
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome
Luuk
Kortekaas
and
Wesley R.
Browne
 *
*
Molecular Inorganic Chemistry, Stratingh institute for Chemistry, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands. E-mail: w.r.browne@rug.nl
First published on 31st May 2019
Abstract
Spiropyrans have played a pivotal role in the emergence of the field of chromism following their discovery in the early 20th century, with almost ubiquitous use in materials applications especially since their photochromism was discovered in 1952. Their versatility continues to lend them to application in increasingly diverse fields not least due to recent discoveries of properties that have expanded their utility extensively. This review provides an overview of their rich history and highlights the contemporary relevance of the spiropyrans.
 Luuk Kortekaas | Luuk Kortekaas received his PhD in Chemistry from the University of Groningen under supervision of Prof. W. R. Browne and Prof B. L. Feringa. Since February 2019, he holds a Humboldt Postdoctoral Fellow in the group of Prof. Bart Jan Ravoo at the University of Münster's Center for Soft Nanoscience. His research interests lie with functional electrode surfaces, multiresponsive molecular systems, dynamic supramolecular adhesion, redox and photochemically switchable polymers, as well as emerging electrochemical applications. |
 Wesley R. Browne | Wesley Browne completed his PhD thesis on the photochemistry and photophysics of Ru(II) polypyridyl complexes at Dublin City University (2002), in the group of Prof J. G. Vos. Following postdoctoral positions at Queen's University Belfast and the University of Groningen, in 2008, he joined the faculty at the University of Groningen as assistant professor and is currently full professor and Chair of Molecular Inorganic Chemistry at the Stratingh Institute for Chemistry. His research interests are in the application of spectroscopy, and especially Raman spectroscopy and electrochemistry to (bio)inorganic catalysis and molecular based materials science. |
Introduction
The field of molecular switches continues to hold great promise in the control of diverse properties and functions in the macroscopic world.1–4 Despite over 100 years of study, new applications both in research and materials (not least in light responsive sun glasses) emerge owing to the wide and diverse range of building blocks available.5–13 The known photochromes include a range of switches that have been proven versatile to chemical modification, enabling changes to, enhancement of, and sometimes even completely novel, functionality. Several classes of molecular switch have featured prominently such as the azobenzenes,14,15 dithienylethenes16,17 and spiropyrans.18 Interfacing molecular switches with the macroscopic world and especially optoelectronic devices has focused heavily on surface immobilization and incorporation in polymer films.19,20 Furthermore, immobilization at electrode surfaces adds the immediate and complete control over redox properties, making electropolymerisable molecular switches an interesting subclass that, if designed correctly, can provide more and new ways to influence the macroscopic function through microscopic manipulation.21 Of the classes of molecular switches available, spiropyrans form a class that exhibits an extraordinarily wide range of responsivity to photons, redox changes, and changes in temperature and pH.13,18,22,23 Here, the characteristics and fundamental properties (i.e., thermodynamics, photophysics, pH and redox response) of spiropyrans in solution and on surfaces will be systematically outlined to provide a comprehensive overview of the mechanisms involved in their switching. The goal is to highlight the range of chromisms that spiropyrans provide and to demonstrate how, in applications, these chromisms should not be treated in isolation. Indeed their combination allows for rapid expansion of the basic set of properties and responses available in applications.Discovery of the spiropyran structure
The versatility of the spiropyran class of photochromes we know today was not immediately apparent when their basic structures were first described. The non-photochromic spiropyrans that preceded the multifunctional indoline–pyran hybrid were reported first by Decker in 1908, who coined the term for the newly reported chiral centre of the double pyran a ‘spiropyran’ (Scheme 1).24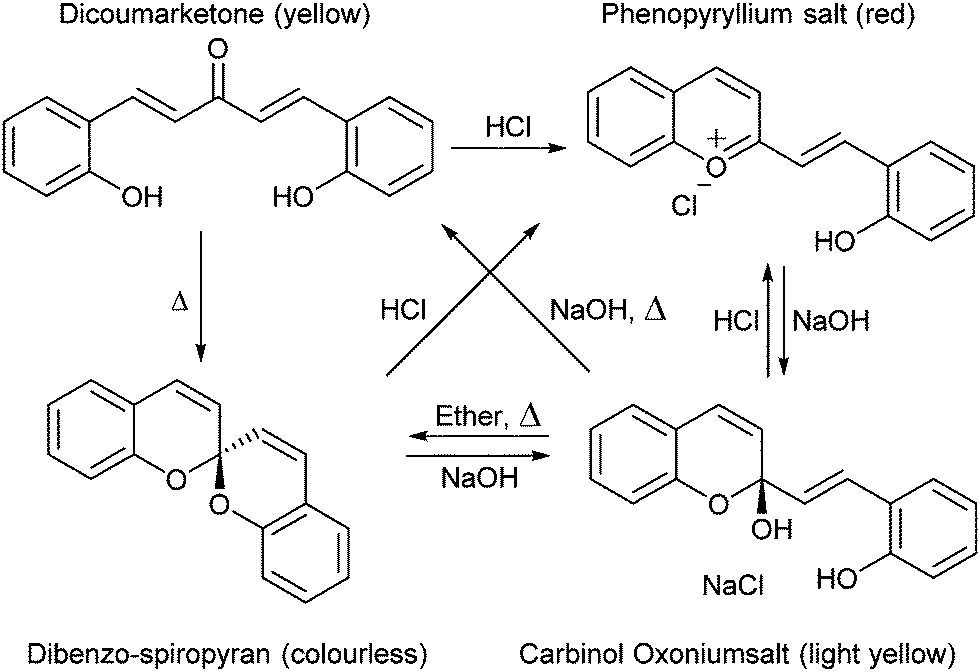 | ||
| Scheme 1 The first report of spiropyran discovered as an anomaly in the synthesis of coumarin derivatives. | ||
Over the following decades many variations of the acidochromic spiropyran were reported, including α- and β-naphthospiropyrans, and the influence of substituents on the heterocycles was explored (vide infra). Though the discovery of the thermochromic spiropyrans attracted early interest, it was the photochromism of a particular form, prepared by condensation of simple Fischer bases (Scheme 2, named for the versatile indole synthesis described by Emil Fischer25) with salicylaldehyde, that led to the steep rise in interest and the immediate association of the term with its base structure.26
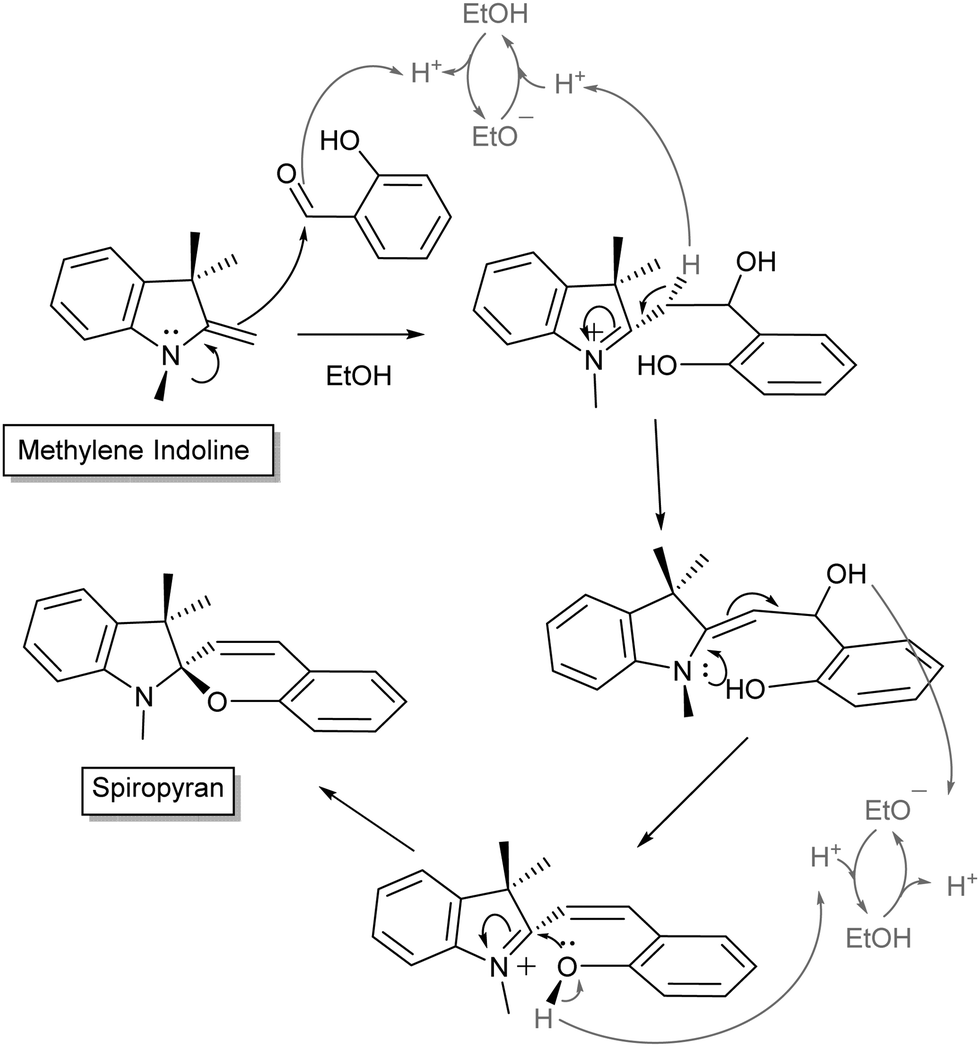 | ||
| Scheme 2 General mechanism for Fischer's indole condensation with salicylaldehyde to form indolinobenzospiropyrans. Variations often include modification of the N-alkyl sidechain or the pyran. Note also that the use of salicylaldehyde in excess promotes formation of doubly condensed spiropyrans.28,29 | ||
Discussion of the non-photochromic spiropyrans provides a background to the reactivity of spiropyrans as a whole. For example, several thermochromic spiropyrans, such as xanthonaphthospiropyran and benzoxanthospiropyran, were reported to be acidochromic depending on the pKa of the corresponding acid.27 As indeed turns out for spiropyrans in general, using this behaviour relies on differences in pKa (vide infra).
Ultimately, however, the widespread interest in the photochromic spiropyrans since their discovery over 65 years ago manifests a bias towards this specific form, and not without reason. The extensive functionality of this class of photochromes is well described in the literature but nevertheless new properties and behaviour are still being reported.30–35 In this review, the functionalities of photochromic spiropyrans reported to date are discussed to provide as full an overview of their versatility as we can and to establish a foundation from which even more of their properties may be revealed.
Thermochromism of spiropyrans
The colour change exhibited by spiropyrans at elevated temperature (vide supra, Scheme 1) was noted in the earliest reports,24 albeit complete thermal reversibility upon cooling was not noted until 1926, when three independent accounts of di-β-naphthospiropyran derivatives appeared concurrently.37 The change in visible colouration made this new class of colour-changing compounds attractive, as they could be studied without the use of optical equipment, their thermochromism becoming perhaps the best studied of thermochromic compounds by the early 1960s.37 Though the original dibenzospiropyran did not exhibit a reversible colour change upon heating, thermochromism was observed commonly in its naphtho- and indolino-derivatives (Scheme 3).†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36,37
36,37
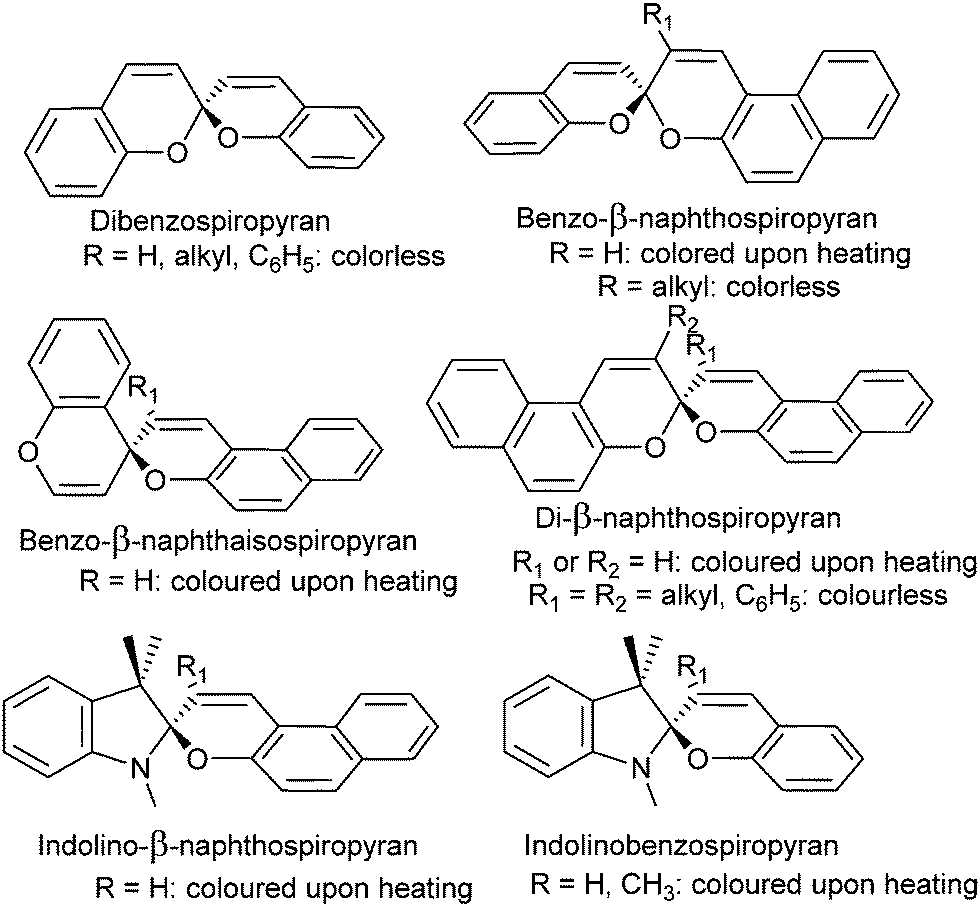 | ||
| Scheme 3 Variations of thermo- and acidochromic spiropyrans developed initially, the response of α-naphthospiropyrans resembles that of their β-isomer.36 Similarly, the isospiropyran class of thermochromes were developed further in derivatives such as di-β-naphtho, di-α,β-naphtho, xantho-, thiaxantho- and acridinospiropyran, with similar thermochromic properties reported.37 | ||
The observation by Wizinger38 that the thermochromism of benzopyrans could be accessed by introducing an auxiliary indolino group to polarize the spiro-centre was seminal. General trends in the quickly broadening family of spiropyrans were swiftly established, such as the colour changes upon heating, and salt formation with acids, which will be discussed in more detail below under acidochromism. Elevated temperatures generated an increasing amount of the coloured form and the mechanism for the thermal ring-opening and accompanying colouration of these spiropyrans was anticipated to involve ionization of the pyran and its counterpart across the spiro-centre (Scheme 4)39 as a radical dissociation pathway was discounted at that time, although Heller et al. reported a weak to medium EPR signal for merocyanine.40
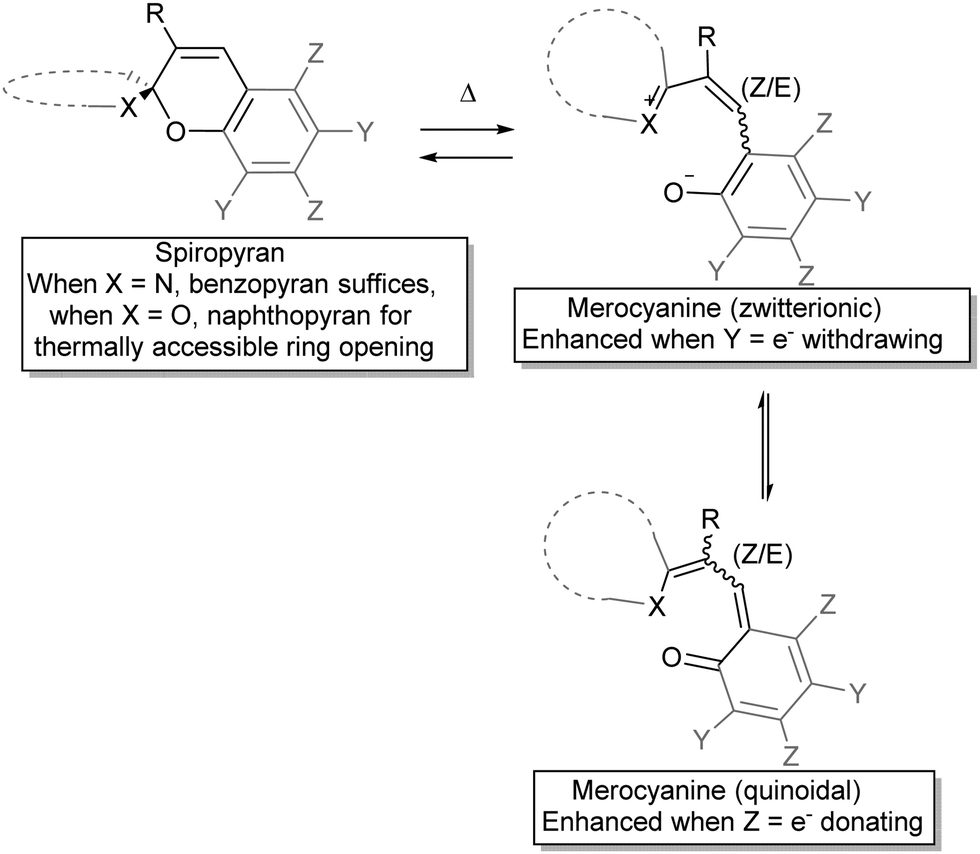 | ||
| Scheme 4 Mechanism for the observed thermochromism in spiropyrans. At elevated temperatures heterocyclic C–O bond cleavage occurs, with zwitterionic and quinoidal resonance forms contributing to merocyanine stabilization. Appropriate substitution at the pyran ring can “stabilize” either resonance form, while substitution at the R-position can hinder the ring opening by steric obstruction in the preferably planar merocyanine form. | ||
The zwitterionic and the quinoidal forms each offer a better description of the true structure depending on molecular structure and conditions, including the nature of the heterocycle, the substituents on the pyran ring and solvent interactions, although experimental properties generally lean towards the ionic form.37 The balance in charge delocalization greatly contributes to the stability of the open form and therefore substituents can have a pronounced influence. The reduced tendency towards ring opening in benzospiropyrans compared to naphthospiropyrans supports this model as formation of the quinoidal structure requires loss of aromaticity in the former, reflected also in the oxidation potential of o-benzoquinone (0.833 V) and that of 6-naphthoquinone (0.576 V).41 Thus, for thermochromism to occur it was established that one of the pyran rings has to be at least a naphthopyran, unless the structure of the heterocyclic ring contributes significantly to the stabilization of delocalized charge in the conjoined spiro-centre, as seen in the indolinobenzospiropyrans reported by Wizinger38 and popularized by Fischer26 (vide infra).
