
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArtificial photosynthesis: opportunities and challenges of molecular catalysts
Biaobiao
Zhang
 a and
Licheng
Sun
*ab
a and
Licheng
Sun
*ab
aDepartment of Chemistry, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, 10044 Stockholm, Sweden. E-mail: lichengs@kth.se
bState Key Laboratory of Fine Chemicals, Institute of Artificial Photosynthesis, DUT-KTH Joint Education and Research Center on Molecular Devices, Dalian University of Technology (DUT), 116024 Dalian, China
First published on 21st March 2019
Abstract
Molecular catalysis plays an essential role in both natural and artificial photosynthesis (AP). However, the field of molecular catalysis for AP has gradually declined in recent years because of doubt about the long-term stability of molecular-catalyst-based devices. This review summarizes the development history of molecular-catalyst-based AP, including the fundamentals of AP, molecular catalysts for water oxidation, proton reduction and CO2 reduction, and molecular-catalyst-based AP devices, and it provides an analysis of the advantages, challenges, and stability of molecular catalysts. With this review, we aim to highlight the following points: (i) an investigation on molecular catalysis is one of the most promising ways to obtain atom-efficient catalysts with outstanding intrinsic activities; (ii) effective heterogenization of molecular catalysts is currently the primary challenge for the application of molecular catalysis in AP devices; (iii) development of molecular catalysts is a promising way to solve the problems of catalysis involved in practical solar fuel production. In molecular-catalysis-based AP, much has been attained, but more challenges remain with regard to long-term stability and heterogenization techniques.
 Biaobiao Zhang | Dr Biaobiao Zhang received his PhD in Applied Chemistry from Dalian University of Technology in 2015 under the guidance of Prof. Licheng Sun and is currently a postdoc at KTH Royal Institute of Technology. His current work focuses on the development of manganese oxide based WOCs, molecular WOCs and molecular WOC engineered materials for artificial photosynthesis devices. |
 Licheng Sun | Prof. Licheng Sun received his PhD in 1990 from Dalian University of Technology. He went to Germany as a postdoc at Max-Planck-Institut für Strahlenchemie with Dr Helmut Görner (1992–1993), and then as an Alexander von Humboldt fellow at Freie Universität Berlin (1993–1995) with Prof. Dr Harry Kurreck. He moved to KTH Royal Institute of Technology, Stockholm, in 1995 and became an assistant professor in 1997, an associate professor in 1999 (at Stockholm University) and a full professor in 2004 (KTH). He is presently also a distinguished professor in DUT. His research interests cover artificial photosynthesis, molecular catalysts for water oxidation and hydrogen generation, functional devices for total water splitting, dye sensitized, quantum dot/rod sensitized solar cells, and perovskite solar cells. |
1. Introduction
Over the last hundred years, approximately 80% of worldwide energy consumption has been based on fossil fuels, including coal, oil, and natural gas.1 However, humankind now has to face the consequences arising from this dependence on fossil fuels. Worldwide energy consumption is expected to increase by over 50% by the mid-2000s.2 Because fossil fuels are finite and regional around the world, it is greatly challenging to ensure that this demand can be met, in the face of possible political tensions and other potential problems with energy supplies. Due to the usage of fossil fuels, large quantities of emissions, e.g., CO2, SO2, and oxide particles, are the predominant reasons for global warming and severe pollution. Recent reports from the Intergovernmental Panel on Climate Change emphasized the necessity of decreasing CO2 emissions on a global scale to the zero level before the next century.3 These arguments make the development of sustainable and carbon-neutral energy technologies one of the most urgent challenges facing humankind all over the world. The most abundant renewable energy source on the planet is solar energy: solar illumination on Earth every hour is greater than the worldwide energy consumption for a whole year.4 Therefore, the conversion and utilization of solar energy is a promising solution for energy problems.The nature of a solar-energy carrier is that of a photon moving at light speed. A fundamental step for using solar energy is to harvest these high-energy particles with light absorbers to generate separated electrons and holes, i.e., charge separation. In photovoltaic cells, these photogenerated electrons and holes can be collected to produce potential and current; in consequence, the harvested solar energy is directly converted into electricity, which is an important energy format in daily life. However, electricity is not readily stored on a large scale, and cannot realize a complete replacement of fossil fuels, because the energy density of batteries is far below that of chemical fuels with respect to both weight and volume.3 Therefore, electricity is not an ideal energy source for applications such as large oversea vessels and long-distance air transport. Inspired by the natural photosynthesis, rather than being collected for electricity, the photo-generated electrons and holes can be used to drive chemical reactions; thus the harvested solar energy can be converted into chemical energy (solar fuel) stored in the form of chemical bonds. This multiple-step process, which mimics the function of natural photosynthesis, is generally named artificial photosynthesis (AP).3,5
Since the proposal of the AP concept and the modeling of AP devices, chemists have believed that developing molecular catalysts is one promising way to solve the problems of catalysis involved in AP, because of the abundant advantages of molecular catalysts.6 Over the past few decades, hundreds of catalysts have been developed and investigated for the reactions involved in AP. However, it seems that the devices assembled from molecular catalysts are less stable and perform less efficiently than those built from material catalysts. On the other hand, it has been found that, under certain experimental conditions, the involved molecular catalysts decomposed into metal-based inorganic materials that were the true catalysts. Because of the underachievement of molecule-based AP devices, the research on development of molecular catalysts has been gradually losing focus over the last five years.
We believe that molecular catalysts are still promising for the future of AP. The real bottleneck problems for realizing applicable AP have obviously not been solved. The achievement of molecule-based AP should include several stages, such as development of efficient molecular catalysts, heterogenization of molecular catalysts, and engineering of applicable devices. In previous decades, scientists have expended much effort on the first stage, and several highly efficient and promising molecular catalysts have been developed for water oxidation, hydrogen evolution and CO2 reduction. However, studies into the second stage, which is equally important and challenging, have largely been inadequate. Many problems and factors have not been researched and have not even been well considered. We have therefore written this review article to summarize previous research on molecular catalysts for AP, to emphasize the significance of molecular catalysts, and to discuss the opportunities, challenges and future of molecular catalysts, with the aim of providing readers with some guidelines on how to choose catalysts when they start to design their systems for AP.
2. Artificial photosynthesis and functional devices inspired by nature
For billions of years, plants and organisms on Earth have provided oxygen and foods for aerobic life cycles via photosynthesis, a process that produces O2 and carbohydrates (e.g., sugar C6H12O6) from CO2 and H2O by utilizing sunlight.7 Consequently, solar energy is converted into chemical energy and stored in carbohydrates. Photosynthesis occurs through the cooperation of photosystem II (PSII) and photosystem I (PSI).8,9 Driven by sunlight, H2O is oxidized in PSII into O2, releasing four protons and four electrons. The electrons and protons are transferred via cytochrome b6f to PSI, in which the separated electrons and protons are finally consumed by CO2 reduction to produce carbohydrates (Fig. 1a).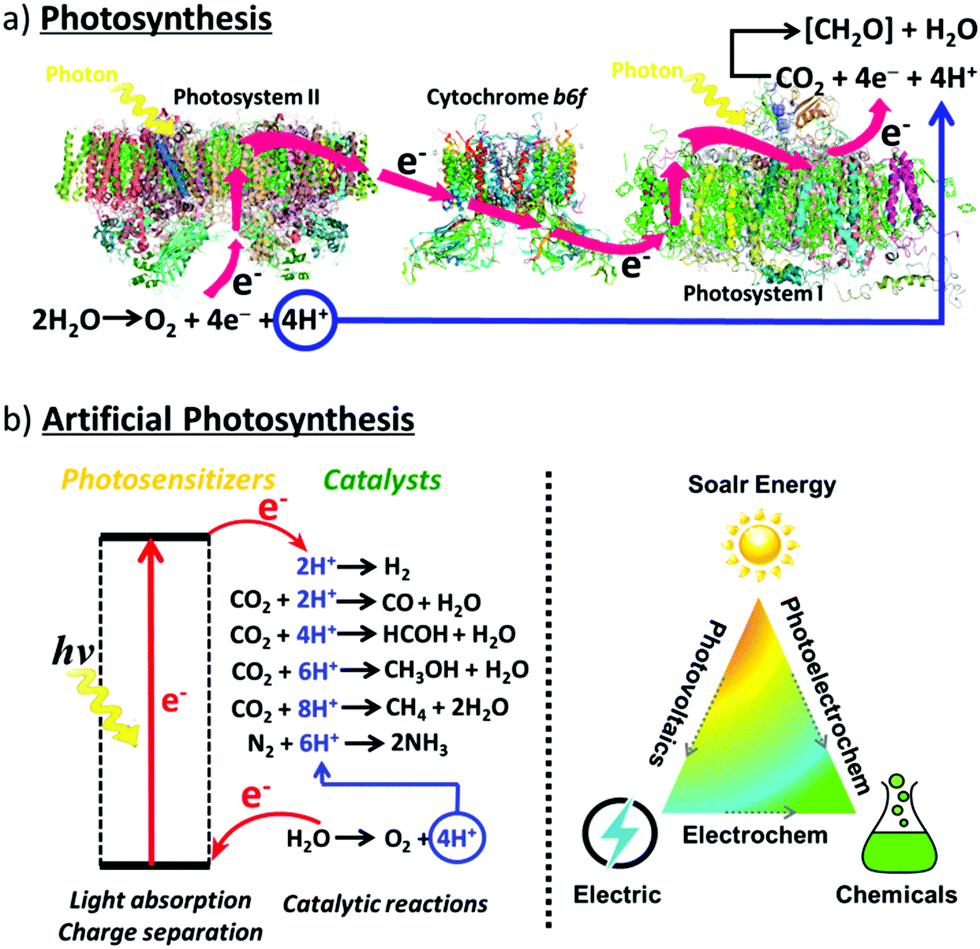 | ||
| Fig. 1 Schematic representation of (a) the natural photosynthetic chain in photosystems; (b) general concept of AP. | ||
The photosystems and photosynthesis are extremely complicated. It is unachievable to exactly mimic their structures and components. However, the functions and chemical processes can be replicated to realize AP to convert solar energy into chemical energy and store it as synthetic chemical fuels (Fig. 1b). Solar energy can be directly converted into chemical fuels via photoelectrocatalytic reactions, or it can be converted into fuels indirectly by using the electricity generated from solar energy to drive electrocatalytic reactions. Water splitting to produce hydrogen fuel (2H2O → O2 + 4e− + 4H+; 2H+ + 2e− → H2) and CO2 reduction to produce carbon-neutral fuel (e.g. CO2 + 6H+ + 6e− → CH3OH + H2O) are widely investigated reactions in artificial photosynthesis. In this review, we mainly focused on water splitting and CO2 reduction reactions. However, it should be noted that the production of other types of fuels and high-value-added chemicals can also be achieved with AP by driving the corresponding reactions,10 such as ammonia production from nitrogen fixation,11 epoxide production from hydrocarbon oxygenation,12–14 hydrogen peroxide production from oxygen reduction,15 and polymer production from biomass related 5-hydroxymethylfurfural (HMF) conversion reactions.16
Two types of devices have been designed for the production of solar fuels, the photoelectrochemical (PEC) cell (Fig. 2a) and the photovoltaic-coupled electrolyzer (PVE) device (Fig. 2b).17 An ideal PEC cell consists of a photoanode and a photocathode. The photoanode can be a molecular dye sensitized TiO2 electrode18–20 or n-type visible-light-absorbing semiconductors21 (e.g., α-Fe2O3,22 WO323 and BiVO424) with enough driving force or valence band potential for water oxidation. The photocathode can be a molecular dye sensitized NiO electrode25 or other p-type visible-light-absorbing semiconductors26 (such as InP27 and Cu2O28) with conduction bands that are negative enough to reduce the reactants to chemical fuels. Under illumination, the generated holes and electrons are directly transferred to the catalysts loaded on the surface of the photoanode and photocathode to drive the photocatalytic reactions and produce fuels (Fig. 2a). In practice, a PEC cell can also be fabricated with a single photoelectrode, either one photoanode or one photocathode. Because of the challenge of assembling full PEC cells, most of the reported PEC cells to date are single-photoelectrode cells. For the PVE devices (Fig. 2b), the light absorption and charge separation are completed by a solar cell, which can be separated from the electrolyzer cell.29 Potentials generated by the solar cell are applied to the electrolyzer to drive the electrochemical water-oxidation reaction on the anode and the electrochemical reduction reactions for fuel production on the cathode. In the PVE device, solar energy is indirectly converted into chemical fuels in two steps.
