
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic self-sufficient hydride transfer processes
Erika
Tassano
 and
Mélanie
Hall
*
and
Mélanie
Hall
*
Department of Chemistry, University of Graz, Heinrichstrasse 28, 8010, Graz, Austria. E-mail: melanie.hall@uni-graz.at
First published on 1st November 2019
Abstract
A number of self-sufficient hydride transfer processes have been reported in biocatalysis, with a common feature being the dependence on nicotinamide as a cofactor. This cofactor is provided in catalytic amounts and serves as a hydride shuttle to connect two or more enzymatic redox events, usually ensuring overall redox neutrality. Creative systems were designed to produce synthetic sequences characterized by high hydride economy, typically going in hand with excellent atom economy. Several redox enzymes have been successfully combined in one-pot one-step to allow functionalization of a large variety of molecules while preventing by-product formation. This review analyzes and classifies the various strategies, with a strong focus on efficiency, which is evaluated here in terms of the hydride economy and measured by the turnover number of the nicotinamide cofactor(s). The review ends with a critical evaluation of the reported systems and highlights areas where further improvements might be desirable.
 Erika Tassano | Erika Tassano, born in 1985, received her undergraduate education in Chemistry and Pharmaceutical Technologies from the University of Genova (Italy), where she graduated in Chemical Sciences and Technologies in 2016 under the supervision of Prof. Luca Banfi. During her PhD studies, she joined the group of Prof. Kurt Faber at the University of Graz (Austria) as an ‘Ernst Mach' fellow, financed by the Austrian Federal Ministry of Education, Science and Research (BMBWF). She then worked as a postdoctoral fellow with Dr Mélanie Hall in the Department of Chemistry at the University of Graz, focusing on the development of novel biocatalytic redox-neutral processes and the enzymatic synthesis of biologically active compounds. |
 Mélanie Hall | Mélanie Hall, born 1980 in Brest (France), obtained her Master's degree in Chemistry from the National Graduate School of Chemistry (ENSCR) in Rennes, France, and received her PhD degree (2007) in Chemistry from the University of Graz, Austria, under the supervision of Prof. Kurt Faber. After conducting postdoctoral research with Prof. Andy Bommarius at the Georgia Institute of Technology in Atlanta, USA, she returned to the University of Graz as a University Assistant (2010). She obtained her Habilitation in Organic Chemistry in 2016 and is currently an independent group leader in the Department of Chemistry. Her research focuses on the development of biocatalysis for organic synthesis. |
1. Introduction
An important challenge for today's synthetic chemists is to design novel synthetic routes that are superior in terms of yield and selectivity and are cost-effective, while displaying low environmental impact. The borrowing hydrogen strategy in this regard is highly attractive as it combines oxidation and hydrogenation in a highly atom-efficient reaction scheme, targeting redox neutrality.1,2 Homogeneous and heterogeneous catalysis have relied on this concept for a large variety of transformations, while the field of biocatalysis only starts to implement more broadly equivalent strategies with added benefits, such as regio- and stereo-selectivity.The borrowing hydrogen strategy, also known as hydrogen autotransfer, implies that hydrogen (H-atom) is first abstracted from a substrate by a catalyst, and returned in a subsequent step to the intermediate previously generated, thereby delivering the final product that eventually (re)incorporates the hydrogen (oxidation–hydrogenation sequence). Ideally, there is no net change in the hydrogen count. Additional steps are usually added between or along the two redox reactions to increase the synthetic value and chemical complexity of the final product. Typical examples are the alkylation of amines by alcohols or amines, the β-functionalization of alcohols, C–C-bond forming reactions from alcohols or net alkane metathesis.1,3–7 Conceptually closely related but mechanistically distinct are intramolecular hydride shifts connected to redox-neutral C–H functionalization, which covers a broad range of reactions. In particular, 1,5-hydride shifts are involved in a number of ring closure reactions via formation of new C–C-, C–O- and C–N-bonds.8 In biocatalysis, only hydride transfer reactions can be practically considered since no enzyme has yet been shown able to catalyze transfer (de)hydrogenation reactions involving net transfer of H2 between two organic molecules. Although biological – mostly photo- – production of H2 could in theory be coupled to biosynthetic reductive processes, such an approach would however not follow a transfer hydrogenation scheme; most hydrogenases indeed transfer hydrogen atoms from H2 indirectly in the form of hydrides via external ‘temporary’ electron acceptors, such as a nicotinamide cofactor.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9–13 Exceptions are [Fe]-hydrogenases, which can directly transfer a hydride from H2 onto the organic molecule N5,N10-methenyl-5,6,7,8-tetrahydromethanopterin.14,15
9–13 Exceptions are [Fe]-hydrogenases, which can directly transfer a hydride from H2 onto the organic molecule N5,N10-methenyl-5,6,7,8-tetrahydromethanopterin.14,15
Systematic analysis of enzymatic strategies for hydride transfer in closed systems,16,17 so-called redox-neutral biotransformations, is currently lacking, although the potential and value of enzymes in synthesis are now well recognized.18,19 In this review, the concept of hydride transfer reactions in biocatalysis will be presented and analyzed, with particular attention to redox neutrality achieved through internal shuffling of hydrides, i.e., between at least two different redox (half-)reactions. This generates redox self-sufficient systems, and usually results in no loss of hydride to the ‘outside’. Briefly, this concept consists in designing a synthetic scheme combining at least two reactions catalyzed by redox enzymes, in which the hydride in focus of the reaction of interest is either abstracted from or donated to the substrate and donated to or abstracted from a hydride shuttle, respectively, in the form of a cofactor (nicotinamide molecule, vide infra). This step is followed by the reverse hydride event (donation or abstraction, respectively) with the formed intermediate product to deliver the final product, either through exchange with the hydride shuttle directly or via an auxiliary molecule necessary as a co-substrate for the reaction. An ideal system relies on catalytic amounts of the hydride shuttle and releases no waste. In most cases, the ‘moving’ pair of electrons formally remains in the starting material, and the system can be considered redox neutral (Scheme 1).
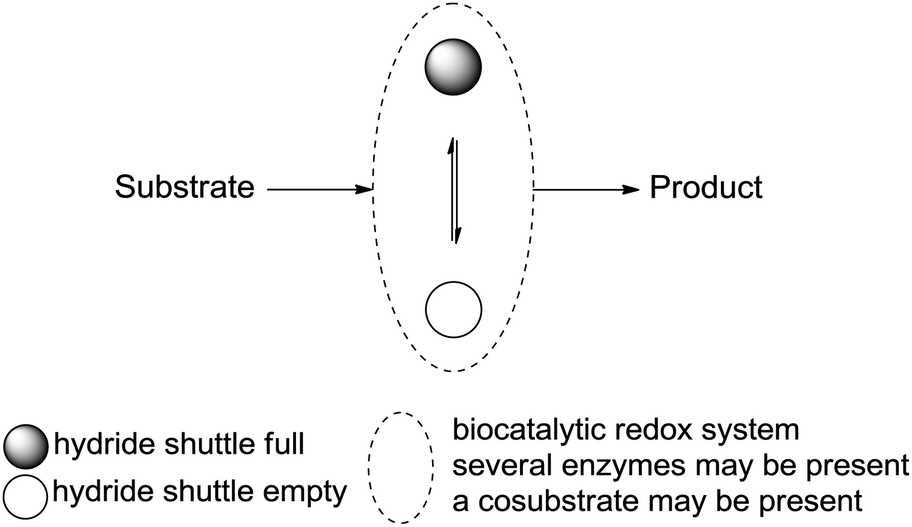 | ||
| Scheme 1 Representation of enzymatic self-sufficient hydride transfer processes. The hydride shuttle used in catalytic quantities is typically a nicotinamide cofactor (full: reduced; empty: oxidized). Ideally, no electron is lost to ‘the outside’ and no waste is produced. | ||
Such reaction schemes are in stark contrast to the nicotinamide cofactor regeneration strategy commonly employed in in vitro biocatalysis, which relies either on the coupled-substrate or the coupled-enzyme approach (Scheme 2).20,21 In the first case, one enzyme is sufficient to carry out the redox reaction of interest as well as the recycling of the nicotinamide cofactor through conversion of a sacrificial substrate. A typical example is the alcohol dehydrogenase-catalyzed reduction of ketones with concurrent oxidation of isopropanol as the co-substrate to acetone. In the latter case, a second enzyme is responsible for the regeneration of the cofactor in the opposite redox direction to that of the reaction of interest and also requires a co-substrate. Exemplary is the use of glucose dehydrogenase for the nicotinamide-dependent oxidation of glucose to gluconolactone, while the substrate of interest is reduced in the first catalytic event. In both cases, a co-product is generated in at least molar equivalent to the targeted reaction product. While these strategies present economical advantages (only catalytic equivalents of the expensive cofactor are needed) and may benefit from favorable thermodynamics when equilibrium reactions are engaged, it may on a large scale pose challenging issues when the product is to be separated from a complex reaction mixture.22 An exception is encountered in the co-product free H2/hydrogenase-based coupled-enzyme strategy, which is attracting renewed interest for atom-efficient nicotinamide cofactor recycling.23,24 Excellent reviews and book chapters on the topic of nicotinamide cofactor regeneration are available to the interested reader and such systems, which generate co-products, are out of scope here.12,21,25–28
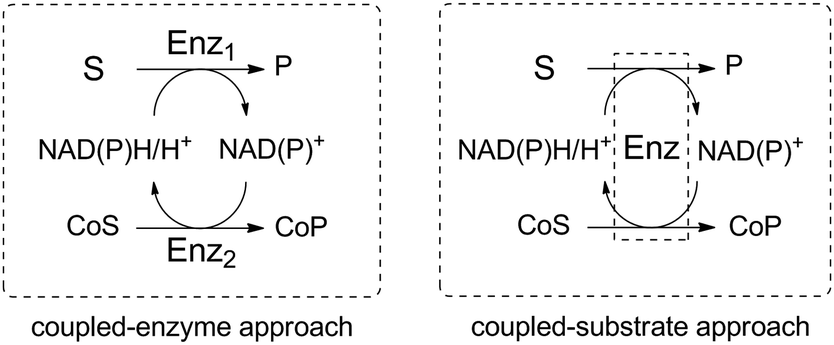 | ||
| Scheme 2 Two main approaches in the nicotinamide (NADP or NAD) cofactor recycling strategy in in vitro biocatalysis. S: substrate; P: product, Enz: enzyme; CoS: cosubstrate; CoP: coproduct. In this depiction, the reaction of interest oxidizes the cofactor, which gets reduced in the regeneration cycle; the same concept can be applied in the opposite redox direction. | ||
Nicotinamide adenine dinucleotide (phosphate) [NAD(P)] is the cofactor of choice for biological redox reactions. It operates by single transfer of two electrons in the form of a hydride (Scheme 3) either directly with the substrate or through redox active mediators, such as flavin coenzymes (flavin mononucleotide FMN or flavin adenine dinucleotide FAD). The standard reduction potential of the pair NAD(P)+/NADPH according to
| NAD(P)+ + H+ + 2e− → NAD(P)H |
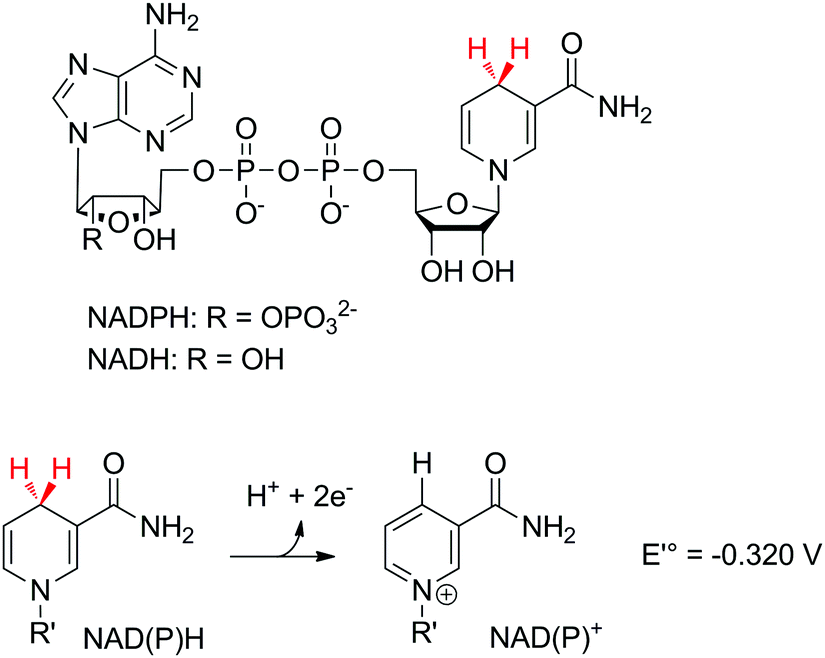 | ||
| Scheme 3 Oxidation of nicotinamide adenine dinucleotide (phosphate) [NAD(P)] and standard redox potential of the pair NAD(P)+/NADPH (at pH 7). H+/2e− are usually abstracted as H− by nicotinamide-dependent enzymes (highlighted in red). | ||
In this review, we shall concentrate on enzymatic redox reactions which utilize NAD(P) as a hydride shuttle, not excluding those enzymes which rely on an additional mediator (flavin, heme-bound iron, iron–sulfur cluster). Importantly, NAD(P) is usually bound non-covalently and can diffuse in and out of the enzymes, depending on the enzyme affinity for the cofactor. For in vitro biocatalytic applications, NAD(P) must therefore be added externally and its prohibitive cost has supported the development of regeneration strategies (vide supra, and Scheme 2). For in vivo biocatalysis, resting cells, which over-express the enzyme(s) of interest, are typically employed. These cells, though not growing anymore, contain a certain amount of nicotinamide as a mixture of NAD+, NADH, NADP+ and NADPH; nevertheless, in such cases, an external cofactor tends to be generally added (see example of Scheme 18), usually to compensate for the difficult-to-balance reaction rates of the several reactions at play. Similarly to the case of nicotinamide regeneration strategies, the use of resting cells on a large scale may be associated with challenging downstream processing operations, in particular when removal of cells hinders product isolation and recovery.22 The case of growing cells will not be considered here since the flux of electrons in the reaction(s) of interest is eventually controlled by the source of nutrients (usually glucose) and does not fit the scope of this review.
The concept of atom economy lately emerged in organic chemistry as a main driver for innovation in reaction design, pushed by the necessity to comply with stricter environmental legislations and economical constraints.33 The concept of atom economy was initially coined to pertinently relate to efficiency in synthesis, which should display high selectivity and economy in atom count.34,35 In the most synthetically efficient cases, what goes in as reactant(s) should come out as product(s), thereby no waste is generated. Only later did redox economy become a point of concern in designing total synthesis.36–38 In this review, we will focus instead on redox neutrality, showcasing artificial biosynthetic schemes with at least two enzymatic redox transformations, which though not complying with strict redox economy are highlighted for their hydride economy. In an ideal case, a reaction sequence is redox self-sufficient and is characterized by a high turnover number for the hydride shuttle; this practically translates into catalytic amounts of nicotinamide and full conversion of the substrate into the targeted product. When pertinent, the atom economy will be discussed, bearing in mind that the most atom economical synthesis does not produce any waste and incorporates all atoms of the reactants in the final product. In biotransformations that run mostly in aqueous media, the release of water as the environmentally benign sole by-product is clearly not a concern. In these cases – typically involving the action of a monooxygenase – internal hydride shuffling is limited to one cycle and the hydride from the substrate ends up in water as the sole by-product through formal reduction of oxygen. While not undergoing strictly redox-neutral transformations, these systems are being reviewed here, as redox self-sufficiency in the presence of air is achieved.
The scope of enzymatic nicotinamide-dependent sequences fulfilling the criteria of self-sufficient hydride transfer is vast and covers racemization and stereoinversion reactions, formal amination of alcohols and α-hydroxy-acids, formal oxo-functionalization of unactivated C–H bonds, redox isomerization of allylic alcohols and γ-oxo-lactols, disproportionation of aldehydes, and formation of cyclic amines and lactones (Scheme 4). In this review, we propose a classification of such enzymatic nicotinamide-dependent self-sufficient hydride transfer systems, which overall distinguishes between processes based on the number (one, two or more) of substrates, enzymes, and/or products (Scheme 5). Special cases will be highlighted, in which the overall process results in a formal intramolecular hydride transfer, such as when the hydride removed from the substrate in the first step ends up in the product through the last step, thereby mimicking the borrowing hydrogen strategy in chemistry. For each scenario, relevant examples will be presented and their applicability discussed in terms of efficiency, represented by the maximum number of turnovers that the cofactor can practically sustain. To be viable, each reported sequence must operate as a one-pot enzymatic cascade,39,40 since the reactions in the sequence are tightly controlled and connected by the hydride shuttle in catalytic amounts. In multi-enzymatic systems, this implies that the biocatalysts must all display sufficient catalytic activity under the chosen reaction conditions (pH, temperature, pressure, buffer salts, co-solvent).