The second major aspect that determines ring-opening, therefore, is the electron releasing ability of the heterocyclic ring (dotted in Scheme 4), which can be complemented by stabilization of the phenolate by appropriate substituents on the pyran ring (Y in Scheme 4).37,38,42 Last but not least, both steric substitution and the presence of an acidic hydrogen at the R-position (Scheme 4) have also been shown to dictate the driving force of isomerization, as substitution of the C3′ hydrogen obstructs ring opening.43 Though the proposed steric blocking of concurrent planarization became the accepted rationalization of this effect eventually, an initially proposed H-bonding stabilizing interaction between the C3′–H and phenol in the ring-open form44 turns out to play a pivotal role as discovered more recently (vide infra).45 The compounds 3,3′-dimethylene- and 3,3′-trimethylenedi-β-naphthospiropyran, with fusing of the naphthopyrans at their 3-positions, despite being disubstituted show ring-opening driven by imposition of a planar orientation in the closed form as well, although 3,3′-tetramethylenedi-β-naphthospiropyran inhibits ring opening due to its restored flexibility.41,44 In parallel this behaviour is also reflected in the preceding stage, as the synthesis of spiropyrans can also be hindered by unfavourable substitution patterns, which push the reactive centre out of plane (Scheme 5), showing the same response to in-plane confinement.
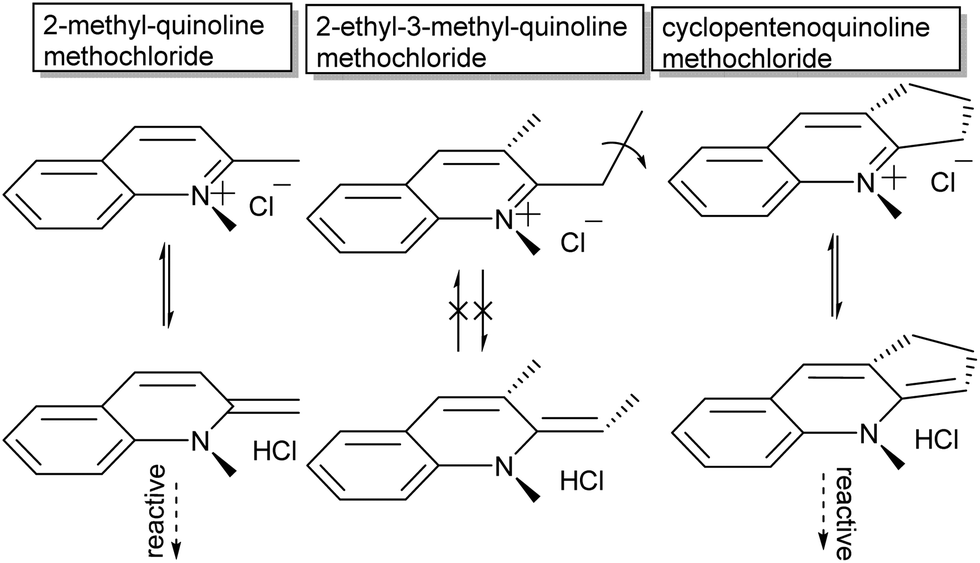 | ||
| Scheme 5 Sterics in spiropyrans influence ease of ionization but also impact their synthesis, in particular conformational restrictions at the reactive centre. Attachment of secondary carbon atoms at the 2-position causes twisting out of plane in their parent Fischer base tautomer thus creating steric hindrance in their reactive form, unless the nature of the substituent forces it into the same plane to begin with. | ||
Thus, the aforementioned energetic favourability of ring opening and the subsequent access to sufficient resonance structures unhindered by sterics and stabilized by intramolecular interactions play a key role in the diversity of conformations and isomers the spiropyran adopts. Considering this driving force for the merocyanine form to adopt a planar structure, though, further rotations around several of its bonds conceivably provide energetically distinct, yet thermally accessible, states. A hint that this was the case appears in a footnote in the report by Koelsch in 1951,41 in which a thermal barrier to ring-closing was demonstrated by plunging a hot deep purple solution of dinaphthospiropyran into dry ice and acetone. A pale blue colour persisted at this temperature, but faded to colourless rapidly when warmed to room temperature (Scheme 6).
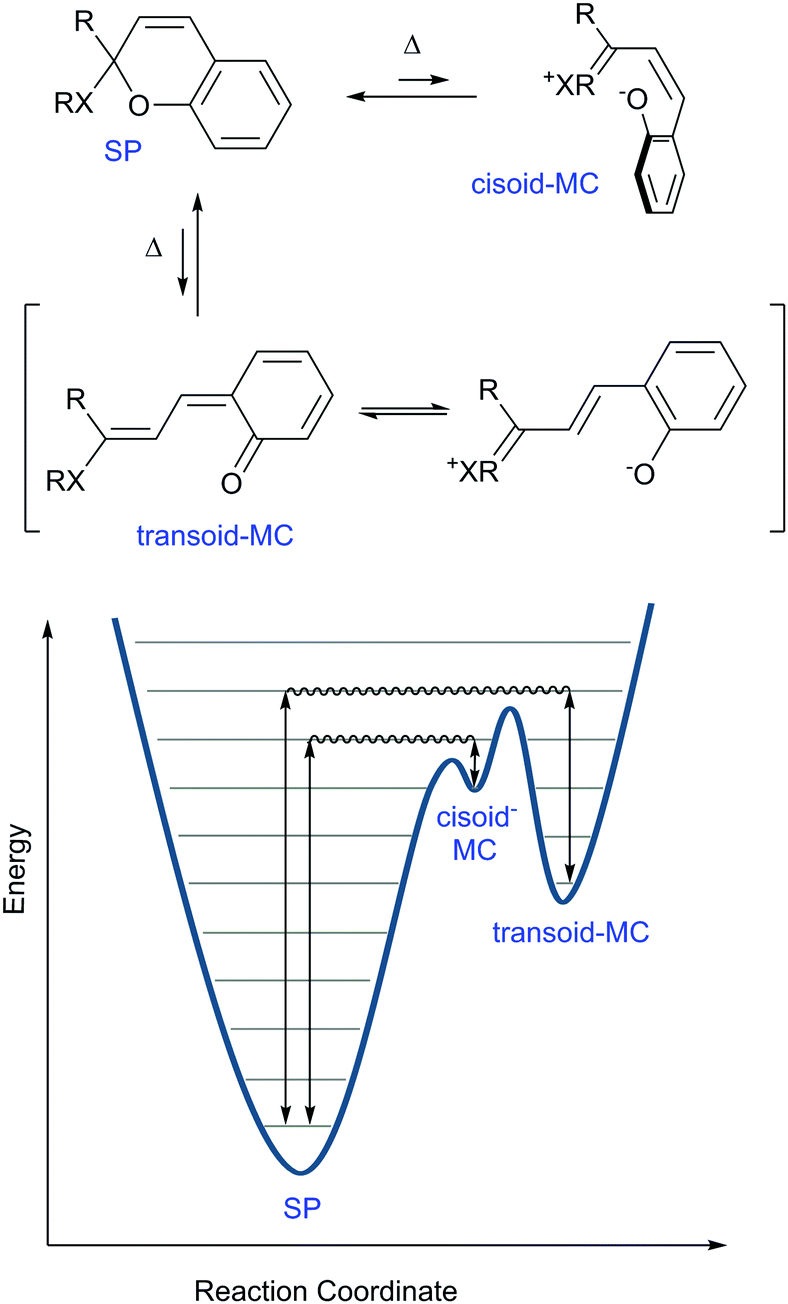 | ||
| Scheme 6 Simplified reaction coordinate diagram for thermochromism in spiropyrans. The nature of X lowers the height of the energy barriers in the order N > O ≫ C, as do the electron releasing ability of the R groups contribute to this lowering in energy. Appropriate substitution of the pyran ring can lower the ground state energy of the MC form (see Scheme 4) and can thereby also cause the barrier to decrease. Note that the separation of charge contributes substantially to the energies of the cisoid and transoid forms. | ||
Only two years later, Hirshberg and Fischer reported the appearance of variously coloured isomers at temperatures starting from 105 K.46 Excitation with UV-light allowed photo-induced access to the merocyanine form (vide infra) of dibenzo-, benzo-β-naphtho- and di-β-naphthospiropyran, albeit at this temperature only a few stable isomers are “frozen-in”47 by the photoexcitation manifested in the unusual colour. Subsequent thermochromism was observed upon raising the temperature gradually, providing thermal access to additional isomers at 123 K and again at 163 K with large changes in colour, and ultimately fading to colourless again when brought to room temperature. These data support the existence of energetically distinct cisoid and transoid isomers, where at any given temperature a weighted average of all thermally accessible coloured states is obtained. Moreover, the higher the temperature, the more isomers that become accessible, leading to the earlier collectively observed further colour changes for some spiropyrans close to solvent boiling points.41 There are eight conformers/isomers that constitute energetically distinct merocyanine forms, identified commonly by combined Z/E configuration about the α, β and γ bonds (Scheme 7).
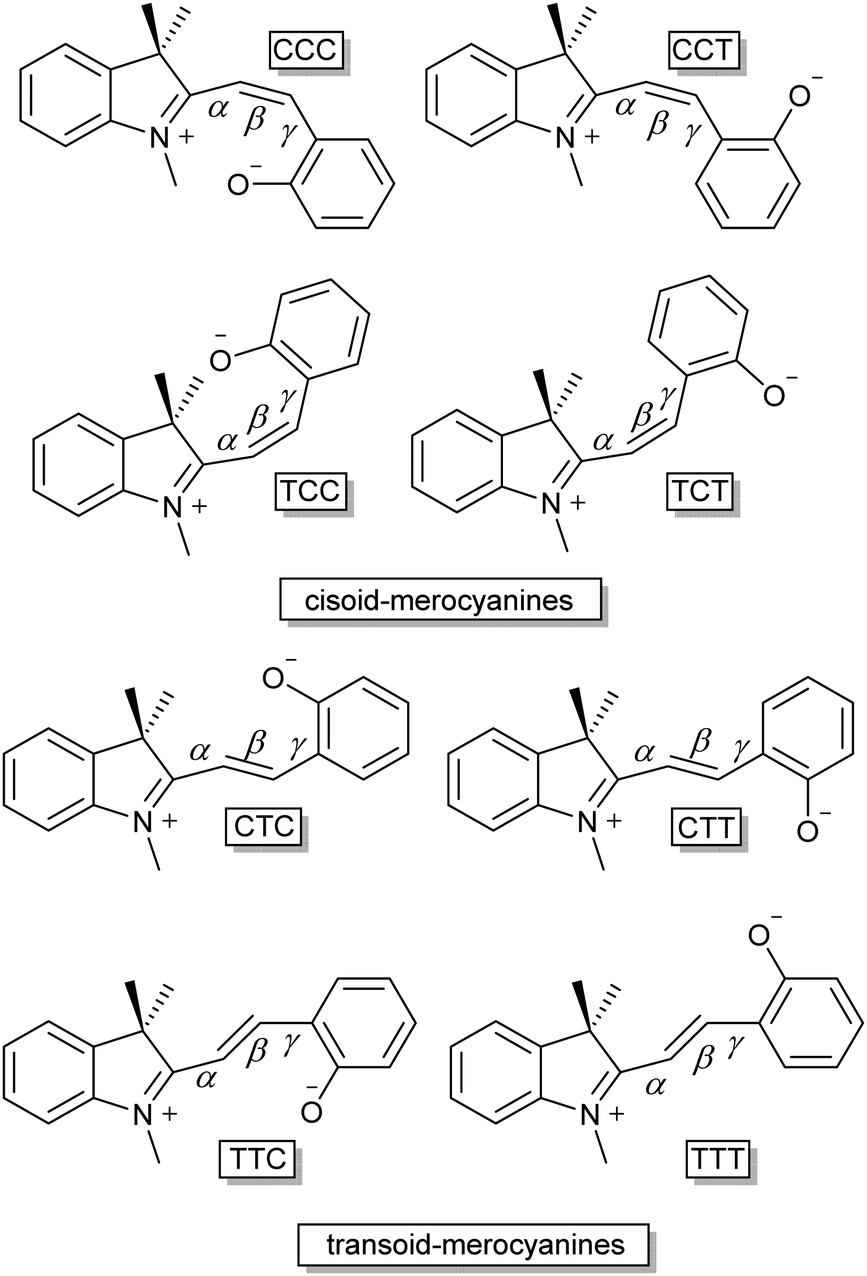 | ||
| Scheme 7 The eight distinct isomers of the ring open merocyanine form. The three lettered nomenclature follows the E/Z orientation over the three adjoining bonds (α, β and γ) between the Cspiro and the phenolate, the double bond (β) governs the categorization into the cisoid (Z) and transoid (E) isomers. | ||
The conformations and the internal rotations involved in converting between the isomers can result in restrictions to the photochemical and thermal isomerizations.48,49 Steric constraints can be controlled environmentally also, e.g. in MOFs, COFs (vide infra), and by solvent, and indeed the kinetics of isomerization are affected by spatial arrangement as well as the polarity of the immediate environment.
Solvatochromism of spiropyrans
In addition to thermal access to several coloured forms, solvent can affect the appearance and nature of their absorption spectra, i.e., solvatochromism, which was noted already in the early 20th century.36 In the potential energy surface (PES) above (Scheme 6) the cisoid and transoid isomers are considered as a single energy minimum since they are close in energy. The observed solvatochromism is consistent with such a landscape,50 as the progressive colouration of spiropyrans with increasing solvent polarity is due to the decrease in the energy of, and thermal barrier to, the polar merocyanine forms. This phenomenon occurs primarily through differences in solvation energies and possible H-bonding interactions and is analogous to lowering in energy by structural modification.42 Typically, this lowering of barrier to ring-opening is accompanied by a blue-shift of up to 40 nm in the merocyanine absorption or even the appearance of a shoulder, indicating the appearance of additional transoid forms.37 Both the changing barrier between the closed and open form and the shift in absorption bands can be rationalized by the increase in energy gap as a result of differences in stabilization of the various ground and excited states (Scheme 8).45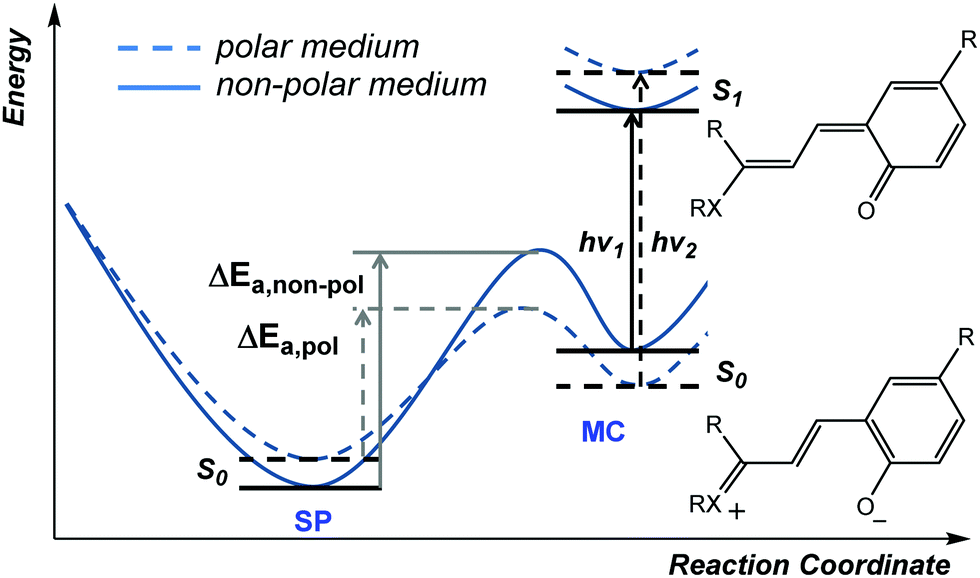 | ||
| Scheme 8 Solvatochromism of merocyanines. For convenience, the cisoid and transoid isomers are considered as a single minimum, although the lowest energy stable isomers are in a transoid configuration. | ||
The ground state of the merocyanine resembles most closely a zwitterionic form in benzospiropyrans, whereas the excited state is better represented by a quinoidal structure.45 Hence, increasing solvent polarity destablises the excited state and stabilises the ground state increasing the S0–S1 gap, manifested in a blue-shift. The measured dipole moments of merocyanines have been shown to decrease upon excitation, which is fully in line with the proposed structural change.51 Notably, the naphthopyrans and related spiro-oxazine family show a reverse behavior and have an essentially quinoidal ground state and zwitterionic excited state, attributed to the shift in specific interactions and absence of the need to breaking aromaticity in quinoidal form. This dependence on the relative energies and energy barriers between the accessible isomers is a core feature that ultimately determines the electronic properties in each individual spiropyran system.
Despite that multiple isomers can be accessed, their similarity in energy is reflected in the similarity of their spectroscopic properties, which renders distinguishing them and confirming their presence challenging. The observation and characterization of all four transoid isomers of some analogues was ultimately achieved by multiple dimensional NMR and Raman spectroscopy starting in the late 1980s.52 Furthermore, over the years several time resolved studies and theoretical calculations have pointed towards the TTC merocyanine isomer being the energetically most stable transoid form closely followed by the TTT isomer, as was first observed for unsubstituted benzoindolinospiropyran by Ernsting et al. in 1990.53 Later studies by Hobley et al. using 1H NMR spectroscopy identified several isomers, e.g., the strongly solvatochromic 6,8-dinitrobenzoindolinospiropyran.54 2D transient absorption spectroscopy by Kullmann and co-workers clarified the dynamics of the system,55 although the non-observed photoisomerization pathway between the TTC and TTT isomers at the time was later found to be a minor reaction channel using fluorescence microscopy (vide infra).30
The relatively energies of the isomers, it should be noted, is not immediately obvious. The greater charge-separation in the TTT isomer should render it energetically more stable that the TTC form. However, as noted above in the discussion of their thermochromism the methine C3′–H is acidic and lowers the barrier to opening of the spiro ring by H-bonding with the electron rich phenolate.44 Although steric interactions in C3′ substituted spiropyrans placed doubt on this rationalisation, more recent NMR spectroscopic studies have revealed the significant contribution played by such interactions in favouring the TTC conformation.45
Aldoshin and co-workers56 proposed that the positive charge on the indoline nitrogen and the negative charge on the phenolic oxygen are delocalized over the bridging methine, with a partial negative charge at the C3′ position and partial positive charge at the C4′. The former is stabilised by electron donation from the phenolate to the attached hydrogen in the stable TTC form, as confirmed by 1H NMR and 13C NMR spectroscopy (Scheme 9).54,57 In the 13C NMR spectra, resonances of C3′ and C4′ carbons at 111 ppm and 152 ppm indicating charge separation, while the carbonyl carbon resonates at 182 ppm, characteristic of a partial double bond, and the C2′ carbon at 169 ppm, indicating a positive charge at the adjacent indoline nitrogen. Furthermore, in its 1H NMR spectrum the C3′–H is downfield from that expected, and is sufficiently acidic to exchange with D2O or MeOD.54 Protonation of the phenol decreases hydrogen bonding with C3′, and hence acidity, reducing the rate of H/D exchange. The chemical shift of the N-methyl protons moves to 4.2 ppm, indicating loss of the quinoidal character and a resulting full positive charge on the indoline nitrogen and through space H-bonding interactions specifically between the C3′–H and phenol are supported by the spectra of the C4′-deuterated isotopologue, in which, despite being closer through bond, reduced isotope shifts were found.57
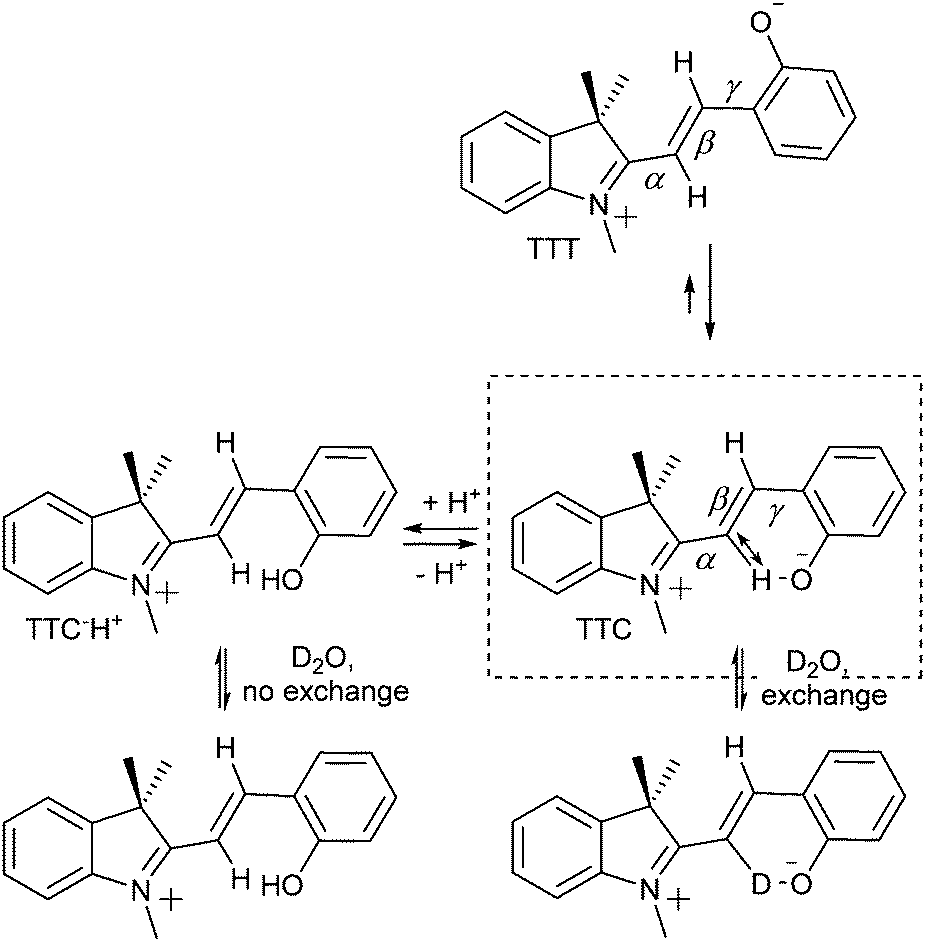 | ||
| Scheme 9 Intramolecular H-bonding stabilization of the TTC merocyanine form between the C3′–H and the phenolate with concurrent bond elongation, and the absence in the TTT form. Protonation of the phenolate diminishes the through space interaction, inhibiting H/D exchange at the C3′ position. | ||
Efforts to define the reactivity of a wide range of spiropyran-analogues is manifested in the extensive literature,‡ however, the focus in this review will for the most part be on the much-studied indolinobenzospiropyran core (Scheme 2). Furthermore, we will focus on properties that are intrinsic to these spiropyrans and closely related analogues, starting with their remarkable photochromism.
