs/PVA are more pronounced as noticed in the Cs/PVA-CuNP spectrum. As demonstrated by other authors, these groups play a leading role in the stabilization of nanoparticles.33,34 Additionally, the new band at 746 cm−1 could be assigned to the loading effect of metallic Cu in the hydrogel matrix, confirming the successful formation of such particles.21
N) formed due to the crosslinking of Cs with glutaraldehyde.30 The band at 1559 cm−1 is assigned to N–H deformation (amide II band) while the band at 1412 cm−1 is attributed to C–N stretching.30 The bands in the 1150–1000 cm−1 region are ascribed to the C–O–C bending and C–OH stretching of Cs and the C–O stretching of PVA.31 In parallel, the Cu2+-containing hydrogel (Cs/PVA-Cu2+) spectrum shows an obvious modification as compared to the spectrum of the pristine hydrogel. In general, the different vibrational modes of the amino and hydroxyl groups in the Cs/PVA-Cu2+ spectrum were shifted to distinct wavenumber regions or they showed different absorbance intensities as a result of the interaction of these groups with Cu2+ ions. For instance, the band at 3423 cm−1 (O–H and N–H stretching modes) was shifted to 3445 cm−1 due to a reduction of intra and intermolecular H-bonds. The bands associated with N–H and C–N deformation modes and C–OH stretching decreased in intensity likely due to the interaction between the amino and hydroxyl groups with Cu2+ ions.21,32 These results were in good agreement with FAAS analysis, which demonstrated that the Cs/PVA hydrogel sample is able to adsorb and retain 1.0 ± 0.2 mg of Cu2+ (n ≈ 6). After the reduction of Cu2+ ions, band changes associated with the amino and hydroxyl groups of Cs/PVA are more pronounced as noticed in the Cs/PVA-CuNP spectrum. As demonstrated by other authors, these groups play a leading role in the stabilization of nanoparticles.33,34 Additionally, the new band at 746 cm−1 could be assigned to the loading effect of metallic Cu in the hydrogel matrix, confirming the successful formation of such particles.21
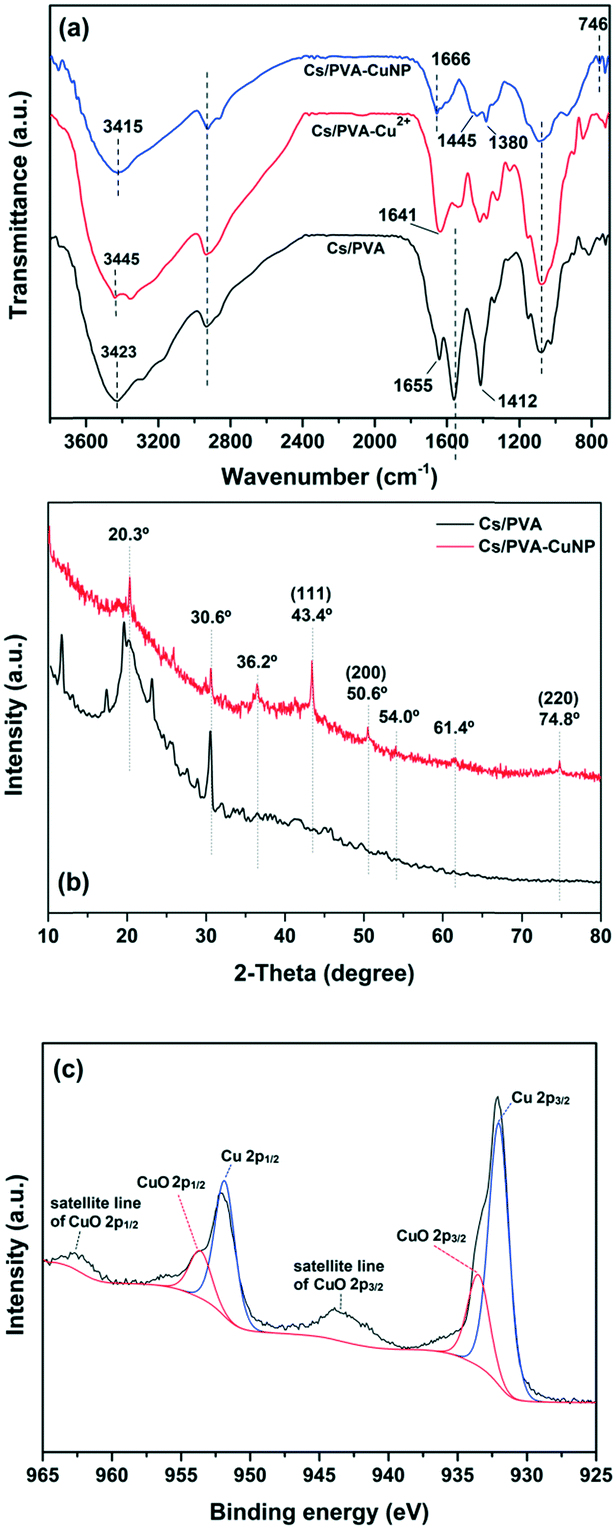 | ||
| Fig. 1 (a) FTIR spectra and (b) XRD patterns recorded from Cs/PVA and Cs/PVA-CuNP hydrogels. (c) XPS spectrum of Cu 2p of the Cs/PVA-CuNP hydrogel. | ||
XRD analysis was performed in order to identify the different crystalline phases present in Cs/PVA and Cs/PVA-CuNP hydrogels. As depicted in Fig. 1b, the XRD pattern of Cs/PVA shows some diffraction peaks at 2 theta ≈ 11.7°, 19.5°, 20.3°, and 30.6°. These diffraction peaks suggest that Cs/PVA shows a semi-crystalline structure likely due to the inter and intramolecular interactions that take place between Cs and PVA chains.35 These interactions were confirmed by FTIR analysis. On the other hand, the XRD patterns recorded for the CuNP-containing hydrogel exhibit an obvious decrease in the diffraction peaks associated with the Cs–PVA interaction probably due to the interaction between the CuNPs and hydrogel matrix. The presence of CuNPs in the hydrogel matrix disrupts the H-bonding between the amino and hydroxyl groups of Cs and PVA. Moreover, the appearance of three peaks at 2 theta ≈ 43.4°, 50.6°, and 74.8° corresponding to the (111), (200), and (220) crystalline planes of Cu was observed. These planes are in agreement with the face-centered cubic (FCC) phase of Cu [JCPDS 04-0836]36 and confirm that the CuNPs were successfully synthesized.