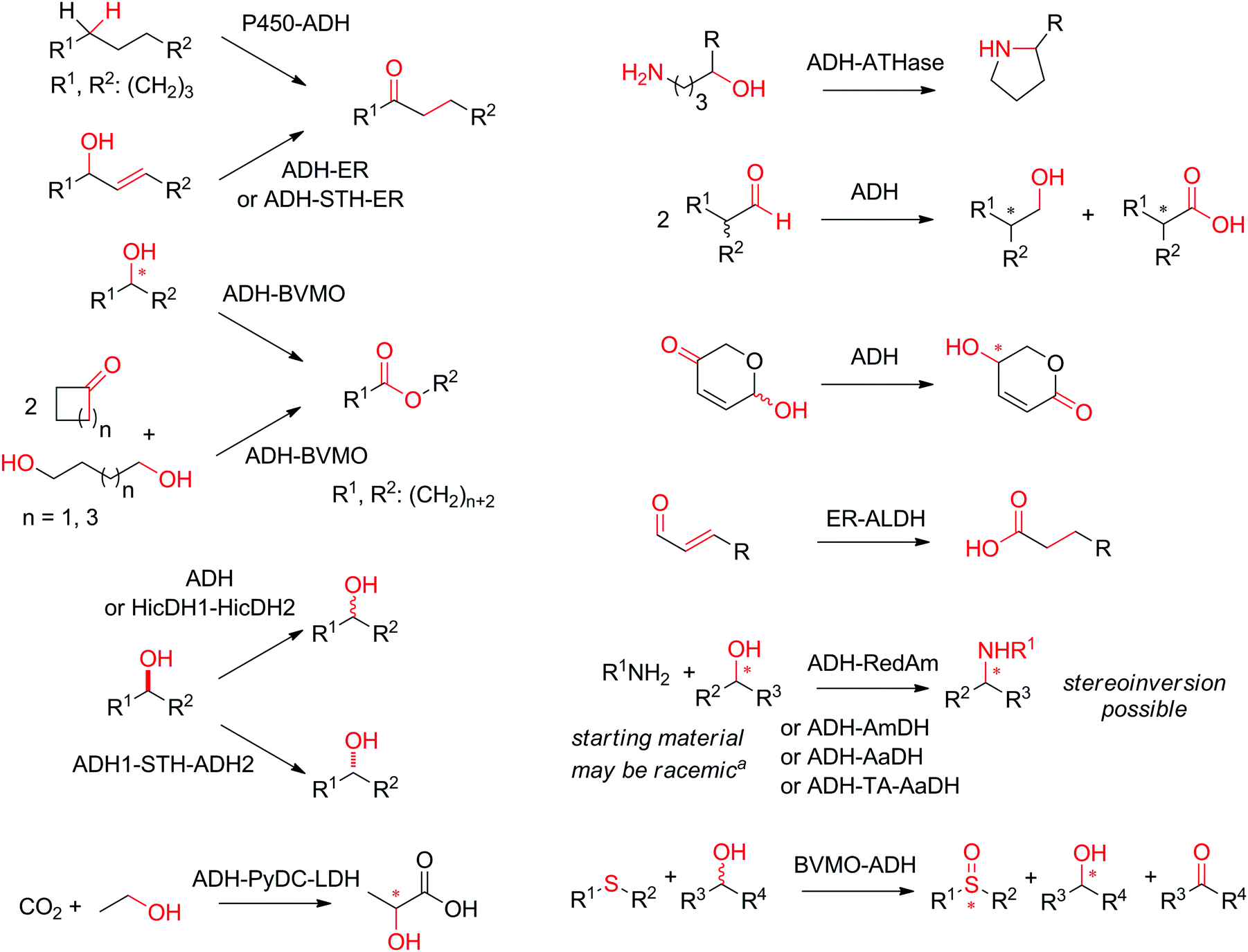 | ||
| Scheme 4 Scope of enzymatic nicotinamide-dependent sequences fulfilling the criteria of self-sufficient hydride transfer. ADH: alcohol dehydrogenase; P450: P450 monooxygenase; ER: ene-reductase; STH: pyridine nucleotide transhydrogenase; BVMO: Baeyer–Villiger monooxygenase; HicDH: α-hydroxyacid dehydrogenase; PyDC: pyruvate decarboxylase; LDH: lactate dehydrogenase; ATHase: artificial transfer hydrogenase; ALDH: aldehyde dehydrogenase; RedAm: reductive aminase; AmDH: amine dehydrogenase; AaDH: amino acid dehydrogenase; TA: transaminase. aIn the case of a racemic alcohol, two stereocomplementary ADHs are employed or a non-stereoselective ADH. * Denotes a chiral center and a fixed absolute configuration. All sequences depend on catalytic NAD(P), not displayed for clarity. | ||
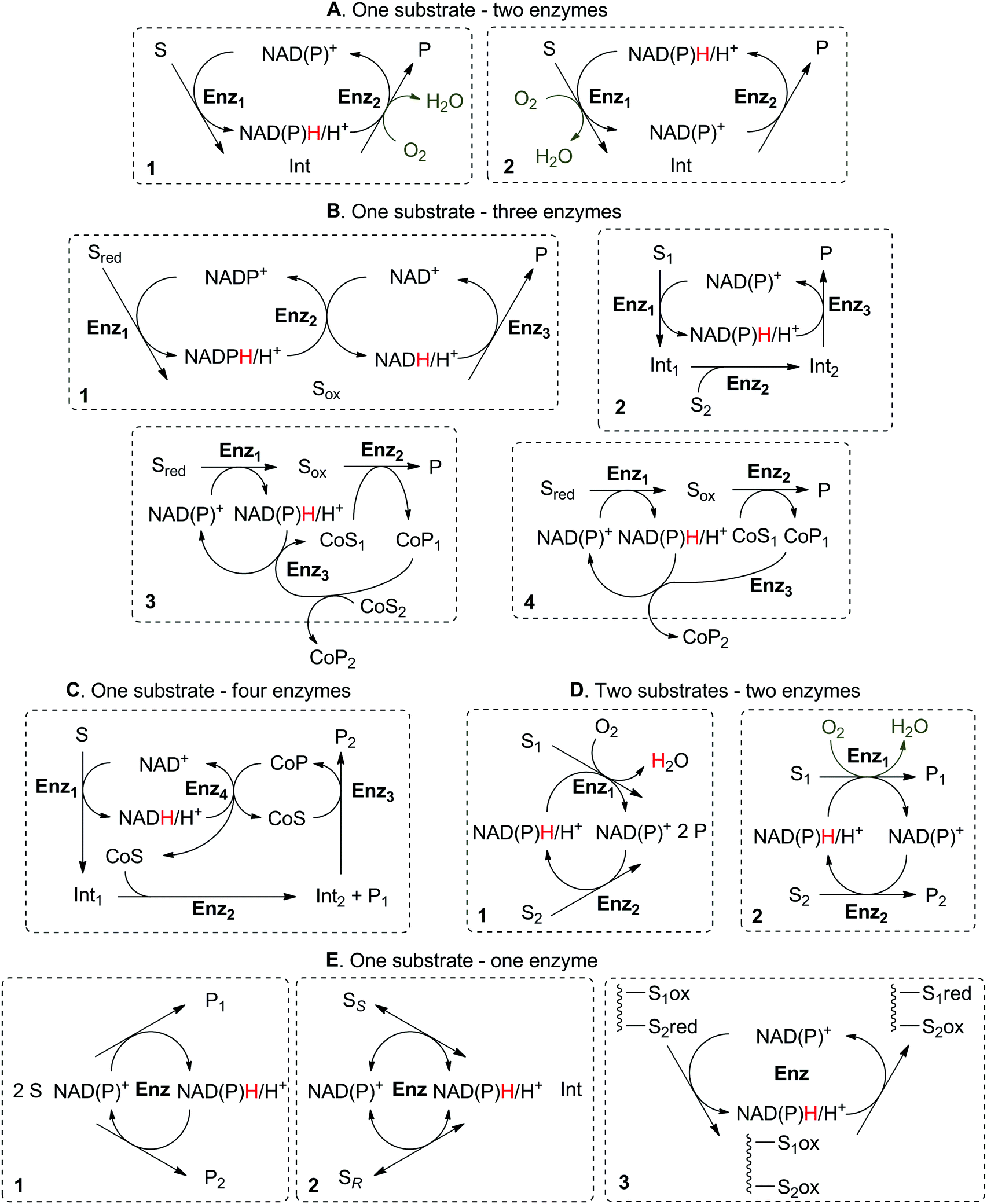 | ||
| Scheme 5 Enzymatic nicotinamide-dependent self-sufficient hydride transfer reactions based on A: two enzymes and A1: cofactor reduction–oxidation sequence or A2: cofactor oxidation–reduction sequence; B: three enzymes and B1: two types of nicotinamide cofactor (the order of both redox states and types of cofactor may be switched); B2: incorporating a third non-redox enzyme for functionalization of the intermediate; B3: co-substrate regeneration by the third enzyme; B4: co-product consumption by the third enzyme; C: four enzymes and one co-substrate; D: two substrates and two enzymes with formation of D1: one product; D2: two products; E: one substrate and one enzyme only for E1: disproportionation reaction; E2: racemization (doubled arrows used for simplification, both reactions are reversible); E3: redox isomerization. S: substrate; P: product; Sred: substrate to be oxidized; Sox: oxidized substrate as (ideally) non-accumulating intermediate; CoS: co-substrate (as reactant); CoP: co-product; Int: intermediate; Enz: enzyme. Half-reactions marked in grey may be coupled to the described system, in which case hydrides are lost to the ‘outside’ as water. | ||
2. One substrate–two enzymes–one product
One substrate can be converted to a single product by the action of two redox enzymes through formation of a non-accumulating intermediate obtained in the first catalytic step, in which the nicotinamide cofactor (hydride shuttle) either gets reduced (2.1) or oxidized (2.2). In a second step, the intermediate thus formed is either reduced (2.1) or oxidized (2.2) to the final product. Despite the use of catalytic amounts of the nicotinamide cofactor in both cases, the first set-up (cofactor reduction–oxidation sequence) is economically more attractive on a large scale, since the cofactor is added in the cheaper oxidized form.2.1. Cofactor reduction–oxidation sequence
The cofactor reduction–oxidation sequence (Scheme 5(A1)) usually corresponds to a formal intramolecular hydride transfer, since the hydride necessary to reduce the cofactor is abstracted from the substrate and reincorporated in the final product in the second catalytic redox event. Here, nicotinamide functions as temporary electron storage, similarly to a metal in the borrowing hydrogen strategy that captures hydrogen atoms. An exception is the case of double oxidation of the substrate, as in the conversion of alcohol to lactone43 (Scheme 11), where hydrides are expelled as water as a by-product of the sequence. This concept in biocatalysis was pioneered by Wandrey and Kula for the asymmetric synthesis of enantiomerically pure amino acids.41 The formal amination of hydroxyacids and alcohols to amino acids and amines, respectively, requires an external nitrogen source (typically ammonium salt) and releases water as the sole by-product. However, in contrast to aerobic di-oxidation sequences (Schemes 11, 14 and 15), the hydride from the substrate remains within the final product. Such sequences represent biocatalytic equivalent to borrowing hydrogen strategies.This concept has been successfully applied to the production of amino acids (Scheme 6), initially relying on three enzymes. In a seminal study by Wandrey and coworkers,41 lactic acid was converted to L-alanine using an immobilized nicotinamide cofactor (PEG-20000-NADH) in a membrane reactor, combining oxidation to pyruvate by lactate dehydrogenases (LDHs) and reductive amination by alanine dehydrogenase (L-AlaDH). Starting from racemic lactic acid, two stereocomplementary LDHs were required for the oxidative step; a space time yield of 134 g L−1 d−1 was achieved in continuous mode and maintained for over 30 days, but required supplementation of pyruvate to ensure efficient hydride shuffling over time. In a subsequent study, a 45 day continuous production of L-leucine with a space time yield of 72 g L−1 d−1 was reported employing a similar strategy.46 An outstanding turnover number for the cofactor (up to 10000) was reported by combining both D- and L-hydroxyisocaproate dehydrogenases (D-HicDH from Lactobacillus casei and L-HicDH from Lactobacillus confusus) with leucine dehydrogenase from Bacillus cereus. More recently, this approach was optimized for the synthesis of non-canonical α-amino acids, as part of a multi-step cascade.42 Using medium chain fatty acids (C6 to C10) as the starting material, peroxygenase P450CLA from Clostridium acetobutylicum catalyzed non-stereoselective and regioselective α-hydroxylation to corresponding α-hydroxyacids at the expense of H2O2. In the subsequent redox self-sufficient reaction module, D-/L-Hic-DHs41 and L-phenylalanine dehydrogenase from Rhodococcus sp. were combined for the production of seven different amino acids with good to high conversion levels (up to 99%) and perfect enantioselectivity (typically >99%), however with low TONNAD (<3) since the cofactor was added in 30 mol%.