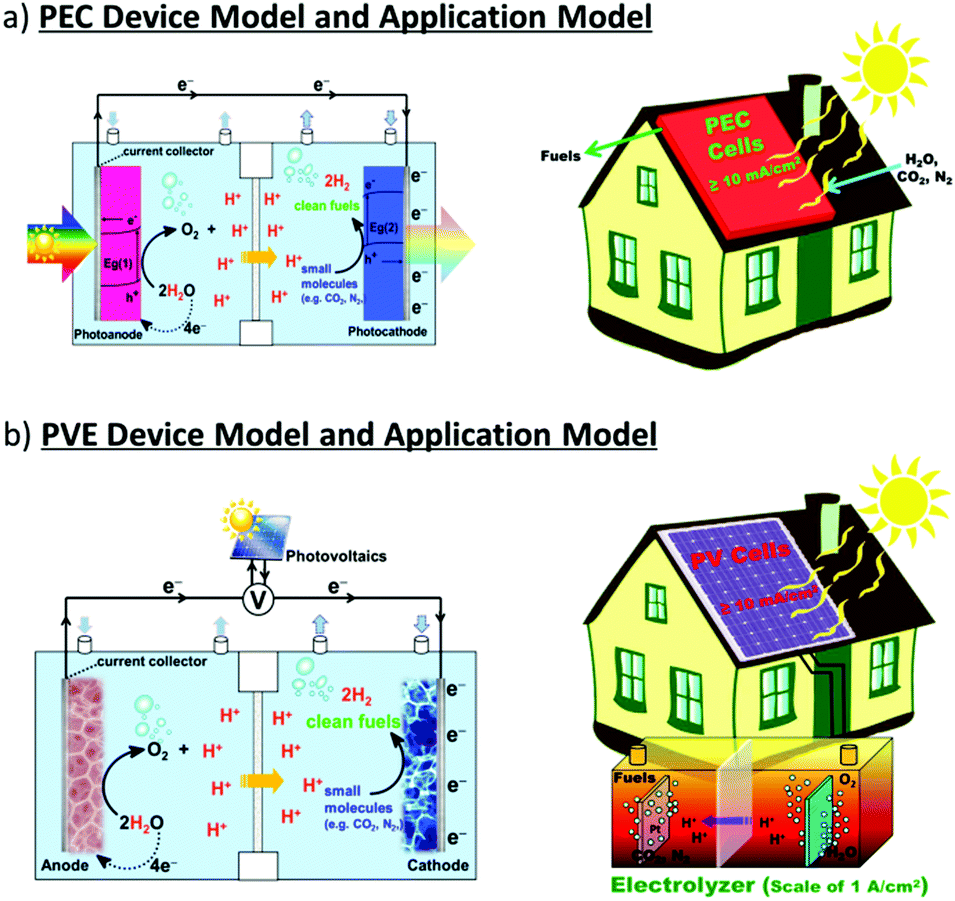 | ||
| Fig. 2 Schematic representation of (a) PEC device model and application model; (b) PVE device model and application model. | ||
Fig. 2 also shows two possible ways to domestically apply PEC and PVE devices in the future. The required performances of each device are marked in the figures.30 To meet the forecasted worldwide annual energy consumption in the year 2050, only 1% of the earth's surface needs to be covered by solar energy converting devices with ≥10% solar-to-fuel efficiencies.30,31 Accordingly, in the PEC device, the photo-generated current densities of water splitting need to be over 10 mA cm−2. In the PVE device, the lowest demand on the ability of the solar cell in general is a photocurrent density over 10 mA cm−2 and a potential high enough to load the electrolyzer. Moreover, the electrolyzer cell needs to be a highly integrated device, which can generally generate a current for electrochemical synthesis with a current density in the region of 1 A cm−2.
Four main processes, namely, light harvesting by light absorbers, charge separation in the light absorbers, charge transfer from the light absorbers to the catalysts, and the occurrence of catalytic reactions, will be involved in complete AP. Accordingly, two fundamental components are necessary for building AP devices. Ideal light absorbers, with broad absorption in the solar spectrum, stable excited states and high charge carrier mobility, and efficient catalysts, with high intrinsic activity and stability, are the two essential components to realize AP. Photosensitizers or light absorbers, such as molecular dyes,19,32,33 semiconductors34 and quantum dots,35,36 are needed to absorb the light and generate the charge separation. Moreover, efficient catalysts are the other key. If the half reactions shown in Fig. 1b are taken as examples, the reactions in AP all involve multiple electron and proton transfers, which make these reactions kinetically sluggish and require high overpotentials. Efficient catalysts are required to overcome the high reaction barriers, lower the requirement for overpotential, and accelerate the reaction rate. To achieve catalysts with high intrinsic activity, low overpotential, and a high catalytic rate, the development of molecular catalysts is the most promising way to go.
3. Why and how to engage molecular catalysts in applicable AP devices
3.1 Advantages of molecular catalysts
Complexes 1, 2, and 3 (Fig. 3a) are well-known examples of Fe-based molecular catalysts for water oxidation, hydrogen evolution, and CO2 reduction, respectively.39–41 The metal centers of 1, 2, and 3 are all iron; however, these complexes can catalyze different reactions because the electronic structures of the iron cores are precisely adjusted by the particular coordination environments created by the ligands. Thus, a molecular catalyst usually comprises a metal center as the active site and specific organic ligand(s) to produce the correct coordination environment for regulating the electronic structure of the active site. Thanks to this configuration, molecular catalyst possesses plenty of advantages as partially listed below: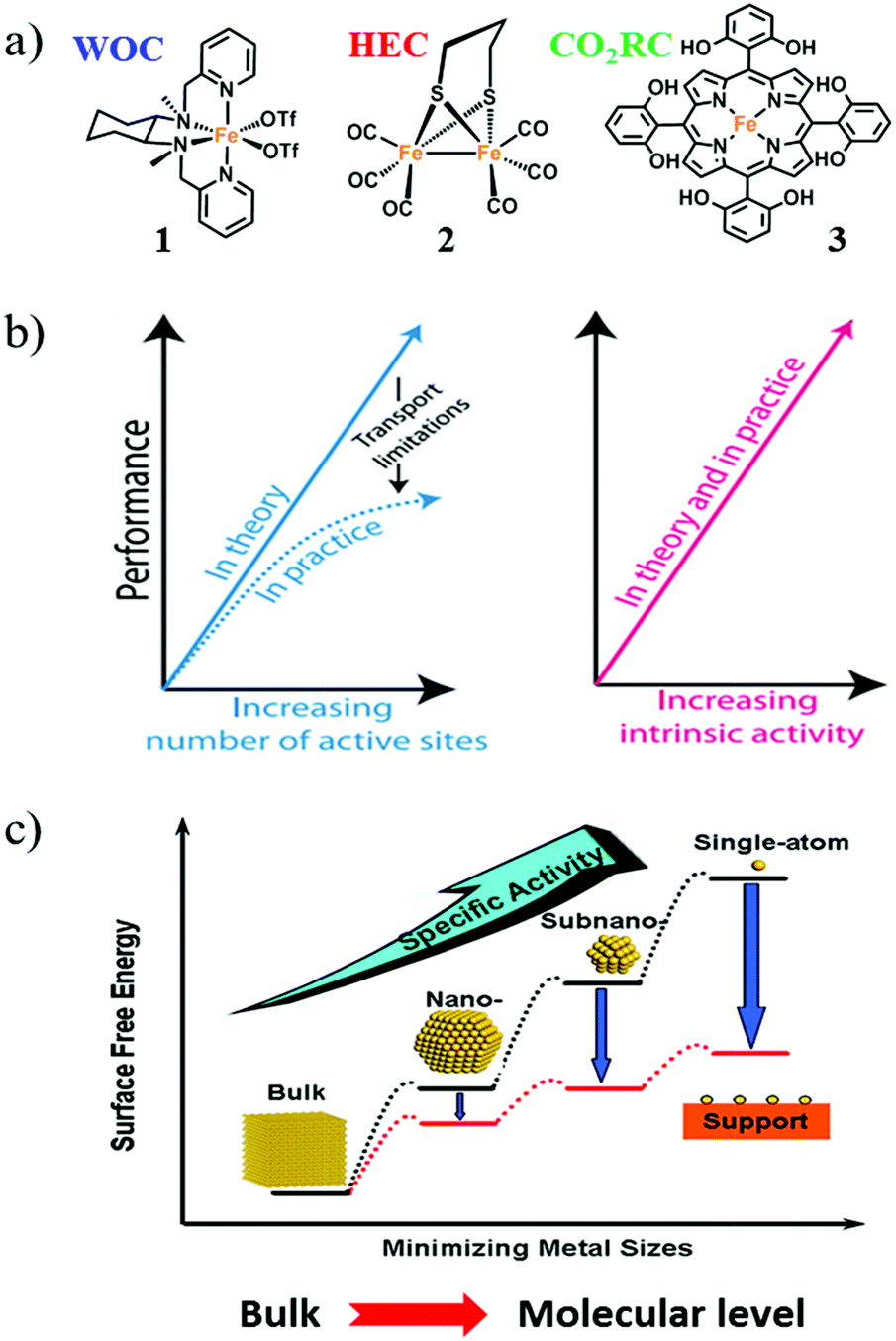 | ||
| Fig. 3 (a) Molecular structures of Fe complexes 1, 2, and 3 as molecular catalysts. WOC: water-oxidation catalyst; HEC: hydrogen-evolution catalyst; CO2RC: CO2 reduction catalyst. (b) The enhancement of catalytic performance by increasing the number of active sites and/or increasing the intrinsic activity of each active site. Adapted with permission from ref. 37. Copyright 2017 American Association for the Advancement of Science (AAAS). (c) The positive effect on catalytic performance obtained by minimizing the size of nanoparticle catalysts. Adapted with permission from ref. 38. Copyright 2013 American Chemical Society. | ||
Jaramillo and co-workers have given a precise statement about the intrinsic activity in their review on material design. They have written that “increasing intrinsic activity leads to direct increases in electrode activity in a manner that mitigates transport issues arising from high catalyst loadings; with improved intrinsic activity, the catalyst loading can be decreased, which also saves on catalyst costs. Moreover, catalyst activity is measured across many orders of magnitude; the difference in intrinsic activity between a good catalyst and a poor catalyst can be more than 10 orders of magnitude, whereas the difference between a high-loading and a low-loading catalyst might only be one to three orders of magnitude” (Fig. 3b).37
Therefore, studies to effectively improve the intrinsic activity of catalysts are important for the advancement of AP. For molecular catalysts, every molecule is a single active site with suitable intrinsic catalytic activity based on its unique structure. In conjunction with the first two advantages of molecular catalysts, the development of molecular catalysts can directly promote their intrinsic activity.