In addition, the diffraction pattern of Cs/PVA-CuNP reveals the presence of impurity peaks at 2 theta ≈ 36.2°, 54.0°, and 61.5° associated with CuO species.37 Considering these diffraction peaks assigned to metallic copper, the average particle size was estimated by using the Debye–Scherrer equation.38 Herein, the in situ synthesized CuNPs exhibited a particle size ranging from 3 to 7 nm. This particle size is comparable to or even smaller than other values reported in previous studies employing similar protocols.21,37,39
Fig. 1c shows the high-resolution XPS spectrum of Cu 2p recorded from the Cs/PVA-CuNP surface. As observed, the spectrum displays peaks corresponding to Cu 2p3/2 and Cu 2p1/2 spin–orbit components.37,40
These peaks were deconvoluted into two peaks each due to the presence of reduced Cu species and CuO species. The peaks centered at 932.1 and 951.8 eV are assigned to reduced Cu species while the peaks centered at 933.8 and 953.6 eV are due to CuO species.37 Moreover, the broad peaks observed between 965–960 eV and 947–940 eV are associated with the shake-up satellite lines of CuO 2p1/2 and CuO 2p3/2, respectively.40 Despite the detection of different valence states of copper, the main species is reduced Cu (90.1%). The presence of CuO species can be credited to the incomplete reduction of Cu2+ ions previously adsorbed on the Cs/PVA matrix. In addition, the possibility related to the re-oxidation of CuNPs due to their exposure to air should be considered as stated by Feng et al.41 No peaks corresponding to Cu2O specimens (529.6 eV) were observed in the XPS spectra of Cu 2p or O 1s (not shown here). The low CuO content in the Cs/PVA-CuNP sample as inferred from XPS analyses is in agreement with the XRD analyses that detected the tiny presence of copper oxidized phases as compared to metallic copper reflections.
The thermal stability of the Cs/PVA, Cs/PVA-Cu2+ and Cs/PVA-CuNP hydrogels was investigated by TGA/DTG analyses (Fig. S1a and b†). The TGA curves recorded for all the hydrogels show an initial stage of weight loss in the temperature range of 50 °C and 130 °C due to the elimination of moisture and volatile compounds. The TGA curve of Cs/PVA exhibits additional stages of weight loss assigned to the elimination of hydroxyl side groups of PVA (140–340 °C), thermal decomposition of the linear and crosslinked moieties of Cs (350–460 °C) and polyene backbone degradation of PVA (above 490 °C).21,42 Although the TGA curves recorded from the Cu-containing hydrogels (Cs/PVA-Cu2+ and Cs/PVA-CuNP) show some similarity, some clear discrepancies can be noticed. For instance, the DTG curve for Cs/PVA-Cu2+ exhibits a sharp peak centered at 237 °C suggesting that Cu2+ ions adsorbed on the hydrogel matrix decrease its thermal stability as compared to the pristine hydrogel (see Fig. S1b†). The presence of Cu2+ ions disrupts the inter and intramolecular H-bonds in the hydrogel matrix reducing the thermal energy required to degrade the Cs/PVA-Cu2+ sample.43 Conversely, after the reduction of Cu2+ ions, the functional groups available in the hydrogel (amino and hydroxyl groups) strongly interact with CuNPs in order to reduce their great surface energy. Therefore, the thermal stability of Cs/PVA-CuNP increases as compared to the other hydrogel samples. As mentioned in other studies, the interactions between nanoparticles and the functional groups of the hydrogel act as additional crosslinking points hampering the thermal degradation of the hydrogel matrix.44,45
The morphology of the Cs/PVA and Cs/PVA-CuNP hydrogels was investigated by means of SEM. The images taken from the hydrogel samples confirm that Cs/PVA has a rough and superporous structure with interconnected pores (Fig. 2a and b). This type of structure can be associated with the protocol used in the hydrogel synthesis.46 Preliminary experiments revealed that the hydrogels prepared using just Cs or PVA did not exhibit this morphology. The Cs/PVA sample possessed irregularly shaped pores with an average size of 177.9 ± 38.7 μm, while the total porosity of this sample was around 87%. The surface area examined by using the BET adsorption isotherm was 17 m2 g−1 for Cs/PVA. On the other hand, although Cs/PVA-CuNP still preserves the morphological characteristics of Cs/PVA (high porosity and interconnected pores), some discrepancies are observed (Fig. 2c and d). As evaluated, the average pore size decreased to 135.8 ± 22.9 μm while the surface area decreased to 12 m2 g−1 for Cs/PVA-CuNP. Moreover, the estimated total porosity of this sample decreases to 77%. This reduction of these morphological parameters can be attributed to the presence of CuNPs, which increases the crosslinking effect in the hydrogel matrix due to interactions that take place between these particles and the polymers. In addition, some granular materials are deposited on the pore surface of Cs/PVA-CuNP, which is not observed in the SEM image taken from Cs/PVA. Obvious visual discrepancies are also noticed between these two hydrogel samples (Fig. S1a and b†). For example, the pristine hydrogel shows a yellowish color proceeding from Cs while the CuNP-containing hydrogel shows a dark color typically observed for hydrogels embedded with metal nanoparticles.16