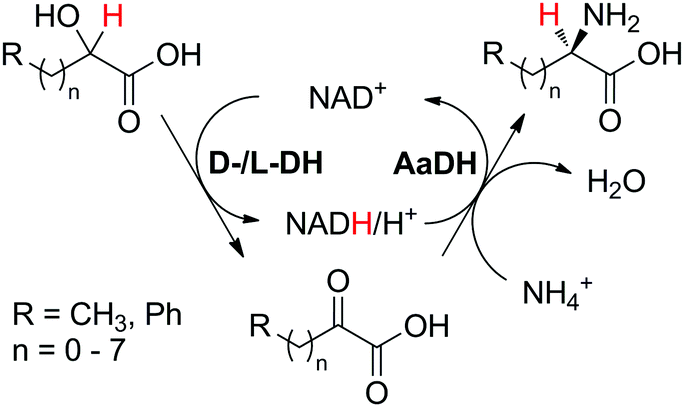 | ||
| Scheme 6 Biocatalytic production of enantiomerically pure amino acids from corresponding α-hydroxyacids via an oxidation–reductive amination sequence. DH: dehydrogenase; AaDH: amino acid dehydrogenase. The starting material may be racemic, in which case two DHs are employed.41,42 | ||
The practical application of this closed-loop cascade dramatically widened in 2015, after two groups independently reported the formal amination of alcohols in one-pot one-step, broadening the substrate scope of the sequence from α-hydroxyacids to primary and secondary alcohols.44,45 In a first study,45 an alcohol dehydrogenase, either a primary ADH from Bacillus stearothermophilus, or a Prelog and/or anti-Prelog ADH from Aromatoleum aromaticum and Lactobacillus brevis, respectively, converted the alcohol into the corresponding ketone; an engineered amine dehydrogenase47 (Ph-AmDH from Bacillus badius or chimeric Ch1-AmDH48) then yielded (R)-configurated amines in the reductive amination step. A wide-ranging pool of primary and secondary alcohols (both as racemic and optically pure material) was tested (Scheme 7). Under optimized reaction conditions at 20 mM substrate concentration, moderate to excellent conversion (generally 80–95%) was obtained with consistently high enantioselectivity (97–99% ee). A detailed investigation of the redox self-sufficiency revealed a TONNAD of up to 76 at 1 mol% NAD+, although the highest conversions were typically obtained at 5 mol% NAD+ (max. TON of 20). The same system was independently developed for secondary alcohols combining an ADH from Streptomyces coelicolor with the double mutant (K77S/N270L) amine dehydrogenase EsLeuDH-DM from Exiguobacterium sibiricum.44 The exploitation of a non-stereoselective ADH in the first oxidation step allowed application of the cascade to racemic alcohols without the need for two stereocomplementary enzymes.45 The redox neutral cascade could be performed on a 50 mM scale with 2 mol% NAD+ on a range of alcohols (up to 48.5 TONNAD), although a max. TONNAD of 192 was reported employing 0.4 mol% NAD+. While only (R)-selective biocatalysts have been available for the reductive amination so far, the recent discovery of naturally occurring amine dehydrogenases with (S)-selectivity should open up the cascade to both amine enantiomeric products.49
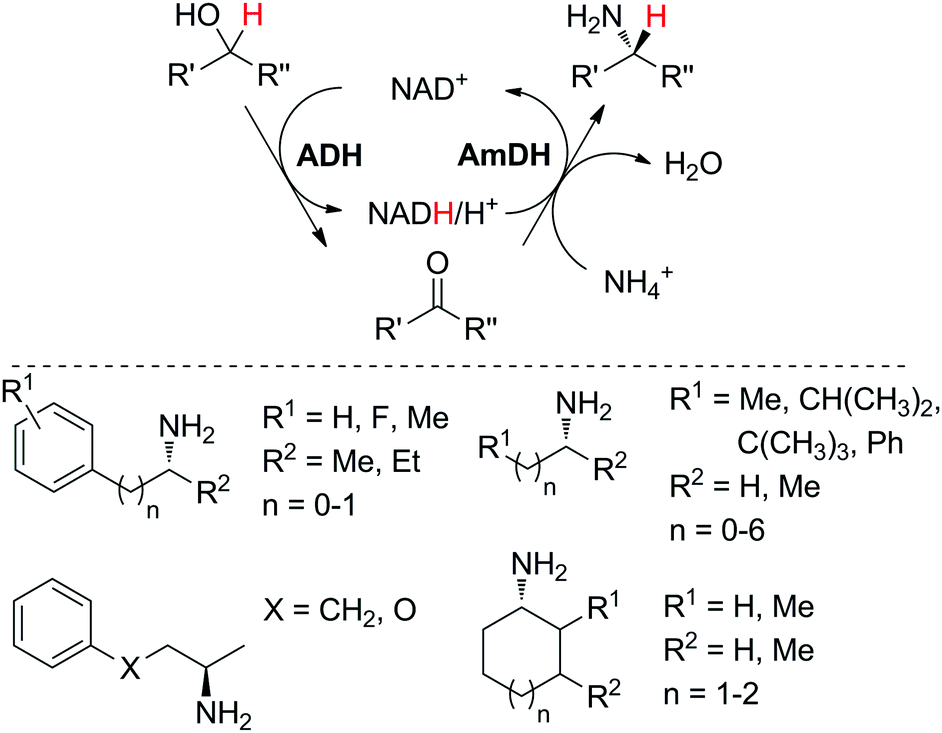 | ||
| Scheme 7 Biocatalytic equivalent to borrowing hydrogen cascade for the formal amination of alcohols (one or two ADHs: alcohol dehydrogenases; AmDH: amine dehydrogenase) and scope of (R)-amines obtained. The alcohol may be primary or secondary (racemic or optically pure).44,45 | ||
To further improve the process, several modifications were later implemented, such as the use of enzyme variants, which were engineered to display reversed cofactor specificity (NADP to NAD) and non-stereoselective behavior,50 or of co-immobilized biocatalysts,51 or by connecting this sequence to additional enzymatic steps to extend the series of catalytic events.52
One main limitation in the described enzymatic system is the requirement for NH3 as a nitrogen source, restricting the scope of products to primary amines. Besides, the employed ammonia concentrations are generally high (∼2 M as ammonium salt) to prevent the more favored reverse reaction (oxidative deamination). The application of a recently discovered reductive aminase (AspRedAm, from Aspergillus oryzae)54 allowed extension of the sequence to secondary amines. The enzyme catalyzes the reductive amination of various carbonyl compounds, using different primary amines as co-substrates, and produces water as the sole by-product. AspRedAm was successfully incorporated into a self-sufficient system for the biocatalytic alkylation of amines with primary and secondary alcohols, in combination with alcohol dehydrogenase (TeSADH W110A from Thermoanaerobacter ethanolicus, ADH-150 or SyADH from Sphingobium yanoikuyae; Scheme 8).53 Notwithstanding low turnover numbers for NADP+ (max. 5 TON at 20 mol% cofactor), the cascade showed a broad variety of accepted substrate pairs, with generally good conversions (up to >99%) and moderate to excellent enantioselectivity (up to >97%). In the case of racemic secondary alcohols, conversions were limited due to the stereopreference of the ADH employed (one ADH only). A major drawback of the current cascade is the requirement for a large excess of the amine donor (5 molar equivalents), which puts a strong burden on atom economy. In a follow-up study, an additional P450-catalyzed hydroxylation step was incorporated prior to this sequence, starting from unfunctionalized cyclic alkanes; production of N-propargylcyclohexylamine was achieved in one-pot two-step with a space-time yield of 2 g L−1 d−1.55
 | ||
| Scheme 8 Biocatalytic alkylation of amines with primary and secondary alcohols (ADH: alcohol dehydrogenase; RedAm: reductive aminase).53 | ||
Related to biocatalytic formal amination strategies, a mixed system composed of an alcohol dehydrogenase and an artificial transfer hydrogenase (ATHase) was designed for the preparation of a pyrrolidine product starting from 4-amino-1-phenyl-1-butanol (Scheme 9). ATHase – a streptavidin-based structure containing a biotinylated Cp*Ir catalyst – catalyzed the reduction of the intermediate cyclic imine obtained from oxidation of the starting material by ADH and subsequent spontaneous cyclization. Under the tested conditions however, the intermediate imine accumulated, likely due to redox imbalance (unfavorable NADP+/substrate ratio). A moderate TON for the cofactor of 3.6 was obtained (based on the final product formed).56
 | ||
| Scheme 9 Combination of alcohol dehydrogenase (ADH) with artificial transfer hydrogenase (ATHase) for preparation of cyclic amine from amino-alcohol (although both substrate and product are chiral, no indication on enantiopurity was provided).56 | ||
The racemization of α-hydroxyacids by a mixture of stereocomplementary α-hydroxyacid dehydrogenases, namely, L- and D-α-hydroxyisocaproate dehydrogenases (HicDHs) from Lactobacillus paracasei and Lactobacillus confusus, respectively, was inspired by the racemization of secondary alcohols by ADHs (see Section 6 and Scheme 25) and relied on concurrent NAD-dependent oxidation–reduction by HicDHs. Running the reaction on 35–45 mM of substrate with a mixture of cofactors (3 mol% NADH and 2 mol% NAD+) allowed racemization within 24 h (Scheme 10).57
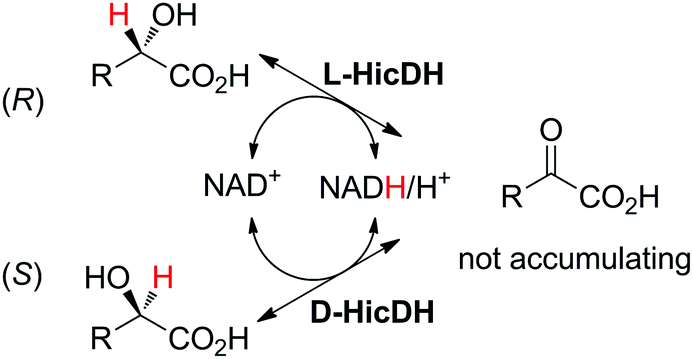 | ||
| Scheme 10 Racemization of (S)- or (R)-α-hydroxyacid by combining L- and D-α-hydroxyisocaproate dehydrogenases (HicDHs; doubled arrows used for simplification, both reactions are reversible).57 | ||
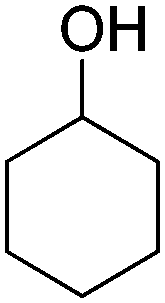
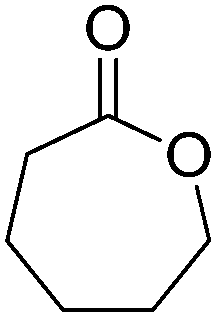
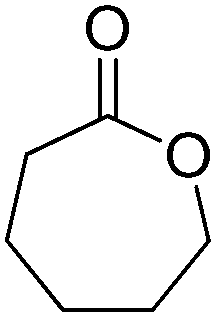
The cofactor reduction–oxidation sequence was adapted to the production of lactones from secondary alcohols, by merging ADH-catalyzed oxidation of secondary alcohols with Baeyer–Villiger monooxygenase (BVMO)-catalyzed oxygenation of intermediate ketones. The double oxidation of secondary cyclic alcohols to lactones was initially developed with bicyclic substrates using whole cells of Acinetobacter sp. NCIMB 9871, and later performed using (partially) purified enzyme preparations of NADP-dependent ADH and BVMO. In this set-up, hydrides abstracted from the substrate end up in water, through formal oxygen reduction by BVMO mediated by the flavin coenzyme. The alcohol oxidation was coupled to the monooxygenation of intermediate ketone through use of sub-stoichiometric amounts of NADP+ (Scheme 11).43 Bicyclic compounds were successfully converted, however the still relatively high cofactor concentration (33 mol%) limited nicotinamide turnover numbers (<3). Additionally, nonperfect regio- or enantio-selectivity in the Baeyer–Villiger oxidation reaction prevented access to a single enantiomeric lactone product. Further improvements (up to 10 TON) were obtained by switching to NAD-dependent enzymes and lowering the cofactor concentration (10 mol%).58 Limitations connected to the poor stability of some monooxygenases were quickly identified,59 highlighting the initial difficulty in generating a protocol applicable to a broad range of substrates. The concept was later applied to simpler structures, granting access to lactones with important industrial applications as monomers. Employing a redesigned polyol dehydrogenase with enhanced thermostability and NADPH-specificity, and CHMO (cyclohexanone monooxygenase from Acinetobacter calcoaceticus), preparative scale synthesis of ε-caprolactone relying on the pair NADPH/NADP+ in 1:1 ratio, each in 6 mol%, resulted in an isolated yield of 55% for the lactone in >99% purity.60 By substituting the ADH, the same cascade was ran at 1 mol% NADP+, providing high conversion levels up to 60 mM substrate (TONNADP of 141).61 This reaction scheme was then linked to the redox self-sufficient formal amination of alcohol (as in Scheme 20) by introducing enzyme-catalyzed methanolysis of ε-caprolactone to generate 6-hydroxyhexanoic acid methyl ester, a key intermediate of the second module.62 High hydride economy was obtained in the double oxidation step, which was scaled up to 100 mg (TONNADP of 120 employing 0.8 mol% NADP+). This sequence has also been extended upstream with the oxidation of cyclohexane by P450 monooxygenase to cyclohexanol; however, an excess of glucose and glycerol were added to support cofactor regeneration.63 The biocatalytic double oxidation was later incorporated in cascades for polymerization reactions to oligo- and poly-ε-caprolactone;64,65 however, here also acetone and glucose were added as co-substrates to support cofactor regeneration.65 Recent improvement in the double oxidation cascade was shown by fusing the two proteins (ADH and CHMO) into a bifunctional biocatalyst,66 while implementing the fed-batch strategy and extending the cascade by lipase-catalyzed hydrolysis of ε-caprolactone to prevent substrate and product inhibition of the biocatalyst, respectively.61,64,65 This resulted in 99% consumption of 200 mM cyclohexanol using 0.1 mol% NADP+.
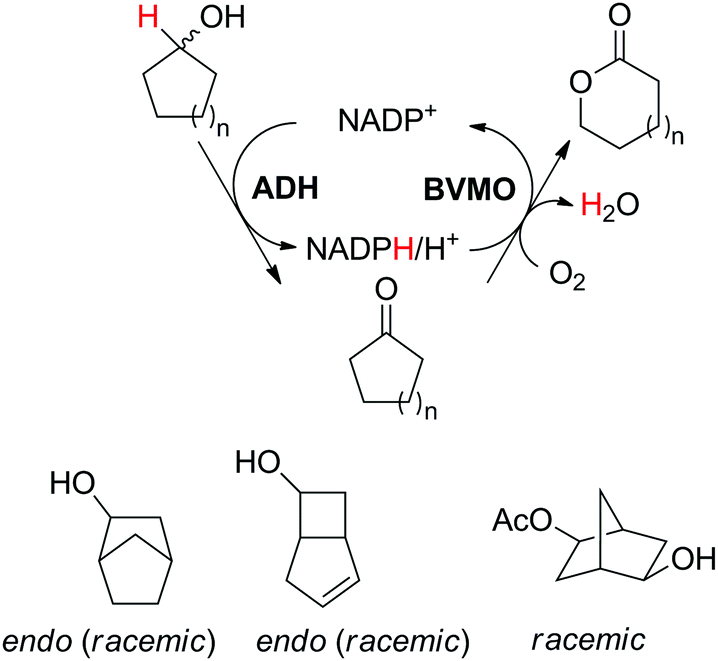 | ||
| Scheme 11 Oxidation–oxygenation sequence for the conversion of cyclic secondary alcohols into lactones by employing alcohol dehydrogenase (ADH) and Baeyer–Villiger monooxygenase (BVMO) as biocatalysts. Representative example with monocyclic compounds (n = 2–4) and additional accepted substrates displayed.43,60 | ||
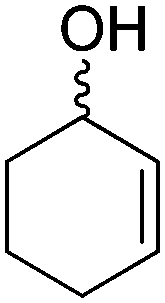
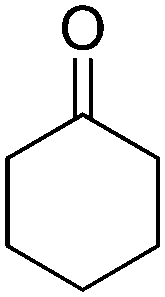
The redox isomerization of allylic alcohols to saturated ketones was designed by coupling NAD+-dependent ADH-catalyzed oxidation of racemic 2-cyclohexenol with NADH-dependent reduction of intermediate cyclohexenone by ene-reductase (ER) to furnish cyclohexanone (Scheme 12).67 Optimization of the reaction conditions was necessary to prevent over-reduction of the final product by ADH and dehydrogenation of the intermediate product by ER. Basic pH values as well as low ADH/ER ratio (∼1:10) were favored and enabled 60% conversion to the final product, with 12 turnovers of the cofactor. The moderate conversion level was suggested to result from the stereopreference of the ADH for one enantiomer of the racemic starting material, resulting in kinetic resolution and incomplete oxidation of the allylic alcohol. Despite limited synthetic interest, this work set the ground for a bi-enzymatic redox isomerization protocol, which was later incorporated into other biosynthetic schemes including further functionalization to lactones68 or saturated alcohols.69
 | ||
| Scheme 12 Redox isomerization of allylic alcohol combining alcohol dehydrogenase (ADH)-catalyzed oxidation of racemic 2-cyclohexenol and ene-reductase (ER)-catalyzed reduction to saturated ketone.67 | ||
2.2. Cofactor oxidation–reduction sequence
In the cofactor oxidation–reduction sequence (Scheme 5(A2)), two enzymes contribute to the hydride transfer, leading to the formation of a single final product. This sequence does not necessarily represent an intermolecular hydride transfer, since the oxidation of the cofactor may be connected to the oxidation of the substrate, in the case where the hydride is required to ‘activate’ the enzymatic reaction (such as in oxidation reactions by P450 enzymes, Schemes 14 and 15). In that case, double oxidation of the substrate is connected to abstraction of one single hydride from the substrate, which is released to the outside in the form of water, concomitant to oxygen reduction. In that case too, the system is redox self-sufficient since molecular oxygen acts as an oxidizing reactant (incorporation of one oxygen atom) and no external source of hydride in theory is required.An example of a system following this sequence of redox events was reported for the preparation of saturated carboxylic acids, using crude cell extracts of E. coli over-expressing the selected biocatalysts.70 Coupling the activity of ene-reductase (ER) and aldehyde dehydrogenase (ALDH), a small set of α,β-unsaturated aldehydes was converted to the corresponding final products on a 10 mM scale (Scheme 13).
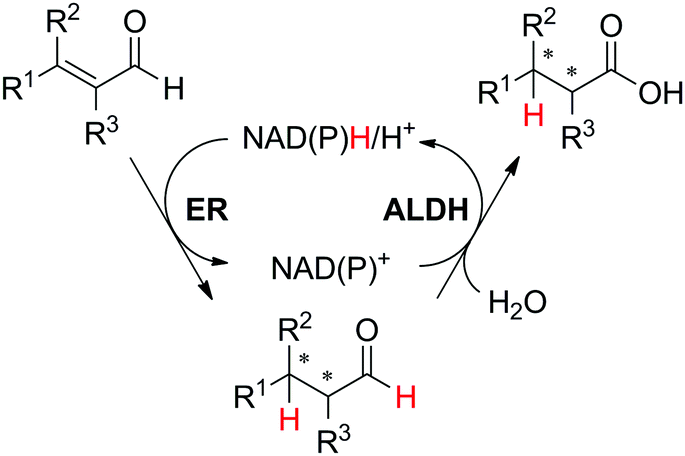 | ||
| Scheme 13 Redox self-sufficient (asymmetric) biocatalytic synthesis of carboxylic acids based on NAD(P)-dependent ene-reductase (ER)-aldehyde dehydrogenase (ALDH) sequence.70 | ||
The main challenge of the system resided in the identification of a suitable ALDH (from Pseudomonas putida KT2440) displaying high chemoselectivity for the saturated aldehyde intermediate. Indeed, competing oxidation of the starting material by ALDH would lead to the formation of the unsaturated carboxylic acid, inert to most ERs,72 and thereby deplete the reaction of the starting material. The cascade was originally optimized using citral – mixture of geranial and neral – as the starting material, which was converted quantitatively with excellent chemo- and stereo-selectivity. Additional substrates were also accepted, and stable turnover numbers of the cofactor were observed, albeit with slightly reduced chemo- or stereo-selectivity (Table 1, entries 1 and 2).