Photochromism of spiropyrans
Although the thermochromism of 1,3,3-trimethylindolinobenzospiropyran (Scheme 2) was reported in 1940 already (vide supra),38 the first account of its photochromism was made by Fischer and Hirshberg only in 1952,26 in which the thermal barrier to the merocyanine form was shown to be overcome by photoexcitation. In 1989 switching induced by two photon absorption with visible and NIR light was demonstrated also.58 A key aspect leading to the discovery and unravelling of the photochromism of spiropyrans, however, is the sufficiently high thermal barrier to reversion to the spiro form in certain spiropyrans (Scheme 6) that allows for photochromism to be observed even where thermochromism is prevented.50,59 The thermally activated nature of the decolouration prompted the use of low temperatures to generate various merocyanine isomers by photo-irradiation.46 It was not until later, owing to the stabilizing effect of certain substituents, that direct observation of the four transoid species through various spectroscopic techniques was made.52 While the photostationary state of spiropyrans is comprised largely of an mixture of the low energy spiro (closed) and the energetically most stable transoid merocyanine (open) forms, the primary step in the photochemical reaction is widely considered to be the dissociation of the Cspiro–O bond in the electronically excited state.5 This step is followed by either recombination to the ring-closed form, or by a “free rotor” effect along the π–π* surface, i.e. a twisting motion to relieve strain guiding the excited state to a geometry that favours radiationless deactivation (Scheme 10).6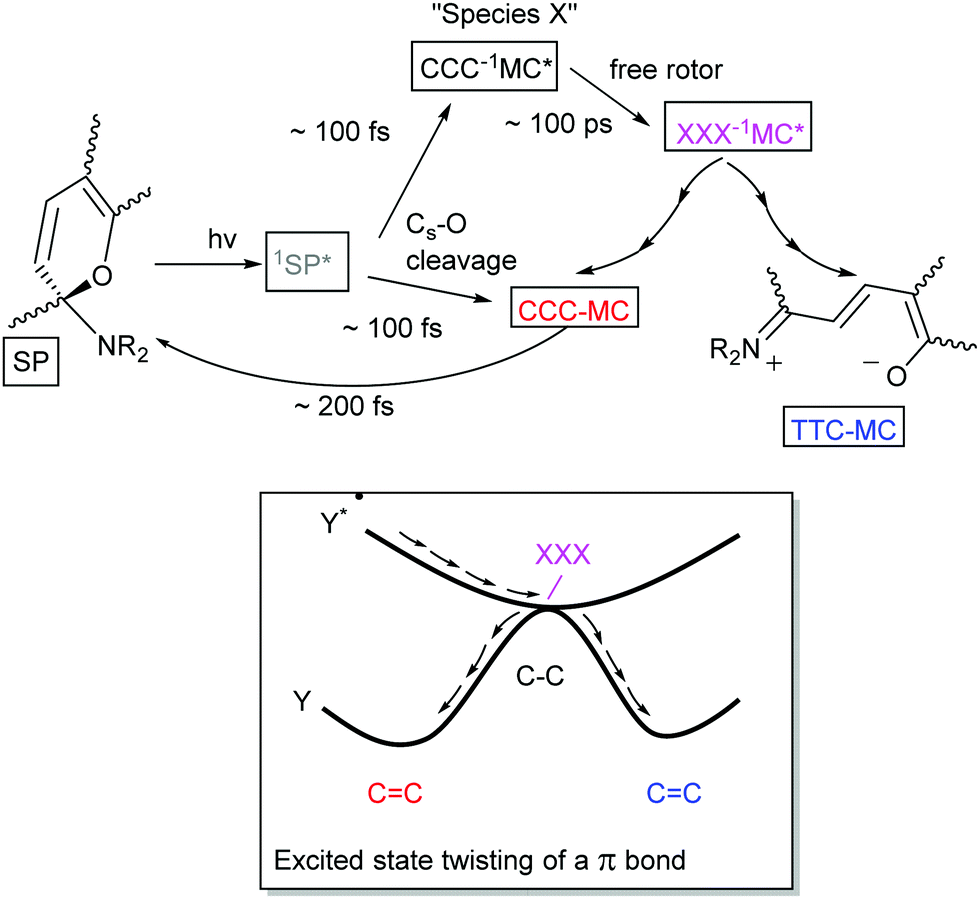 | ||
| Scheme 10 Singlet excited state pathway of (unsubstituted) spiropyrans to the ring open merocyanine form. After excitation to 1SP*, pericyclic recombination with concurrent ring opening (bottom pathway) yields the ground state CCC-MC which relaxes rapidly back to the ring closed form, while the perpendicular oriented CCC-1MC* form (“species X”, top pathway) undergoes a ‘free rotor’ rotation while traversing its PES. Upon arrival at a conical intersection (with XXX geometry) radiationless conversion to the ground state yields either cis or trans geometry. | ||
Unsubstituted spiropyrans have been shown to access only this singlet manifold energy surface, while certain substitution patterns enable a triplet manifold pathway that also enhances ring opening (vide infra). The singlet manifold pathway involves excitation of (unsubstituted) spiropyran (SP) to 1SP*, causing it to lose its double bond character through a formally π–π* transition and to increase its energy upon the spontaneous change in hybridization. Since electronic motion is up to 104 times faster than nuclear motion60 bond breaking in the excited state lowers the energy before rotation can occur, with radiationless intersystem crossing to a species typically referred to as “species X”.5 This metastable species X, later identified to have the structure (but not necessarily the stable planar orientation yet) of the cis-cisoid isomer (CCC in Scheme 7) by time-resolved spectroscopic techniques, can either re-attain a ground state through a pericyclic rearrangement, i.e., reformation of the broken Cspiro–O bond, or move across the excited state surface and undergo rotation around the π–π* system. If nuclear movement following excitation to the Frank–Condon state leads to a conical intersection (CI) then radiationless relaxation to the a ground state surface can occur. In either case, relaxation to the ground state surfaces results in formation of both thermally unstable cisoid and thermally stable transoid merocyanine.
Remarkably, nitro-substitution on the pyran ring at 6- or 8-positions overrides this mechanism by facilitating access to triplet states through formally n–π*-transitions (Scheme 11, right), analogous to the enhancement of S1 to T1 intersystem crossing in stilbenes with NO2-substitution.61 Nitro-substitution also increases the quantum efficiency of ring opening by typically 2-fold, owing in part to the elongated Cspiro–O bond,62 though photodegradation by reaction with triplet oxygen is substantially increased compared to spiropyrans that do not bear substituents with low-lying π*- and/or n-orbitals.5
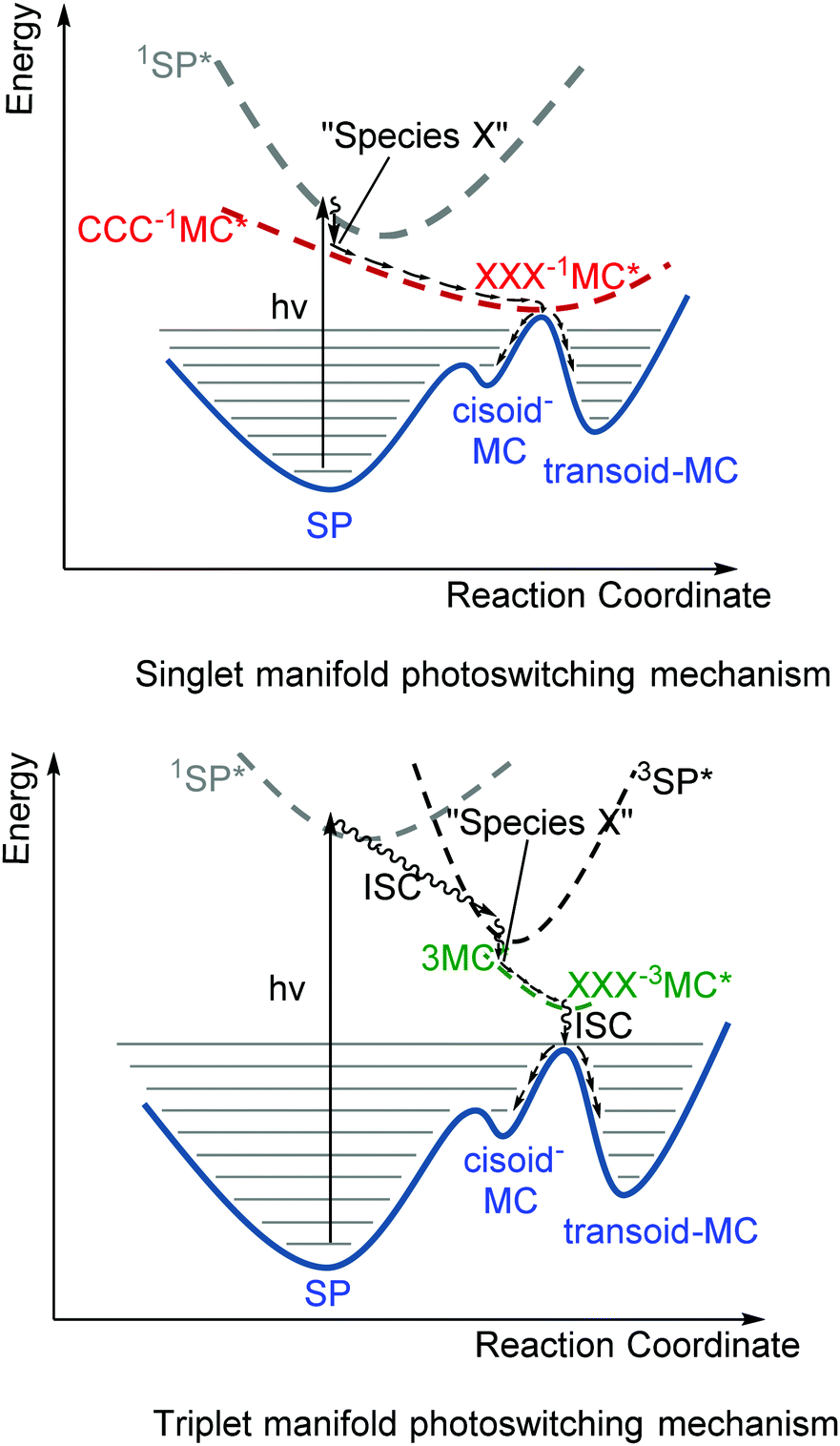 | ||
| Scheme 11 Photochemical ring opening through a funnel (upper) in (non-substituted) spiropyrans on the singlet manifold and (lower) via the triplet state. Traversing the PESs is more likely to occur through a conical intersection, a point on a hypersurface that does not correspond with local minima and maxima on either surface. | ||
The triplet excited state shows absorption at 430 nm,45 typically with a shoulder at 630 nm ascribed initially to aggregation of the transoid merocyanine and species X (the perpendicular CCC-1MC*), but later assigned to aggregates of the transoid form alone.63,64
Visible light excitation of non-substituted indolinobenzomerocyanines leads to radiationless relaxation and fluorescence. Indolinonaphthomerocyanines were the first for which recovery of the spiro form was observed upon irradiation with visible light.65 Later, photochemical ring closing was observed in (nitro-)substituted merocyanines of both the naphtho- and benzo-type, and it was shown that reversion to the ring closed spiro forms proceeds via the singlet excited state manifold.5 Notably, the quantum yields for ring-opening and ring-closing in spiropyrans can be tuned by variation in their substituents. A disubstituted 6,8-dinitroindolinospiropyran, for example, has a greater tendency to ring-open than 6-nitroindolinospiropyran, which in turn ring-opens more easily than unsubstituted indolinospiropyrans.66 Increasing the quantum efficiency of one of these processes, however, results in a concomitant decrease in the yield in the other direction. Nevertheless, the quantum efficiency for ring-closing is typically higher than that for ring-opening and while reaction rates can be decreased by triplet state pathways to the open form, they can also lead to degradation.5
Fluorescence from spiropyrans
Spiropyran itself does not fluoresce significantly in solution, nor does the photoaccessible cisoid merocyanine at 105 K.46 The transoid merocyanine isomer formed upon warming from 105 K to 123 K is fluorescent, which competes with radiationless conversion (trans to cis) and intersystem crossing pathways. The transoid merocyanine forms do not fluoresce appreciably in water but do fluoresce in polar organic solvents.52,67–70 The photophysics is analogous to that of cis- and trans-stilbenes, the former being non-fluorescent and the latter weakly fluorescent (Φf = 0.05).6 While the steric interactions present in cis-stilbene favour twisting along the π–π*excited state surface (vide supra), trans-stilbene does not. Furthermore, when the cis and trans stilbene isomers are locked in their geometry, e.g., by alkyl tethering at the benzyl 2-position approximately quantitative fluorescence (Φf ≈ 1.0) is observed.6Analogously, where there are alternative (and competitive) pathways available to the excited state of the spiropyran–merocyanine system, the fluorescence quantum yield is shown to be considerably reduced and vice versa enhanced when E/Z-isomerisation is precluded by low temperatures or (steric) hindrance to isomerization (Scheme 12).46,71–73 Notably, shielding of the spiropyran–merocyanine system from electron transfer induced by collisions with solvent molecules can also considerably improve the fluorescence intensity.34,74–80
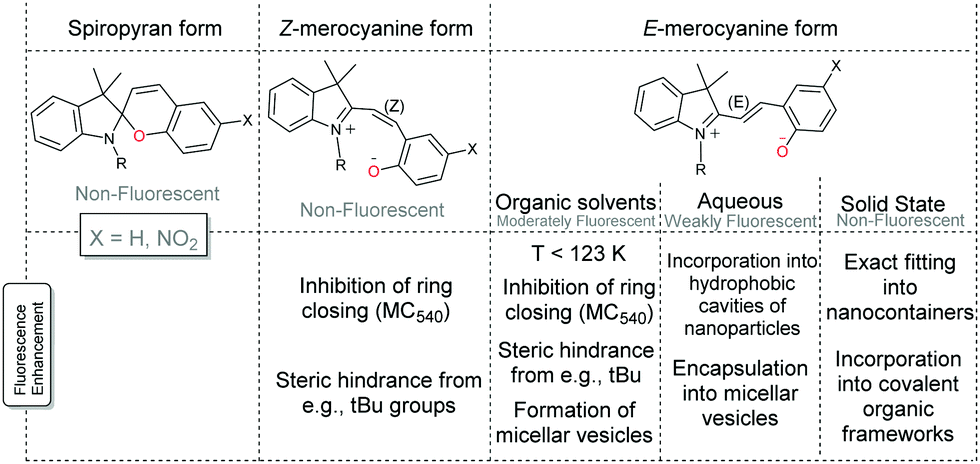 | ||
| Scheme 12 Fluorescence of spiropyran and merocyanine. The fluorescence of the Z-merocyanine form (in organic solvents) can be increased by precluding ring-closure, e.g., fully as with the Merocyanine 540 dye71 or partially by steric encumberment.72,73 The fluorescence of the E-merocyanine form in organic solvents can also thus be increased in micellar aggregates74 and vesicles75 and by lowering the temperature to 123 K.46 The fluorescence of the E-merocyanine form in aqueous solutions can be increased by incorporation into hydrophobic cavities of nanoparticles, i.e., nanocages,34,76–79 or in micellar vesicles,80 and in the solid state when the nanocage fits the E-merocyanine only.34 Additionally, incorporation into a covalent organic framework with orientation into the cavities of the porous framework is also shown to increase fluorescence.81 | ||
The fluorescent dye Merocyanine 540, in which extension of the alkene bridge requires formation of an eight-membered ring to form the spiro structure, highlights the enhancement of fluorescence possible through geometric or steric constraints.71 Partial hindrance, however, can also result in an appreciable barrier to isomerization by stabilizing the ring open form through substituents such as correctly placed electron withdrawing and donating groups (Scheme 4), or sterically cumbersome groups, e.g., tert-butyl.72,73 In fact, the tert-butyl groups raise the energy barriers between the merocyanine forms to the extent that fluorescence from the cisoid isomer is observed also. Howlader and co-workers have shown that palladium nanocontainers that fit the shape of the E-merocyanine only, restrict them to this form regardless of the stimuli used.34 Hence the fluorescence observed in solution is obtained in the solid state also, and applied as the active component of an invisible ink that is revealed upon UV-irradiation to form the merocyanine form, which is in situ encapsulated (Scheme 13).