Indeed, SACs and DHCs are similar to molecular catalysts, because they catalyze on a molecular level. The difference between them is that the chemical environment of the active metal center of molecular catalysts is provided by molecular ligands, whereas it is provided by the anchoring supports for SACs and DHCs. It is clear that the development of material catalysts is now focused deeply at the molecular level. In addition, nature also chose molecular catalytic centers for various enzymes to complete the catalysis of water oxidation,9 proton reduction,51 and CO2 reduction reactions,52 indicating that molecular catalysis is no doubt also promising for AP. Consequently, the development of molecular catalysts is one of the effective ways to achieve practical AP.
3.2 Stages prior to achieving applicable molecular AP devices
AP is a major project to solve the sustainable energy problem, which is a big issue facing all human beings. In this context, although research into molecule-based AP has been ongoing for over 30 years, we are still at the early stages, and many more stages have not been researched or have not even been well proposed. There are at least three stages involved in producing a final AP device based on molecular catalysts: development of efficient molecular catalysts, heterogenization of molecular catalysts, and engineering of applicable AP devices (Fig. 4). These stages are correlated but they also have their own totally different challenges. For the first stage, the research community in this field has accumulated a lot of methods, strategies, and principles. There are also several successful WOCs, HECs, and CO2RCs in hand. However, for the remaining two stages, the investigations are far from complete.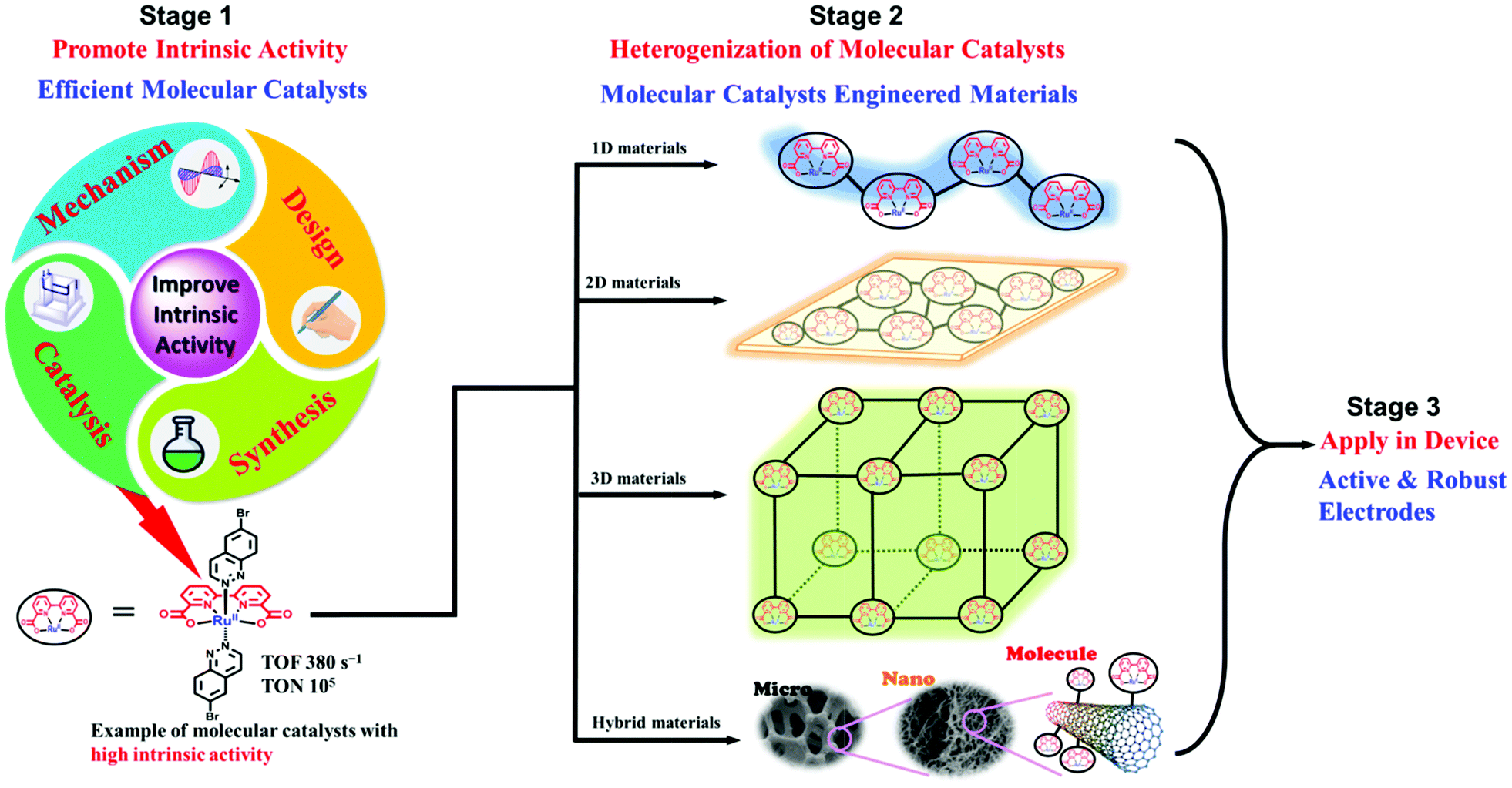 | ||
| Fig. 4 Schematic representation of the stages involved in achieving applicable molecular AP devices. TOF: turnover frequency; TON: turnover number. The Ru complexes shown here are highly efficient molecular WOCs. | ||
Heterogenization of molecular catalysts, that is, from molecules to materials and from a homogeneous solution to a heterogeneous surface, is even more essential and challenging. However, there has been far from enough research on the heterogenization of molecular catalysts, in comparison with research on the first stage. Effective strategies for the heterogenization of molecular catalysts are needed to preserve the intrinsic high activity and selectivity of the homogeneous catalysts after heterogenization. In addition, the properties of the resultant heterogeneous surface or material, including the morphology (e.g., particle size and surface area), density of active sites, hydrophobicity/hydrophilicity, and mass transfer limitations, require elaborate optimization. Without adequate methods, the molecular catalysts cannot be employed by the device effectively and stably. Indeed, this is the reason for the current poor situation of molecular catalyst based AP. Although several molecular catalysts with extremely efficient activities have been obtained, AP devices based on these molecular catalysts did not show any apparent superiority in performance; on the contrary, these devices were not comparable with material-based devices in terms of bulk current density and stability.
On the basis of success at the first stage, it is currently urgent and essential to explore applicable strategies for the heterogenization of molecular catalysts, as illustrated in the general representation in Fig. 4. In contrast to catalysts made of inorganic materials, the most prominent advantage of the materials obtained from the heterogenization of molecular catalysts is that each metal has the same coordination environment and is able to be the active site.
Obviously, the development of applicable AP devices based on molecular catalysts is now in the middle of the first stage, at the very beginning of the second stage, and far away from the third stage. Therefore, it is not reasonable to reject this promising pathway at such an early stage of the research and to miss the potential opportunities of AP devices with molecular catalysts.
4. Developing efficient molecular catalysts for AP
4.1 Overview of molecular water-oxidation catalysts (WOCs)
Water is the only ideal source of protons and electrons, which are necessary for the fuel-production reactions. No other alternative reducing substrate has the capacity to meet the heavy demands for fuel production on a global scale, while releasing no waste.56 Therefore water oxidation is key for AP. However, it is also the bottleneck for the development of AP because it requires a high thermodynamic potential (ΔG ≈ 237 kJ mol−1 and E0 ≈ 1.23 V) and a high overpotential to overcome the kinetic barrier involved in the transfer of 4 H+ and 4 e−.55 Efficient catalysts can reduce the kinetic barrier by forming low-activation-energy intermediates, and consequently, accelerate the rates of water oxidation. Due to the advantages of molecular catalysts mentioned above, development of molecular WOCs has attracted considerable attention in the field of AP, and major progress has been achieved.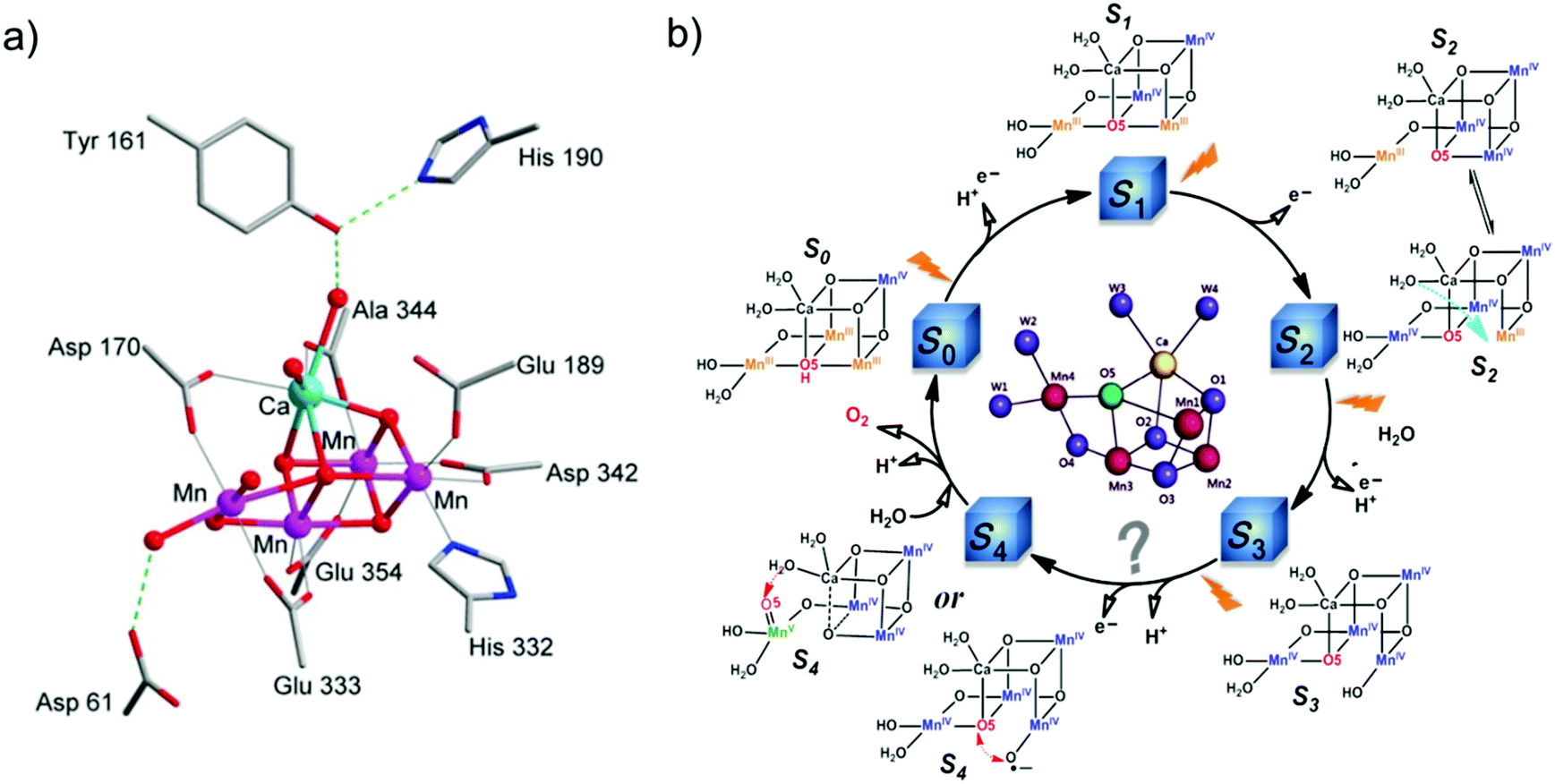 | ||
| Fig. 5 (a) Structure of the Mn4CaO5 cluster of the OEC in PSII.9 (b) Classic Kok cycle for water oxidation in PSII, with widely-proposed structures of the intermediate “S” states, electron transfer, and proton release. | ||
Prior to the revelation of the OEC structure, it was only known that water oxidation at the OEC proceeds via the Kok cycle involving five intermediates, referred to as the Si (i = 0–4) states, but the details of the catalytic mechanism were unknown (Fig. 5b).61 Based on the clear crystal structure and the advanced physical techniques used in recent years, PSII scientists are in general agreement on the oxidation states62 and structures of the metastable S0, S1, and S2 intermediates (Fig. 5b).9,63–65 Nevertheless, the mechanistic details of O–O bond formation remain unclear.66–68 Currently, two main O–O bond formation mechanisms are widely considered for PSII: the oxo–oxyl radical coupling mechanism69 and the nucleophilic attack mechanism,68,70 involving a MnIV–oxyl radical and a MnV–oxo electrophile, respectively, as active S4 intermediates. However, the existence of a MnIV–oxyl radical or a MnV–oxo electrophile has not been experimentally confirmed; the actual mechanism of water oxidation by the Mn4CaO5 cluster in PSII remains to be determined. Very recently, we proposed a new water oxidation mechanism involving a charge-rearrangement-induced MnVII–dioxo intermediate state.71–73 Elucidating the catalytic mechanism in PSII will not only uncover the secret of water oxidation in nature, but also provide a blueprint for developing more efficient water-oxidation catalysts for AP.