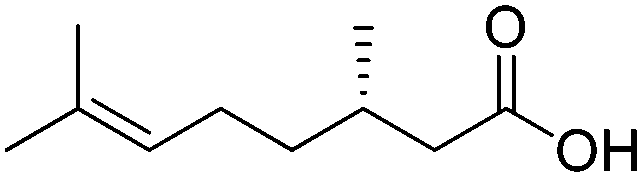
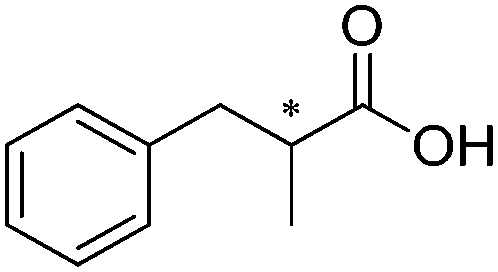

A further optimization of the system was later performed by Scrutton et al.,71 following a systematic analysis of the different parameters involved in order to maximize both chemo- and stereo-selectivity. To identify the best performing pair of enzymes for each substrate, the two redox half-reactions were examined separately, and later combined in the optimization step. The best suitable combination of ene-reductase and aldehyde dehydrogenase allowed the production of different α-substituted carboxylic acids (Table 1, entries 3–5) with high chemo- (81–95%) and stereo-selectivity (91–99% ee). Moreover, for the conversion of α-methylcinnamaldehyde, the applied systematic methodology led to the identification of a matching pair of enzymes (OYE2 from Saccharomyces cerevisiae and ALDH from horse), which acted with remarkable efficiency in closed loop on the 5 mM substrate (470 TONNADP, Table 2, entry 5).
| Entry | Substrate(s) | Product(s) | Enzyme(s)a | [S] (mM) | [cofactor] (mol%)b | TONNAD(P)c | Ref. |
|---|---|---|---|---|---|---|---|
| a ADH: alcohol dehydrogenase; AmDH: amine dehydrogenase; RedAm: reductive aminase; BVMO: Baeyer–Villiger monooxygenase; ER: ene-reductase; ALDH: aldehyde dehydrogenase; P450: cytochrome P450 monooxygenase; FdeH: 5-exo-hydroxycamphor dehydrogenase; MDH: morphine dehydrogenase; STH: soluble pyridine nucleotide transhydrogenase; MR: morphinone reductase; TA: transaminase; AlaDH: alanine dehydrogenase; for source of enzymes, see text and corresponding references. b In relation to the substrate starting concentration; the redox state and type of cofactor is indicated (may be added as pair). c TON: turnover number, each turnover converts one molecule of the substrate to the final product (except entries 9g and 13h); the cofactor goes through double this amount of half-reactions (on substrate and generated intermediate, respectively). d 50 mM of ketone and 25 mM of diol. e 15 mM of sulfide and 30 mM of racemic alcohol. Corresponding ketone is formed (14.1 mM). f 11.3 mM of each substrate. g Two turnovers for each molecule of difunctionalized product formed. h Each turnover converts two molecules of the substrate. i Calculated half-reactions for NAD+ and NADH respectively (for racemization of 26 mM, only 13 mM of substrate are effectively converted). | |||||||
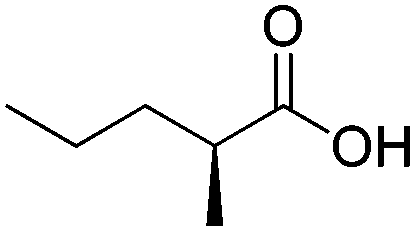
| 1 |

|
|
ADH, AmDH | 20 | 5 (NAD+) | 19 | 45 |
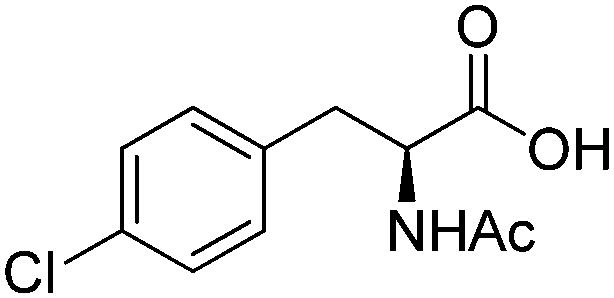
| 2 |

|
|
ADH, RedAm | 5 | 20 (NADP+) | 5 | 53 |
| 3 |
|
|
ADH, BVMO | 200 | 0.8 (NADP+) | 120 | 62 |
| 4 |
|
|
ADH, ER | 10 | 5 (NAD+) | 12 | 67 |
| 5 |
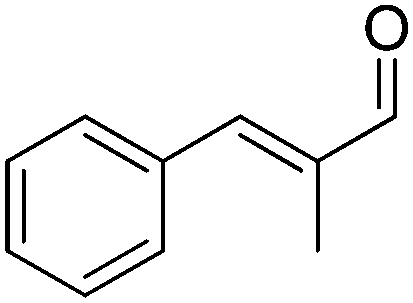
|
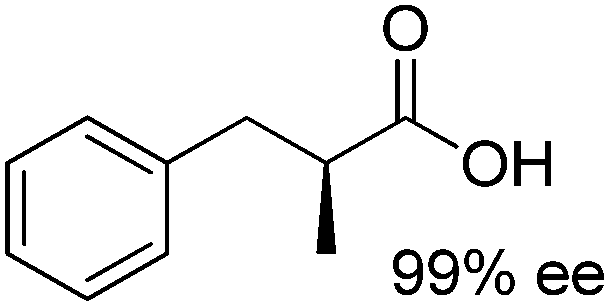
|
ER, ALDH | 5 | 0.2 (NADPH) | 470 | 71 |
| 6 |
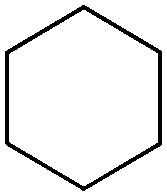
|
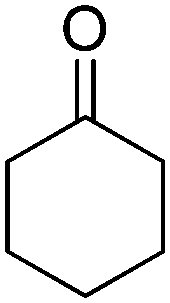
|
P450, ADH | 100 | 10 (NADPH) | 0.42 | 73 |
| 7 |
|
|
P450, FdeH | 1 | 10 (NADPH) | 9.8 | 79 |
| 8 |
|

|
MDH, STH, MR | 20 | 1:1 (NADPH/NAD+) |
78 each | 84 |
| 9 |
|
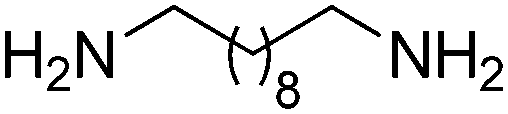
|
ADH, TA, AlaDH | 50 | 1.5 (NAD+) | 96g | 87 |
| 10 |
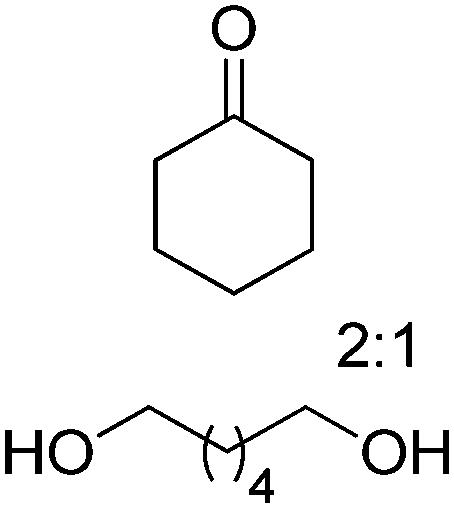
|
|
BVMO, ADH | 75d | 0.2 based on [diol] (NADP+) | 915 | 96 |
| 11 |
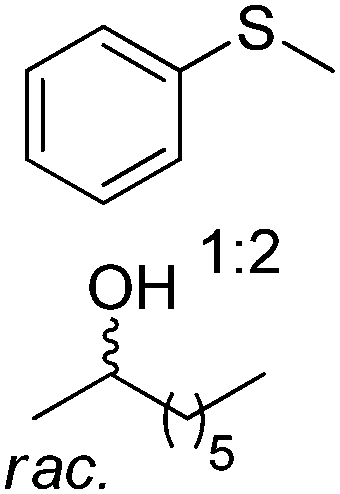
|
|
BVMO, ADH | 45e | 1.3 based on [sulfide] (NADPH) | 73 | 104 |
| 12 |
|
|
BVMO, ADH | 22.6f | 0.009 based on [ketone] (NADPH) | ∼5500 | 104 |
| 13 |
|
|
ADH | 75 | 1.3 (NAD+) | 26.5h | 106 |
| 14 |
|

|
ADH | 26 | 2.7:4.6 (NAD+/NADH) |
18.6/6.8i | 109 |
| 15 |

|
|
ADH | 7 | 7.1 (NAD+) | 10.2 | 110 |
Biocatalytic one-pot oxidation of cyclohexane to cyclohexanone was established in a set-up combining two oxidation steps.73 Alkane hydroxylation catalyzed by a variant from P450 BM3 from Bacillus megaterium (19A12) was coupled to alcohol dehydrogenation catalyzed by ADH from Lactobacillus kefir (Scheme 14). As alluded earlier, in this set-up, the hydride abstracted from the substrate ends up in water through reduction of oxygen by P450 monooxygenase. 10 mol% NADPH were used on 100 mM cyclohexane, eventually leading to a final product concentration of 0.41 g L−1 (4.2 mM). As pointed out by the authors, only low efficiency was achieved with this reaction set-up (TONNADP < 1, Table 2, entry 6). Slightly higher cycloalkanone concentrations (up to 0.80 g L−1, 8.2 mM) were reached by supplementing the system with 0.25 vol% isopropanol (∼33 mM). The alcohol acted as an auxiliary co-substrate that could regenerate NADPH via concomitant ADH-catalyzed oxidation to acetone. This compensated for the loss of NADPH due to strong uncoupling toward H2O2 production, which P450 enzymes are plagued with.74 In follow-up studies, this concept was applied to the double oxidation of n-heptane and cyclooctane in a whole-cell biocatalyst and/or using purified enzymes.75–77 Since redox self-sufficiency could not be achieved due to uncoupling in the P450-catalyzed hydroxylation reaction, glucose was used as a sacrificial co-substrate. Following a similar strategy, the combination of P450 monooxygenase and ADH was applied to the conversion of (+)-valencene to (+)-nootkatone, a grapefruit flavor sesquiterpenoid ketone.78 Although P450 BM3 shows preference for NADPH as a cofactor, the selected variant BM3-AI (F87A/A328I) showed activity also with NADH, which is the only cofactor accepted by the second enzyme (c-LEcta ADH-21). To overcome the problem of uncoupling with P450 enzyme, 2-butanol was added as a co-substrate, thus preventing self-sufficiency, and butanone was generated as an oxidized by-product from ADH. The biocatalytic production of ketoisophorone from α-isophorone was similarly described, although in a whole-cell system co-expressing a variant of the chimeric self-sufficient P450cam-RhFRed and Cm-ADH110 (ADH from Candida magnoliae).80 Here too, glucose was added externally.
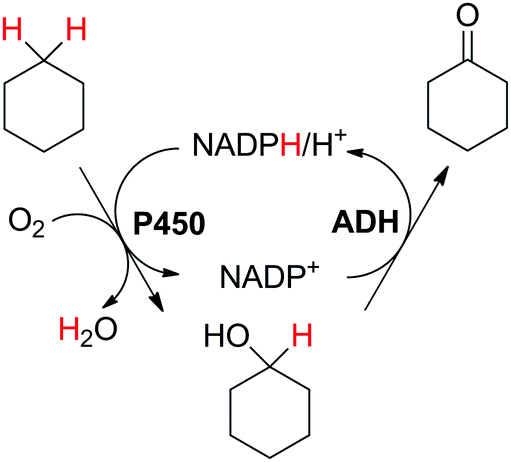 | ||
| Scheme 14 Double oxidation of cyclohexane to cyclohexanone using P450 monooxygenase BM3 from Bacillus megaterium and alcohol dehydrogenase from Lactobacillus kefir (ADH).73 | ||
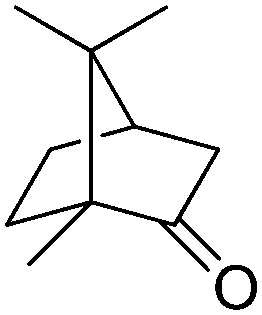
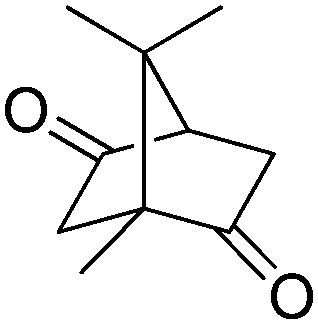
The P450cam system, composed of P450cam monooxygenase, putidaredoxin PdX, and putidaredoxin reductase PdR, and FdeH, a 5-exo-hydroxycamphor dehydrogenase, were successfully combined for the regioselective hydroxylation–oxidation sequence of camphor (Scheme 15).79 Using purified enzyme preparations under optimized reaction conditions, 1 mM of camphor was fully converted to 2,5-diketobornane in 2 h, with a modest cofactor turnover number of 4. By extending the incubation time to 4 h, the cofactor amount could be lowered to 0.1 mM, with a conversion of 98% and 2.5-fold increase in TONNADP. Remarkably, the optimized system ran efficiently in a closed-loop fashion, since the P450-driven cascade was reported to proceed without noticeable uncoupling (Table 2, entry 7).
 | ||
| Scheme 15 Conversion of camphor through double oxidation of 2,5-diketobornane by P450 monooxygenase P450cam and 5-exo-hydroxycamphor dehydrogenase FdeH (P450cam supplemented with redox partner putidaredoxin PdX and putidaredoxin reductase PdR). Enantioselectivity of P450cam not reported (abstracted hydrogen nonrepresentative).79 | ||
2.3. Special cases
The requirement for two stereocomplementary dehydrogenases in the formal amination of racemic α-hydroxyacids to enantiopure amino acids41 (see Section 2.1 and Scheme 6) can be circumvented by supplementing the sequence with a racemase, overall rendering a sequence with three enzymes to convert one substrate into one product. This approach was implemented in the redox-neutral biotransformation of rac-mandelic acid into L-phenylglycine (Scheme 16).81 Given the high stereoselectivity of D-mandelate dehydrogenase (D-MDH from Rhodotorula graminis), only one of the two enantiomers of the α-hydroxy acid was consumed; mandelate racemase from Pseudomonas putida was therefore introduced to allow almost full consumption of the racemic starting material (conversion up to 94%). The reductive amination step was performed using different commercial preparations of L-AaDHs, which yielded the final product with excellent enantioselectivity (commonly >97%) and cofactor turnover number up to 14 at 6.7 mol% NAD+. In a separate study, a combination of newly identified and engineered enzymes was tested for the optimization of the redox self-sufficient cascade (D-MDH from Lactobacillus brevis, an engineered mandelate racemase from Pseudomonas putida and leucine dehydrogenase from Exiguobacterium sibiricum).82 Impressive cofactor turnover numbers of up to 3165–3185 were obtained at 50–500 mM substrate loading and 0.02 mol% NAD+. The reaction performed at 200 mM substrate and 0.05 mol% NAD+ led to 87% isolated yield of L-phenylglycine in perfect enantiomeric excess. In addition, four different substituted amino acid derivatives were successfully produced (Scheme 16), with TONNAD between 145 and 483.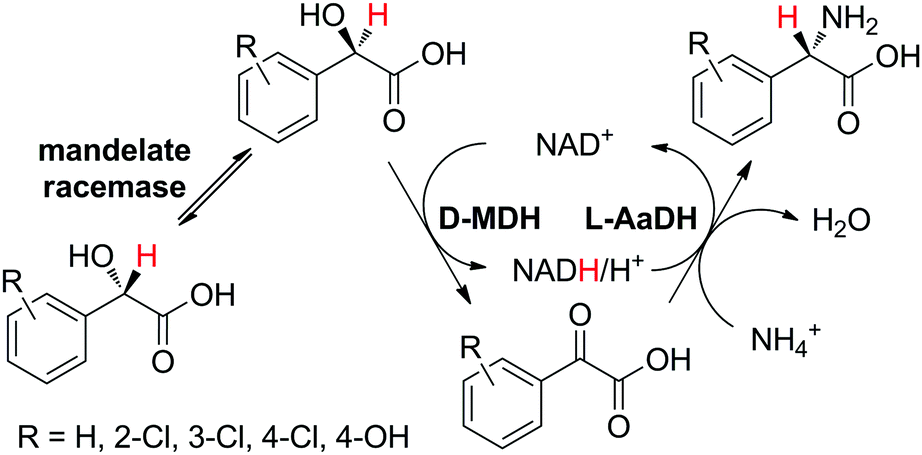 | ||
| Scheme 16 Redox-neutral production of substituted L-phenylglycine from racemic mandelic acid derivatives using mandelate racemase, D-mandelate dehydrogenase (D-MDH) and L-amino acid dehydrogenase (L-AaDH).81 | ||
The self-sufficient formal amination cascade from Scheme 6 was operated in the reverse direction for the simultaneous production of (S)-3-fluoroalanine and (R)-3-fluorolactic acid.83 Kinetic resolution of rac-3-fluoroalanine by oxidative deamination catalyzed by L-alanine dehydrogenase from Bacillus subtilis led to isolation of (S)-3-fluoroalanine in 60% yield and 88% ee. Concurrently, rabbit muscle L-lactate dehydrogenase converted intermediate 3-fluoropyruvate to (R)-3-fluorolactic acid in 80% yield and perfect enantioselectivity (>99% ee). The sequence was performed on a 100 mg scale with 1.25 mol% NADH and generated two products from one substrate using two enzymes.