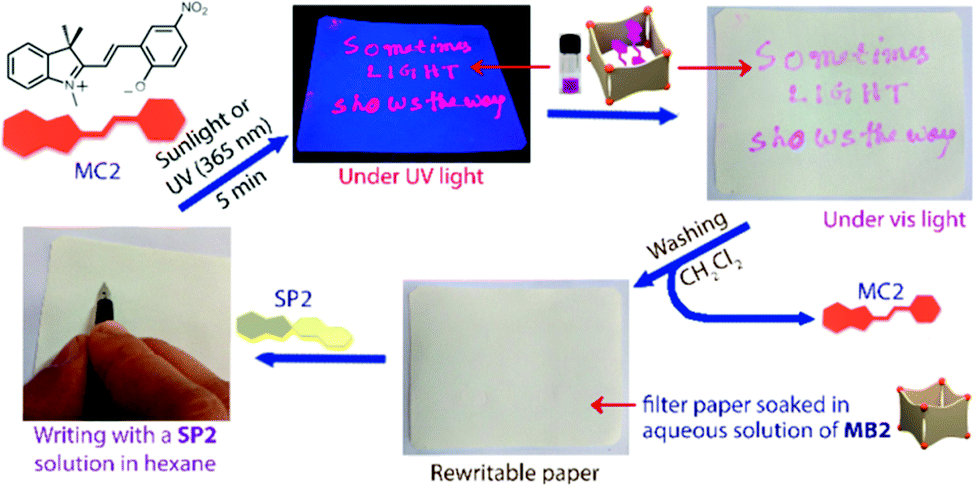 | ||
| Scheme 13 Writing with a colourless hexane solution of SP2 on a filter paper soaked with an aqueous solution of nanobarrel MB2 yields no visible markings. Subsequent UV-irradiation results in conversion of SP2 to MC2, which is immediately encapsulated by the MB2 nanobarrels to reveal the inscription, which can be erased by washing with dichloromethane. Adapted with permission from Howlader et al., copyright American Chemical Society (2018).34 | ||
An alternative to caging spiropyrans is to constrain their motion through self-assembly into nanostructures or densely packed films to enhance fluorescence.30,74–77,82 Kim et al., for example, recently applied Spectrally Resolved STochastic Optical Reconstruction Microscopy (SR-STORM) to solvated 6-nitro benzospiropyrans to investigate the time dependent dynamics of ring-opening and ring-closing using fluorescence on/off switching.30 Upon simultaneous excitation of nitrospiropyran to the ring-open form at 405 nm and 560 nm excitation of nitrospiropyran to induce fluorescence or ring-closing, ring-opening to the fluorescent and distinguishable TTC (λmax 590 nm) and TTT (λmax 635 nm) merocyanine forms was observed, with assignment based on earlier studies.83 The decrease in the relative contribution of the latter in increasingly polar solvents was in agreement with previously calculated polarities of these forms, and though the order of assignment is opposite to that made earlier,84 recent interpretations concur with the TTC-isomer absorbing and emitting at shorter wavelengths.83 Furthermore, by monitoring the positions of single molecules rapid photobleaching was observed, as well as TTT to TTC photoconversion (Fig. 1), which complement earlier ultrafast spectroscopic studies in which TTC to TTT isomerization was observed only. As the authors noted, immobilization of the molecules may impact isomerization dynamics.
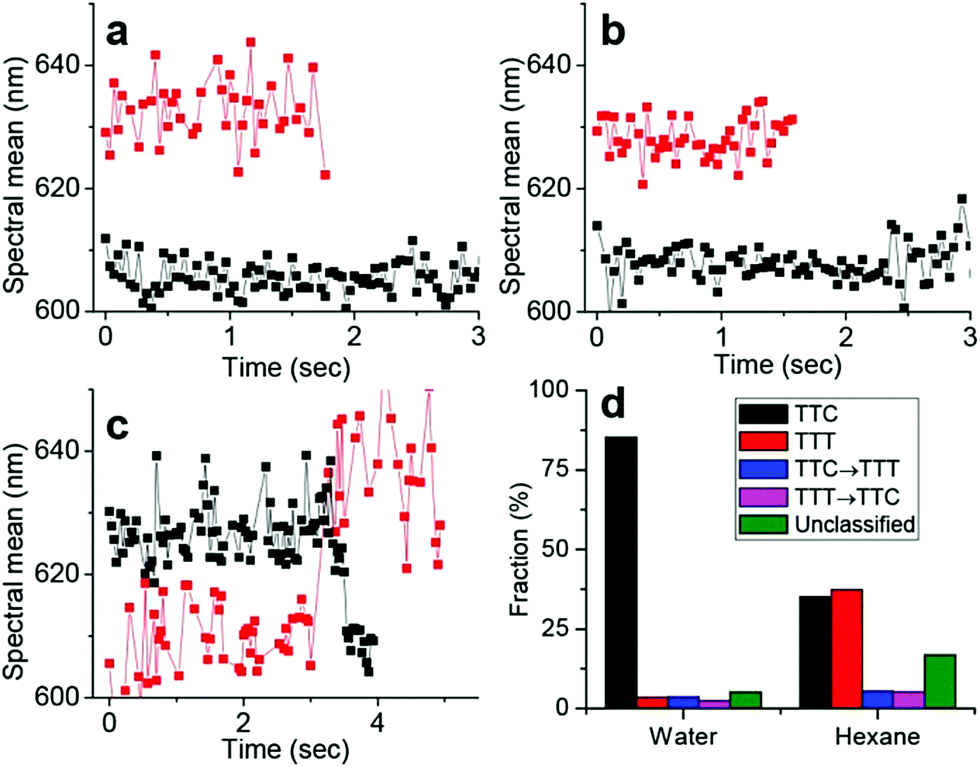 | ||
| Fig. 1 Dynamics of isomerization in single merocyanine molecules through spectral time traces. (a and b) Tracking two individual molecules corresponding to the TTC (black) and TTT (red) isomers, respectively, in water (a) and n-hexane (b). (c) Two individual time traces showing examples of TTC → TTT isomerization (red) and TTT → TTC isomerization (black) of single molecules in n-hexane. (d) Statistical analysis of ∼1000 of such single-molecule spectral time traces in water and hexane. The “unclassified” fraction exhibited complex switching/scattering with noise. Reproduced with permission from Kim et al., copyright American Chemical Society (2017).30 | ||
Acidochromism and pH-gated Z/E-isomerization of spiropyrans
Acidochromism is the change in colour of a compound due to charge-induced change in ionization, a phenomenon which is already observed in the solvatochromism of spiropyrans (vide supra). The acidochromism mentioned below is analogous to metallochromism reported also for spiropyrans.85 In the presence of any charged species, protons in the case of acidochromism, the polar merocyanine form can be stabilized relative to the apolar spiropyran form.‡ Prior to the discovery of the photochromism of spiropyrans, formation of coloured compounds upon addition of acid was observed to be a general phenomenon for non-photochromic spiro compounds.36 Indeed studies of the coloured phenopyryllium salt led to the discovery of spiropyrans and their pH-dependent colouration (vide supra, Scheme 1). As with thermochromism, the pH dependence of acidochromism is dependent on the ease with which the molecule can planarise. Obstructing planarisation at the spiro-centre by appropriate substitution at the 3- and 3′-positions reduces the tendency to undergo protonation driven ring opening to the merocyanine form. Constraining the configuration to planarity with a cyclopentane linker renders the open form accessible also (Scheme 14), despite the absence of an acidic C3′–H capable of H-bonding with the phenol (vide supra). Notably, with a six-membered ring there is sufficient flexibility to prefer the non-planar ring-closed isomer, analogous to effects observed in the study of spiropyran thermochromism. | ||
| Scheme 14 The dependence of pH-driven ring closing on the ease with which a molecule can become planar. | ||
Importantly, basicity is not a reliable predictor of thermochromism. Salt formation with, e.g., benzospiropyrans does not involve serious disturbance of the aromatic system and instead forms a stable phenolate salt. For instance, while 3′-methylbenzo-β-naphthospiropyran is not thermochromic, the driving force for salt formation is as strong as its thermochromic 3-methylbenzo-β-naphthospiropyran.41
A more important consideration in driving ring opening of spiropyrans by protonation is that the acid employed has sufficient strength to protonate the spiropyran and induce concomitant ring-opening. This was noted as early as 1929 by Irving and co-workers for xanthonaphthospiropyran and benzoxanthospiropyran, neither of which exhibited acidochromism with acetic acid, but did with trichloroacetic acid.27 This effect in the contemporary indolinobenzospiropyrans was noted much later (1995) both by Roxburgh and Sammes.86 The intermediate species observed by 1H NMR spectroscopy was assigned as either the protonated or non-protonated short-lived cisoid-species. At the same time, Zhou and co-workers assigned an intermediate observed upon protonation of 6′,8′-dinitrospiropyran to a protonated form with a broken Cspiro–O bond that had not yet achieved planarisation (“species X”).87 The proposed intermediacy of the protonated cisoid form was supported by Shiozaki shortly thereafter,88 who showed a better-defined response to protonation of spiropyran in ethanol with sulfuric acid than with the more commonly used trifluoroacetic68,69,89–97 and hydrochloric acids.18,98–109 These observations indicate that distinct protonated cisoid and transoid structures differ in pKa. Indeed, the dependence of the acidochromic response on the acid employed enables access to pH-gated photochromism in both indolinobenzospiropyran as well as 6-nitroindolinobenzospiropyran when the acids are stronger than the phenolates formed upon ring opening (Scheme 15 and Fig. 2).32
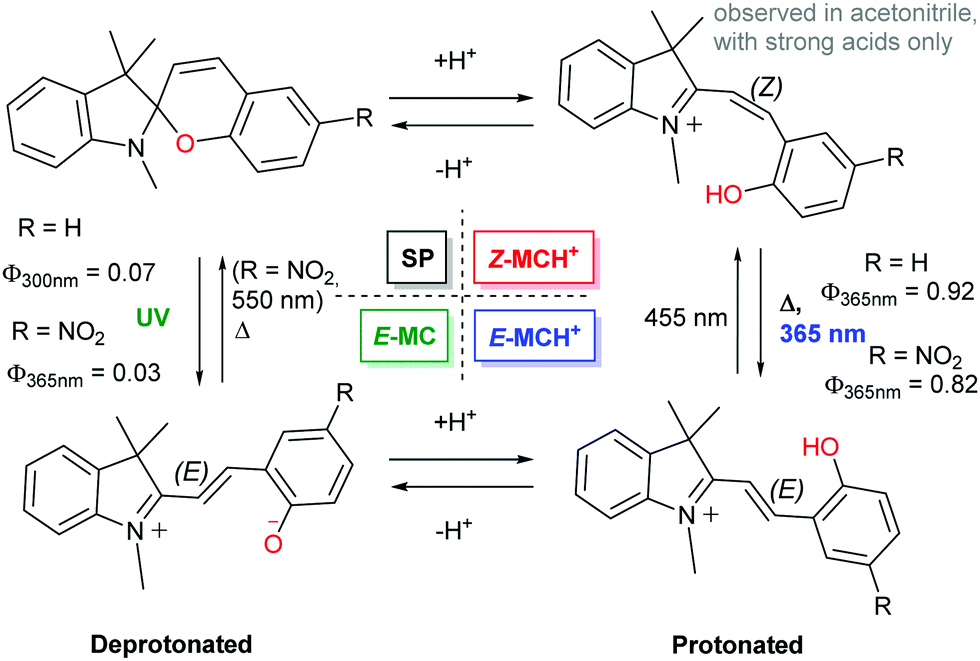 | ||
| Scheme 15 The 4-state pH-gated photochromism of indolinobenzospiropyran (R = H) and 6-nitroindolinobenzospiropyran (R = NO2) in acetonitrile when acids of sufficient strength (low enough pKa) are used, i.e., H2SO4 (pKa 8.7), CF3SO3H (pKa 0.7) and HClO4 (pKa −0.7).32 In the presence of weak acids, i.e., CF3CO2H, CCl3CO2H and HCl, poorly defined acidochromism was observed with limited access to the protonated Z-form and only partial formation of the E-form upon UV-irradiation. | ||
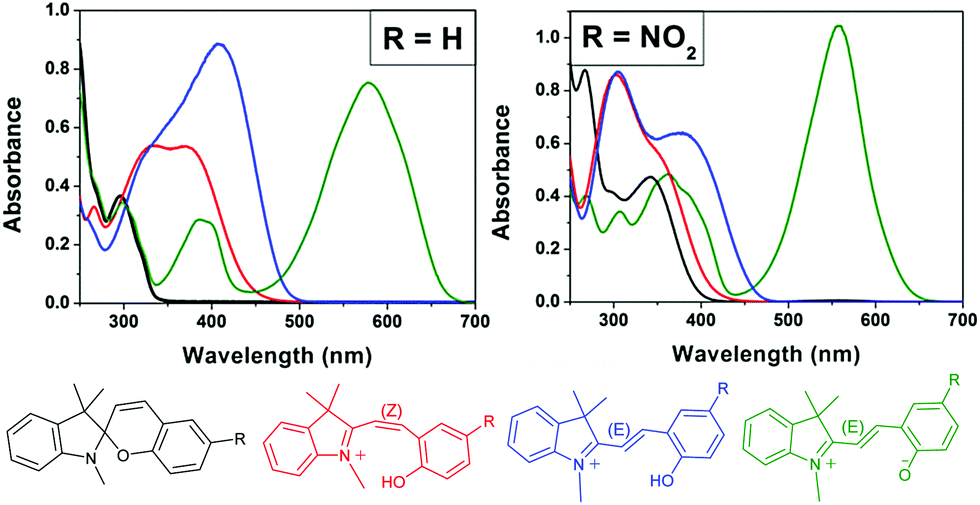 | ||
| Fig. 2 The acido- and photochromic response of indolinobenzospiropyran (R = H) and 6-nitroindolinobenzospiropyran (R = NO2) in acetonitrile in which the acid used has a pKa less than that of the protonated Z-form. Spontaneous protonation induced ring opening to the protonated Z-form (red) is observed followed by irradiation with UV light to generate the protonated E-form (blue). The spiro (black) and zwitterionic E-form (green) are shown also. Adapted with permission from Kortekaas et al. copyright American Chemical Society (2018).32 | ||
The use of stronger acids to enable bistable Z/E photo-switching of the ring-opened merocyanine opens up opportunities in the application of spiropyrans. It is of note that the UV absorption bands of the protonated Z-merocyanine are absent between pH 2 and 8 in PBS buffer, and instead only absorption of the protonated E-form at 422 nm is observed.104 The higher pKa and concomitant greater thermal stability of the E-form, which has been isolated,110 is consistent with the infrequent observation of the Z-form. The lower pKa of the Z-merocyanine has, however, enabled their application as photoacids23 since the first report on photochemical deprotonation in 1967.18,111,112
The water soluble alkyl-sulfonate spiropyran (Fig. 3) can induce changes in pH over the range of ca. pH 4 to 6, and has seen many applications.35,96,99,112–121 Although aqueous environments are known to accelerate hydrolysis of especially the merocyanine form (within hours), hydrolysis is generally slower at lower pH.122 Notably, incorporating a pyridinium in the chromene moiety of similar water-soluble spiropyrans greatly increases aqueous thermal stability to weeks by favouring the more stable ring-closed spiropyran form, while still operating as a photoacid.109,123,124
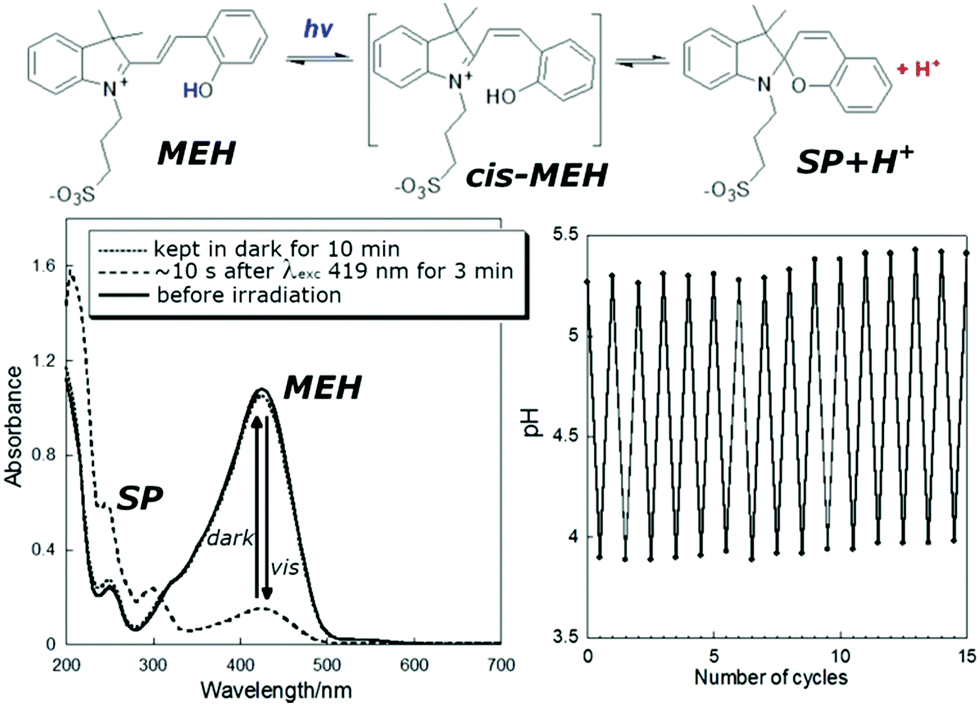 | ||
| Fig. 3 (lower left) UV/vis absorption of a solution of photoacid MEH before irradiation, 10 s after irradiation for 3 min, and kept in the dark for 10 min after irradiation, and (lower right) the pH under cycles of irradiation and darkness. Adapted with permission from Liao, copyright American Chemical Society (2017).112 | ||
The application of spiropyrans, typically but not only the sulfonate form, as a photoacid requires that the apparent pKa of the acid to be deprotonated is greater than that of the ring-closed spiropyran, but less than that of the ring-opened E-form (Scheme 16). Thus, UV-irradiation provides access to the proton-scavenging E-form, while subsequent visible light induced E to Z isomerization (i.e., 455 nm, see Scheme 15) leads to deprotonation of the Z-merocyanine with concomitant spontaneous ring-closing. Note that electron donating groups on the phenol ring raise the pKa of the E-form and increase the rate of reversion to the photogenerated spiropyran form.112
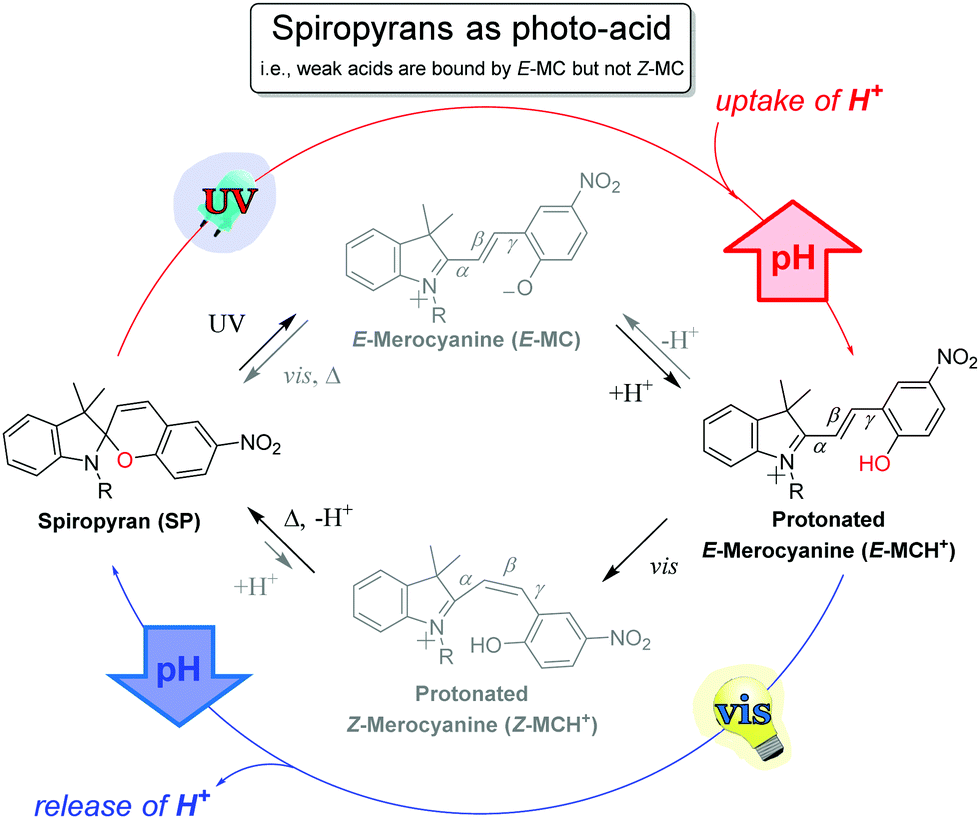 | ||
| Scheme 16 Photoacid behaviour in spiropyrans that are matched with the pKa of the acid employed, i.e., the acid used is selected such that its pKa lies between that of the Z-merocyanine and E-merocyanine form. In such systems, the spiropyran form possesses a pKa lower than the employed acid and therefore does not undergo protonation. Upon UV-irradiation the ring-open merocyanine form is accessed, which possesses a higher affinity (higher pKa) to the proton than the present acid, effecting a proton uptake and hence raising the pH. Visible light irradiation stimulates E to Z isomerization, lowering the pKa, and thereby causing release of the proton and ring-closure, concomitant with lowering in pH. | ||
Although 6-nitroindolinobenzospiropyrans are commonly used as photoacids, visible light-induced ring-closing is not restricted to nitro-substituted spiropyrans as protonation enables E- to Z-isomerization of indolinobenzospiropyrans as well (see Scheme 15).35,68,96,99,101,112,116,118,125,126 Near-stoichiometric amounts of phosphoric acid in acetonitrile, e.g., allow the non-substituted form to act as a photoacid (Fig. 4).32
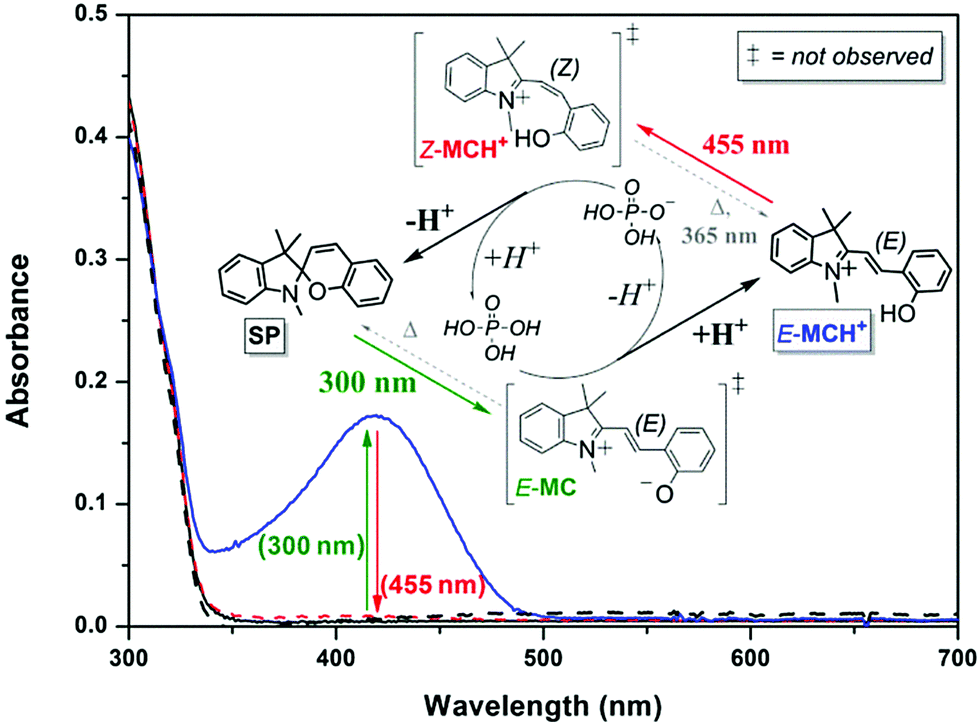 | ||
| Fig. 4 The UV/vis absorption of the ring-closed unprotonated spiropyran form SP (62 μM in acetonitrile, black solid line) is unchanged upon addition of 1 equiv. H3PO4 (red dashed line), signifying that the pKa of phosphoric acid is higher than that of the conjugate acid of the spiropyran, i.e., the protonated Z-merocyanine (Z-MCH+). Subsequent irradiation at 300 nm to induce ring-opening to the E-merocyanine (E-MC) instead results in the appearance of absorption of the protonated E-merocyanine (E-MCH+, blue solid line), the pKa of which thus exceeding that of phosphoric acid as opposed to the protonated Z-form. Inducing E to Z photoisomerisation of the protonated merocyanine at 455 nm therefore favours deprotonation of the Z-MCH+ form with concomitant ring-closing back to the spiropyran SP form (black dashed line). Adapted with permission from Kortekaas et al., copyright American Chemical Society (2018).32 | ||
Adventitious protons can catalyse such an effect of tipping the balance even when no appreciable amount of the protonated E-form is observed, as exemplified in encaged spiropyrans, which were recently found to undergo visible light induced ring-closing in water despite not bearing a nitro-substituent.35 Klajn and co-workers proposed that since targeting the absorption of the unprotonated merocyanine form did not decolourize the solution but blue light did, ring-closing through the protonated form had to have been involved at 460 nm (Scheme 17), which is consistent with more recent studies of the acid/base photochemistry of spiropyrans (Fig. 2).