 | ||
| Fig. 2 SEM images recorded from Cs/PVA hydrogel (a and b) and Cs/PVA-CuNP hydrogel samples (c and d) at different magnifications. | ||
SEM/EDS-analysis was used to investigate the elemental composition of Cs/PVA-CuNP. According to the EDS spectrum recorded from this sample, the presence of signals related to the elements that compose the polymers (Cs and PVA) and signals confirming the presence of Cu in the hydrogel composition is observed (Fig. S3a†). Additionally, the EDS mapping image confirms that the CuNPs are homogeneously distributed throughout the sample (Fig. S3b†). The red dots represent the elemental map distribution of the CuNPs. This is a remarkable result since it confirms that the Cs/PVA matrix is an efficient device to immobilize CuNPs, preventing agglomeration and other undesirable results.
Swelling properties
The ability to absorb and retain large amounts of liquid is the most iconic property of hydrogels. This ability is particularly interesting in catalytic systems engineered from metal nanoparticles and hydrogels because the liquid uptake profile of this set will regulate the contact between the catalysts embedded in the hydrogel matrix and the starting materials.16 Herein, the liquid uptake capacity of the Cs/PVA and Cs/PVA-CuNP hydrogels was examined by a swelling experiment. The swelling curves presented in Fig. 3 reveal that at short immersion times (<15 min), both hydrogels showed fast swelling rates likely due to the hydrophilic groups distributed throughout the hydrogel surface. The large pore sizes presented by these hydrogels also favor the liquid uptake.47 After that, the swelling curves exhibited completely different patterns. For Cs/PVA, the swelling rate slowed down and the equilibrium was achieved close to 100 min after immersion. As time goes on, the liquid uptake process ends and the maximum swelling rate calculated for Cs/PVA is 382%. In contrast, the swelling curve of Cs/PVA-CuNP achieves the equilibrium more quickly (20 min of immersion) and the maximum swelling rate calculated for this sample is 208%. As demonstrated by FTIR and thermal analyses, CuNPs interact with the hydrophilic groups of the Cs/PVA matrix and, as a consequence, the availability of such groups to interact with water molecules is impaired. Additionally, the interaction between the particles and the hydrogel matrix acts as an extra crosslinking point reducing the expansion of the polymeric network and limiting the liquid diffusion into the hydrogel. These effects explain why the Cs/PVA-CuNP hydrogel shows lower swelling ability as compared to the pristine hydrogel. Other similar studies also report this effect caused by filler materials on hydrogel swelling properties.39,45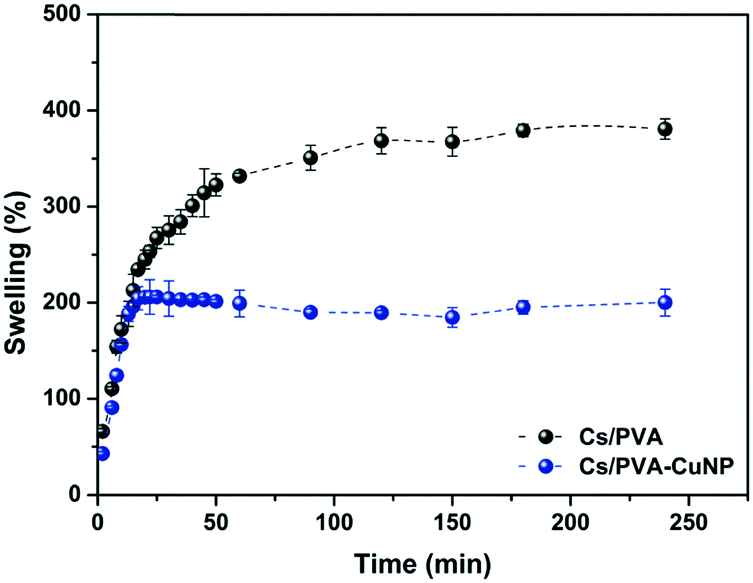 | ||
| Fig. 3 Swelling kinetics curves of Cs/PVA and Cs/PVA-CuNP hydrogels in distilled water at room temperature. | ||
The effects of the pH and temperature of the swelling medium were also investigated. Overall, these two factors had a clear influence on the maximum swelling of both hydrogels (Cs/PVA and Cs/PVA-CuNP). Under strong acidic conditions (pH < 2), both hydrogels showed a high swelling rate due to protonation of the amino groups of Cs, a typical behavior reported in different studies focused on Cs-based hydrogels.48,49 The positively charged groups in the hydrogel bulk phase cause the expansion of the polymeric network, increasing liquid uptake (Fig. S4a†). The increase in the pH of the medium results in a decrease of the swelling rate (4 < pH < 8) for both hydrogels likely due to the reduction of the protonated amino groups. In this stage, the expansion of the polymeric network is less pronounced and at neutral pH conditions, it no longer occurs. As a result, the amount of liquid that diffuses into the hydrogel matrix decreases considerably. Under alkaline pH conditions (pH ≥ 8), no significant variation in the swelling rate was observed for Cs/PVA. However, for the Cs/PVA-CuNP hydrogel, a decrease in the maximum swelling rate was observed at pH 12, which can be assigned to the low availability of functional groups on the hydrogel matrix to interact with water molecules due to interactions between these groups and the CuNPs. Importantly, the CuNP-containing hydrogel exhibited a low swelling ability as compared to the pristine hydrogel. These results strengthen the discussion that the CuNPs enhance the crosslinking density of the hydrogel matrix because of the coordination of the particles with the functional groups of Cs and PVA.