3. One substrate–three enzymes
A few systems require the presence of three enzymes to operate according to the redox self-sufficiency principle. Two enzymes are collectively responsible for the transformation of the substrate into the product of interest while a third enzyme is necessary for the hydride shuffling between two types of cofactor or between one cofactor and one co-product. Alternatively, the third enzyme may be responsible for additional (non-redox) functionalization.3.1. Two cofactors
In the case where two distinct hydride shuttles (NADP and NAD) in complementary redox states are necessary for two distinct catalytic events, a third enzyme allows the crucial hydride transfer as a connector between the two catalytic loops and results in formal intramolecular hydride transfer. This necessitates high chemo-selectivity of the enzymes to prevent unnecessary consumption of one cofactor in the wrong loop. Practically, this translates into use of a NADP-specific enzyme for one reaction and a NAD-specific enzyme for the second reaction of interest (or vice versa) to ensure ideal redox balance. The two enzymes responsible for the transformation of the substrate into the final product should catalyze consumption of both nicotinamide cofactors in complementary redox directions. The key transfer of electrons between the two cofactor types is catalyzed by a pyridine nucleotide transhydrogenase (Scheme 5(B1)).This strategy was elegantly applied to the production of the opiate drug hydromorphone starting from morphine.84 In this example, the substrate contains two functional groups, which react independently through the action of two distinct enzymes: morphine dehydrogenase catalyzes the oxidation of the secondary alcohol in a NADP+-dependent manner, while morphinone reductase catalyzes the subsequent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-bond reduction of the intermediate enone at the expense of NADH (Scheme 17). The cofactors were added as a pair of NADPH/NAD+ in a 1:1 ratio (1 mol% each), so that the first catalytic event was the hydride exchange catalyzed by the soluble pyridine nucleotide transhydrogenase STH from Pseudomonas fluorescens. Morphine (20 mM) was converted in 78% to the final product with little accumulation of undesired side-product dihydromorphine (8.4%). Slight improvement of hydromorphone formation up to 84% was observed when changing the composition of the cofactor mixture closer to that likely present in the cell (2 mM NAD+, 0.15 mM NADH, 0.25 mM NADP+ and 0.2 mM NADPH).
C-bond reduction of the intermediate enone at the expense of NADH (Scheme 17). The cofactors were added as a pair of NADPH/NAD+ in a 1:1 ratio (1 mol% each), so that the first catalytic event was the hydride exchange catalyzed by the soluble pyridine nucleotide transhydrogenase STH from Pseudomonas fluorescens. Morphine (20 mM) was converted in 78% to the final product with little accumulation of undesired side-product dihydromorphine (8.4%). Slight improvement of hydromorphone formation up to 84% was observed when changing the composition of the cofactor mixture closer to that likely present in the cell (2 mM NAD+, 0.15 mM NADH, 0.25 mM NADP+ and 0.2 mM NADPH).
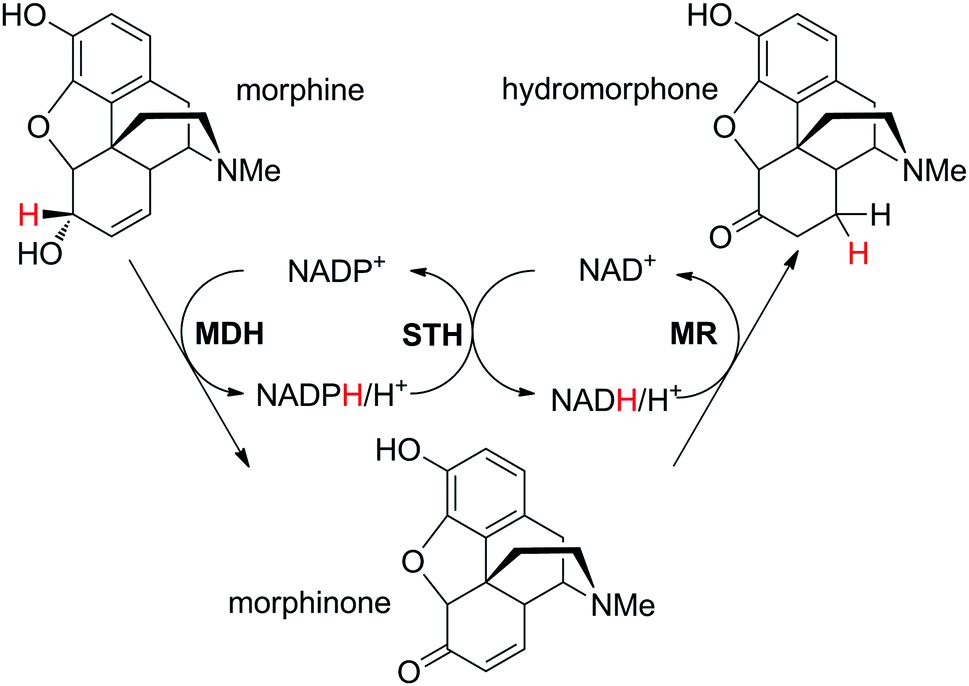 | ||
| Scheme 17 Cofactor reduction–oxidation sequence for conversion of morphine to hydromorphone using a combination of NADP-selective morphine dehydrogenase (MDH), NAD-dependent morphinone reductase (MR) and soluble pyridine nucleotide transhydrogenase (STH).84 | ||
A similar enzyme orchestration was applied to the stereoinversion of enantiopure chiral secondary alcohols, in which two stereocomplementary ADHs from Candida parapsilosis with opposite cofactor preferences (NAD and NADP, respectively) acted in opposite redox directions with high selectivity (Scheme 18).85 The substrate possessed only one functional group reactive to the enzyme (i.e., the primary alcohol functionality remained untouched). Pyridine nucleotide transhydrogenase from E. coli was employed to connect the two loops. Interestingly, although whole cells co-expressing all enzymes were used as biocatalyst preparations, which naturally contain nicotinamide, addition of external cofactors in the form of NAD+/NADPH at a 3:1 ratio (1–2 mM each) was necessary to reach the highest conversion and optical purity of the (S)-enantiomeric product (95% and 97%, respectively) starting from 100 mM of (R)-substrate. One explanation proposed by the authors was that the variation in cofactor concentration compensated for the unequal over-expression of the two ADHs and resulting unmatched enzymatic rates.
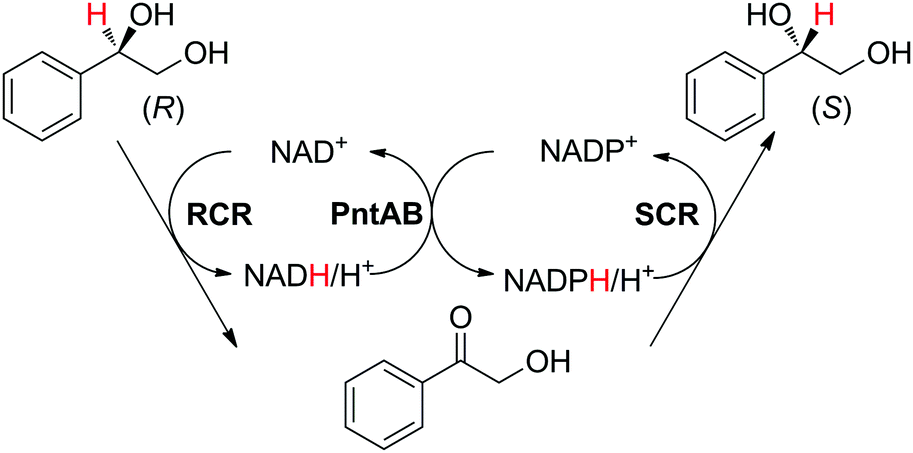 | ||
| Scheme 18 Stereoinversion of (R)-1-phenyl-1,2-ethanediol by stereocomplementary alcohol dehydrogenases (ADHs). RCR: NAD+-dependent (R)-selective ADH from Candida parapsilosis CCTCC M203011; SRS: NADPH-dependent (S)-selective ADH from Candida parapsilosis CCTCC M203011; PntAB: pyridine nucleotide transhydrogenase from E. coli.85 | ||
3.2. One co-substrate–no co-product
A self-sufficient hydride transfer can be integrated into a three-enzyme cascade, in which one enzyme is responsible for nicotinamide-independent functionalization of the intermediate product (Scheme 5(B2)), such as reported for the production of L-lactate starting from ethanol and carbon dioxide.86 In the first step, ethanol was oxidized to acetaldehyde by NAD-dependent alcohol dehydrogenase; then PyDC (pyruvate decarboxylase) catalyzed the C–C-bond forming reaction that incorporated CO2 to form pyruvic acid, which was subsequently reduced to L-lactic acid by lactate dehydrogenase (Scheme 19). After a 4 day reaction, 8.7 TONNAD and 41% conversion to L-lactic acid were achieved, employing 10 mol% cofactor and using a substrate feeding strategy to keep the concentration of ethanol constant throughout the reaction (100 μM).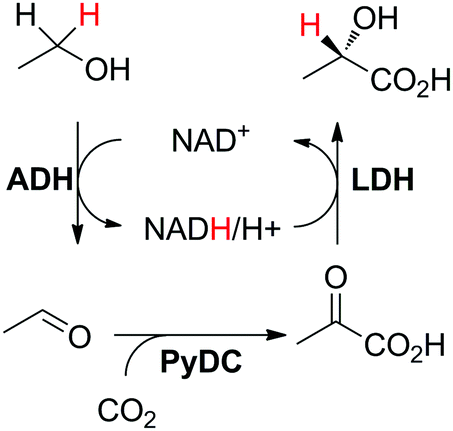 | ||
| Scheme 19 Enzymatic production of L-lactic acid by CO2 fixation coupled with a redox self-sufficient system (ADH: alcohol dehydrogenase; LDH: lactate dehydrogenase).86 | ||
3.3. One/Two co-substrate(s)–one co-product
In a given enzymatic sequence, an additional co-substrate may be required to perform the transformation of the intermediate into the product of interest, thereby acting as a reactant and possibly releasing a co-product. In the case of two consecutive reactions with one NAD(P)-dependent and one NAD(P)-independent catalytic event, internal regeneration of the cofactor can be coupled to (i) recycling of the co-product formed in the second step (Scheme 5(B3)) or (ii) consumption of the co-product, thereby generating an end co-product (Scheme 5(B4)). In both cases, the hydride shuffling connects the two loops in a formal intramolecular hydride transfer.An enzymatic amination sequence was designed to allow conversion of primary (di)alcohols to corresponding (di)amines.87 Oxidation of the substrate to the aldehyde intermediate by ADH at the expense of NAD+ was followed by nicotinamide-independent transaminase-catalyzed amination to the corresponding amine. L-Alanine was employed as an amine donor and converted to pyruvate (Scheme 20). To retain the hydride released in the first oxidation reaction in a closed loop, a third enzyme, nicotinamide-dependent alanine dehydrogenase AlaDH, was added for the regeneration of alanine from pyruvate. AlaDH-catalyzed reductive amination, through addition of an ammonium salt as an amine source, could capture the hydride from NADH, thereby regenerating NAD+ for the first step and ensuring redox neutrality of the system. Despite efficient internal nicotinamide regeneration (addition of only 1.5 mol% of NAD+, with up to 96 turnovers on diols), the system suffers from unfavorable equilibrium in the amination step with products being generally less energetically stable than the substrates.88,89 A large excess of alanine as an amine donor is required in order to drive the amination toward product formation, since the transaminase-catalyzed reaction is reversible. The atom economy is eventually affected from a dual excess of amine donor (ammonium salt and alanine). Overall, the cascade is redox-neutral. Noteworthily, the hydride abstracted from the alcohol substrate in the first step is eventually transferred in the transamination reaction in two distinct steps as a proton and electron pair to give the amine through formal intramolecular hydride transfer. In practice, pyruvate does not accumulate, and water is generated as the sole by-product.
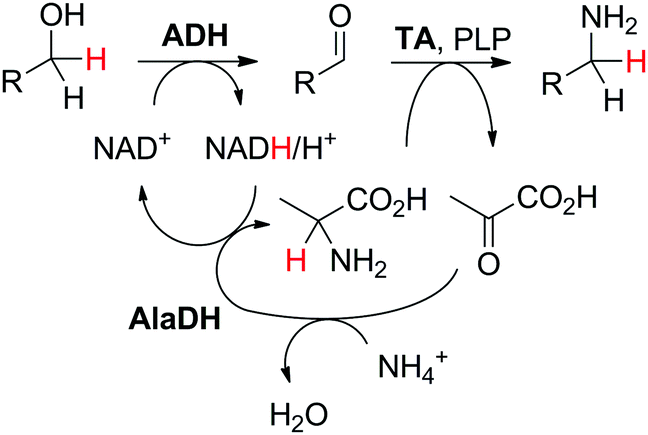 | ||
| Scheme 20 Biocatalytic cascade for redox-neutral amination of primary alcohols employing alcohol dehydrogenase (ADH) and transaminase (TA). L-Alanine, although regenerated, and the ammonium salt are provided in excess. AlaDH: alanine dehydrogenase; PLP: 5′-pyridoxal phosphate (catalytic amounts). Diols are also accepted and form diamines.87 | ||
This system was later adapted to the production of secondary amines by selecting ADHs that convert (enantiopure) secondary alcohols with high enantioselectivity and applying stereoselective transaminases.90 A second strategy was also evaluated (Scheme 5(B4)), in which the pyruvate co-product formed in the transamination reaction was further converted through the action of NADH-dependent lactate dehydrogenase.88 This in situ co-product removal is a strategy commonly used for shifting equilibria of thermodynamically unfavorable reactions,89 and in the reported set-up, rendered an orthogonal cascade.91 Again, redox neutrality was ensured; however, this set-up led to accumulation of lactate as a by-product in molar equivalent to the targeted amine product, thereby impacting strongly the atom economy of the reaction.