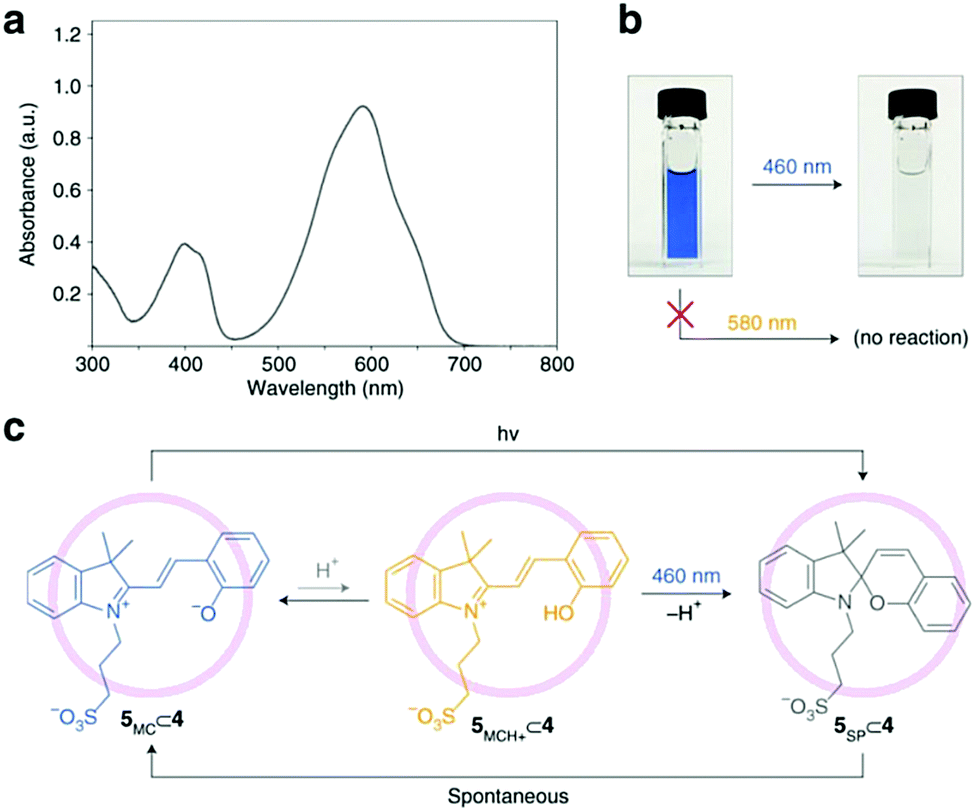 | ||
| Scheme 17 Photochromism of merocyanine 5 in the cavity of self-assembled palladium coordination cage 4. (a) UV/vis absorption spectrum of a dilute (0.5mM) aqueous solution of merocyanine 5 after subtracting the background due to cage 4. (b) Efficient decolouration of the blue solution of 5⊂4 at 460 nm, but not at 580 nm. (c) Proposed mechanism of the light-induced decolouration of 5⊂4. Reproduced with permission from Samanta et al., copyright Springer Nature (2018).35 | ||
Indeed when the protonated E-merocyanine is observed, the pKa of the protonated Z-merocyanine plays a vital role in its photoacid activity as it has to lack proton-affinity against the complementary component. This point is demonstrated further in the following examples in which the blue-shift in absorption due to the protonated Z-merocyanine with respect to the protonated E-merocyanine is not observed either.118,127 The spiropyran/merocyanine photoacid can be utilized to trigger pH switching when the pKa of the complementary pH-switch happens to be intermediate to that of the Z-merocyanine and the protonated E-merocyanine form, such as in the combination with a hydrazone fluorescence switch96 or an osmium(II) polypyridine complex97 (Scheme 18).
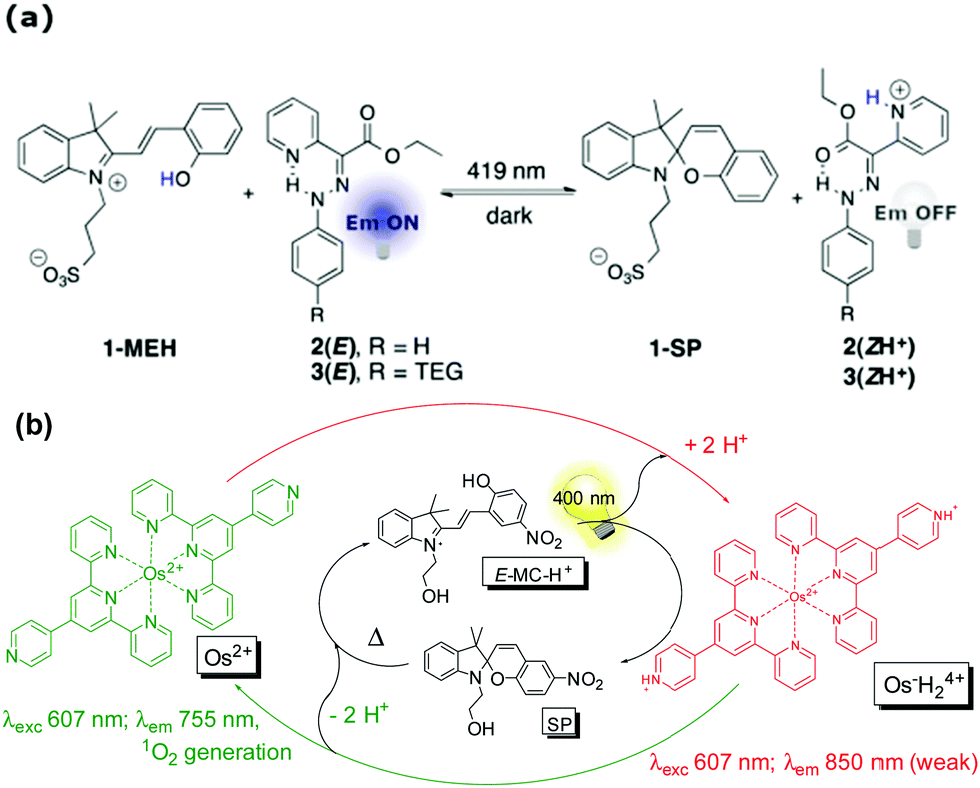 | ||
| Scheme 18 (a) Photoinitiated cycling of hydrazone fluorescence switches by proton transfer from the merocyanine photoacid 1-MEH, affording multistep interlinked switching (adapted with permission from Tatum et al., copyright American Chemical Society, 2014).96 (b) Photocontrol of luminescence and photosensitized singlet oxygen generation of an Osmium pH-switch through coupling its response with a merocyanine photoacid. Adapted with permission from Silvi et al., copyright Royal Society of Chemistry (2009). | ||
New levels of interconnected complexity can thus be achieved when the pKa of a complementary pH sensitive compound is matched appropriately with the spiropyran photoacid,35,96,97,105,128 including but not limited to reversible polymerization,106,107 permeability,120,129–131 photogelation121 and swelling,118,126,132–135 photocontrolled chemopropulsion,115 photocurrent generation,113,136 and reversible photoassembly of nanoparticles125,137,138 (Scheme 19).
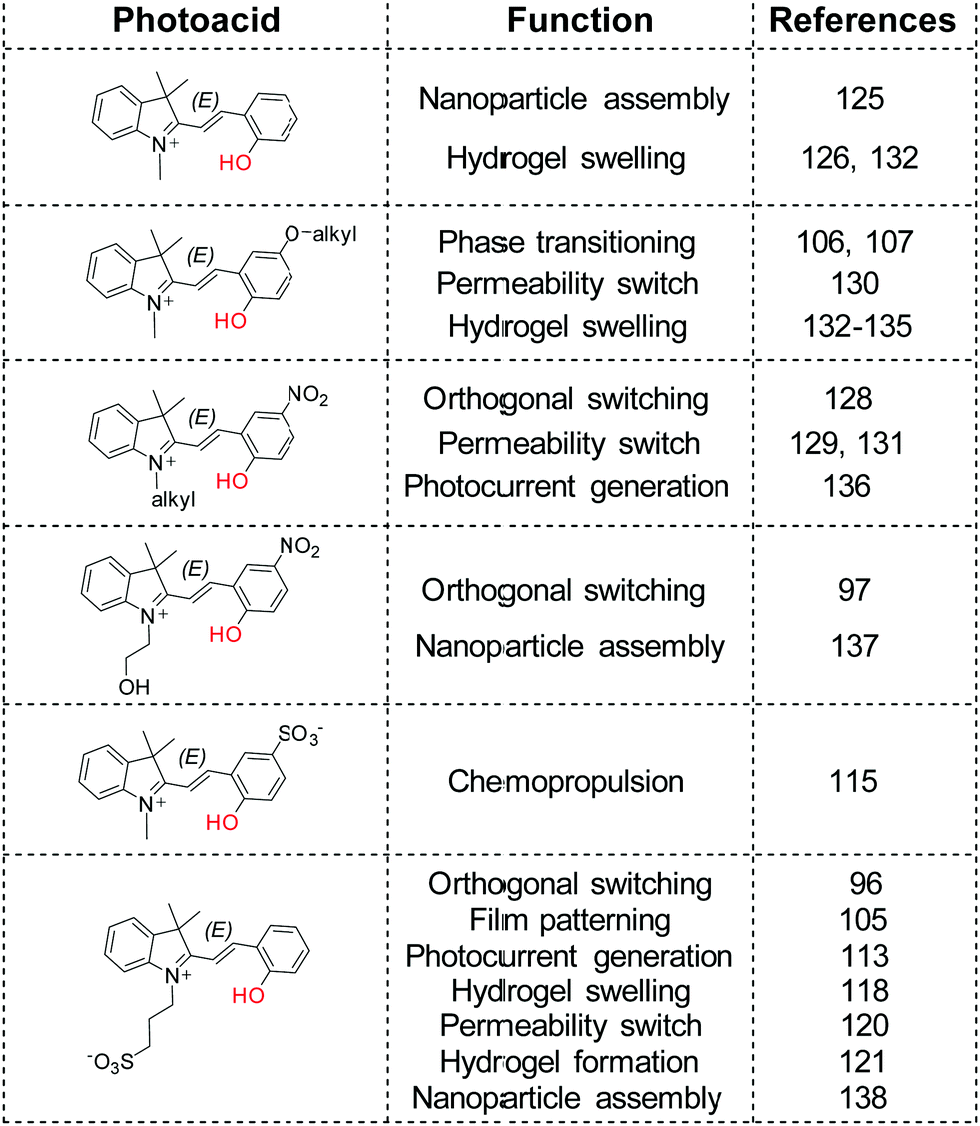 | ||
| Scheme 19 Spiropyrans used as photoacids in various applications. | ||
Redox-properties of spiropyrans
The redox-chemistry of nitro-indolinospiropyrans was not reported until 1993, when it was shown that the electrochemical reduction of the naphthospiropyrans was fully reversible and the radical anion was characterized by EPR spectroscopy after chemical reduction with butoxide.139 Shortly after, Zhi and co-workers reported the reductive reversible colouring of the nitrospiropyrans on several occasions and noted electrochemical ring-opening through reduction (Fig. 5).140,141 Notably, the voltammetry does not correlate linearly with the generation of the radical anionic closed form and its conversion to the neutral open form, indicating that another process such as redox-gated photochromism (vide infra) may be involved as the sample is under constant irradiation of the spectrometer.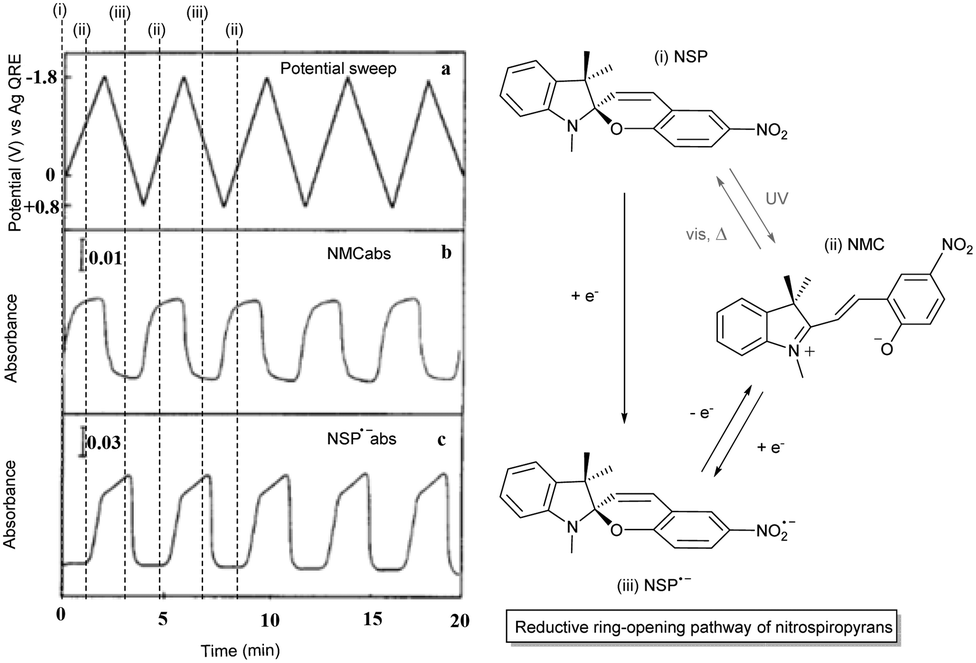 | ||
| Fig. 5 (left) Reversible electrochromism of 6-nitroindolinobenzospiropyran in DMF at −42 °C (platinum working, Ag/AgCl reference and platinum counter electrode, TBAPF6 supporting electrolyte) as a function of (a) the potential sweep between −1.8 V and 0.8 V for absorption at (b) 556 nm and (c) 445 nm. Reproduced with permission from Zhi et al., copyright Elsevier Science S. A. (1995).140 (right) Observed photoelectrochemical operation of the reductive ring-opening process in 6-nitroindolinospiropyrans. | ||
Although Zhi et al. examined the reductive chemistry of nitrospiropyrans extensively the oxidative voltammetry was reported only later by Campredon and co-workers, whom noted the oxidation of three nitro-indolinonaphthospiropyrans at potentials >0.8 V vs. SCE (Fig. 6). Prior studies by the group using the chemical oxidant nitric oxide indicated that the initially formed radicals could not be trapped when the indoline moiety was absent, indicating its involvement in the redox chemistry.142 In the presence of both an indoline moiety and a nitro substituent, though, Campredon et al. observed irreversible oxidation of nitrospiropyrans at >1 V, even with sweep rates of >10 V s−1. A cathodic response, though small and manifested in two reduction waves, was apparent for 8′-nitroindolinonaphthospiropyran (Fig. 6, middle entry).