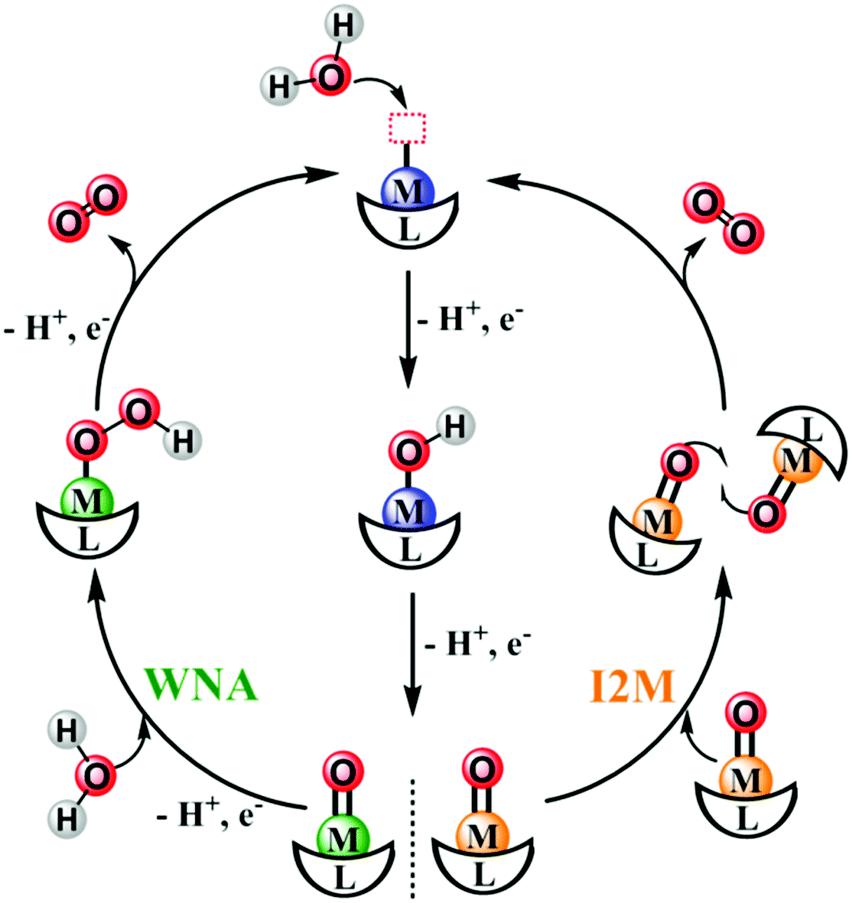 | ||
| Fig. 6 Schematic representation of the water nucleophilic attack (WNA) and the interaction between two M–O intermediate (I2M) pathways to form O–O bonds catalyzed by molecular catalysts. | ||
Fig. 6 shows that after binding with a substrate H2O molecule and several steps of proton-coupled electron transfer (PCET), the WOC generates a high-valent M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate, which is essential for the next crucial O–O bond formation step. Depending on the nature of this MO intermediate, there are two classic pathways for O–O bond formation: (i) water nucleophilic attack (WNA) and (ii) interaction of two M–O units (I2M).76–79 In the WNA pathway, a second H2O molecule, which acts as the nucleophile, attacks the electrophilic O on the MO intermediate, resulting in a two-electron reduction of the metal center and O–O bond formation on a metal hydroperoxide intermediate (M–OOH). Next, O2 evolution occurs after further oxidation of the M–OOH. In the I2M pathway, the O on the MO intermediate may have higher electron spin and have more radical character. In this context, the O–O bond forms via coupling of two radical-like species, leading to an M–O–O–M species, which can release O2 upon further oxidization.
O intermediate, which is essential for the next crucial O–O bond formation step. Depending on the nature of this MO intermediate, there are two classic pathways for O–O bond formation: (i) water nucleophilic attack (WNA) and (ii) interaction of two M–O units (I2M).76–79 In the WNA pathway, a second H2O molecule, which acts as the nucleophile, attacks the electrophilic O on the MO intermediate, resulting in a two-electron reduction of the metal center and O–O bond formation on a metal hydroperoxide intermediate (M–OOH). Next, O2 evolution occurs after further oxidation of the M–OOH. In the I2M pathway, the O on the MO intermediate may have higher electron spin and have more radical character. In this context, the O–O bond forms via coupling of two radical-like species, leading to an M–O–O–M species, which can release O2 upon further oxidization.
Currently, the WNA mechanism is the most widely proposed mechanism for molecular catalysis and metal-oxide-based material catalysis, but the state-of-the-art catalyst Ru-bda (bda = 2,2′-bipyridine-6,6′-dicarboxylate) catalyzes water oxidation via the I2M mechanism.80–83 Moreover, both these mechanisms have been considered as the possible water oxidation mechanism in PSII. We cannot conclude which mechanism is superior; however, there is no doubt that: (i) the catalytic mechanism of a WOC is dominated by its configuration and electronic structure, which are closely controlled by its coordination environment, for example, the ligands, and (ii) a better understanding of the catalytic mechanism of a WOC catalyst will significantly enhance catalytic activity.
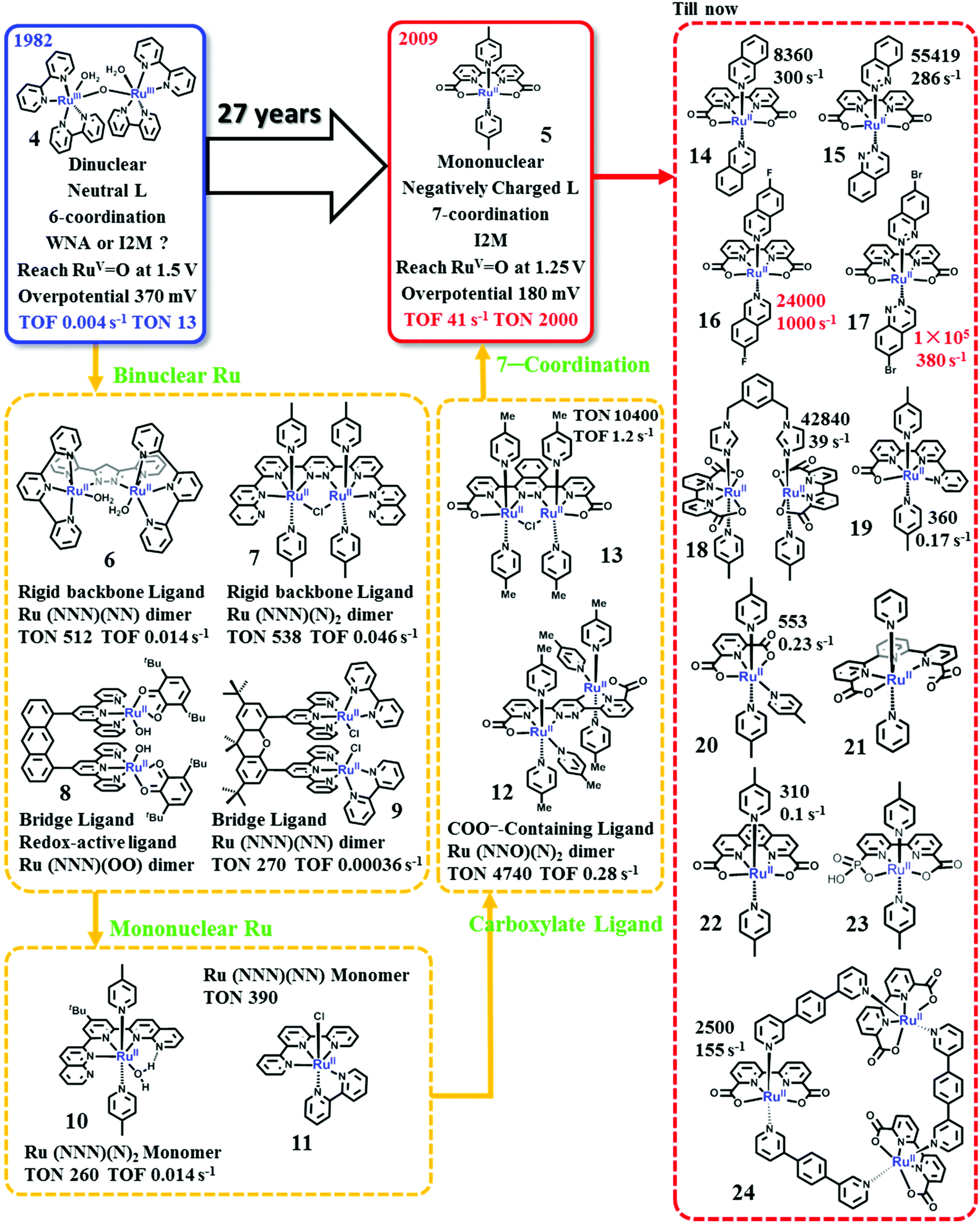 | ||
| Fig. 7 Development history and recent status of Ru-based WOCs. Labelled TONs and TOFs are obtained from experiments using Ce4+ as the chemical oxidant. | ||
The blue dimer (BD). Although the BD showed modest catalytic activity for water oxidation, studies on the BD led to the development of various theories for the evolution of molecular WOCs. First, the mechanistic details of water oxidation catalyzed by transition metal complexes were established. The O
RuV–RuVO state is proved to be the final necessary state prior to oxygen evolution.88–93 Three different mechanisms have been proposed for O–O bond formation by the BD: the intramolecular/intermolecular oxo–oxo coupling, the WNA pathway and a pathway involving the oxidation of the bpy-ligand.93,94 Although several critical reaction steps remain open to interpretation, the WNA mechanism is preferred for the BD, and has been extensively studied by using various experimental techniques92,95–97 and theoretical calculations.98 It was subsequently proved that this general O–O bond formation mechanism is involved in the water oxidation catalysis by many different kinds of catalysts, including molecular catalysts4,55,78 and metal-oxide-based material catalysts.99,100 Second, these early studies emphasized the essential role of proton-coupling electron-transfer (PCET) processes for water oxidation catalysis.84,101–104 In the PCET processes, oxidation of the metal center in a catalyst leads to a reduction in the pKa of the H2O binding at the active site; this leads to the release of one proton, resulting in stronger donor ligands like hydroxo and oxo. This in turn stabilizes the high valent metal state and facilitates further oxidation of the metal center, thus advancing the multi-electron oxidation, which is necessary for water oxidation catalysis. Third, the success of the BD as a water oxidation catalyst also confirmed the suitability of polypyridine ligands, which are capable of tolerating harsh oxidation conditions and are stable towards hydrolysis, as a scaffold to build molecular WOCs.103 Research studies on the BD have inspired the development of both binuclear and mononuclear Ru-based WOCs and molecular catalysts based on other transition metal complexes.4,55
Ru(NNN)(NN) dimers with rigid polypyridyl-based backbone ligands. Following the studies on the BD, an obvious step forward is the development of Ru dimer catalysts with a rigid polypyridyl-based backbone ligand. To prevent the deactivation of μ-O-linked Ru dimer catalysts, in 2004, Llobet and coworkers reported the first ruthenium dimer WOC (complex 6) without a μ-O bridge ligand by using a rigid backbone ligand, 2,6-bis(pyridyl)pyrazole (Hbpp).105 Under similar experimental conditions, the oxygen evolution catalyzed by 6 is more than three times faster than that by the BD. Moreover, since the turnover and the variation of the intermediates are slow, the mechanistic studies of this catalyst have been validated with details.106 The O–O bond formation catalyzed by 6 occurs via the I2M pathway. The Llobet group further investigated the performances of Ru-bpp analogues107–111 and the Ru-bpp catalysts anchored onto solid supports.112,113
Ru(NNN)(N)2 dimers with rigid polypyridyl-based backbone ligands. Later, the Thummel group designed and synthesized a series of Ru dimer catalysts, containing a rigid polypyridyl-based backbone ligand and two pyridine axial ligands, e.g. complex 7.114,115 One advance made by the Thummel group is the introduction of monodentate axial ligands, instead of another polypyridyl ligand, into the catalyst structure. It produced a new family of Ru WOCs, the backbone-ligand-linked Ru(NNN)(N)2 type. Thanks to the steric structure of the Ru(NNN)(N)2-type catalysts, the open sites of catalysts became larger, facilitating the binding of the substrate H2O molecule as an equatorial ligand and optimization of the catalysts. The Ru(NNN)(N)2-type catalysts developed by Thummel showed significantly higher TONs (up to 600) than the BD catalysts and Ru-bpp catalysts.