4. One substrate–four enzymes
A single case of hydride self-sufficient biocatalytic cascade has been reported employing 4 enzymes for the breakage of a small lignin model dimer into two monomeric products (Scheme 5(C)). From the four reactions involved, two are nicotinamide-dependent redox reactions, but only one targets the actual substrate. Glutathione is used as a co-substrate and acts as a redox mediator, shuffling electrons in the nicotinamide-independent reactions. The designed cascade aimed at cleavage of the ether bond of 1-(4-hydroxy-3-methoxyphenyl)-2-(2-methoxyphenoxy)-1,3-propanediol, typical for ether linkages found in lignin, to liberate lignin monomeric constituents (Scheme 21).92 The first step consisted in LigD-catalyzed oxidation of the secondary hydroxy moiety at the expense of NAD+, followed by glutathione-dependent cleavage of the ether bond by β-etherase LigF, in which 2-methoxy-phenol is released along with a glutathionylated intermediate. The third enzyme, glutathione lyase LigG, can liberate glutathione (GSH) from this intermediate at the expense of a second glutathione equivalent, thereby generating glutathione disulfide (GSSG) and releasing the second product 3-hydroxy-1-(4-hydroxy-3-methoxyphenyl)-1-propanone. Finally, redox self-sufficiency is ensured by reducing GSSG back to GSH using NAD-dependent glutathione reductase from Allochromatium vinosum. With 10 mol% NAD+ and 23% mol% GSSG, 35% conversion of 30 mM substrate was achieved, indicating recycling of both NAD+ and GSSG, though with a low turnover number. This was partly explained by the stereoselectivity of LigF, translating into kinetic resolution of the first intermediate generated in the first step of the cascade, which limits the overall conversion to 50%.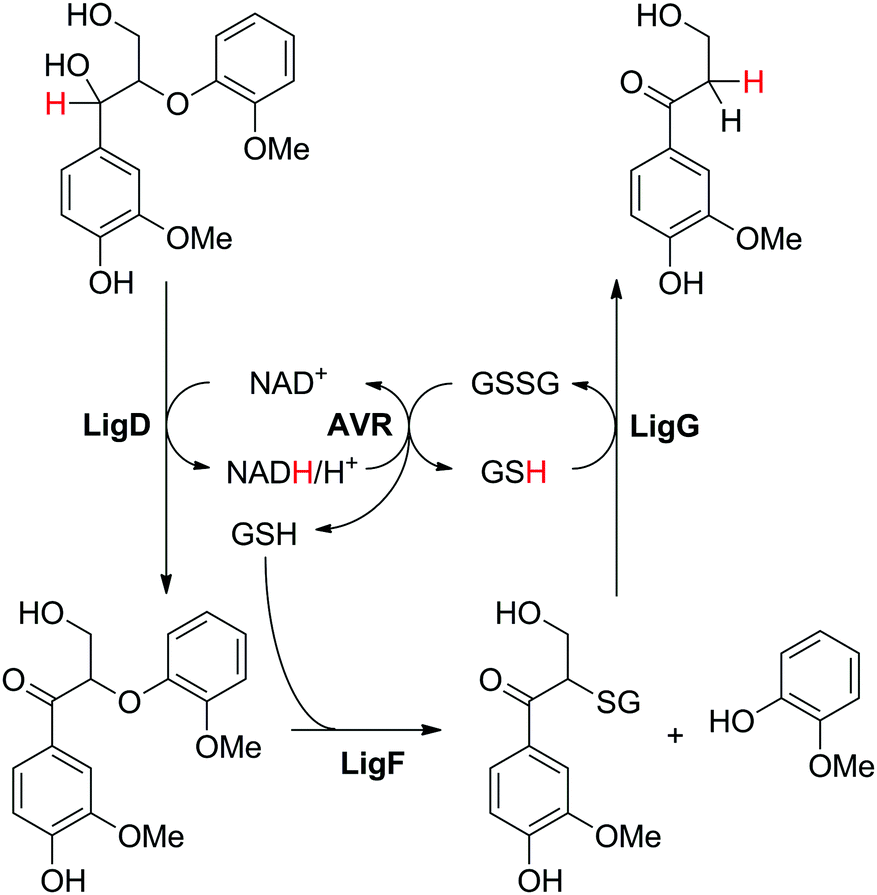 | ||
| Scheme 21 Redox-neutral cleavage of lignin-type dimer employing four enzymes. LigD: dehydrogenase; LigF: β-etherase, LigG: glutathione lyase (LigDFG from Sphingobium sp. SYK-6); AVR: reductase from Allochromatium vinosum.92 | ||
5. Two substrates–two enzymes
5.1. One product
The use of two substrates can result in the formation of a single product via a convergent process and requires one enzyme specific to each substrate (Scheme 5(D1)). Given the difficulty in identifying two pairs of substrate/enzyme leading to the same product through consumption of nicotinamide in opposite redox directions, such redox self-sufficient processes are rare.Inspired by the ‘smart co-substrate’ approach, in which 1,4-butanediol§ is used as a hydride source for cofactor regeneration,93 hexane-1,6-diol was converted to ε-caprolactone in a sequential double oxidation by NADPH-dependent alcohol dehydrogenase (ADH from T. ethanolicus).94 Concurrently, the generated two hydride equivalents were consumed by Baeyer–Villiger monooxygenase (CHMO from Acinetobacter sp. NCIMB 9871) for the aerobic oxidation of two molecules of cyclohexanone, yielding two additional molecules of the desired final lactone product (Scheme 22). Therefore, the cascade theoretically yields three equivalents of product, starting from two molecules of ketone and one of diol. Since the BVMO-catalyzed reaction consumes oxygen as an oxidant, the hydrides originating from the diol are eventually lost to the outside in the form of water; the system is still redox self-sufficient. Unfortunately, hydrolysis95 and/or polymerization of the final lactone product prevented calculations of the overall efficiency of the system. Notwithstanding, a maximum turnover number for the cofactor (2 mol%) of 23 was reached when 34.7 mM of ε-caprolactone were produced (considering that two cofactor cycles are needed to generate three molecules of product).
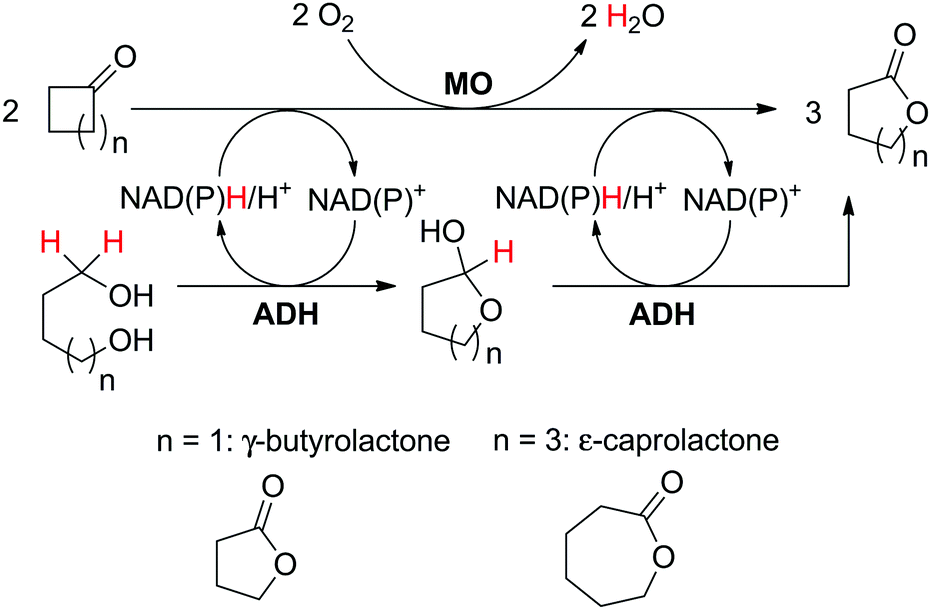 | ||
| Scheme 22 Convergent oxidative cascade toward lactone formation from two different substrates, coupling a monooxygenase (MO, Baeyer–Villiger monooxygenase or flavin-monooxygenase) and an alcohol dehydrogenase (ADH).94,98 | ||
Two follow-up studies were performed targeting improvement of the redox self-sufficiency.96,97 A two-step design-of-experiment approach and a biphasic reaction system were successfully implemented, leading to a final optimized ε-caprolactone yield of 71%, combined with 915 TONNADP (Table 2, entry 10). Such a high number was later attributed to the relatively low temperature used in the reaction (20 °C), which prevented considerable auto-hydrolysis of the final product.97
Based on the same concept, a recently discovered NADH-dependent type II flavin-containing monooxygenase (FMO-E from R. jostii RHA1) was combined with horse liver ADH (HLADH from E. caballus) for the convergent production of γ-butyrolactone (Scheme 22).98 Starting from 100 mM cyclobutanone and 50 mM 1,4-butanediol, 134 mM of final product was obtained at 2 mol% NAD+, corresponding to 89 TONNAD. In a recent update, the cascade was also performed in low-water organic media using a single biocatalyst obtained by fusion of the two aforementioned enzymes.99 No external nicotinamide was added and the system relied on the cofactor already present in the lyophilized cell extracts employed; however, addition of nicotinamide was shown to have a positive effect on the moderate conversion level (<30%).
5.2. Two products
Alternatively, two products of value are formed by the action of two enzymes running in opposite redox directions on two different substrates, resembling the coupled-enzyme cofactor regeneration strategy (Scheme 5(D2)). In contrast to linear or convergent cascades,39 in which all the steps lead to the formation of a single compound, the two redox reactions are combined in a one-pot (anti)-parallel process, generating concurrently two (or more) desired final products in a redox-neutral fashion. The nicotinamide, acting as a hydride shuttle, interconnects the two biotransformations, which ideally should display comparable reaction rates for maximum efficiency. Exemplary is the case of parallel interconnected kinetic asymmetric transformation (PIKAT).100This concept was initially reported as a tandem concurrent process for the preparation of enantiopure secondary alcohols using a single alcohol dehydrogenase,101 coupling stereoselective reduction of prochiral α-haloketones with kinetic resolution of racemic secondary alcohols (Scheme 23A, ADH1 = ADH2).102 Several ketones were reduced by either Prelog (ADH-A from Rhodococcus ruber or ADH-T from Thermoanaerobacter sp.) or anti-Prelog (LB-ADH from Lactobacillus brevis) ADH with good to excellent conversions (57–>99%), perfect enantioselectivity (typically >99% ee for (R)- or (S)-enantiomers) and TONNAD(P) between 29 and 50. Simultaneously, kinetic resolution of the second racemic substrate yielded corresponding enantio-enriched alcohol (up to >99% ee). Redox self-sufficiency and optimum hydride economy relied on a ketone/alcohol substrate ratio of 1:2 and benefited from impeded reverse oxidation of the formed halohydrin through intramolecular hydrogen bonding between the alcohol and the halogen, which stabilizes the product.103 Noteworthily, half of the alcohol substrate is wasted by concurrent formation of the corresponding ketone as a by-product, thereby severely impacting the atom economy of this approach.
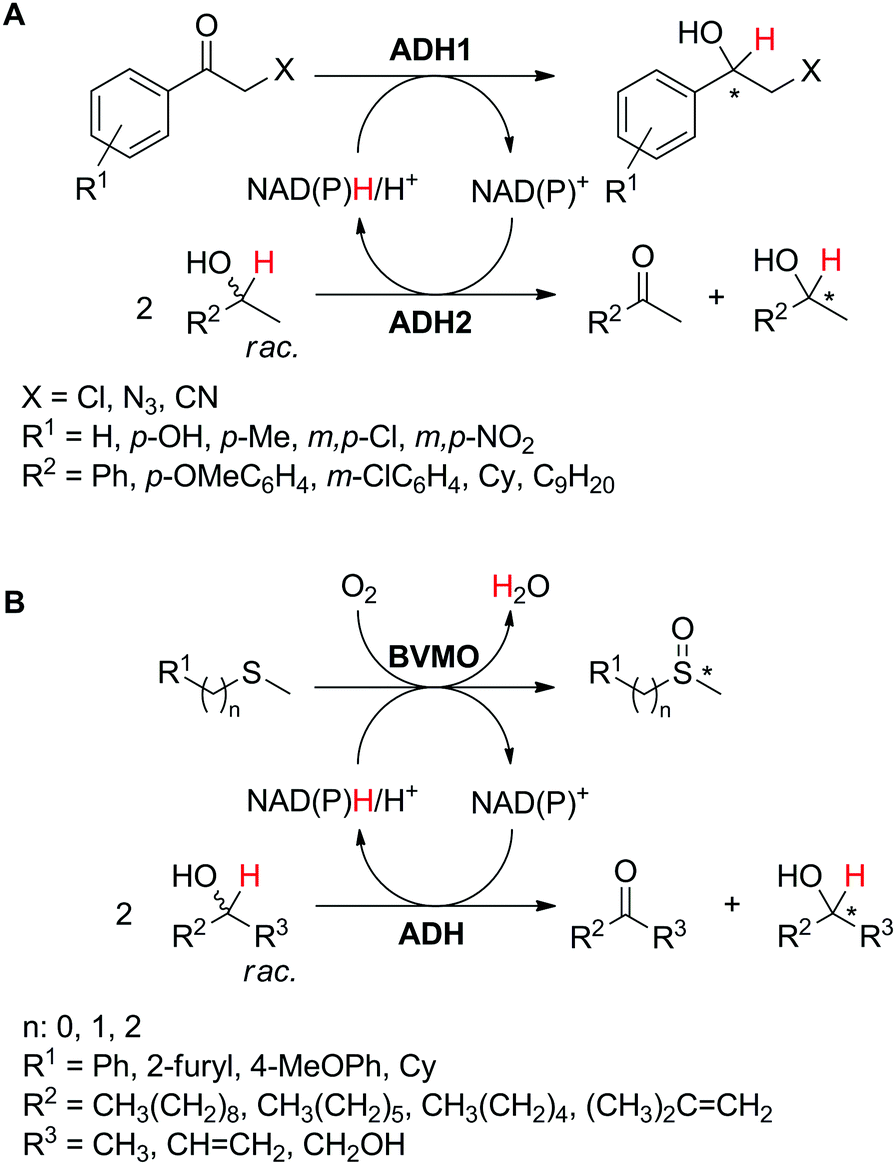 | ||
| Scheme 23 Parallel interconnected kinetic asymmetric transformations (PIKAT), coupling (A) two alcohol dehydrogenases (ADH) or (B) one ADH and a Baeyer–Villiger monooxygenase (BVMO).100,102,104 | ||
In the actual PIKAT process employing two enzymes, ADH-catalyzed kinetic resolution of racemic alcohols was combined with monooxygenase-catalyzed asymmetric oxidation of sulfides or enantioselective Baeyer–Villiger oxidation of racemic α-substituted ketones (Scheme 23B).100 To reach high conversion levels and enantiomeric excess values for the target products, a matching pair of biocatalysts was selected. Using monooxygenase PAMO M446G (a variant of phenylacetone monooxygenase from Thermobifida fusca) or HAPMO (4-hydroxyacetophenone monooxygenase from Pseudomonas fluorescens), and LB-ADH or ADH-T, with matching or opposite enantioselectivity, all four possible combinations of the enantio-enriched pair of products (sulfoxide/secondary alcohol or ester/secondary alcohol) were obtained. In both cases, the hydride abstracted from the secondary alcohol substrate ends up in water formed as a by-product of the monooxygenase-catalyzed reaction. Importantly, in the case of the Baeyer–Villiger oxidation, two kinetic resolutions take place, thereby generating four products, in ideal case in equivalent molar amounts: resolved α-substituted ketone, corresponding ester, resolved secondary alcohol and corresponding ketone. For best redox self-sufficiency, the sulfide/alcohol and ketone/alcohol pairs, respectively, must be provided in 2:1 and 1:1 ratio, respectively.
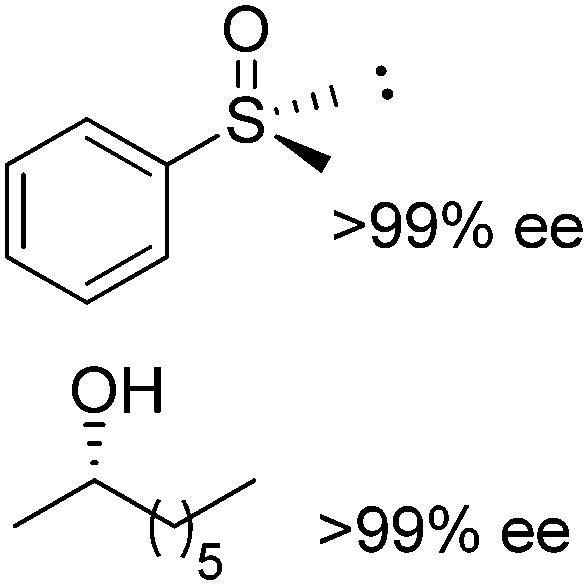
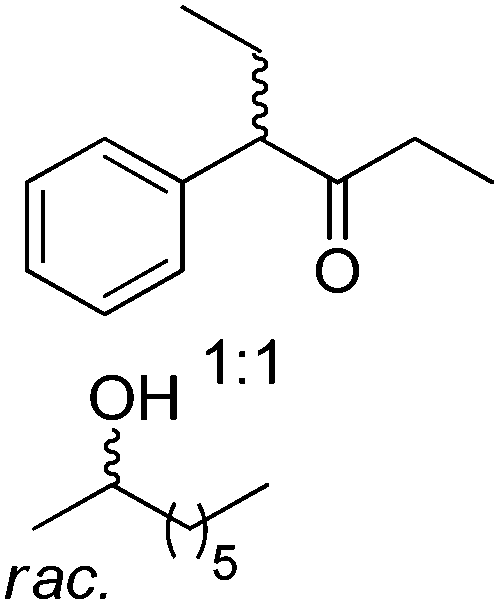
Exploiting the PIKAT approach, the substrate scope of the reaction was expanded to the concurrent production of aromatic and aliphatic chiral sulfoxides and different enantiopure secondary alcohols.104 In particular, efforts were dedicated to improving the cofactor turnover number, which increased beyond 300 when NADPH was provided in low molar concentration (∼0.03 mol% referenced to the sulfide). In the same study, a remarkable TONNAD (∼5500) was achieved in the concurrent oxidation of (±)-4-phenylhexan-3-one and kinetic resolution of (±)-2-octanol using PAMO and LB-ADH.