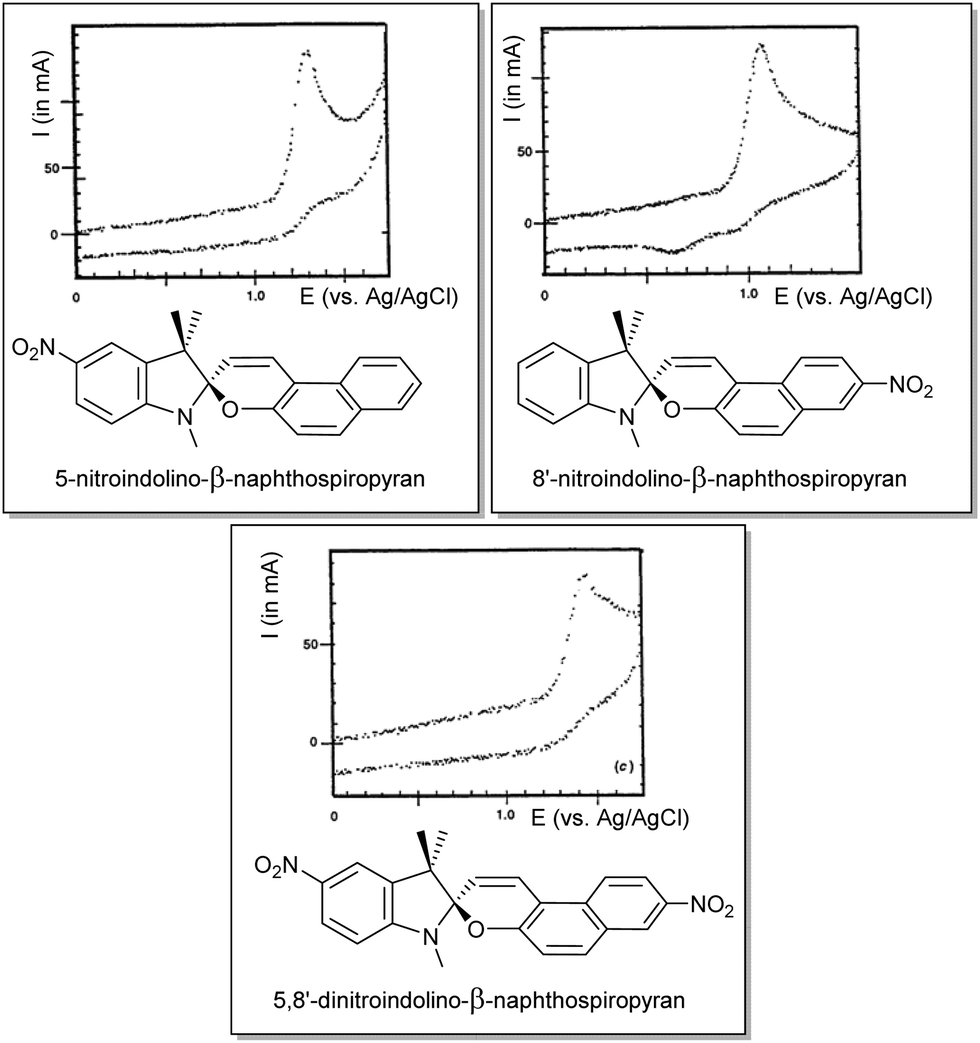 | ||
| Fig. 6 The first reported oxidative electrochemistry of spiropyrans in acetonitrile (versus SCE, at 1 V s−1), showing some cathodic response in the spiropyran that was unsubstituted in its indolinic fragment. Reproduced with permission from Campredon et al., copyright Royal Society of Chemistry (1993).143 | ||
This response was confirmed in a report by Zhi and co-workers of concurrent electrochromism in various spirobenzopyrans, including non-nitro substituted forms.144 The nature of the species formed upon oxidation was not immediately apparent however (Scheme 20). Preigh et al.145 proposed a quinoidal dimeric structure, while Doménech et al.146 also proposed coupling through the benzopyran albeit with a peroxide species as a result. More recently, Wagner and co-workers suggested that the product of oxidation would be of the ring-opened merocyanine form.147
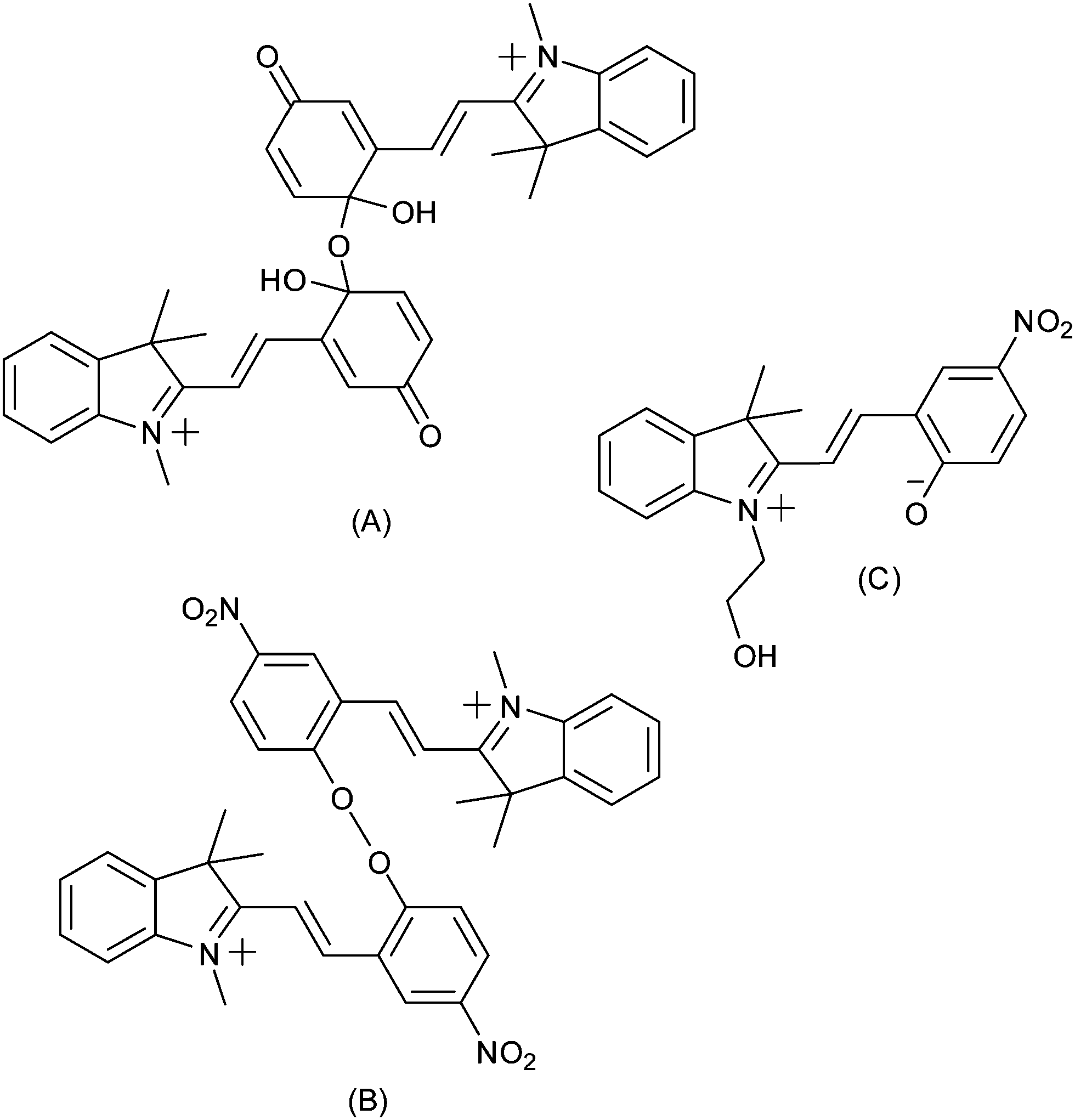 | ||
| Scheme 20 Tentative assignment of the products of oxidation of indolinospiropyrans, made by (A) Preigh et al.,145 (B) Doménech et al.146 and (C) Wagner et al.147 | ||
Subsequently it was shown that the oxidation chemistry was centreed on the indoline rather than the benzopyran moiety. Natali and Giordani first described the isolation of two spiropyran (indoline coupled) dimers upon addition of Cu(II) salts to solutions of spiropyrans when studying metal ion binding with transition metal cations.148 The series of reactions that lead to the coupling are analogous to the oxidative coupling of anilines. Ivashenko et al. showed subsequently that blocking coupling by methyl-substitution in the para-position prevents dimerization and the oxidative cyclic voltammetry thus becomes reversible (Scheme 21).149 The blocking of the oxidative coupling by the methyl group demonstrates that the radical character lies predominantly at this position and not the chromene unit (Scheme 22).148 Dimerization by oxidative coupling was shown later in self-assembled monolayers of nitrospiropyran also.150
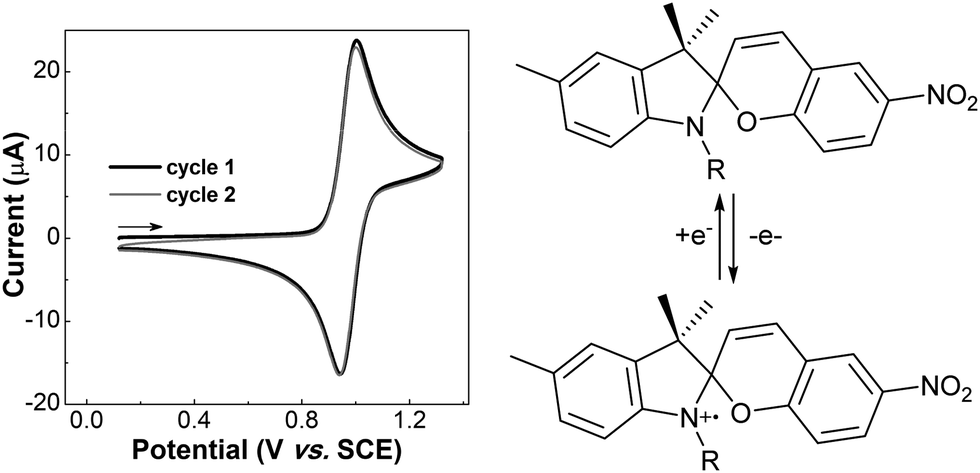 | ||
| Scheme 21 Methyl-substitution at the para-position in the indoline unit blocks C–C coupling, rendering the electrochemistry chemically reversible.149 | ||
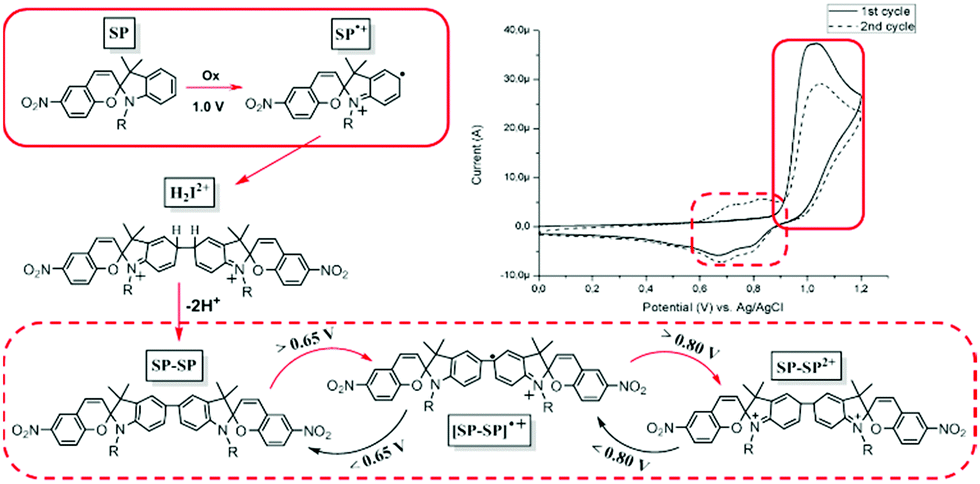 | ||
| Scheme 22 Mechanism of oxidative dimerization in spiropyrans to form the spiropyran dimer photochrome. The initial oxidation of spiropyrans cause aryl–aryl coupling of the generated radical cations (SP to H2I2+), followed by spontaneous double deprotonation to recover the energetically favoured diaryl SP–SP form. Since the redox potential of the dimer lies at approximately 0.65 V for its first and 0.80 V for its second oxidation, the newly generated spiropyran dimer immediately oxidized to its dicationic state (SP–SP to SP–SP2+). These series of reactions (red arrows) result in a current that is twice that expected for a one electron oxidation. | ||
This feature of spiropyrans, which has emerged in the past decade only, has important implications with regard to earlier studies and to more recent contributions in which the fabrication process of the spiropyran embedded materials is redox-driven, e.g., forming polymers from acrylamides and acrylates.126,130,133–135,151 However, this electrochemically driven coupling reaction alone can also be used to generate polymers at the electrode when tethering spiropyrans together through their alkyl side-chain.31 In addition to the retention of pH-stimulated switching, this approach enables observation and utilization of redox-gated ring-opening with visible light, yielding a multi-stimulus switchable spiropyran polymer (Scheme 23).