Ru-Dimers with rigid bridge ligands. Another strategy to obtain a dimer catalyst with cis geometry between the two Ru centers is to use a rigid bridge to link two Ru monomers.116,117 One typical example is the Ru dimer [Ru2(OH)2(3,6-tBu2qui)2(btpyan)]2+ (8, 3,6-tBu2qui = 3,6-di-t-butyl-1,2-benzoquinone; btpyan = 1,8-bis(2,2′:6′,2′-terpyridyl)anthracene) reported by the Tanaka group.116 Interestingly, complex 8 is a unique water oxidation catalyst because it contains a redox-active ligand, which plays a fundamental role in the catalytic activity of 8.118–120 Molecular WOCs involving redox-active ligands have recently attracted increasing attention.121,122 The Berlinguette group has developed three Ru dimer catalysts with various distances between the Ru centers by linking two Ru(tpy)(bpy) units using different rigid bridge ligands (9).123 However, no remarkable improvement in activity was obtained for the rigid-bridge-ligand-linked dimer catalysts.
From binuclear to mononuclear. Considering the multi-metallic Mn4CaO5 core of the OEC, it was long believed that synthetic molecular WOCs must also have multiple metal centers (at least two) to allow the accumulation of four oxidizing equivalents needed for catalytic water oxidation. In 2005, the Thummel group reported the first series of mononuclear Ru WOCs, e.g., complex 10, suggesting that the single metal center is active enough for catalyzing water oxidation.115 Later, Meyer and co-workers emphasized that one site is enough for water oxidation by revealing a well-defined mechanism involving the Ru(V) centers of two monomeric Ru-based WOCs, [Ru(tpy)(bpm)(OH2)]2+ (bpm = 2,2′-bipyrimidine) and [Ru(tpy)(bpz)(OH2)]2+ (bpz = 2,2′-bipyrazine).124,125 The concept of a single metal site catalyzing water oxidation represented a paradigm shift for the field and opened the door for developing a new series of catalyst systems with synthetic flexibility. With only one metal site, reaction mechanisms could also be deeply studied using much simpler platforms. Thus, the catalog of WOCs based on transition metal complexes has been significantly expanded. The paradigm shift from binuclear molecular WOCs to mononuclear ones is an essential step forward for developing efficient molecular WOCs.
The Ru(NNN)(N)2 and Ru(NNN)(NN) type mononuclear catalysts. Mononuclear catalysts like complexes 10 and 11 can be classified as the Ru(NNN)(N)2- and Ru(NNN)(NN)-type mononuclear catalysts.115,126 One of the important differences between these two different types of catalysts is the binding of the substrate H2O. The substrate H2O molecule binds on the Ru site as an equatorial ligand and as an axial ligand in the Ru(NNN)(N)2-type and the Ru(NNN)(NN)-type catalysts, respectively. The Ru(NNN)(N)2-type catalysts were extensively investigated by the Thummel group.115,127–129 They studied the effect of the axial ligands on the properties and catalytic performances of these catalysts.127 Moreover, a great variety of equatorial ligands were designed and synthesized for these Ru catalysts. These studies inspired the design of ligands for Ru-based WOCs.
Ru(NNN)(NN)-type complexes constitute a major family of promising mononuclear WOCs. A typical representative of Ru(NNN)(NN)-type WOCs is the [Ru(tpy)(bpy)(X)]n+ (X = H2O or halogen atom), which, together with various derivatives, has been extensively researched.126,130–134 The effect of the electronic properties on the activity of the [Ru(tpy)(bpy)(X)]n+ catalysts was carefully studied by the groups of Berlinguette,133–135 Thummel,126,127 Sakai130,131 and Yagi136 by introducing a range of substituents to the periphery of both the tpy and bpy ligand frameworks. Instead of bpy or tpy, Meyer and co-workers have incorporated several strong σ-donating bidentate or tridentate ligands into complexes of the [Ru(tpy)(bpy)(X)]n+ motif.132 The electrochemistry and spectroelectrochemistry of these catalysts have been studied extensively to elucidate the reaction kinetics and mechanism.124,125,137,138 Based on these studies, Meyer and co-workers proposed an important strategy to accelerate the O–O bond formation, i.e., the concerted atom-proton transfer (APT).137,138
Introduction of carboxylate-containing ligands. The Ru catalysts developed before 2009 are mainly based on neutral polypyridyl ligands. The studies on these complexes as WOCs have provided us considerable knowledge about how to design a WOC and how to evaluate the catalytic performance and catalytic mechanism. However, significant improvement in the catalytic activity for water oxidation was not achieved until the introduction of carboxylate-containing ligands into the catalyst structure.87
Inspired by the OEC in PSII containing several carboxylate ligands, the Sun group employed carboxylate ligands to synthesize Ru-based complexes that can efficiently catalyze water oxidation. The first carboxylate-containing Ru WOC reported by the Sun group was an unexpected trans-dimeric complex 12 with TON 4700 and TOF 0.28 s−1 (Ce4+ was used as the oxidant).139,140 Later, a modified ligand 1,4-bis(6′-COOH-pyrid-2′-yl)phthalazine was synthesized to obtain a cis dimeric Ru complex 13. By using Ce4+ as the oxidant, the cis complex 13 showed a catalytic TON of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 and a TOF of 1.2 s−1, far superior to those of the trans complex 12 and other reported catalysts at the time.141 Moreover, the cis complex 13 showed a low onset potential of 1.15 V vs. NHE for water oxidation; therefore, light-driven water oxidation was also successfully achieved in the presence of [Ru(bpy)3]2+ as the photosensitizer. The outstanding activities of complexes 12 and 13 obviously indicate that introduction of carboxylate groups is an effective and essential strategy to obtain promising WOCs with low overpotentials, because the carboxylate groups could make the higher oxidation states more accessible and more stable.
400 and a TOF of 1.2 s−1, far superior to those of the trans complex 12 and other reported catalysts at the time.141 Moreover, the cis complex 13 showed a low onset potential of 1.15 V vs. NHE for water oxidation; therefore, light-driven water oxidation was also successfully achieved in the presence of [Ru(bpy)3]2+ as the photosensitizer. The outstanding activities of complexes 12 and 13 obviously indicate that introduction of carboxylate groups is an effective and essential strategy to obtain promising WOCs with low overpotentials, because the carboxylate groups could make the higher oxidation states more accessible and more stable.
Ru(bda)(pic)2, founder of a new era for efficient molecular WOCs. Based on the concept of introducing carboxylate groups into the catalyst structure, the Sun group also designed a dianionic ligand, the 2,2′-bipyridine-6,6′-dicarboxylate (bda), to coordinate with Ru, producing a mononuclear Ru-bda complex, which is one of the most efficient catalytic center units known thus far.80,83,87,142,143 Using Ce4+ as the driving force at pH 1, the parent [Ru(bda)(pic)2] complex (complex 5) catalyzes water oxidation via a bimolecular I2M pathway with a TON of 2000 and a TOF of 41 s−1.144 An electrochemical study of 5 indicates that it needs an overpotential of only 180 mV to initiate water oxidation.145
Because of the outstanding activity, Ru-bda-type WOCs have received extensive research attention worldwide and have been widely investigated by the Sun group among many other groups; researchers have studied the effect of axial ligands,146–149 catalytic mechanisms,80,81,150–156 development of Ru-bda analogues157–160 and applications of Ru-bda in AP devices.161–166 The studies on the effect of axial ligands of Ru-bda-type WOCs were very fruitful. Electron-withdrawing and hydrophobic substituents on the axial ligands had positive influence on the water oxidation activity, e.g. Ru-bda catalysts with both 4-(EtOOC)-pyridine (TON = 4800 and TOF = 120 s−1) and 4-bromopyridine (TON = 4500 and TOF = 115 s−1) showed excellent water oxidation activities.148 Ru-bda catalysts (e.g. complexes 14 and 15) with π-extended axial ligands, such as isoquinoline and phthalazine, showed dramatically enhanced activities because the π–π stacking and hydrophobic effects between the axial ligands reduce the energy barrier for O–O bond formation via radical coupling.143 Introduction of halogen substituents into the π-extended axial ligand further improved the catalytic activity of the Ru-bda catalysts, complex 16 with record TOF > 1000 s−1 and complex 17 with record TON > 100000. To obtain higher catalytic activity, based on the bimolecular I2M mechanism of Ru-bda catalysts, various strategies have been tested to enhance the intermolecular interaction167–170 or to create facile intramolecular reactions by linking two or more Ru-bda units,171,172e.g., complex 18.
Revealing the reason behind the outstanding catalytic performance of Ru-bda catalysts is essential to inspire further design of more efficient WOCs. The question has not been satisfactorily answered thus far; however, at least two dominant factors have been proved essential to the breakthrough activity of these catalysts. First, the carboxylate groups in the structure lower the potential needed to generate active Ru-oxo species for O–O bond formation. Second, the equatorial bda ligand accesses a strongly distorted octahedral geometry of [Ru(bda)(L)2] with a unique O–Ru–O angle of ∼124° for Ru(bda)(pic)2, which is 34° larger than an ideal rectangular arrangement; consequently, it opens up a crucial seventh coordination site for the substrate water molecule to bind.80,83,143 Further theoretical investigations concluded that the RuVO intermediate is hydrophobic and shows a distinct oxyl radical character, e.g. RuIV–O˙, facilitating the bimolecular I2M pathway.153,173 In complex 19, one of the carboxylate groups was replaced by pyridine. Although the distorted octahedral geometry of the complexes was retained, the catalytic performance became several orders of magnitude lower and O–O bond formation proceeded via the WNA pathway.174 The Ru complexes 20 and 21, with a tridentate dianionic ligand 2,6-pyridine dicarboxylate (pdc)175,176 and a pentadentate dianionic ligand [2,2′:6′,2′-terpyridine]-6,6′′-dicarboxylate (tda),159,177,178 respectively, also showed greater catalytic activities than the relatively neutral complex. Without the open site for hepta-coordination, these catalysts adopt the WNA pathway and the overpotentials are much higher than those of the Ru-bda catalysts, indicating that hepta-coordination is crucial for the I2M mechanism.