In parallel, the effect of the temperature of the swelling medium was also investigated for both hydrogel samples (Fig. S4b†). As noticed, an increase of temperature from 25 to 75 °C increases the maximum swelling rate of Cs/PVA, particularly from 55 °C. In general, the increase of the temperature causes an increment in the kinetic energy of the polymer chains that form the hydrogel matrix and disrupts some inter and intramolecular interactions among them.50 Thus, free hydrophilic groups can interact with the water molecules, increasing the swelling rate. In contrast, the effect due to the increase of the temperature of the swelling medium is more discrete for Cs/PVA-CuNP. Overall, slight variations were observed in the maximum swelling rates of the Cs/PVA-CuNP hydrogel with the increase of temperature. The additional crosslinking points that result from the coordination of CuNPs and the hydrogel matrix impair the mobility of the polymer chains even at high temperatures. At high temperature (75 °C), a slight decrease in the swelling rate was observed for the CuNP-containing hydrogel. It can be suggested that under such conditions, the catalytic nature of Cu damages the crosslinking that keeps the hydrogel matrix stable and also can promote the loss of fragments of polymer chains.42 These experiments reveal that both hydrogels show pH- and temperature-sensitive swelling, which are very attractive features particularly in catalysis applications.
Catalytic activity: synthesis of 1,4-disubstituted-1,2,3-triazoles
Cs/PVA-CuNP was subsequently examined as the catalytic system to promote azide–alkyne cycloaddition reactions. Inspired by the pioneering report of Huisgen11 on the thermal 1,3-dipolar cycloaddition of azides with terminal alkynes, a number of catalytic strategies employing transition metals, especially copper (CuAAC),51,52 have been employed to selectively synthesize 1,2,3-triazoles. However, the search for efficient methods using appropriate and environmentally catalytic systems for the selective synthesis of 1,2,3-triazoles still remains a challenge in synthetic chemistry. Thus, we started our studies using phenyl azide (1a) and phenylacetylene (2a) as model substrates in reactions carried out in an ultrasonic bath (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time (h) | Temperature (°C) | Yield of 3ab (%) |
| a Reactions were performed with phenyl azide 1a (0.157 mmol), phenylacetylene 2a (0.157 mmol), and Cs/PVA-CuNP (10 mol%, 1 mg of Cu per hydrogel sample) under an air atmosphere using 1.5 mL of solvent and an ultrasonic bath. b Yields are given for isolated products. c Different extraction processes (see in the text). d Reaction was performed without Cs/PVA-CuNP. e Reaction was performed with the chitosan hydrogel in the absence of copper. f n.d. = not detected. | ||||
| 1 |
tBuOH/H2O (1:1) |
6 | 50 | 65 |
| 2c |
tBuOH/H2O (1:1) |
6 | 50 | 83 |
| 3d |
tBuOH/H2O (1:1) |
6 | 50 | n.d.f |
| 4e |
tBuOH/H2O (1:1) |
6 | 50 | n.d.f |
| 5c | EtOH/H2O (1:1) |
6 | 50 | 92 |
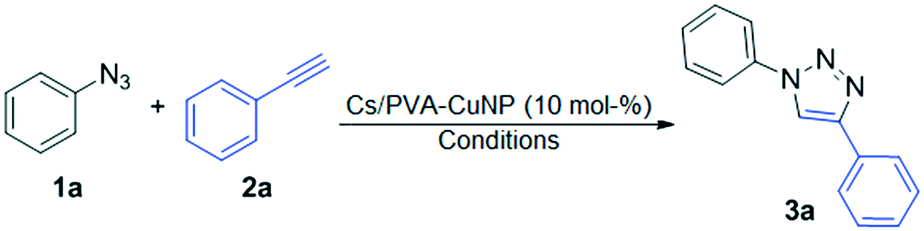
In the initial experiment, phenyl azide (1a), phenylacetylene (2a) and Cs/PVA-CuNP (10 mol%, 1 mg of Cu per hydrogel sample) were mixed together in a mixture of tBuOH and H2O (1:1) and sonicated under an air atmosphere for 6 h. This reaction was carried out in a vial and monitored by TLC until the total disappearance of the starting materials. After this time, the mixture was diluted in water (3 mL) and washed 3 times with ethyl acetate (3 × 3 mL). The obtained product was purified by flash chromatography on silica gel affording the desired compound 3a in 65% yield (Table 1, entry 1).