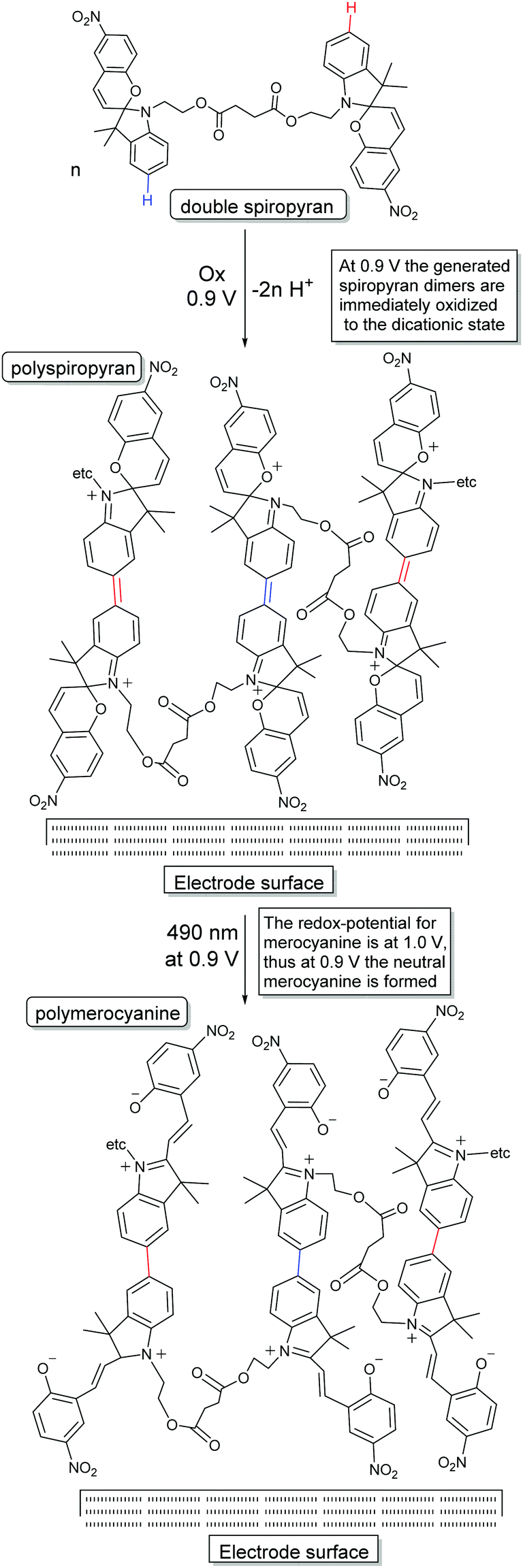 | ||
| Scheme 23 Approach to use electrochemical dimerization (i.e., single coupling) of spiropyrans to achieve polymerization by having each building block able to couple twice, and the novel reactivity of the spiropyran dimers toward visible light irradiation in the dicationic state.31 | ||
Notably, although this method creates opportunities for the preparation of multiphotochromes in general to form multifunctional polymers, it is particularly attractive when two spiropyrans are incorporated due to their inherent oxidative dimerization avoids the need for additional functional units.152
Mechanochromism of spiropyrans
Spiropyrans undergo changes in planarity, acidity, reactivity, polarity and dipole moment, polarizability, and electronic structure upon ring-opening to the merocyanine form. Using environments that favour (stabilize) the merocyanine form stimulates switching from the spiropyran form to it, e.g., solvatochromism in highly polar solvents or acidochromism under sufficiently acidic conditions. Similarly, a forced change in molecular shape can also induce ring-opening of the spiropyran, specifically when stress is applied to a spiropyran-embedded material which requires ring opening to its coloured merocyanine form to relieve strain, i.e., mechanochromism. A vital requirement for spiropyran embedded materials is that the stress is exerted on both sides of the spiro centre, such that the indoline and chromene components being pulled apart causes breaking of the Cspiro–O bond (Scheme 24), although the strain induced by ultrasound or grinding can also cause a mechanochromic response.22,153–155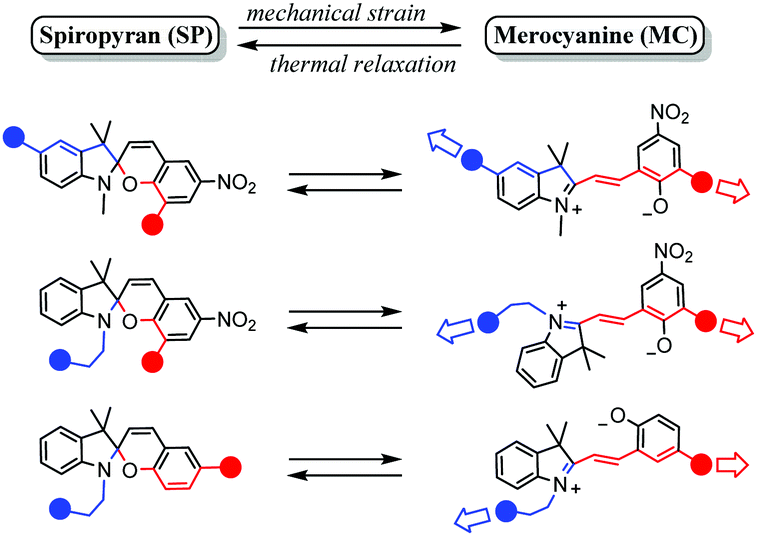 | ||
| Scheme 24 Substitution patterns in spiropyran materials (the blue and red spheres represent continuation patterns in the spiropyran material) that have shown to facilitate mechanochromism through enabling mechanical strain about the Cspiro-centre, as reported in the recent review of Li et al.22 Adapted from Li et al. with permission, copyright Elsevier Science S. A. (2018). | ||
Although this requires modification to the basic spiropyran structure, the phenomenon provides considerable additional opportunities in materials applications of spiropyrans. Indeed, mechanochromism in spiropyran-containing materials is now well established, reviewed recently by Li et al. in 2017.22 More recent fundamental contributions include tuning of the robustness of the chromophore by introducing varying degrees of crosslinking156 and tuning of the colouration threshold over a wide range of tensile strain,157 while practical applications are found in visible cues for pending failure in soft electronics158 and a method for electrospinning fibers capable of orientation dependent mechanochromism.159
Switching of spiropyrans in the solid state
Although quenching of photochromism in the solid state can be put to use for reversible activation of spiropyran features upon wetting and drying,35 overcoming this fundamental challenge to photochromes in general opens up many alternative opportunities in materials science. Switching functions have been shown to be restricted in thin films, monolayers and microstructures incorporating the spiropyran photochrome, and often an additional stimulus e.g., pH or temperature change, is needed.31,105,160,161 When moving toward closely packed ensembles such as crystals and thick layers, however, the changes that need to occur collectively upon ring-opening are strongly hindered both spatially and ionically. By default, the solid state is not an ideal environment in which to undergo changes in their charge and spatial properties, let alone the drastic changes in shape (elongation and planarization) and charge (densely packed zwitterionic charges without a solvation sphere) the spiropyran–merocyanine system undergoes. Nevertheless, switching in the solid state in spiropyrans has been achieved by circumventing the spatial restriction. Two strategies have been applied to avoid dense solid state packing: utilization of nanocavities in, e.g., nanocages,34 covalent organic81 and metal organic frameworks,162–164 or, alternatively, free volume generation by controlling the packing through, e.g., columnar stacking.33 Note that restriction of conformational freedom, and thereby the ability to undergo photochemical switching, results in other quenching pathways, i.e., fluorescence (vide supra). This aspect is especially apparent in the nanocages of Howlader et al. that enable efficient fluorescence in solution when fitted to the shape of the merocyanine form only (Scheme 13), but also allow for solid state switching when the size of the cage was increased.34 Similarly, the utilization of free space in two synergetic approaches towards spiropyran frameworks left one, though able to undergo solid state acidochromism and reversible metal-binding, unable to switch (Scheme 25a) but the other free to undergo solid state switching with on/off control over the fluorescence in the merocyanine form (Scheme 25b).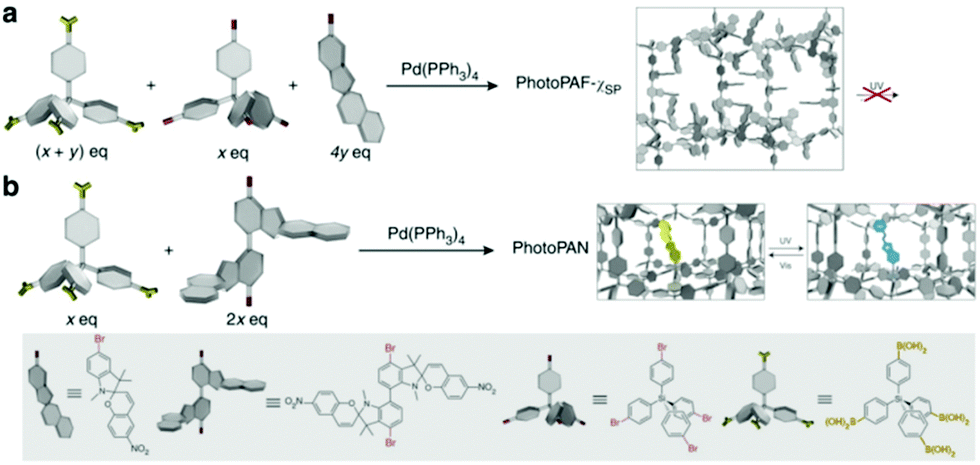 | ||
| Scheme 25 (a) The preparation of porous aromatic frameworks (PAFs), exhibiting solid state acidochromism and reversible metal-binding but no solid state photoswitching to the merocyanine form. (b) Preparation of porous aromatic networks (PANs) which retain their ability to photoswitch in the solid state with accompanying on/off control of fluorescence. Adapted with permission from Kundu et al., copyright Springer Nature 2014.81 | ||
The sensitiveness of embedding frameworks with spiropyran was underlined by Williams et al., who show that the cavities in a metal organic framework that enable switching can be tuned by varying the size of the TNDS pillars to provide control over isomerization kinetics, achieving, as done by Schwartz and co-workers earlier,164 switching rates close to those in solution (Scheme 26).162
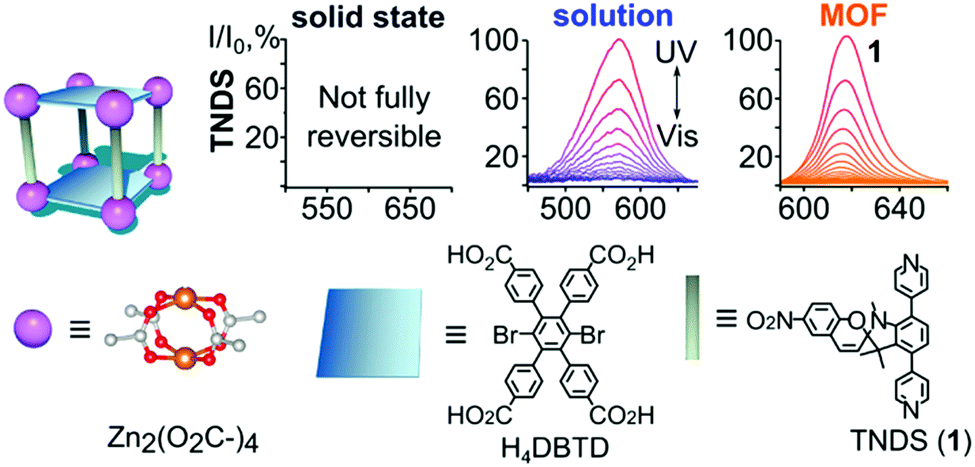 | ||
| Scheme 26 TNDS linkers were used to synthesize the displayed MOF in the presence of H4DBTD. At the top, normalized absorption plots are shown of TNDS in the solid state, in solution, and coordinatively immobilized inside a MOF upon irradiation with UV and visible light. Adapted with permission from Williams et al., copyright American Chemical Society 2018.162 | ||
In another strategy, Wu and co-workers show how a simple design is able to recover the multi-responsiveness of spiropyran by controlling the solid state packing (Scheme 27).33 A tetraphenylethene core is effective in directing π–π stacking such that columnar phases are formed in the solid state, controlling the intermolecular packing and restoring photophysical properties.117 The free volume around the pendant spiropyran arms in these stacks are sufficient to allow for the former restrictions to be lifted, tolerating gradual and reversible colour change, as well as reversible acidochromism when gaseous HCl and Et3N are blown over.
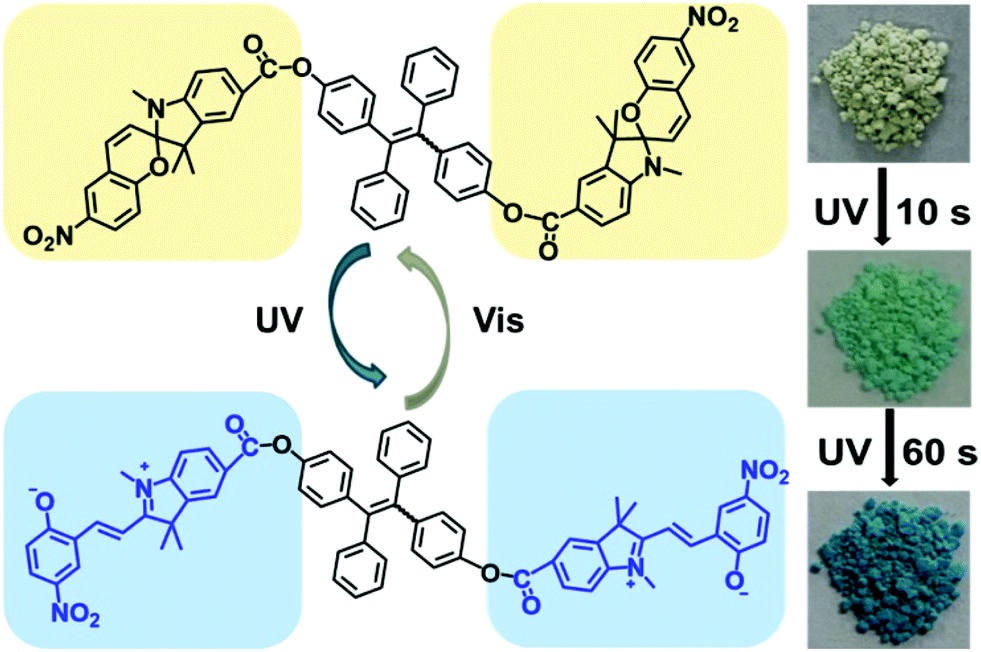 | ||
| Scheme 27 A spiropyran–tetraphenylethene–spiropyran hybrid capable of solid state photochromism. Reproduced with permission from Wu et al., copyright American Chemical Society 2018.33 | ||
Conclusions
Over the last century, the spiropyran family has proven to be an exceptionally diverse class of functional compounds, manifested in a myriad of interconnected properties (Scheme 28). Although the unravelling of their versatility started over a century ago, we are still discovering more of this widely used photochrome. More importantly, with an ever-increasing understanding of their fundamental properties, the number of possibilities increases also. Despite the thousands of reports on spiropyrans already, it is important to reflect on older work in light of more recent discoveries as in many cases new functionality could be explored in slight variations to or even directly within reported spiropyrans, since it continues to reveal new aspects of its complex chromism. However, the widespread use as a photochrome can result in the breadth of the functionality available being overlooked. This aspect is important given that the multifunctionality of spiropyrans has proven a major asset in building functional responsive materials already, and continues to play a pivotal role in discoveries that are still being made over 100 years after the discovery of spiro compounds.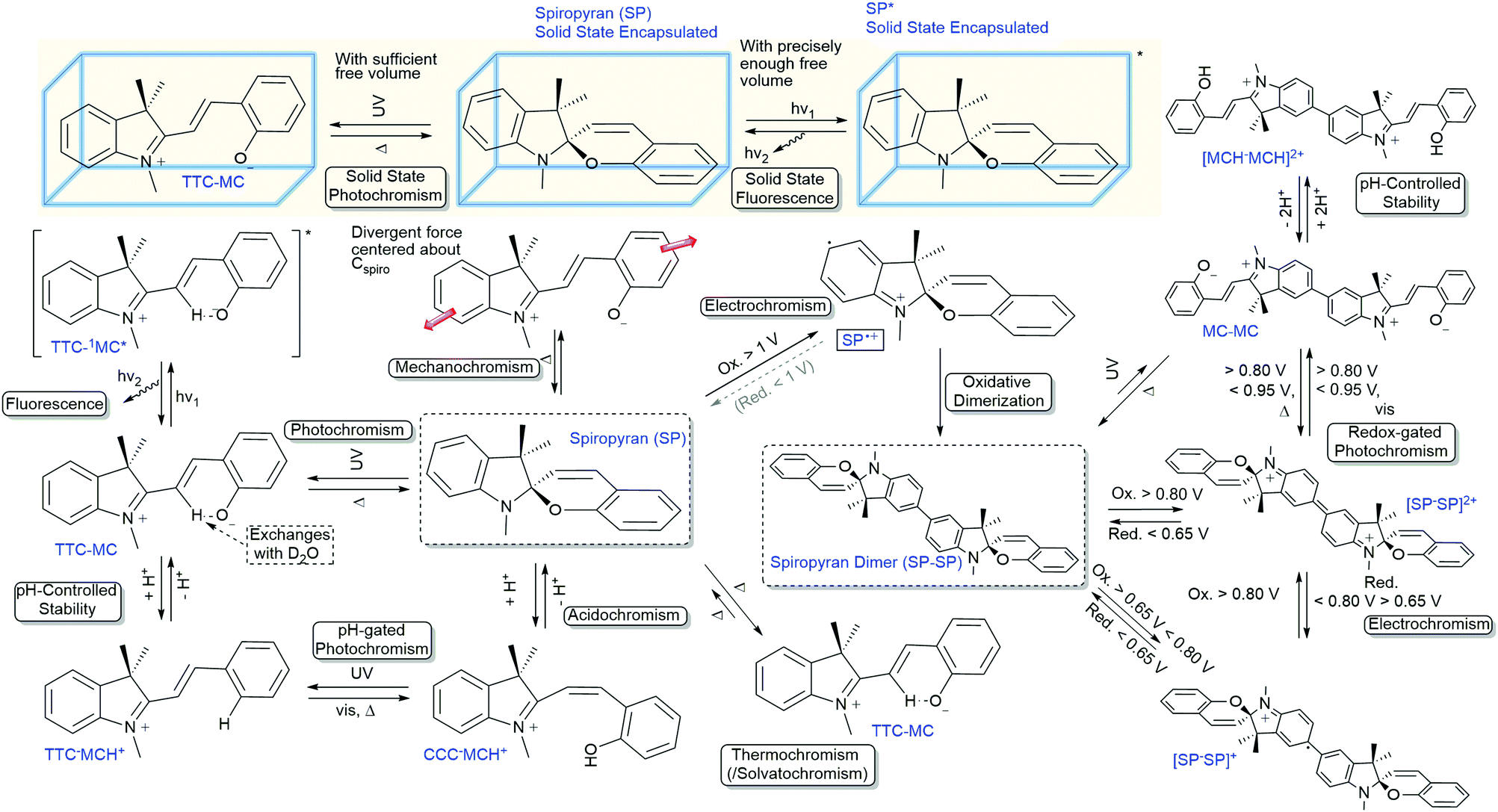 | ||
| Scheme 28 Overview of functionalities of spiropyrans and their relation. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by The Netherlands Ministry of Education, Culture and Science (Gravity Program 024.001.035 to L. K. and W. R. B.).Notes and references
- R. Yerushalmi, A. Scherz, M. E. van der Boom and H.-B. Kraatz, J. Mater. Chem., 2005, 15, 4480 RSC.
- X. Zhang, L. Chen, K. H. Lim, S. Gonuguntla, K. W. Lim, D. Pranantyo, W. P. Yong, W. J. T. Yam, Z. Low, W. J. Teo, H. P. Nien, Q. W. Loh and S. Soh, Adv. Mater., 2019, 31, 1804540 CrossRef PubMed.
- M. Li, J. Zhao, H. Chu, Y. Mi, Z. Zhou, Z. Di, M. Zhao and L. Li, Adv. Mater., 2018, 1804745 CrossRef PubMed.
- Z. L. Pianowski, Chem. – Eur. J., 2019, 25, 5128–5144 CrossRef CAS PubMed.
- B. L. Feringa and W. R. Browne, Molecular Switches, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, 2009 Search PubMed.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 1807333 CrossRef PubMed.
- Y.-M. Zhang, Y.-H. Liu and Y. Liu, Adv. Mater., 2019, 1806158 CrossRef PubMed.
- J. Boelke and S. Hecht, Adv. Opt. Mater., 2019, 1900404 CrossRef.
- C. Sun, C. Wang and R. Boulatov, ChemPhotoChem, 2019 DOI:10.1002/cptc.201900030.
- W. Francis, A. Dunne, C. Delaney, L. Florea and D. Diamond, Sens. Actuators, B, 2017, 250, 608–616 CrossRef CAS.
- D. Y. Tam, X. Zhuang, S. W. Wong and P. K. Lo, Small, 2019, 1805481 CrossRef PubMed.
- G. Moncelsi and P. Ballester, ChemPhotoChem, 2019, 1–15 Search PubMed.
- R. J. Mart and R. K. Allemann, Chem. Commun., 2016, 52, 12262–12277 RSC.
- X. Xia, H. Yu, L. Wang and Z. ul-Abdin, RSC Adv., 2016, 6, 105296 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- H. Logtenberg and W. R. Browne, Org. Biomol. Chem., 2013, 11, 233–243 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- T. P. Russell, Science, 2002, 297, 964–967 CrossRef CAS PubMed.
- Q. Zhang, D.-H. Qu and H. Tian, Adv. Opt. Mater., 2019, 1900033 CrossRef.
- C. P. Harvey and J. D. Tovar, Polym. Chem., 2011, 2, 2699 RSC.
- M. Li, Q. Zhang, Y. N. Zhou and S. Zhu, Prog. Polym. Sci., 2018, 79, 26–39 CrossRef CAS.
- C. J. Martin, G. Rapenne, T. Nakashima and T. Kawai, J. Photochem. Photobiol., C, 2018, 34, 41–51 CrossRef CAS.
- H. Decker and H. Felser, Ber. Dtsch. Chem. Ges., 1908, 41, 2997–3007 CrossRef.
- E. Fischer and O. Hess, Ber. Dtsch. Chem. Ges., 1884, 17, 559–568 CrossRef.
- E. Fischer and Y. Hirshberg, J. Chem. Soc., 1952, 4522–4524 CAS.
- F. Irving, J. Chem. Soc., 1929, 1093–1095 RSC.
- S. R. Keum, S. M. Ahn, S. J. Roh, S. J. Park, S. H. Kim and K. Kon, Magn. Reson. Chem., 2006, 44, 90–94 CrossRef CAS PubMed.
- S. R. Keum, B. S. Ku, M. H. Lee, G. Y. Chi and S. S. Lim, Dyes Pigm., 2009, 80, 26–29 CrossRef CAS.
- D. Kim, Z. Zhang and K. Xu, J. Am. Chem. Soc., 2017, 139, 9447–9450 CrossRef CAS PubMed.
- L. Kortekaas, O. Ivashenko, J. T. van Herpt and W. R. Browne, J. Am. Chem. Soc., 2016, 138, 1301–1312 CrossRef CAS PubMed.
- L. Kortekaas, J. Chen, D. Jacquemin and W. R. Browne, J. Phys. Chem. B, 2018, 122, 6423–6430 CrossRef CAS PubMed.
- Z. Wu, K. Pan, S. Mo, B. Wang, X. Zhao and M. Yin, ACS Appl. Mater. Interfaces, 2018, 10, 30879–30886 CrossRef CAS PubMed.
- P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952–7960 CrossRef CAS PubMed.
- D. Samanta, D. Galaktionova, J. Gemen, L. J. W. Shimon, Y. Diskin-Posner, L. Avram, P. Král and R. Klajn, Nat. Commun., 2018, 9, 2–10 CrossRef PubMed.
- A. Mustafa, Chem. Rev., 1948, 43, 509–523 CrossRef CAS PubMed.
- J. H. Day, Chem. Rev., 1963, 63, 65–80 CrossRef CAS.
- R. Wizinger and H. Wenning, Helv. Chim. Acta, 1940, 23, 247–271 CrossRef CAS.
- A. Löwenbein and W. Katz, Ber. Dtsch. Chem. Ges., 1926, 59, 1377–1383 CrossRef.
- C. A. Heller, D. A. Fine and R. A. Henry, J. Phys. Chem., 1961, 65, 1908–1909 CrossRef CAS.
- C. F. Koelsch, J. Org. Chem., 1951, 16, 1362–1370 CrossRef CAS.
- E. B. Knott, J. Chem. Soc., 1951, 3038–3047 RSC.
- H. Decker and T. v. Fellenberg, Justus Liebigs Ann. Chem., 1909, 364, 1–44 CrossRef CAS.
- W. Dilthey and H. Wübken, Ber. Dtsch. Chem. Ges., 1928, 61, 963–968 CrossRef.