In addition to the above-mentioned ones, there must be other factors governing the special catalytic performance of Ru-bda catalysts. For instance, the [Ru(pda)(pic)2] (complex 22, pda = phenanthroline dicarboxylate) complex possesses a coordination environment highly similar to that of [Ru(pda)(pic)2], including both dianionic groups and the large open site for hepta-coordination. However, the activity of 22 is much lower than that of 5 (TON = 310; TOF = 0.1 s−1), and the kinetics of catalytic water oxidation switches to first order.179 DFT calculations suggest that the WNA pathway is more favored over the I2M pathway for 22. The flexibility of the bda ligand may be one reason for this change. Further investigations are necessary to obtain a better understanding as to why Ru-bda WOCs are highly active and why they go through the uniquely effective I2M pathway; consequently, it will guide the design of more efficient WOCs.
Fast water oxidation has also been achieved by Ru WOCs bearing dianionic ligands via the WNA pathway. Electrochemical assessment by Llobet and co-workers yielded a TOF as high as 8000 s−1 at pH 7.0 for the [Ru(tda)(pic)2] (complex 21).159 Concepcion and co-workers investigated water oxidation catalyzed by Ru WOCs obtained by replacing one or two carboxylate groups of the Ru-bda catalysts with phosphate groups.158,160 Both catalysts adopt the WNA pathway. The diphosphate catalyst showed very low activity (TOF = 0.3 s−1).158 Due to the lability and basicity of the bipyridine-carboxylate-phosphonate ligands, complex 23 exhibited remarkable catalytic activity via the WNA pathway (TOF = 100 s−1).160 Kunz et al. prepared the trinuclear macrocycle complex 24 by linking three Ru-bda units with ditopic axial bridging ligands.157,180–182 The rigid cyclic structure prevents the coupling of the two Ru-bda units. Thus complex 24 efficiently catalyzes water oxidation (TON = 7400 and TOF = 150 s−1) via the WNA mechanism, having benefited from an ordered hydrogen bonding network inside the macrocyclic cavity, demonstrating that the negatively charged ligand and the open coordination site can also enhance the oxygen evolution rate via the WNA pathway.157
After over 35 years of research, scientists nowadays have very detailed understanding of Ru-based molecular WOCs. Moreover, the catalytic activities have been dramatically improved with a low overpotential of 200 mV and TOF over 1000 s−1, which is highly promising for fabricating efficient AP devices.
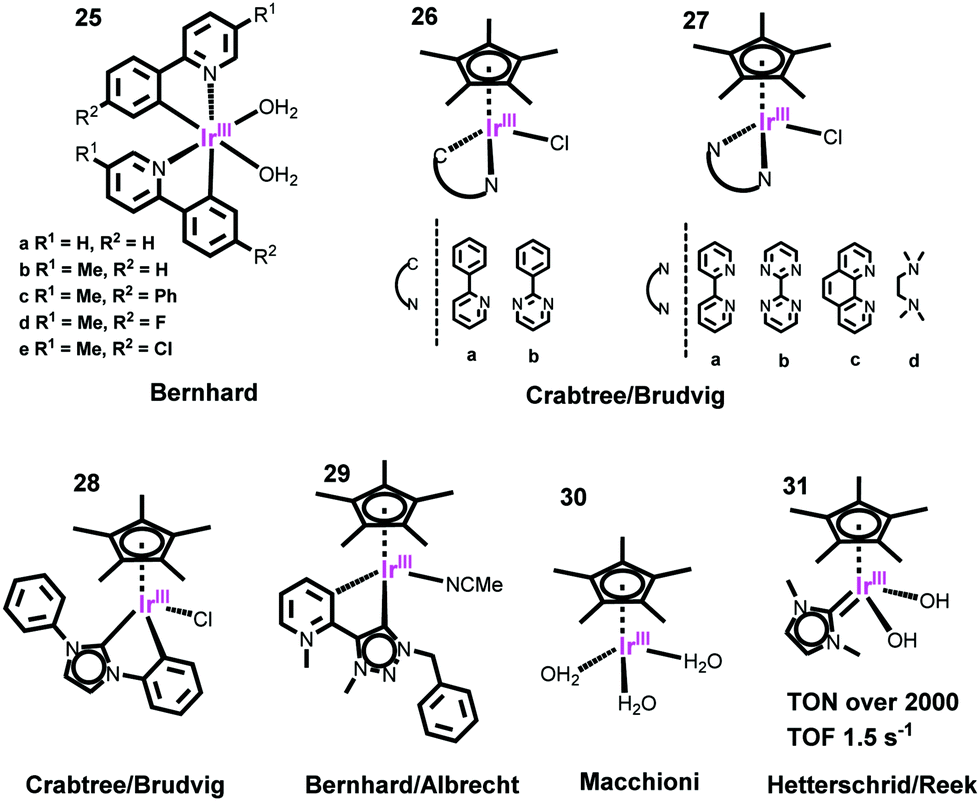 | ||
| Fig. 8 Representative structures of Ir-based WOCs. | ||
To improve their stability, a Cp*Ir complex bearing a strongly σ-donating N-heterocyclic carbine (NHC) ligand was developed, which effectively stabilizes the IrIV intermediates.187 By the same strategy, Bernhard and Albrecht employed an abnormal pyridinium-carbene ligand to coordinate with the half-sandwich Cp*Ir (complex 29), achieving a new benchmark for TON: 10000 after 5 days. Very recently, they investigated the promoting effect of incorporating MeO– groups.188,189 Macchioni and co-workers found that the simple Cp*Ir complex [Ir(Cp*)(H2O)3]2+ (30) showed faster catalysis than those bearing auxiliary chelating ligands.190 Hetterscheid and Reek reported a series of NHC-ligand-coordinated half-sandwich Cp*Ir complexes (31) with both high TOF (1.5 s−1) and high TON (>2000) by using Ce4+ as the oxidant.191 For the half-sandwich Cp*Ir WOCs, it should be noted that many initially obtained structures are precatalysts, with the actual catalyst losing the Cp* ligand to form a mono-μ-oxo dimer that is quite robust and active.183,192,193
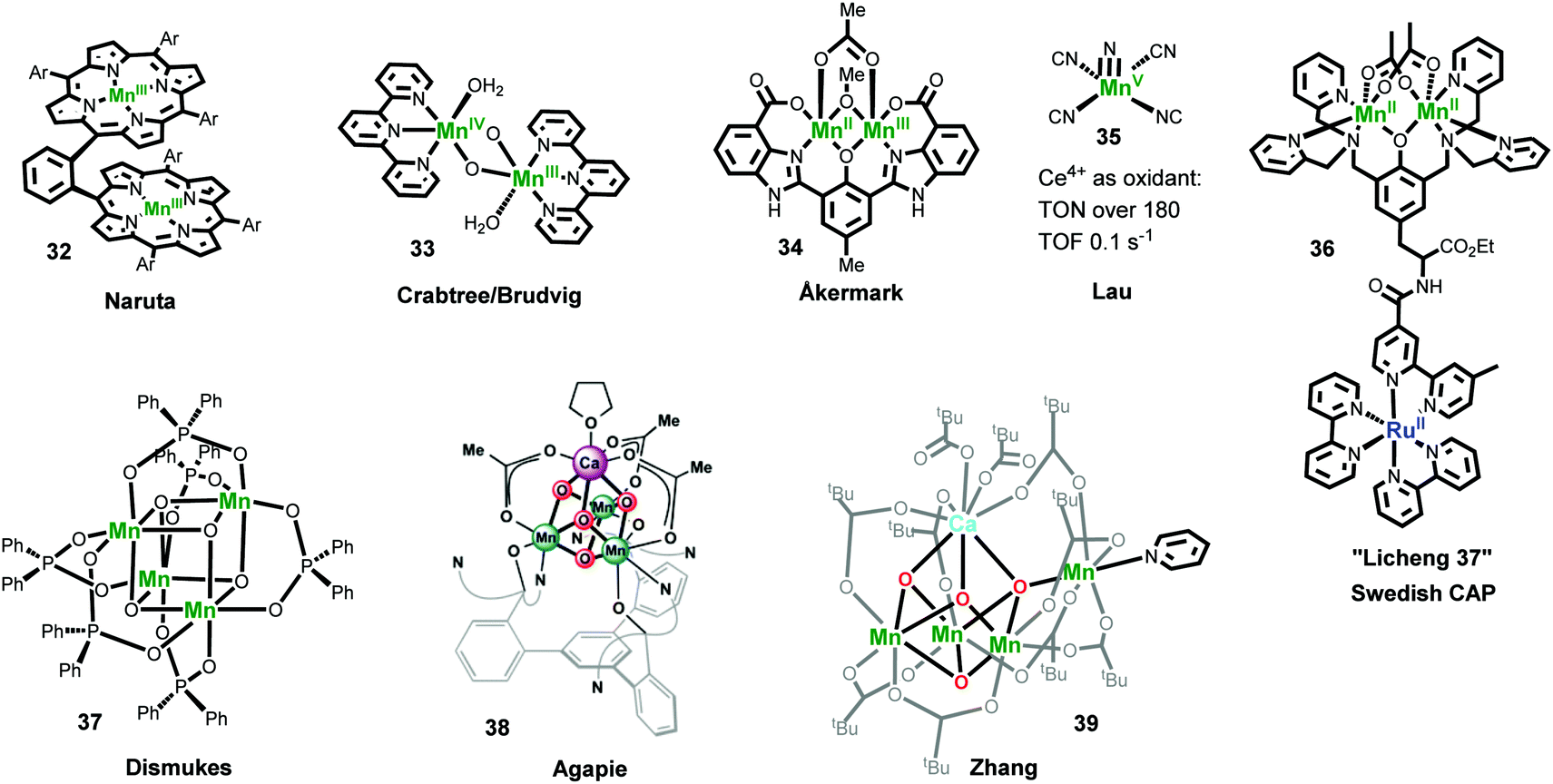
Naruta and co-workers repoted the first family of Mn-based molecular WOCs: three analogous dimeric manganese triphenylporphyrin complexes linked by an o-phenylene bridge (Fig. 9, complex 32).195 They found that these catalysts catalyze water oxidation with a TON of 9.2 and a TOF of 0.0018 s−1 under electrochemical conditions. In a further mechanistic study, the dimeric MnVO species was identified as a key intermediate by using meta-chloroperoxybenzoic acid (mCPBA) as an oxidant.196 In 1999, Crabtree and Brudvig reported another important Mn-based molecular WOC, [(H2O)(tpy)MnIII(μ-O)2MnIV(tpy)(OH2)]3+ (complex 33). When the catalytic performance of 33 was researched by using NaClO (0.07 M) as an oxidant, it gave a TON of 4 and an initial TOF of 0.0038 s−1.57 By using another chemical oxidant oxone (KHSO5), the catalytic performance was further promoted to TON > 50 and TOF of 0.034 s−1.197,198 A MnV−MnVO species was proposed as a key intermediate for O−O bond formation by 33, and the formation of MnO4−via oxidation of 33 was suggested as the deactivation pathway.57 Complex 33 showed no catalytic oxygen evolution with the classical oxidant Ce4+ or with electrochemical oxidation.199 However, complex 33 adsorbed on heterogeneous materials, such as clay compounds,200–202 TiO2,203,204 or even metal–organic frameworks (MOFs) showed obvious water oxidation activity using Ce4+ as the oxidant.205 Since the reports of the two above examples, many Mn-based catalysts have been explored for oxygen evolution.4,58,206,207 However, a Mn-based molecular WOC, which can catalyze water oxidation without involving oxygen transfer reagents, was first reported by Åkermark and co-workers in 2011.208 Complex 34 showed acceptable catalytic water oxidation activity by using [Ru(bpy)3](PF6)3 oxidant, with a TON of 25 and an initial TOF of 0.027 s−1. In 2015, a different class of Mn-based catalysts, viz. manganese(V)–nitrido complexes, were explored by Lau and co-workers. Complex 35, [MnV(N)(CN)4]2−, turned out to be an active molecular WOC using Ce4+ as the oxidant, with a TON exceeding 180 and a maximum TOF of 0.1 s−1, suggesting that active Mn-based WOCs may be constructed based on the MnV(N) platform.209
 | ||
| Fig. 9 Representative structures of Mn-based WOCs. | ||
Mn complexes have also been designed and synthesized to mimic the electron transfer reactions in PSII and the structures of the OEC.210,211 Groups of the Swedish Consortium for Artificial Photosynthesis (CAP) did some pioneering research and have prepared several Mn complexes covalently coupled to [Ru(bpy)3]2+-type chromophores as functional models of PSII to investigate photoinduced electron transfer of binuclear Ru–Mn assemblies.212–215 For example, complex 36 (also named as Licheng-37 synthesized by professor Sun in CAP) showed excellent photoinduced redox properties because it could transfer three electrons from the Mn centers via the Ru photosensitizer to the acceptor, resulting in a MnIIIMnIV state with accumulation of three charges.214 The studies on mimicking the key structural motif of the Mn4CaO5 cluster have yielded fruitful results in recent years.211 The Dismukes group synthesized the first example the Mn4O4 cubane core (complex 37). They found that under UV light irradiation, complex 37 efficiently released an O2 molecule by coupling two of the corner oxos in the Mn4O4 cubane. An artificial Mn3CaO4 cluster coordinated by a multi-pyridylalkoxide ligand (complex 38) was reported by Agapie and co-workers as the first representative model of the Mn4CaO5 cluster in PSII.216,217 Although this cluster closely mimicked most of the key structural features of the Mn4CaO5 cluster of the OEC, the unique dangling Mn4 in the Mn4CaO5 cluster was missed. A breakthrough to overcoming this challenge was made by the Zhang group.218 They reported the first artificial Mn4Ca cluster (complex 39) that entirely mimicked the structural features of the OEC. Although complex 39 is similar in many ways to native OEC, it did not display efficient water oxidation catalytic activity, indicating that the secondary coordination environment surrounding the Mn4CaO5 cluster in the OEC is also significant. These mimicking studies can enhance our understanding of the OEC, thereby facilitating the development of artificial systems. For instance, Maayan and co-workers recently reported a soluble manganese-oxo cluster with an exceptionally low overpotential of only 334mV for electrochemical water oxidation operating at pH 6.219