Subsequently, the reaction was carried out under identical conditions, although with a change in the extraction procedure. After the total disappearance of the starting materials, ethyl acetate (3 mL) was added and the reaction mixture was additionally sonicated for 5 minutes. The reaction mixture was then separated from Cs/PVA-CuNP using a Pasteur pipette. This procedure was then repeated four times and the combined extracts were dried over MgSO4 and concentrated under vacuum. Using this extraction procedure, the desired product 3a was obtained in 83% yield after flash chromatography on silica gel (Table 1, entry 2).
In order to prove the importance of the catalyst in this reaction, blank runs (in the absence of Cs/PVA-CuNP) were performed, providing no formation of the desired product 3a (Table 1, entry 3). We observed the same result when the reaction was performed using only the chitosan hydrogel in the absence of copper (Table 1, entry 4). In both cases, the starting materials were fully recovered (Table 1, entries 3 and 4). An excellent result was achieved using a mixture of EtOH/H2O (1:1) as the solvent, which gave 3a in 92% yield after purification (Table 1, entry 5). From Table 1, the optimum reaction conditions to obtain 1,4-diphenyl-1H-1,2,3-triazole (3a) are presented in entry 5, using 1a (0.157 mmol), 2a (0.157 mmol) and 10 mol% of Cs/PVA-CuNP in a mixture of EtOH and H2O (1:1) (1.5 mL) as the solvent, sonicated in an ultrasonic bath under air for 6 h.
After the reaction optimization, a study regarding the recovery and reuse of the catalyst was performed. In order to investigate the reusability of the catalyst, six consecutive reactions were performed using the same Cs/PVA-CuNP sample. Upon product 3a formation (1st reaction), we added ethyl acetate (3 mL) to the reaction mixture which was sonicated for 5 min, followed by separation from the catalyst using a Pasteur pipette. This procedure was repeated five times and the extracted products were dried over MgSO4 and concentrated under vacuum. The recovered Cs/PVA-CuNP was dried under vacuum and reused directly in the next reaction run. The progress of this reaction was monitored visually as the product crystallized out from the reaction medium. Fig. 4 illustrates each experimental step. The simplicity of this process should be highlighted.
 | ||
| Fig. 4 Photographic images taken from the reaction system after each step: (A) the as-prepared catalyst placed in the vial; (B) the catalyst in the presence of azide and alkyl substrates (t ≈ 0 h); (C) at the end of the cycloaddition reaction (t ≈ 6 h); and (D) the recovered and regenerated catalyst placed in the vial ready for the next reaction run. | ||
According to the experimental data collected after each reaction, it was observed that a good level of efficiency was maintained even after the catalyst was reused for six consecutive times. Overall, the 1,4-diphenyl-1H-1,2,3-triazole (3a) yield varied from 92% to 70% after five successive reaction runs, which suggests that the catalytic efficiency of Cs/PVA-CuNP is slightly affected after reuses (Fig. 5a). At the sixth reaction, the efficiency of the catalyst showed a noticeable reduction and, as consequence, the yield of compound 3a dropped to 55%.
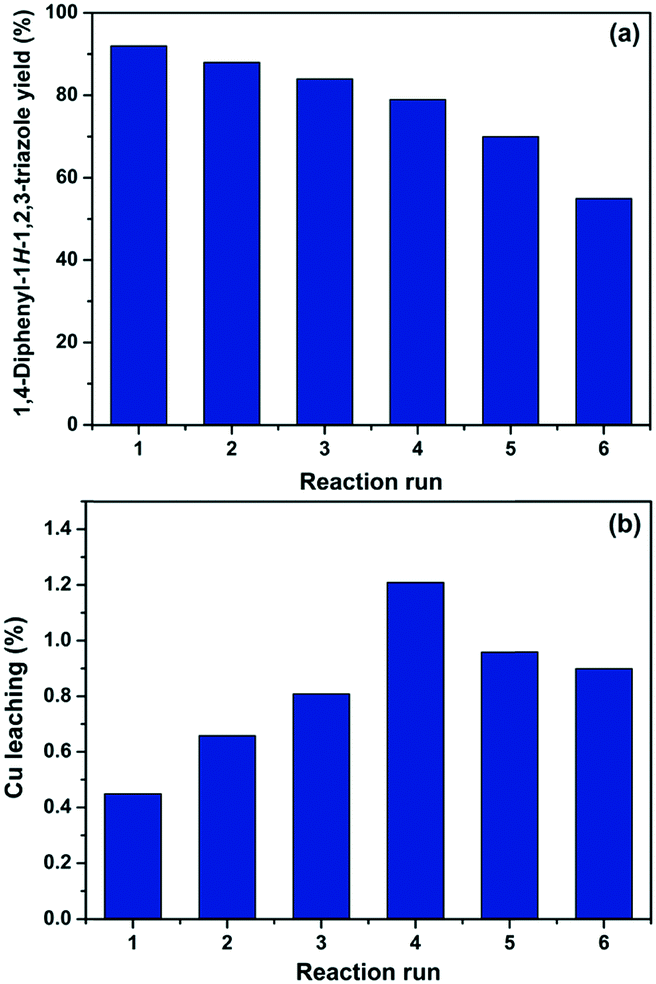 | ||
| Fig. 5 1,4-Diphenyl-1H-1,2,3-triazole (3a) yield (a) and percentage of copper leaching (b) after each consecutive reaction run. | ||