- V. Ramamurthy and K. S. Schanze, Photochemistry of Organic Molecules in Isotropic and Anisotropic Media, Marcel Dekker, Inc., New York, 2003 Search PubMed.
- Y. Hirshberg and E. Fischer, J. Chem. Phys., 1953, 1619–1620 CrossRef CAS.
- Y. Hirshberg and E. Fischer, J. Chem. Soc., 1953, 629–636 RSC.
- J. Sunamoto, K. Iwamoto, M. Akutagawa, M. Nagase and H. Kondo, J. Am. Chem. Soc., 1982, 104, 4904–4907 CrossRef CAS.
- V. Krongauz and A. Golinelli, Polym. Bull., 1982, 6–6, 119–126 Search PubMed.
- Y. Hirshberg and E. Fischer, J. Chem. Soc., 1954, 3129–3137 RSC.
- M. Bletz, U. Pfeifer-Fukumura, U. Kolb and W. Baumann, J. Phys. Chem. A, 2002, 106, 2232–2236 CrossRef CAS.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed.
- N. P. Ernsting, B. Dick and T. Arthen-Engeland, Pure Appl. Chem., 1990, 62, 1483–1488 CAS.
- J. Hobley, V. Malatesta, R. Millini, L. Montanari and W. O Neil Parker, Jr, Phys. Chem. Chem. Phys., 1999, 1, 3259–3267 RSC.
- M. Kullmann, S. Ruetzel, J. Buback, P. Nuernberger and T. Brixner, J. Am. Chem. Soc., 2011, 133, 13074–13080 CrossRef CAS PubMed.
- S. M. Aldoshin, L. O. Atovmyan, O. A. D’yachenko and M. A. Gal’bershtam, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1981, 30, 2262–2270 CrossRef.
- J. Hobley, V. Malatesta, W. Giroldini and W. Stringo, Phys. Chem. Chem. Phys., 2000, 2, 53–56 RSC.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CrossRef CAS PubMed.
- Y. Hirshberg and E. Fischer, J. Chem. Soc., 1954, 297–303 RSC.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, 2006 Search PubMed.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, 1991 Search PubMed.
- S. M. Aldoshin and L. O. Atovmyan, Izv. Akad. Nauk SSSR, Ser. Khim., 1985, 180–182 Search PubMed.
- P. Uznanski, Synth. Met., 2000, 109, 281–285 CrossRef CAS.
- Y. Shiraishi, E. Shirakawa, K. Tanaka, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Appl. Mater. Interfaces, 2014, 6, 7554–7562 CrossRef CAS PubMed.
- Y. Hirshberg, E. H. Frei and E. Fischer, J. Chem. Soc., 1953, 2184–2185 CAS.
- J. Buback, M. Kullmann, F. Langhojer, P. Nuernberger, R. Schmidt, F. Würthner and T. Brixner, J. Am. Chem. Soc., 2010, 132, 16510–16519 CrossRef CAS PubMed.
- J. L. Bahr, G. Kodis, L. De la Garza, S. Lin, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2001, 123, 7124–7133 CrossRef CAS PubMed.
- T. Stafforst and D. Hilvert, Chem. Commun., 2009, 287–288 RSC.
- A. Fissi, O. Pieroni, N. Angelini and F. Lenci, Macromolecules, 1999, 32, 7116–7121 CrossRef CAS.
- C. Fleming, S. Li, M. Grøtli and J. Andréasson, J. Am. Chem. Soc., 2018, 140, 14069–14072 CrossRef CAS PubMed.
- J. Widengren and C. A. M. Seidel, Phys. Chem. Chem. Phys., 2000, 2, 3435–3441 RSC.
- A. V. Metelitsa, I. V. Dorogan, B. S. Lukyanov, V. I. Minkin, S. O. Besugliy and J. C. Micheau, Mol. Cryst. Liq. Cryst., 2005, 430, 45–52 CrossRef CAS.
- V. I. Minkin, A. V. Metelitsa, I. V. Dorogan, B. S. Lukyanov, S. O. Besugliy and J. C. Micheau, J. Phys. Chem. A, 2005, 109, 9605–9616 CrossRef CAS PubMed.
- L. Ma, J. Li, D. Han, H. Geng, G. Chen and Q. Li, Macromol. Chem. Phys., 2013, 214, 716–725 CrossRef CAS.
- C. Q. Huang, Y. Wang, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2011, 32, 1174–1179 CrossRef CAS PubMed.
- L. Zhu, W. Wu, M. Q. Zhu, J. J. Han, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 3524–3526 CrossRef CAS PubMed.
- M. Q. Zhu, L. Zhu, J. J. Han, W. Wu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 4303–4309 CrossRef CAS PubMed.
- M. Q. Zhu, G. F. Zhang, Z. Hu, M. P. Aldred, C. Li, W. L. Gong, T. Chen, Z. L. Huang and S. Liu, Macromolecules, 2014, 47, 1543–1552 CrossRef CAS.
- M. Q. Zhu, G. F. Zhang, C. Li, M. P. Aldred, E. Chang, R. A. Drezek and A. D. Q. Li, J. Am. Chem. Soc., 2011, 133, 365–372 CrossRef CAS PubMed.
- J. Chen, F. Zeng, S. Wu, Q. Chen and Z. Tong, Chem. – Eur. J., 2008, 14, 4851–4860 CrossRef CAS PubMed.
- P. K. Kundu, G. L. Olsen, V. Kiss and R. Klajn, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- T. Minami, N. Tamai, T. Yamazaki and I. Yamazaki, J. Phys. Chem., 1991, 95, 3988–3993 CrossRef CAS.
- P. Nuernberger, S. Ruetzel and T. Brixner, Angew. Chem., Int. Ed., 2015, 54, 11368–11386 CrossRef CAS.
- C. J. Wohl and D. Kuciauskas, J. Phys. Chem. B, 2005, 109, 22186–22191 CrossRef CAS PubMed.
- S. V. Paramonov, V. Lokshin and O. A. Fedorova, J. Photochem. Photobiol., C, 2011, 12, 209–236 CrossRef CAS.
- C. J. Roxburgh and P. G. Sammes, Dyes Pigm., 1995, 27, 63–69 CrossRef CAS.
- J. Zhou, Y. Li, Y. Tang, F. Zhao, X. Song and E. Li, J. Photochem. Photobiol., A, 1995, 90, 117–123 CrossRef CAS.
- H. Shiozaki, Dyes Pigm., 1997, 33, 229–237 CrossRef CAS.
- A. Sugahara, N. Tanaka, A. Okazawa, N. Matsushita and N. Kojima, Chem. Lett., 2014, 43, 281–283 CrossRef CAS.
- S. B. Schmidt, F. Kempe, O. Brügner, M. Walter and M. Sommer, Polym. Chem., 2017, 5407–5414 RSC.
- F. M. Raymo and S. Giordani, J. Am. Chem. Soc., 2001, 123, 4651–4652 CrossRef CAS.
- F. M. Raymo, S. Giordani, A. J. P. White and D. J. Williams, J. Org. Chem., 2003, 68, 4158–4169 CrossRef CAS PubMed.
- M. E. Genovese, E. Colusso, M. Colombo, A. Martucci, A. Athanassiou and D. Fragouli, J. Mater. Chem. A, 2017, 5, 339–348 RSC.
- M. E. Genovese, A. Athanassiou and D. Fragouli, J. Mater. Chem. A, 2015, 3, 22441–22447 RSC.
- J. T. C. Wojtyk, A. Wasey, N. N. Xiao, P. M. Kazmaier, S. Hoz, C. Yu, R. P. Lemieux and E. Buncel, J. Phys. Chem. A, 2007, 111, 2511–2516 CrossRef CAS.
- L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 17438–17441 CrossRef CAS PubMed.
- S. Silvi, E. C. Constable, C. E. Housecroft, J. E. Beves, E. L. Dunphy, M. Tomasulo, F. M. Raymo and A. Credi, Chem. Commun., 2009, 1484 RSC.
- S. R. Keum, K. B. Lee, P. M. Kazmaier and E. Buncel, Tetrahedron Lett., 1994, 35, 1015–1018 CrossRef CAS.
- J. Vallet, J. C. Micheau and C. Coudret, Dyes Pigm., 2016, 125, 179–184 CrossRef CAS.
- C. Liu, D. Yang, Q. Jin, L. Zhang and M. Liu, Adv. Mater., 2016, 28, 1644–1649 CrossRef CAS PubMed.
- K. Sumaru, T. Takagi, T. Satoh and T. Kanamori, Macromol. Rapid Commun., 2018, 39, 1–6 CrossRef PubMed.
- Z. Zhao and J. Tian, J. Appl. Polym. Sci., 2017, 134, 1–9 Search PubMed.
- P. Remon, S. M. Li, M. Grotli, U. Pischel and J. Andreasson, Chem. Commun., 2016, 52, 4659–4662 RSC.
- L. Cui, H. Zhang, G. Zhang, Y. Zhou, L. Fan, L. Shi, C. Zhang, S. Shuang and C. Dong, Spectrochim. Acta, Part A, 2018, 202, 13–17 CrossRef CAS PubMed.
- T. Khalil, A. Alharbi, C. Baum and Y. Liao, Macromol. Rapid Commun., 2018, 39, 1–5 CrossRef PubMed.
- K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 4949–4955 CrossRef CAS.
- K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 7854–7856 CrossRef CAS.
- T. Satoh, K. Sumaru, T. Takagi, K. Takai and T. Kanamori, Phys. Chem. Chem. Phys., 2011, 13, 7322–7329 RSC.
- C. Kaiser, T. Halbritter, A. Heckel and J. Wachtveitl, ChemistrySelect, 2017, 2, 4111–4123 CrossRef CAS.
- M. Colaço, A. Carletta, M. Van Gysel, K. Robeyns, N. Tumanov and J. Wouters, ChemistryOpen, 2018, 7, 520–526 CrossRef PubMed.
- I. Shimizu, H. Kokado and E. Inoue, J. Soc. Chem. Ind., Jpn., 1967, 2344–2348 CAS.
- Y. Liao, Acc. Chem. Res., 2017, 50, 1956–1964 CrossRef CAS PubMed.
- H. Bao, F. Li, L. Lei, B. Yang and Z. Li, RSC Adv., 2014, 4, 27277–27280 RSC.
- V. K. Johns, P. K. Patel, S. Hassett, P. Calvo-Marzal, Y. Qin and K. Y. Chumbimuni-Torres, Anal. Chem., 2014, 86, 6184–6187 CrossRef CAS PubMed.
- L. Florea, K. Wagner, P. Wagner, G. G. Wallace, F. Benito-Lopez, D. L. Officer and D. Diamond, Adv. Mater., 2014, 26, 7339–7345 CrossRef CAS PubMed.
- V. K. Johns, Z. Wang, X. Li and Y. Liao, J. Phys. Chem. A, 2013, 117, 13101–13104 CrossRef CAS PubMed.
- Q. Yu, X. Su, T. Zhang, Y.-M. Zhang, M. Li, Y. Liu and S. X.-A. Zhang, J. Mater. Chem. C, 2018, 6, 2113–2122 RSC.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed.
- Y. Luo, C. Wang, P. Peng, M. Hossain, T. Jiang, W. Fu, Y. Liao and M. Su, J. Mater. Chem. B, 2013, 1, 997–1001 RSC.
- Z. Wang, V. K. Johns and Y. Liao, Chem. – Eur. J., 2014, 20, 14637–14640 CrossRef CAS PubMed.
- C. Maity, W. E. Hendriksen, J. H. Van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998–1001 CrossRef CAS PubMed.
- N. Abeyrathna and Y. Liao, J. Phys. Org. Chem., 2017, 30, 1–5 CrossRef.
- J. Kohl-Landgraf, M. Braun, C. Özçoban, D. P. N. Gonçalves, A. Heckel and J. Wachtveitl, J. Am. Chem. Soc., 2012, 134, 14070–14077 CrossRef CAS PubMed.
- T. Halbritter, C. Kaiser, J. Wachtveitl and A. Heckel, J. Org. Chem., 2017, 82, 8040–8047 CrossRef CAS PubMed.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS.
- J. E. Stumpel, D. Liu, D. J. Broer and A. P. H. J. Schenning, Chem. – Eur. J., 2013, 19, 10922–10927 CrossRef CAS.
- F. M. Raymo, R. J. Alvarado, S. Giordani and M. A. Cejas, J. Am. Chem. Soc., 2003, 125, 2361–2364 CrossRef CAS.
- C. W. Lee, Y. H. Song, Y. Lee, K. S. Ryu and K. W. Chi, Chem. Commun., 2009, 6282–6284 RSC.
- L. Chen, X. Yao, Z. Gu, K. Zheng, C. Zhao, W. Lei, Q. Rong, L. Lin, J. Wang, L. Jiang and M. Liu, Chem. Sci., 2017, 8, 2010–2016 RSC.
- J. ter Schiphorst, M. van den Broek, T. de Koning, J. N. Murphy, A. P. H. J. Schenning and A. C. C. Esteves, J. Mater. Chem. A, 2016, 4, 8676–8681 RSC.
- M. Zhang, X. Hou, J. Wang, Y. Tian, X. Fan, J. Zhai and L. Jiang, Adv. Mater., 2012, 24, 2424–2428 CrossRef CAS PubMed.
- T. Satoh, K. Sumaru, T. Takagi and T. Kanamori, Soft Matter, 2011, 7, 8030–8034 RSC.
- B. Ziółkowski, L. Florea, J. Theobald, F. Benito-Lopez and D. Diamond, J. Mater. Sci., 2016, 51, 1392–1399 CrossRef.
- J. E. Stumpel, B. Ziółkowski, L. Florea, D. Diamond, D. J. Broer and A. P. H. J. Schenning, ACS Appl. Mater. Interfaces, 2014, 6, 7268–7274 CrossRef CAS PubMed.
- B. Ziółkowski, L. Florea, J. Theobald, F. Benito-Lopez and D. Diamond, Soft Matter, 2013, 9, 8754 RSC.
- X. Xie, G. A. Crespo, G. Mistlberger and E. Bakker, Nat. Chem., 2014, 6, 202–207 CrossRef CAS.
- S. Silvi, A. Arduini, A. Pochini, A. Secchi, M. Tomasulo, F. M. Raymo, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2007, 129, 13378–13379 CrossRef CAS PubMed.
- J. Guo, H.-Y. Zhang, Y. Zhou and Y. Liu, Chem. Commun., 2017, 53, 6089–6092 RSC.
- M. Campredon, G. Giusti, R. Guglielmetti, A. Samat, G. Gronchi, A. Alberti and M. Benaglia, J. Chem. Soc., Perkin Trans. 2, 1993, 2089 RSC.
- J. F. Zhi, R. Baba, K. Hashimoto and A. Fujishima, J. Photochem. Photobiol., A, 1995, 92, 91–97 CrossRef CAS.
- J. F. Zhi, R. Baba, K. Hashimoto and A. Fujishima, Ber. Bunsen-Ges., 1995, 99, 32–39 CrossRef CAS.
- M. Campredon, A. Samat, R. Guglielmetti and A. Alberti, Gazz. Chim. Ital., 1993, 261 CAS.
- M. Campredon, G. Giusti, R. Guglielmetti, A. Samat, G. Gronchi, A. Alberti and M. Benaglia, J. Chem. Soc., Perkin Trans. 2, 1993, 2089 RSC.
- J. F. Zhi, R. Baba and A. Fujishima, Ber. Bunsen-Ges., 1996, 1802–1807 CrossRef CAS.
- M. J. Preigh, M. T. Stauffer, F.-T. Lin and S. G. Weber, J. Chem. Soc., Faraday Trans., 1996, 92, 3991 RSC.
- A. Doménech, H. García, I. Casades and M. Esplá, J. Phys. Chem. B, 2004, 108, 20064–20075 CrossRef.
- K. Wagner, R. Byrne, M. Zanoni, S. Gambhir, L. Dennany, R. Breukers, M. Higgins, P. Wagner, D. Diamond, G. G. Wallace and D. L. Officer, J. Am. Chem. Soc., 2011, 133, 5453–5462 CrossRef CAS PubMed.
- M. Natali and S. Giordani, Org. Biomol. Chem., 2012, 10, 1162–1171 RSC.
- O. Ivashenko, J. T. van Herpt, P. Rudolf, B. L. Feringa and W. R. Browne, Chem. Commun., 2013, 49, 6737 RSC.
- O. Ivashenko, J. T. van Herpt, B. L. Feringa, P. Rudolf and W. R. Browne, J. Phys. Chem. C, 2013, 117, 18567–18577 CrossRef CAS.
- T. Bai, A. Sinclair, F. Sun, P. Jain, H.-C. Hung, P. Zhang, J.-R. Ella-Menye, W. Liu and S. Jiang, Chem. Sci., 2016, 7, 333–338 RSC.
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC.
- L. Huang, C. Wu, L. Zhang, Z. Ma and X. Jia, ACS Appl. Mater. Interfaces, 2018, 10, 34475–34484 CrossRef CAS PubMed.
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS.
- M. Sommer and H. Komber, Macromol. Rapid Commun., 2013, 34, 57–62 CrossRef CAS.
- C. Xing, L. Wang, L. Xian, Y. Wang, L. Zhang, K. Xi, Q. Zhang and X. Jia, Macromol. Chem. Phys., 2018, 219, 1–8 CrossRef.
- Y. Jia, W. J. Wang, B. G. Li and S. Zhu, Macromol. Mater. Eng., 2018, 1800154, 1–10 Search PubMed.
- M. H. Barbee, K. Mondal, J. Z. Deng, V. Bharambe, T. V. Neumann, J. Adams, N. Boechler, M. D. Dickey and S. L. Craig, ACS Appl. Mater. Interfaces, 2018, 10, 29918–29924 CrossRef CAS PubMed.
- M. Raisch, D. Genovese, N. Zaccheroni, S. B. Schmidt, M. L. Focarete, M. Sommer and C. Gualandi, Adv. Mater., 2018, 1802813 CrossRef PubMed.
- L. Florea, S. Scarmagnani, F. Benito-Lopez and D. Diamond, Chem. Commun., 2014, 50, 924–926 RSC.
- J. Harada, Y. Kawazoe and K. Ogawa, Chem. Commun., 2010, 46, 2593–2595 RSC.
- D. E. Williams, C. R. Martin, E. A. Dolgopolova, A. Swifton, D. C. Godfrey, O. A. Ejegbavwo, P. J. Pellechia, M. D. Smith and N. B. Shustova, J. Am. Chem. Soc., 2018, 140, 7611–7622 CrossRef CAS PubMed.
- H. A. Schwartz, U. Ruschewitz and L. Heinke, Photochem. Photobiol. Sci., 2018, 17, 864–873 RSC.
- H. A. Schwartz, S. Olthof, D. Schaniel, K. Meerholz and U. Ruschewitz, Inorg. Chem., 2017, 56, 13100–13110 CrossRef CAS PubMed.
- I. M. Heilbron, D. H. Hey and A. Lowe, J. Chem. Soc., 1936, 1380–1383 RSC.
- Y. Shiraishi, K. Adachi, M. Itoh and T. Hirai, Org. Lett., 2009, 11, 3482–3485 CrossRef CAS.
Footnotes |
| † It should be noted that the non-photochromic spiropyrans in both reviews from 194836 and 196337 overlooked a correction to two of the structures and their associated derivatives,165 the misinterpretation leading to the incorrect assumption that they were exceptions to the general rules. |
| ‡ Although not discussed here, attack of the electrophilic Cspiro centre in merocyanine by a nucleophile, such as cyanide, can also result in stabilization of the ring-open form.166 |
| This journal is © The Royal Society of Chemistry 2019 |