Despite merging of two synthetically relevant reactions, the main disadvantage of the PIKAT methodology is the challenging separation of the formed products.101,105
6. One substrate–one enzyme
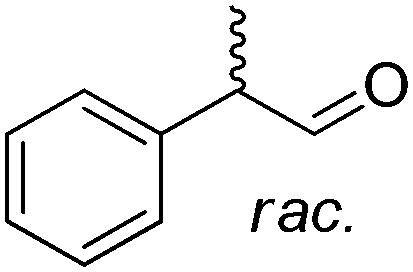
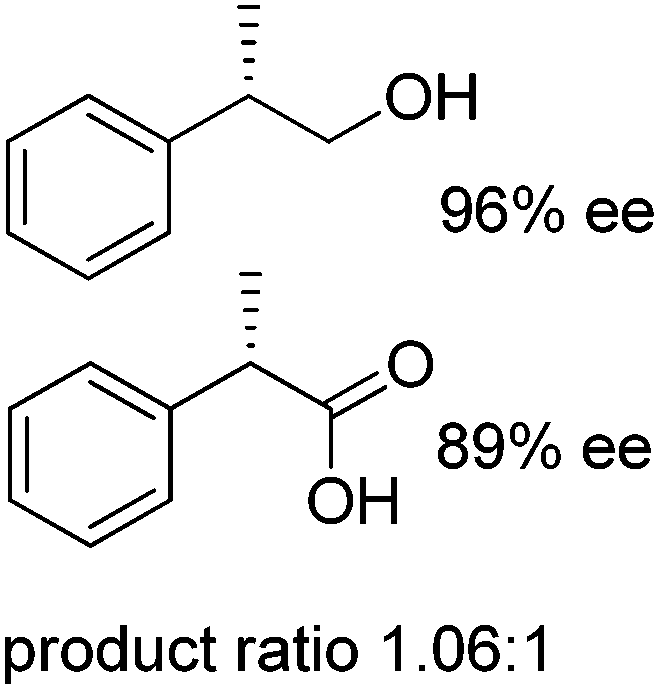
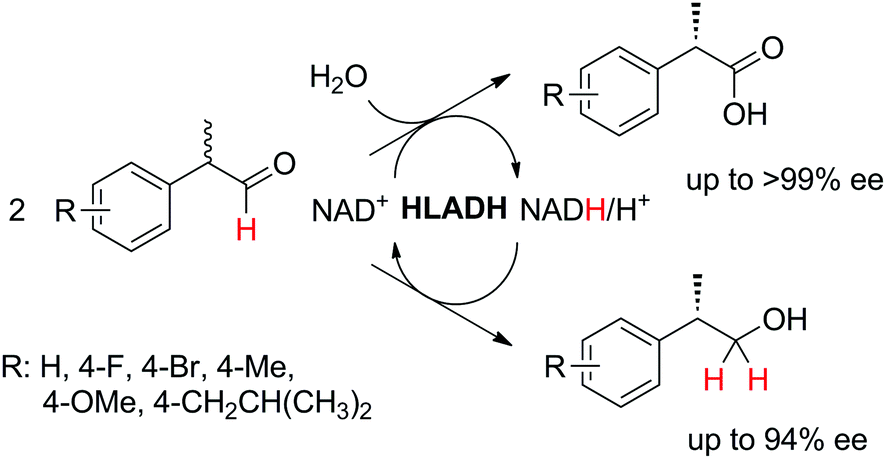
In order to simplify further the above-mentioned systems, the number of enzymes can be reduced to the strict minimum of one. This sets the major requirement on the selected biocatalyst, which should concurrently catalyze the same redox reaction in both directions, via (i) a disproportionation reaction, furnishing two products (Scheme 5(E1)), (ii) racemization of enantiomerically pure molecules via formation of a prochiral intermediate (Scheme 5(E2)), or (iii) a redox isomerization reaction, proceeding through formal intramolecular ‘swap’ of two functional groups (Scheme 5(E3)).Exemplary biocatalytic synthetic disproportionation was reported with the case of parallel interconnected dynamic asymmetric transformation (PIDAT), practically taking place concurrently between two molecules of the same substrate.106 Here, the enzyme concurrently catalyzes with high enantioselectivity the same redox reaction on the same functional group in opposite redox directions. The reported biocatalytic Cannizzaro-type reaction107 was applied to a series of substituted rac-2-arylpropanals employing purified horse liver alcohol dehydrogenase (HLADH) and allowed the formation of enantioenriched (S)-profens and profenols (up to 99% ee) in a high atom-efficient manner (Scheme 24). Only 1.3 mol% oxidized nicotinamide was necessary, and no co-product was generated. The reaction, which tolerated up to 75 mM substrate concentration, allowed up to 2100 TTN of the enzyme and 26.5 turnovers of the oxidized cofactor, with one turnover transforming two molecules of the substrate. The intermolecular hydride transfer reaction could be scaled up to 100 mg of 2-phenylpropanal. In contrast to the PIKAT approach,108 no by-product was generated, owing to dynamic kinetic resolution of α-substituted aldehydes via spontaneous racemization in the buffer.
 | ||
| Scheme 24 Biocatalytic parallel interconnected dynamic asymmetric transformation (PIDAT) catalyzed by horse liver alcohol dehydrogenase (HLADH) applied to rac-2-arylpropanals.106 | ||
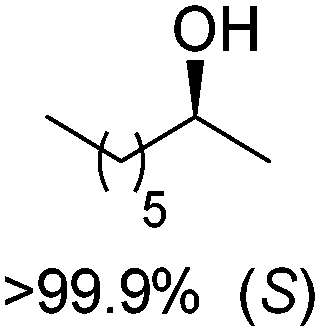
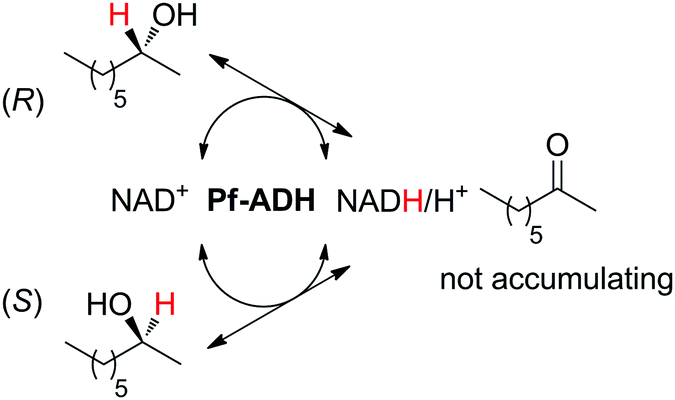
The biocatalytic racemization of enantiopure chiral secondary alcohols by ADH was initially developed using two enzymes of opposite stereopreference, and proceeded through the formation of intermediate prochiral ketone (for a similar concept applied to the racemization of α-hydroxyacids, see Section 2.1 and Scheme 10).109 The whole process relies on the reversibility of the reaction. Substituting the two enzymes with non-selective ADH from Pseudomonas fluorescens (Pf-ADH) was possible, and the enzyme thus catalyzes the same reaction back and forth. While slower (effective racemization observed after 50 h), Pf-ADH could be applied to the racemization of both enantiomers of model substrate 2-octanol (Scheme 25). Nicotinamide was implemented in catalytic amounts as a NAD+/NADH pair in a 1:1.7 ratio (2.7 and 4.6 mol%, respectively).
 | ||
| Scheme 25 Racemization of 2-octanol (of either (S)- or (R)-enantiomer) by alcohol dehydrogenase from Pseudomonas fluorescens (Pf-ADH; doubled arrows used for simplification, both reactions are reversible).109 | ||
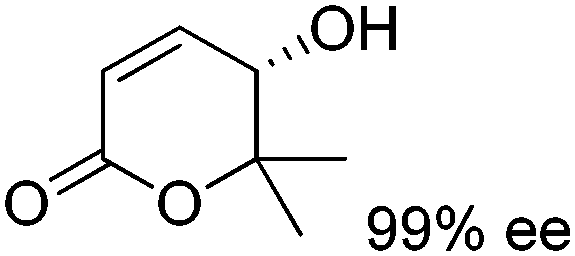
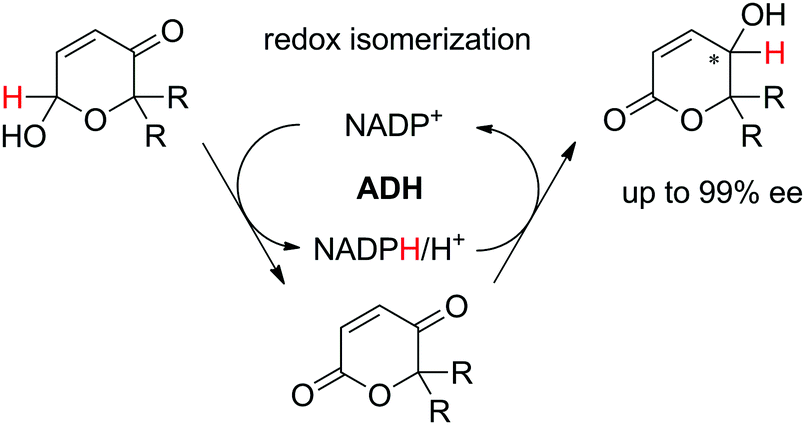
In a recent study, a closely related concept was applied to a redox isomerization protocol applied to Achmatowicz pyranones, which present two functional groups accepted by ADHs.110 The reaction scheme relied on a single ADH to catalyze the overall stereoselective oxidation–reduction sequence to yield corresponding γ-hydroxylated δ-lactones† in high enantiopurity via formation of intermediate γ-oxo-lactones (Scheme 26). The overall reaction is not reversible since ADHs are not known to catalyze the reduction of ester functionality and the enzyme catalyzes the same reaction in opposite redox directions on two distinct functional groups in different redox states. The formal 1,4-intramolecular shift of hydride on the pyranones (max. 8.8 mM starting concentration) via oxidized nicotinamide was moderately efficient, with turnover numbers for NADP+ ranging between 2.2–12.4, corresponding to 4.4–24.8 half-reactions per mol of cofactor. Finally, by employing E. coli resting cells expressing a suitable ADH, no external addition of NADP+ was necessary and this in vivo approach with 100:1 mass ratio of fresh cells to substrate allowed 71% conversion of 7 mM of a pyranone substrate.
 | ||
| Scheme 26 Redox isomerization reaction catalyzed by alcohol dehydrogenase (ADH) to enantioenriched γ-hydroxylated δ-lactones.110 | ||
7. Conclusions and outlook
By evaluating enzymatic nicotinamide-dependent self-sufficient hydride transfer processes reported in the literature, a scale of complexity clearly appears – ranging from a combination of one or two substrates with several enzymes, to one substrate and one enzyme – along with different levels of efficiency. This efficiency can be best represented by the turnover number (TON) of the hydride shuttle – typically a nicotinamide cofactor – as TON accurately represents the number of cycles a hydride can go through before getting lost to the ‘outside’, which automatically translates into a dead end. Exceptions are the enzymatic aerobic sequences such as those described in Sections 2.1–2.2 and 5.1–5.2, in which the overall transformation is a double oxidation of the substrate(s) tightly controlled by hydride loss from either the substrate or the intermediate product of the sequence. Formally, oxygen, as an external oxidizing agent, is reduced to water as a by-product, so that these sequences are not strictly redox-neutral (substrates are oxidized by external oxygen and a reduced by-product is generated). For all other cases, the loss of hydride can be only permitted as long as this loss does not exceed the amount of available/added shuttle. Since this amount is catalytic, errors are only poorly tolerated.Ultimately, the simplest system to implement consists of one enzyme, one cofactor and one substrate and releases one single product and no waste. Such minimized construction has only been reported for the redox isomerization of particular pyranone compounds, during which two functional groups are exchanged within the same molecule in a redox-neutral fashion, rendering an ideal case of intramolecular hydride transfer.110
Comparing the various strategies reviewed herein (Table 2), it is however striking that the enzymatic sequence which contains three biocatalysts and a pair of cofactors displays relatively high efficiency in terms of cofactor turnovers (78 each). The reaction was not studied at substrate concentrations higher than 20 mM and as highlighted in the introduction, the higher the number of enzymes, the narrower the operational window that can accommodate all enzymes (e.g., optimum pH and temperature, substrate tolerance). The highest value (TON ∼5500) was obtained in the PIKAT approach (Table 2, entry 12); unfortunately, the system is plagued by the formation of unwanted ketone by-products, severely impacting the atom economy of the sequence. The conversion of unsaturated aldehydes to saturated carboxylic acids shows efficient cycling of the cofactor (470); however, the sequence (Table 2, entry 5) was performed at a low substrate concentration (5 mM). Importantly, not all systems described here seem to have been optimized for the highest TONNAD(P) and several cases report full conversion of the substrate at a pre-determined cofactor concentration. Noteworthily, when targeting synthetic applications and scale-up of reactions, parameters other than hydride economy are equally important, such as the total turnover number (TTN) of enzymes or concentration of substrates and products. All these need to be taken into account when evaluating the final economical and environmental impact of a given biocatalytic process.22,111 A look behind the curtain also reveals that some biocatalysts can only be poorly combined for efficient internal hydride shuffling, and P450s in general do not appear to be suitable for nicotinamide-dependent self-sufficient hydride transfers. In most cases, the strong uncoupling typical for such oxygen-dependent enzymes depletes the system in reducing equivalents via formal reduction of oxygen to hydrogen peroxide; only an excess of co-substrate as a hydride donor can yield satisfactory turnover numbers for the nicotinamide, however going in hand with by-product formation (coupled-enzyme nicotinamide regeneration strategy).73,78
Incorporating redox self-sufficient enzymatic sequences in larger bio-synthetic schemes also pinpoints the difficulty of handling complex reaction mixtures, kinetics and thermodynamics when multiple reactions are at play simultaneously. Although displaying excellent hydride economy when run individually, some of the sequences depicted here are often complemented with co-substrate as an external hydride source to support cofactor regeneration in multi-step synthetic schemes via a coupled-substrate or coupled-enzyme approach, in particular at high substrate concentrations.63,65 In several cases, reaction engineering techniques are necessary to allow successful realization of the cascades in one-pot.22,64,112
Importantly, the strategies described herein are not limited to the conversion of a single redox functional group but can be nicely implemented on bi-functionalized substrates (Schemes 12, 13, 17 and 26). Although no example of redox self-sufficient systems has been reported on molecules presenting more than two redox functionalities, the tools developed by the community113 appear mature to tackle the next level of complexity and approach the efficiency of natural cell factories. Remarkably, the transfer of electrons between the substrate or intermediate product and nicotinamide can occur indirectly, involving additional mediators such as flavin (e.g., with BVMO, FMO, P450 monooxygenases), a metal ion (P450 monooxygenase), or even oxygen. This feature highlights the robustness of in vitro biocatalysis and the relevance of designing artificial biocatalytic cascades by coupling different enzyme classes.
Many more examples of reactions embracing the general principles of borrowing hydrogen exist in chemistry. Despite high synthetic potential due to appealing hydride and atom economy, these have yet to be adapted to enzymatic variants. With the rapidly growing number of newly identified enzymes (either as homologues complementary to existing proteins or as novel biocatalysts)114 and the impressive development of biocatalytic artificial pathways,39 it can be foreseen that more of these enzymatic self-sufficient hydride transfer reactions will be developed in an attempt to mimic the intrinsic efficiency of cells, with the added benefit of higher product titer – typical to in vitro systems – and the highly practical modular character of combining several enzymes in sequence. In turn, corresponding biosynthetic applications could fulfill some of the strict economical and ecological requirements arising from current industrial settings, where both hydride and atom economy are becoming decisive, and which aim at reducing process costs in, and environmental footprint115 of, the chemical industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by the Austrian Science Fund (FWF) is gratefully acknowledged (project P30519-N36). The authors want to dedicate this paper to the fruitful and influential carrier of Prof. Kurt Faber, who will continue to explore the world on two manpowered wheels.Notes and references
- M. G. Edwards and J. M. J. Williams, Angew. Chem., Int. Ed., 2002, 41, 4740 CrossRef CAS PubMed.
- A. Labonne, L. Zani, L. Hintermann and C. Bolm, J. Org. Chem., 2007, 72, 5704 CrossRef CAS.
- G. Guillena, D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358 CrossRef CAS.
- G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS.
- Y. Zhang, C. S. Lim, D. S. B. Sim, H. J. Pan and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 1399 CrossRef CAS.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS.
- M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef CAS.
- K. Schneider and H. G. Schlegel, Biochim. Biophys. Acta, 1976, 452, 66 CrossRef CAS.
- D. H. Kim and M. S. Kim, Bioresour. Technol., 2011, 102, 8423 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS PubMed.