In order to investigate the reduction efficiency, copper leaching from Cs/PVA-CuNP into the reaction medium was measured. For this, just after each reaction, an aliquot of the reaction medium was collected and the amount of copper leached was quantified by FAAS. The percentage of copper leached from Cs/PVA-CuNP after each reaction run is depicted in Fig. 5b. It should be noticed that these percentages were calculated considering the initial copper content in the Cs/PVA-CuNP sample after each reaction. These results revealed that insignificant amounts (less than 1.2% w/w per reuse step) of copper were leached from the catalyst to the reaction medium. Overall, the Cs/PVA matrix showed a remarkable supporting ability for copper, losing ca. 4 wt% Cu after six reuses. Leaching of the metal into the reaction medium can result in some drawbacks related to product purification.53 Conversely, the loss of small amounts of copper may explain the decrease observed for the 3a yield after consecutive reaction runs performed. We hypothesize that the leaching can be attributed to the removal of copper species immobilized on the catalyst surface and/or due to the experimental procedures carried out for the recovery of the catalyst.
The scope of our methodology was subsequently extended to different aryl azides (1a–e) and terminal alkynes (2a–d) to check the versatility of Cs/PVA-CuNP in azide–alkyne cycloaddition reactions. The results presented in Table 2 unequivocally confirm that our experimental protocol worked well for different substituted azides and alkynes, providing the desired products in moderate to good yields.
|
|
||||
|---|---|---|---|---|
| Entry | Aryl azide 1 | Alkyne 2 | Product 3 | Yieldb (%) |
|
a Reactions were performed with aryl azides 1 (0.157 mmol), terminal alkynes 2 (0.157 mmol), Cs/PVA-CuNP (10 mol%, 1 mg of CuNP per sample) at room temperature for 6 h under an air atmosphere using a mixture of EtOH/H2O (1:1) (1.5 mL). All the reactions were performed using an ultrasonic bath.
b Yields are given for isolated products.
|
||||
| 1 |
 1a
1a
|
 2a
2a
|
 3a
3a
|
92 |
| 2 |
 1b
1b
|
 2a
2a
|
 3b
3b
|
84 |
| 3 |
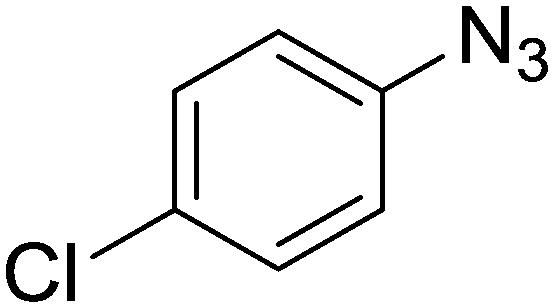 1c
1c
|
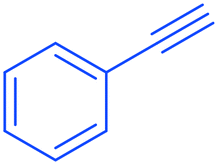 2a
2a
|
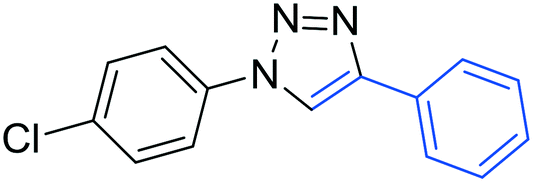 3c
3c
|
65 |
| 4 |
 1d
1d
|
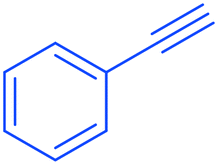 2a
2a
|
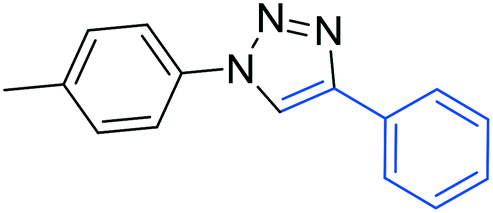 3d
3d
|
86 |
| 5 |
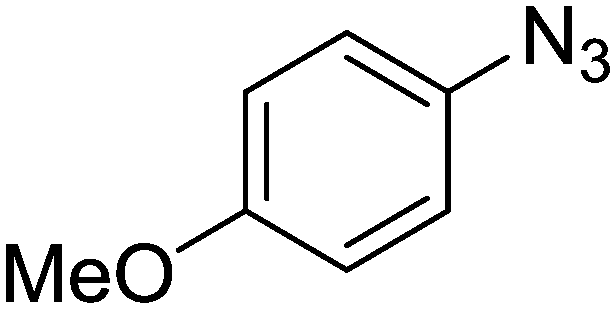 1e
1e
|
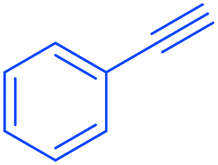 2a
2a
|
 3e
3e
|
85 |
| 6 |
 1a
1a
|
 2b
2b
|
 3f
3f
|
82 |
| 7 |
 1a
1a
|
 2c
2c
|
 3g
3g
|
54 |
| 8 |
 1a
1a
|
 2d
2d
|
 3h
3h
|
55 |
The reactions of phenylacetylene (2a) with aryl azides containing electron-withdrawing (EWG) substituents 1b–c and electron-donating groups (EDG) 1d–e at the aryl moiety yielded the corresponding products 3b–e in 65–86% yields (Table 2, entries 2–5). A little decrease in yields was observed using 4-chloro-phenyl azide as a substrate, which provided the desired product 3c in 65% yield (Table 2, entry 3). The reactivity of phenyl azide (1a) with different terminal aryl alkynes (2b–c), under otherwise identical reaction conditions, was subsequently investigated. In general, the reactions were found to be sensitive to the electronic conditions in the aryl ring of the terminal alkynes, in which an alkyne containing an EDG gave a better yield when compared to an alkyne containing an EWG on the aromatic ring (Table 2, compare entry 6 with 7). The reaction performed with 1-hexyne (2d) furnished exclusively the respective 4-butyl-1-phenyl-1H-1,2,3-triazole (3h) in 55% yield (Table 2, entry 8).