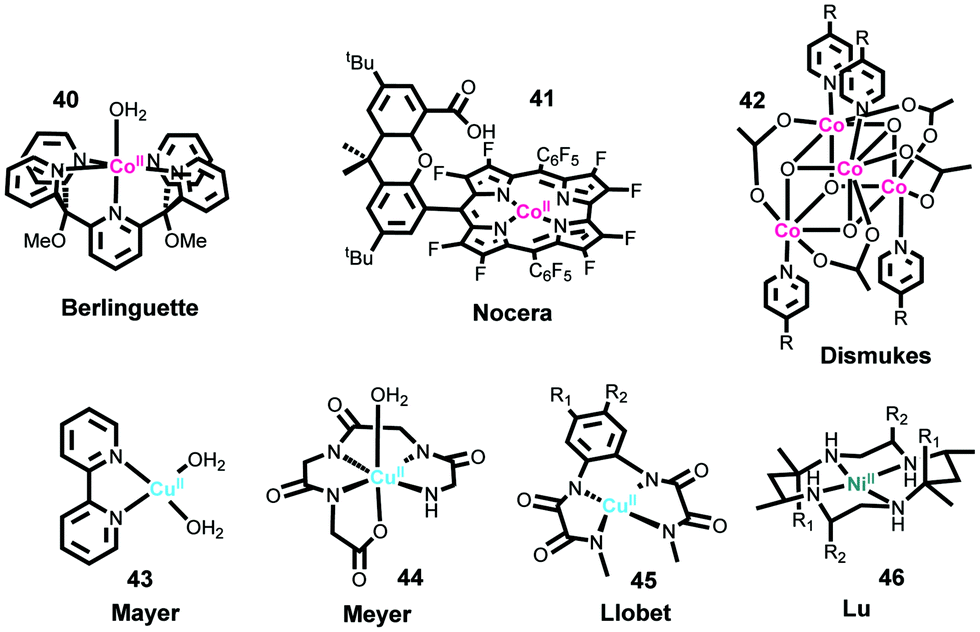
Cobalt ions have been known to catalyze water oxidation since the 1960s;220 however, cobalt complexes were not developed as molecular WOCs, until the Berlinguette group reported the first Co-based WOC (Fig. 10, complex 40) in 2011, using the well-known oxidatively stable pentadentate ligand 2,6-(bis(bis-2-pyridyl)-methoxymethane)-pyridine (PY5).221 Complex 40 showed remarkable activity for electrochemical water oxidation and was also a pioneer example of well-defined PCET at a first-row transition metal aqua site, i.e. M–OH2. Subsequently, Nocera and co-workers reported that a perflurorinated “hangman” porphyrin complex (complex 41) could electrochemically catalyze water oxidation and the hangman carboxylic group was essential in that it functioned as a secondary coordination site.222 Since these two studies, developing Co-based molecular catalysts has attracted great research interest. Many different Co complexes, e.g., Co porphyrins and Co salophens, have been studied as catalysts or precatalysts for water oxidation.121,223–229
 | ||
| Fig. 10 Representative structures of Co-based, Cu-based and Ni-based WOCs. | ||
One extensively studied family of noble-metal-free molecular catalysts comprises tetranuclear Co4O4 cubane compounds, such as complex 42. Dismukes and co-workers were the first to report photocatalytic water oxidation by these Co4O4 cubane catalysts.230 Later Bonchio and co-workers investigated the photocatalytic activities of a series of isostructural analogs of Co4O4 cubane complexes.231,232 They revealed that the primary step of photoinduced electron transfer obeys the Hammett linear free energy relationship. To improve photocatalytic performance, several supramolecular assemblies were produced by coupling Co4O4 cubane cores with transition metal photosensitizers.233,234 The Sun group studied the electrocatalytic properties of Co4O4 cubane catalysts by immobilizing them on ITO electrodes, and for the first time proved that Co4O4 cubane catalysts coupled well with visible-light-absorbing semiconductor α-Fe2O3 to form molecule–semiconductor hybrid photoanodes.235 Based on this work, Co4O4 cubane catalysts have been widely employed as cocatalysts for semiconductor photoanodes, e.g., BiVO4.236–238 Since the Co4O4 cubane complexes are coordinatively saturated, identifying the catalytic site and elucidating the mechanism of O–O bond formation are interesting. Dismukes and co-workers reported that water oxidation by the Co4O4 cubane complex is initiated by OH− addition.239 Mechanistic investigations by the Tilley group indicated the formation of highly oxidized intermediate CoIV–oxo or CoV–oxo complexes and the exclusive terminal oxo participation.240 Although the Nocera group proposed that the activity of Co4O4 cubane catalysts may be derived from the CoII impurities in the synthesized Co4O4 cubane samples, they revealed in their further studies the involvement of the high-valent intermediate CoIV–oxo species in the catalytic mechanism.241,242 In addition to the above fruitful results on the Co4O4 cubane catalysts, several other types of cobalt cubanes have also been investigated as WOCs.243
It is well known that copper-based metalloenzymes, where copper is the essential metal core, can activate oxygen by forming various Cu–oxo intermediate species, which can function as intermediates for O–O bond formation, indicating that Cu-based complexes can be used as WOCs.244 The first molecular Cu-based WOC [Cu(bpy)(OH)2] (complex 43) was reported by the Mayer group in 2012.245 Complex 43 displayed remarkable TOF (∼100 s−1, value calculated from electrocatalytic plot) for electrocatalytic water oxidation; however, this catalyst requires strong basic conditions and high overpotentials (∼750 mV). To overcome these drawbacks, Lin et al. and Papish et al. designed and prepared several analogs of [Cu(bpy)(OH)2] using a 6,6′-dihydroxy-2,2′-bipyridine (6,6′-dhbpy) ligand to assist proton transfer by the intramolecular base group.246,247 Meyer and co-workers reported a Cu-based WOC bearing a polypeptide-type ligand (complex 44).82 The onset potential of complex 44 for water oxidation was 1.10 V at pH 11, corresponding to an overpotential of 520 mV. Cu-Based WOCs similar to complex 44, bearing a polypeptide-type ligand, were also reported by Pap and Szyrwiel et al.248 Recently, the Llobet group synthesized a series of copper complexes (complex 45) containing redox-active tetradentate amidate acyclic ligands with various electron-donating substituents.122 They found that the overpotentials of these catalysts for water oxidation dramatically decreased with increasing electron-donating ability of the substituent on the backbone ligand. An extremely low overpotential of 170 mV was achieved for the complex with two methoxy groups. They also proposed a catalytic mechanism involving oxidation of the ligand. Brudvig and co-workers investigated the water oxidation property and mechanism of a robust copper complex CuII(pyalk)2 (pyalk = 2-pyridyl-2-propanoate).249,250 In addition to the above-mentioned Cu-based catalysts, all of which required strong basic conditions, Zhang and co-workers reported the first Cu-based WOC that can operate under neutral pH conditions.251 These previous results demonstrate that efficient and stable electrochemical Cu-based WOCs can be developed.
Developing Ni-based molecular WOC was initiated by the Lu group with a Ni macrocyclic catalyst (complex 46) in 2014.252,253 Complex 46 showed an electrocatalytic current of 0.9 mA cm−2 at an overpotential of 750 mV in a neutral sodium phosphate buffer (NaPi). They further investigated the catalytic mechanism of this catalyst.253,254 Cao and co-workers reported the use of a water-soluble nickel porphyrin complex as a molecular electrocatalyst for water oxidation under neutral conditions.255 The Sun group studied the water oxidation property of the well-known Ni–PY5 complex, and molecular catalysis was well proved.256 Several Ni complexes based on well-known non-heme-iron ligands, such as N,N′-dimethyl-N,N′-bis(pyridin-2-ylmethyl)-1,2-diaminoethane (mep), N,N′-dimethyl-N,N′-bis(pyridin-2-ylmethyl)-1,2-diaminocyclohexane (mcp), and 2-((bis(pyridin-2-ylmethyl)amino)methyl)phenol, have been synthesized and used to assess the performance of catalytic water oxidation.254,257–259 Some other Ni-based complexes were reported as precursors to generate active Ni oxide films for water oxidation.260–262 Because of the fewer redox states of Ni ions, development of Ni-based WOCs with low overpotentials remains challenging.