- X. D. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. F. Shi, Chem, 2017, 2, 621 CAS.
- H. A. Reeve, P. A. Ash, H. Park, A. L. Huang, M. Posidias, C. Tomlinson, O. Lenz and K. A. Vincent, Biochem. J., 2017, 474, 215 CrossRef CAS PubMed.
- J. Schleucher, C. Griesinger, B. Schworer and R. K. Thauer, Biochemistry, 1994, 33, 3986 CrossRef CAS PubMed.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572 CrossRef CAS.
- W. Hummel and H. Groger, J. Biotechnol., 2014, 191, 22 CrossRef CAS.
- T. Knaus and F. G. Mutti, Chem. Today, 2017, 35, 34 CAS.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305 CrossRef CAS PubMed.
- H. K. Chenault, E. S. Simon and G. M. Whitesides, Biotechnol. Genet. Eng. Rev., 1988, 6, 221 CrossRef CAS PubMed.
- K. Faber, Biotransformations in Organic Chemistry, Springer Verlag, Berlin Heidelberg, 7th edn, 2018 Search PubMed.
- J. M. Woodley, in Modern Biocatalysis: Advances Towards Synthetic Biological Systems, ed. G. Williams and M. Hall, The Royal Society of Chemistry, 2018, p. 516 Search PubMed.
- L. Lauterbach and O. Lenz, Curr. Opin. Chem. Biol., 2019, 49, 91 CrossRef CAS.
- L. Lauterbach, O. Lenz and K. A. Vincent, FEBS J., 2013, 280, 3058 CrossRef CAS PubMed.
- S. Kara, J. H. Schrittwieser, F. Hollmann and M. B. Ansorge-Schumacher, Appl. Microbiol. Biotechnol., 2014, 98, 1517 CrossRef CAS PubMed.
- A. Weckbecker, H. Groger and W. Hummel, in Biosystems Engineering I. Advances in Biochemical Engineering/Biotechnology, ed. C. Wittmann and R. Krull, Springer, Berlin Heidelberg, 2010, vol. 120, p. 195 Search PubMed.
- H. Wu, C. Y. Tian, X. K. Song, C. Liu, D. Yang and Z. Y. Jiang, Green Chem., 2013, 15, 1773 RSC.
- W. A. van der Donk and H. M. Zhao, Curr. Opin. Biotechnol, 2003, 14, 421 CrossRef CAS PubMed.
- N. S. Punekar, Enzymes: Catalysis, Kinetics and Mechanism, Springer Nature Singapore, 2018 Search PubMed.
- M. Richter, Nat. Prod. Rep., 2013, 30, 1324 RSC.
- C. Nowak, A. Pick, P. Lommes and V. Sieber, ACS Catal., 2017, 7, 5202 CrossRef CAS.
- I. Zachos, C. Nowak and V. Sieber, Curr. Opin. Chem. Biol., 2019, 49, 59 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 7th edn, 1998 Search PubMed.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed.
- J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938 CrossRef CAS PubMed.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270 CrossRef CAS PubMed.
- J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249 CrossRef CAS PubMed.
- C. Wandrey, E. Fiolitakis, U. Wichmann and M. R. Kula, Ann. N. Y. Acad. Sci., 1984, 434, 91 CrossRef CAS PubMed.
- A. Dennig, F. Blaschke, S. Gandomkar, E. Tassano and B. Nidetzky, Adv. Synth. Catal., 2019, 361, 1348 CrossRef CAS.
- A. J. Willetts, C. J. Knowles, M. S. Levitt, S. M. Roberts, H. Sandey and N. F. Shipston, J. Chem. Soc., Perkin Trans. 1, 1991, 1608 RSC.
- F. F. Chen, Y. Y. Liu, G. W. Zheng and J. H. Xu, ChemCatChem, 2015, 7, 3838 CrossRef CAS.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525 CrossRef CAS PubMed.
- B. Bossow and C. Wandrey, Ann. N. Y. Acad. Sci., 1987, 506, 325 CrossRef CAS PubMed.
- M. J. Abrahamson, E. Vazquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969 CrossRef CAS PubMed.
- B. R. Bommarius, M. Schürmann and A. S. Bommarius, Chem. Commun., 2014, 50, 14953 RSC.
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg, J. L. Petit, A. Mariage, A. Debard, V. Pellouin, A. Perret, V. de Berardinis, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, Nat. Catal., 2019, 2, 324 CrossRef CAS.
- M. P. Thompson and N. J. Turner, ChemCatChem, 2017, 9, 3833 CrossRef CAS.
- W. Böhmer, T. Knaus and F. G. Mutti, ChemCatChem, 2018, 10, 731 CrossRef PubMed.
- H.-L. Yu, T. Li, F.-F. Chen, X.-J. Luo, A. Li, C. Yang, G.-W. Zheng and J.-H. Xu, Metab. Eng., 2018, 47, 184 CrossRef CAS PubMed.
- S. L. Montgomery, J. Mangas-Sanchez, M. P. Thompson, G. A. Aleku, B. Dominguez and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 10491 CrossRef CAS PubMed.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961 CrossRef CAS PubMed.
- M. Tavanti, J. Mangas-Sanchez, S. L. Montgomery, M. P. Thompson and N. J. Turner, Org. Biomol. Chem., 2017, 15, 9790 RSC.
- Y. Okamoto, V. Kohler and T. R. Ward, J. Am. Chem. Soc., 2016, 138, 5781 CrossRef CAS PubMed.
- A. Bodlenner, S. M. Glueck, B. M. Nestl, C. C. Gruber, N. Baudendistel, B. Hauer, W. Kroutil and K. Faber, Tetrahedron, 2009, 65, 7752 CrossRef CAS.
- G. Grogan, S. Roberts and A. Willetts, Biotechnol. Lett., 1992, 14, 1125 CrossRef CAS.
- R. Gagnon, G. Grogan, S. M. Roberts, R. Villa and A. J. Willetts, J. Chem. Soc., Perkin Trans. 1, 1995, 1505 RSC.
- H. Mallin, H. Wulf and U. T. Bornscheuer, Enzyme Microb. Technol., 2013, 53, 283 CrossRef CAS PubMed.
- S. Staudt, U. T. Bornscheuer, U. Menyes, W. Hummel and H. Gröger, Enzyme Microb. Technol., 2013, 53, 288 CrossRef CAS PubMed.
- J. H. Sattler, M. Fuchs, F. G. Mutti, B. Grischek, P. Engel, J. Pfeffer, J. M. Woodley and W. Kroutil, Angew. Chem., Int. Ed., 2014, 53, 14153 CrossRef CAS PubMed.
- A. Pennec, F. Hollmann, M. S. Smit and D. J. Opperman, ChemCatChem, 2015, 7, 236 CrossRef CAS.
- C. Scherkus, S. Schmidt, U. T. Bornscheuer, H. Gröger, S. Kara and A. Liese, ChemCatChem, 2016, 8, 3446 CrossRef CAS.
- S. Schmidt, C. Scherkus, J. Muschiol, U. Menyes, T. Winkler, W. Hummel, H. Groger, A. Liese, H. G. Herz and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2015, 54, 2784 CrossRef CAS PubMed.
- S. Elleuche, Appl. Microbiol. Biotechnol., 2015, 99, 1545 CrossRef CAS PubMed.
- S. Gargiulo, D. J. Opperman, U. Hanefeld, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2012, 48, 6630 RSC.
- N. Oberleitner, C. Peters, J. Muschiol, M. Kadow, S. Saß, T. Bayer, P. Schaaf, N. Iqbal, F. Rudroff, M. D. Mihovilovic and U. T. Bornscheuer, ChemCatChem, 2013, 5, 3524 CrossRef CAS.
- S. Reich, B. M. Nestl and B. Hauer, ChemBioChem, 2016, 17, 561 CrossRef CAS PubMed.
- T. Winkler, H. Gröger and W. Hummel, ChemCatChem, 2014, 6, 961 CrossRef CAS.
- T. Knaus, F. G. Mutti, L. D. Humphreys, N. J. Turner and N. S. Scrutton, Org. Biomol. Chem., 2015, 13, 223 RSC.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203 CrossRef CAS PubMed.
- S. Staudt, E. Burda, C. Giese, C. A. Müller, J. Marienhagen, U. Schwaneberg, W. Hummel, K. Drauz and H. Gröger, Angew. Chem., Int. Ed., 2013, 52, 2359 CrossRef CAS PubMed.
- P. J. Loida and S. G. Sligar, Biochemistry, 1993, 32, 11530 CrossRef CAS PubMed.
- C. A. Müller, B. Akkapurathu, T. Winkler, S. Staudt, W. Hummel, H. Gröger and U. Schwaneberg, Adv. Synth. Catal., 2013, 355, 1787 CrossRef.
- C. A. Müller, A. Dennig, T. Welters, T. Winkler, A. J. Ruff, W. Hummel, H. Gröger and U. Schwaneberg, J. Biotechnol., 2014, 191, 196 CrossRef PubMed.
- C. A. Müller, A. M. Weingartner, A. Dennig, A. J. Ruff, H. Gröger and U. Schwaneberg, J. Ind. Microbiol. Biotechnol., 2016, 43, 1641 CrossRef PubMed.
- S. Schulz, M. Girhard, S. K. Gaßmeyer, V. D. Jäger, D. Schwarze, A. Vogel and V. B. Urlacher, ChemCatChem, 2015, 7, 601 CrossRef CAS.
- M. Hofer, H. Strittmatter and V. Sieber, ChemCatChem, 2013, 5, 3351 CrossRef CAS.
- M. Tavanti, F. Parmeggiani, J. R. G. Castellanos, A. Mattevi and N. J. Turner, ChemCatChem, 2017, 9, 3338 CrossRef CAS.
- V. Resch, W. M. F. Fabian and W. Kroutil, Adv. Synth. Catal., 2010, 352, 993 CrossRef CAS.
- C.-W. Fan, G.-C. Xu, B.-D. Ma, Y.-P. Bai, J. Zhang and J.-H. Xu, J. Biotechnol., 2015, 195, 67 CrossRef CAS PubMed.
- L. P. B. Goncalves, O. A. C. Antunes, G. F. Pinto and E. G. Oestreicher, Tetrahedron: Asymmetry, 2000, 11, 1465 CrossRef CAS.
- B. Boonstra, D. A. Rathbone, C. E. French, E. H. Walker and N. C. Bruce, Appl. Environ. Microbiol., 2000, 66, 5161 CrossRef CAS PubMed.
- R. Z. Zhang, Y. Xu, R. Xiao, B. T. Zhang and L. Wang, Microb. Cell Fact., 2012, 11, 167 CrossRef CAS PubMed.
- X. Tong, B. El-Zahab, X. Zhao, Y. Liu and P. Wang, Biotechnol. Bioeng., 2011, 108, 465 CrossRef CAS PubMed.
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156 CrossRef CAS PubMed.
- J. S. Shin and B. G. Kim, Biotechnol. Bioeng., 1999, 65, 206 CrossRef CAS PubMed.
- R. Abu and J. M. Woodley, ChemCatChem, 2015, 7, 3094 CrossRef CAS.
- K. Tauber, M. Fuchs, J. H. Sattler, J. Pitzer, D. Pressnitz, D. Koszelewski, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Chem. – Eur. J., 2013, 19, 4030 CrossRef CAS PubMed.
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129 CrossRef CAS.
- J. Reiter, H. Strittmatter, L. O. Wiemann, D. Schieder and V. Sieber, Green Chem., 2013, 15, 1373 RSC.
- S. Kara, D. Spickermann, J. H. Schrittwieser, C. Leggewie, W. J. H. van Berkel, I. W. C. E. Arends and F. Hollmann, Green Chem., 2013, 15, 330 RSC.
- A. Bornadel, R. Hatti-Kaul, F. Hollmann and S. Kara, ChemCatChem, 2015, 7, 2442 CrossRef CAS.
- S. Kara, D. Spickermann, J. H. Schrittwieser, A. Weckbecker, C. Leggewie, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2013, 3, 2436 CrossRef CAS.
- A. Bornadel, R. Hatti-Kaul, F. Hollmann and S. Kara, Tetrahedron, 2016, 72, 7222 CrossRef CAS.
- J. Engel, K. S. Mthethwa, D. J. Opperman and S. Kara, Mol. Catal., 2019, 468, 44 CrossRef CAS.
- L. Huang, E. Romero, A. K. Ressmann, F. Rudroff, F. Hollmann, M. W. Fraaije and S. Kara, Adv. Synth. Catal., 2017, 359, 2142 CrossRef CAS.
- L. Huang, W. Tang, R. Röllig, F. S. Aalbers, M. W. Fraaije and S. Kara, ChemBioChem, 2019, 20, 1653 CrossRef CAS PubMed.
- A. Rioz-Martinez, F. R. Bisogno, C. Rodriguez, G. de Gonzalo, I. Lavandera, D. E. T. Pazmino, M. W. Fraaije and V. Gotor, Org. Biomol. Chem., 2010, 8, 1431 RSC.
- O. Bortolini, G. Fantin, M. Fogagnolo, P. P. Giovannini, A. Guerrini and A. Medici, J. Org. Chem., 1997, 62, 1854 CrossRef CAS.
- F. R. Bisogno, I. Lavandera, W. Kroutil and V. Gotor, J. Org. Chem., 2009, 74, 1730 CrossRef CAS PubMed.
- I. Lavandera, A. Kern, V. Resch, B. Ferreira-Silva, A. Glieder, W. M. F. Fabian, S. de Wildeman and W. Kroutil, Org. Lett., 2008, 10, 2155 CrossRef CAS PubMed.
- F. R. Bisogno, A. Rioz-Martinez, C. Rodriguez, I. Lavandera, G. de Gonzalo, D. E. T. Pazmino, M. W. Fraaije and V. Gotor, ChemCatChem, 2010, 2, 946 CrossRef CAS.
- L. Martinez-Montero and I. Lavandera, in Modern Biocatalysis: Advances Towards Synthetic Biological Systems, ed. G. Williams and M. Hall, The Royal Society of Chemistry, 2018, p. 351 Search PubMed.
- E. Tassano, K. Faber and M. Hall, Adv. Synth. Catal., 2018, 360, 2742 CrossRef CAS PubMed.
- C. Wuensch, H. Lechner, S. M. Glueck, K. Zangger, M. Hall and K. Faber, ChemCatChem, 2013, 5, 1744 CrossRef CAS.
- A. Rioz-Martínez, F. R. Bisogno, C. Rodríguez, G. de Gonzalo, I. Lavandera, D. E. Torres Pazmiño, M. W. Fraaije and V. Gotor, Org. Biomol. Chem., 2010, 8, 1431 RSC.
- C. C. Gruber, B. M. Nestl, J. Gross, P. Hildebrandt, U. T. Bornscheuer, K. Faber and W. Kroutil, Chem. – Eur. J., 2007, 13, 8271 CrossRef CAS PubMed.
- Y. C. Liu, C. Merten and J. Deska, Angew. Chem., Int. Ed., 2018, 57, 12151 CrossRef CAS PubMed.
- A. Illanes, L. Wilson and C. Vera, in Modern Biocatalysis: Advances Towards Synthetic Biological Systems, ed. G. Williams and M. Hall, The Royal Society of Chemistry, 2018, p. 473 Search PubMed.
- C. Scherkus, S. Schmidt, U. T. Bornscheuer, H. Groger, S. Kara and A. Liese, Biotechnol. Bioeng., 2017, 114, 1215 CrossRef CAS PubMed.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409 CrossRef.
- A. Zaparucha, V. R. de Berardinis and C. Vaxelaire-Vergne, in Modern Biocatalysis: Advances Towards Synthetic Biological Systems, ed. G. Williams and M. Hall, The Royal Society of Chemistry, 2018, p. 1 Search PubMed.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082 RSC.
- B. Danielsson, F. Winqvist, J. Y. Malpote and K. Mosbach, Biotechnol. Lett., 1982, 4, 673 CrossRef CAS.
- A. J. Irwin and J. B. Jones, J. Am. Chem. Soc., 1977, 99, 556 CrossRef CAS PubMed.
Footnotes |
| † Hydrogenases have been used to regenerate NAD(P)H at the expense of H2, as demonstrated in the seminal work by Mosbach et al. for the amination of pyruvate by alanine dehydrogenase.116 |
| ‡ While the pair FAD/FADH2 redox couple displays a standard reduction potential of −0.180 V in a protein-free environment, this value significantly varies upon the active site architecture of a given protein, from −0.450 to +0.150 V.29 |
| § The field of enzymatic oxidation of lactols by alcohol dehydrogenases was pioneered by B. Jones et al.117 |
| This journal is © The Royal Society of Chemistry 2019 |