Taken together, these findings further confirm that Cs/PVA-CuNP is a promising reusable catalyst for the synthesis of 1,2,3-triazoles via cycloaddition reactions.
Conclusions
Copper nanoparticles (CuNPs) were synthesized and stabilized in a chitosan/poly(vinyl alcohol) (Cs/PVA) hydrogel matrix using a simple two-step adsorption/reduction protocol. As assessed by different techniques, the CuNPs embedded into the Cs/PVA matrix influenced the thermal, morphological and swelling properties of the final materials as compared to the Cs/PVA hydrogel. XRD and XPS analyses confirmed the metallic nature of the CuNPs, with nanoparticle sizes estimated to be in the 3–7 nm range. SEM/EDS analyses demonstrated that the Cs/PVA-CuNP hydrogel exhibits a superporous structure and the CuNPs are homogeneously distributed throughout the hydrogel surface. Catalytic experiments proved that Cs/PVA-CuNP is an excellent catalyst for the synthesis of 1,2,3-triazole using phenyl azide and phenylacetylene as model substrates (92% yield). Reuse studies revealed that this original CuNP-containing catalyst can be employed in at least 5 consecutive cycloaddition reactions without a significant loss of efficiency. FAAS analysis demonstrated that the Cu leaching from the hydrogel matrix was minor (ca. 4 wt%) during these consecutive reactions. Finally, the Cs/PVA-CuNP hydrogel also efficiently catalyzed the cycloaddition reactions involving different substituted azide and alkyne precursors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support and scholarships from the Brazilian agencies CNPq (Grants. number 310943/2016-7 and 305974/2016-5) and FAPERGS (PRONEM – Grant. number 16/2551-0000240-1). CNPq is also acknowledged for the fellowship to D. A. and A. R. F. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. The publication has been prepared with support from RUDN University Program 5-100.Notes and references
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed
.
- I. T. Horvath, Chem. Rev., 2018, 118, 369–371 CrossRef CAS PubMed
- C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed
- Y. X. Liu, G. F. Zhao, D. S. Wang and Y. D. Li, Natl. Sci. Rev., 2015, 2, 150–166 CrossRef CAS
- N. Li, P. X. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS
- S. Chaturvedi, P. N. Dave and N. K. Shah, J. Saudi Chem. Soc., 2012, 16, 307–325 CrossRef CAS
- L. Zhu, X. L. Guo, Y. Y. Liu, Z. T. Chen, W. J. Zhang, K. B. Yin, L. Li, Y. Zhang, Z. M. Wang, L. T. Sun and Y. H. Zhao, Nanotechnology, 2018, 29, 1–12 Search PubMed
- P. C. Chen, A. P. Periasamy, S. G. Harroun, W. P. Wu and H. T. Chang, Coord. Chem. Rev., 2016, 320, 129–138 CrossRef
- N. Barot, S. B. Patel and H. Kaur, J. Mol. Catal. A: Chem., 2016, 423, 77–84 CrossRef CAS
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 33 Search PubMed
- M. Bakherad, F. Rezaeimanesh and H. Nasr-Isfahani, ChemistrySelect, 2018, 3, 2594–2598 CrossRef CAS
- S. Maddila, R. Pagadala and S. B. Jonnalagadda, Lett. Org. Chem., 2013, 10, 693–714 CrossRef CAS
- P. C. Ray, H. T. Yu and P. P. Fu, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2009, 27, 1–35 CrossRef CAS PubMed
- C. Parmeggiani, C. Matassini and F. Cardona, Green Chem., 2017, 19, 2030–2050 RSC
- N. Sahiner, Prog. Polym. Sci., 2013, 38, 1329–1356 CrossRef CAS
- Y. H. Hou, S. Ogasawara, A. Fukuoka and H. Kobayashi, Catal. Sci. Technol., 2017, 7, 6132–6139 RSC
- S. Zhang, Y. R. Lee, H. J. Jeon, W. S. Ahn and Y. M. Chung, Mater. Lett., 2018, 215, 211–213 CrossRef CAS
- Z. Y. Xiang, Y. Chen, Q. G. Liu and F. C. Lu, Green Chem., 2018, 20, 1085–1094 RSC
- M. Tukhani, F. Panahi and A. Khalafi-Nezhad, ACS Sustainable Chem. Eng., 2018, 6, 1456–1467 CrossRef CAS
- J. F. de Souza, G. T. da Silva and A. R. Fajardo, Carbohydr. Polym., 2017, 161, 187–196 CrossRef CAS PubMed
- K. Karakas, A. Celebioglu, M. Celebi, T. Uyar and M. Zahmakiran, Appl. Catal., B, 2017, 203, 549–562 CrossRef CAS
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS
- S. Brase, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef CAS PubMed
- E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368 CrossRef CAS
- G. Labbe, Chem. Rev., 1969, 69, 345–362 CrossRef CAS
- Q. L. Loh and C. Choong, Tissue Eng., Part B, 2013, 19, 485–502 CrossRef CAS PubMed
- N. O. Alves, G. T. da Silva, D. M. Webber, C. Luchese, E. A. Wilhem and A. R. Fajardo, Carbohydr. Polym., 2016, 148, 115–124 CrossRef CAS PubMed
- I. K. D. Dimzon and T. P. Knepper, Int. J. Biol. Macromol., 2015, 72, 939–945 CrossRef CAS PubMed
- C. Liu, E. Thormann, P. M. Claesson and E. Tyrode, Langmuir, 2014, 30, 8878–8888 CrossRef CAS PubMed
- E. G. Crispim, J. F. Piai, A. R. Fajardo, E. R. F. Ramos, T. U. Nakamura, C. V. Nakamura, A. F. Rubira and E. C. Muniz, eXPRESS Polym. Lett., 2012, 6, 383–395 CrossRef CAS
- D. L. Kong, N. Wang, N. Qiao, Q. Wang, Z. Wang, Z. Y. Zhou and Z. Q. Ren, ACS Sustainable Chem. Eng., 2017, 5, 7401–7409 CrossRef CAS
- M. Adlim, M. Abu Bakar, K. Y. Liew and J. Ismail, J. Mol. Catal. A: Chem., 2004, 212, 141–149 CrossRef CAS
- A. Kyrychenko, D. A. Pasko and O. N. Kalugin, Phys. Chem. Chem. Phys., 2017, 19, 8742–8756 RSC
- L. Y. Lu, F. B. Peng, Z. Y. Jiang and J. H. Wang, J. Appl. Polym. Sci., 2006, 101, 167–173 CrossRef CAS
- M. Gholinejad, F. Saadati, S. Shaybanizadeh and B. Pullithadathil, RSC Adv., 2016, 6, 4983–4991 RSC
- D. E. Diaz-Droguett, R. Espinoza and V. M. Fuenzalida, Appl. Surf. Sci., 2011, 257, 4597–4602 CrossRef CAS
- B. D. Hall, D. Zanchet and D. Ugarte, J. Appl. Crystallogr., 2000, 33, 1335–1341 CrossRef CAS
- M. E. Villanueva, A. M. D. Diez, J. A. Gonzalez, C. J. Perez, M. Orrego, L. Piehl, S. Teves and G. J. Copello, ACS Appl. Mater. Interfaces, 2016, 8, 16280–16288 CrossRef CAS PubMed
- S. P. Prakash and K. R. Gopidas, ChemistrySelect, 2016, 1, 4803–4813 CrossRef CAS
- X. Feng, P. P. Lv, W. Sun, X. Y. Han, L. F. Gao and G. X. Zheng, Catal. Commun., 2017, 99, 105–109 CrossRef CAS
- N. A. El-Zaher and W. G. Osiris, J. Appl. Polym. Sci., 2005, 96, 1914–1923 CrossRef CAS
- X. P. Xia, S. Z. Cai and C. S. Xie, Mater. Chem. Phys., 2006, 95, 122–129 CrossRef CAS
- K. J. De France, T. Hoare and E. D. Cranston, Chem. Mater., 2017, 29, 4609–4631 CrossRef CAS
- F. L. Zhao, D. Yao, R. W. Guo, L. D. Deng, A. J. Dong and J. H. Zhang, Nanomaterials, 2015, 5, 2054–2130 CrossRef CAS PubMed
- C. M. Hassan and N. A. Peppas, Macromolecules, 2000, 33, 2472–2479 CrossRef CAS
- O. Okay, Prog. Polym. Sci., 2000, 25, 711–779 CrossRef CAS
- B. C. Melo, F. A. A. Paulino, V. A. Cardoso, A. G. B. Pereira, A. R. Fajardo and F. H. A. Rodrigues, Carbohydr. Polym., 2018, 181, 358–367 CrossRef CAS PubMed
- B. Mandal and S. K. Ray, Mater. Sci. Eng., C, 2014, 44, 132–143 CrossRef CAS PubMed
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2693 CrossRef CAS PubMed
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed
- C. R. Tubio, J. Azuaje, L. Escalante, A. Coelho, F. Guitian, E. Sotelo and A. Gil, J. Catal., 2016, 334, 110–115 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy01796d |
| This journal is © The Royal Society of Chemistry 2019 |