Iron is the most abundant, non-toxic, and inexpensive transition metal. Additionally, it has a rich redox chemistry, and thus many iron-based complexes can generate reactive valent states for oxidation reactions in both natural263,264 and artificial systems.265 Due to these advantages, we strongly agree that iron complexes are ideal and promising candidates for WOCs.266
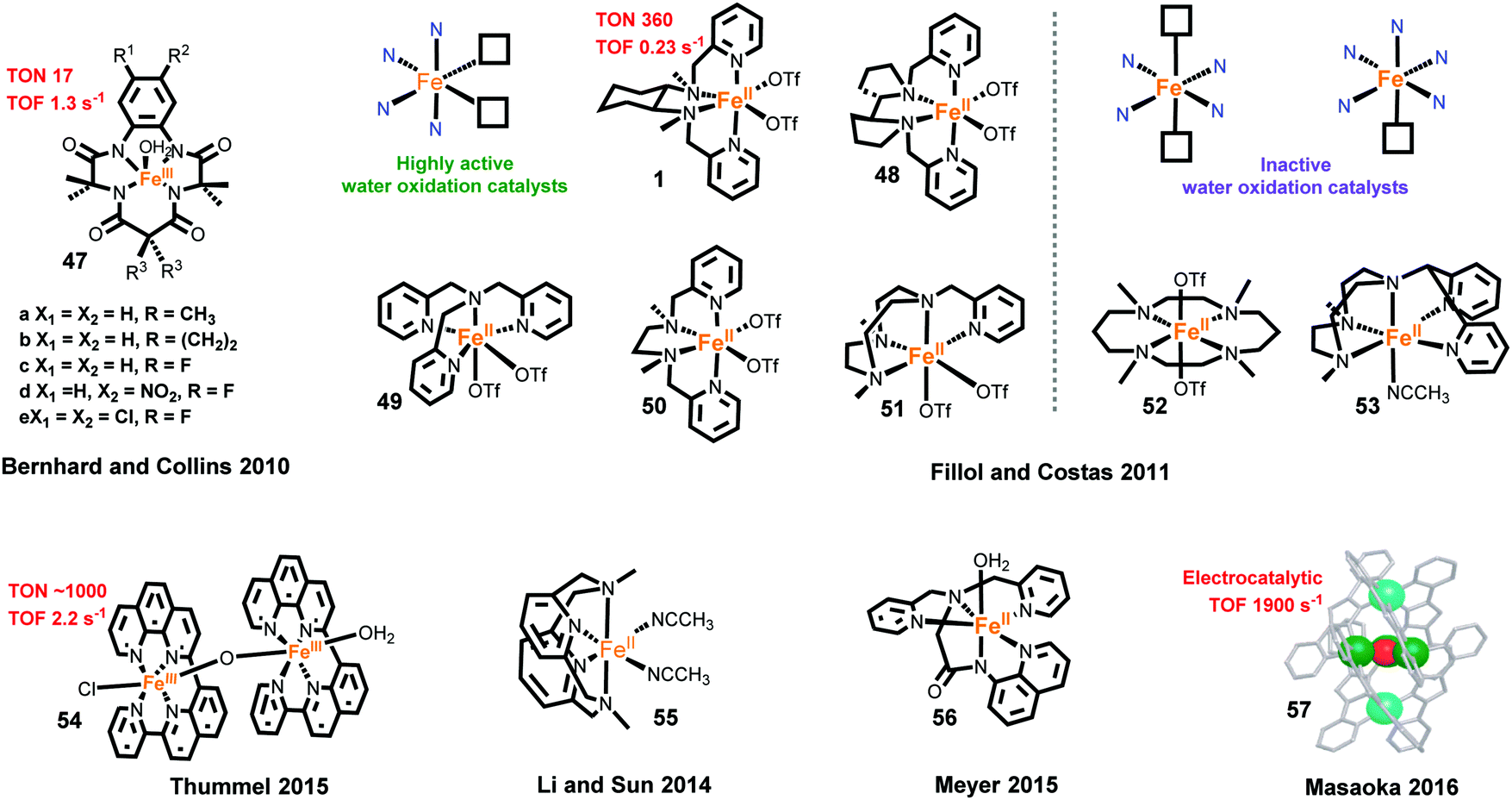
The first family of Fe-based molecular WOCs, iron(III)–tetraamido macrocyclic ligand complexes (FeIII–TAMLs, complex 47) (Fig. 11), was reported by the Bernhard and Collins groups in 2010.267 Using Ce4+ as the oxidant, fast O2 evolution was observed for these FeIII–TAMLs. The catalytic performances of the FeIII–TAMLs were dominated by the substituent groups on the macrocyclic ligands. The highest TOF of 1.3 s−1 was achieved for a FeIII–TAML with a strong electron-withdrawing group; however, these FeIII–TAMLs have low stabilities under the testing conditions, and remain active for only 20 s. Subsequently, this family of Fe-based WOCs was further investigated for electrochemical water oxidation,268 photocatalytic water oxidation268 and catalytic mechanisms.268–270
 | ||
| Fig. 11 Representative structures of Fe-based WOCs. | ||
One year later, a second class of Fe-based WOCs was reported by Fillol, Costas and co-workers. The authors studied catalytic water-oxidation properties of several classical non-heme iron complexes (complexes 1 and 48–53).40 They reported that non-heme iron complexes with two cis labile sites may have catalytic activity for water oxidation, whereas the complexes bearing two trans labile sites or only one labile site are not active for water oxidation. Complex 1 is the most efficient WOC reported by them with a TON of 360 and a TOF of 0.23 s−1 using Ce4+ as the oxidant. This type of Fe-based WOC has attracted widespread research attention, making it one of the most studied molecular noble-metal-free WOCs. Effects of electronic modification and of pendant bases have also been explored.271–273 Several studies have been conducted to elucidate the catalytic mechanism.269,274–277 Recently, a FeIV(O)(OCe4+) motif is proved to be a key intermediate in catalyzed water oxidation by 1 under acidic conditions.276
Recently, a highly active binuclear Fe-based WOC [(ppq)(H2O)Fe(μ-O)(Cl)(ppq)]3+ (complex 54, ppq = 2-(pyrid-2′-yl)-8-(1′′,10′′-phenanthrolin-2′′-yl)-quinoline) was reported by Thummel and co-workers.278 Using Ce4+ as the oxidant, complex 54 showed much higher activity than a mononuclear analog, affording a TON of ∼1000 and a TOF of 2.2 s−1, which are the corresponding records for molecular noble-metal-free WOCs under similar conditions. The excellent catalytic performance may be attributed to the easy disproportionation of the intermediate [FeIVFeIV–OH2] to a key intermediate [FeIIIFeVO]. Moreover, binuclear Fe-based complexes [(tpa)(H2O)Fe(μ-O)(H2O)(tpa)]4+,279 [(tpa)Fe(μ-O)(μ-SO4)(H2O)(tpa)]2+ and [(tpa)(Cl)Fe(μ-O)(Cl) (tpa)]2+ were also reported to have higher activities than the mononuclear analog.280,281 These results suggest that binuclear Fe-based complexes can generate more efficient molecular catalysts for water oxidation. To explore a better coordination environment for efficient Fe-based WOCs, the Sun group examined the activities of eleven separated iron complexes and nine in situ-generated iron complexes towards catalytic water oxidation using Ce4+ as the oxidant.282 Complex 55 was obtained as a promising catalyst for water oxidation, and its catalytic mechanism was further investigated by the Che group.283
Fe-Based molecular electrocatalysts for water oxidation have also been developed in recent years. Meyer and co-workers reported the electrocatalytic properties of an iron complex bearing a pentadentate ligand, [FeIII(dpaq)(H2O)]2+ (complex 56, dpaq = 2-[bis(pyridine-2-ylmethyl)]amino-N-quinolin-8-yl-acetamido) in 2015. Complex 56 showed only moderate performance with a low faradaic efficiency of 45% and a low TOF of 0.15 s−1.284 One year later, Masaoka and co-workers reported the most active Fe-based electrocatalyst, a pentanuclear iron complex surrounded by six 3,5-bis(2-pyridyl)pyrazole ligands and linked by one μ3-O bridging ligand (complex 57).285 The calculated electrocatalytic TOF is as high as 1900 s−1, and the Faradaic efficiency obtained from the controlled potential electrolysis is 96%. A plausible catalytic cycle was constructed based on the promising characterization of intermediate species and quantum chemical calculations. However, it should be noted that the two above-mentioned catalysts work only in special organic solvent systems, due to the instability of the high-valent iron intermediate in aqueous solution.
 | ||
| Fig. 12 Representative structures of POM WOCs. Reproduced from ref. 56 with permission from The Royal Society of Chemistry. | ||
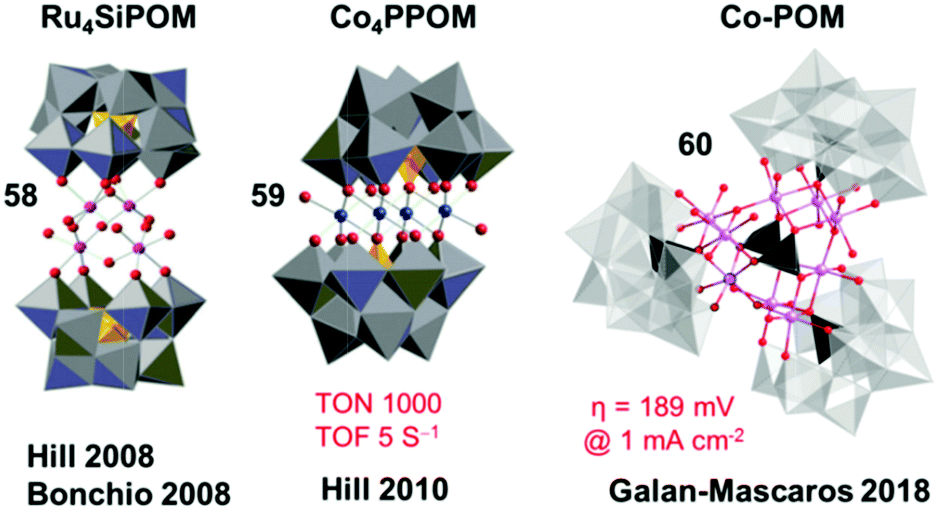
Another important improvement in developing POM WOCs was the report of a tetracobalt-substituted polytungstate, [Co4(H2O)2(PW9O34)2]10− (complex 59, Co4PPOM), which catalyzed water oxidation with a high TON per active site metal of 1000 and a TOF of 5 s−1 using [Ru(bpy)3]3+ as a chemical oxidant in an aqueous Pi buffer solution at pH 8.0.288 Following these two pioneering works, extensive research has been conducted in recent years, on the variation in POM structures289,290 and different metals,291–293 mechanistic evaluation,294–296 application on electrodes,297,298 and the stability of POM WOCs.221,299–303 There are also several recent reports of POM WOCs; for example, the report on the development of Mn-based POM WOCs.304,305 Very recently, the excellent catalytic activity of a water-insoluble Co-POM (complex 60) under acidic conditions was reported by the Galan-Mascaros group in 2018.306 POM WOCs, as a unique molecular system, exhibit considerable promise in the development of AP devices.
WOCs based on noble metals Ru and Ir have been well developed in the past thirty years, and catalysts with promising activity have been achieved. However, from the viewpoint of practical application, an earth-abundant transition-metal-based catalyst will be necessary for AP devices. Although some progress has been made toward the development of WOCs based on earth-abundant transition metals,4,55,244,307 including Mn, Co, Ni, Cu, and Fe, the activity of available earth-abundant transition-metal-based catalysts is still very low. A high overpotential and strong basic condition are required, under which conditions some of the catalysts decompose into metal oxides to serve as the true catalysts.308–310 The unsatisfactory progress in the development of earth-abundant transition-metal-based WOCs signifies the magnitude of the challenge and the efforts needed to overcome it. Research on the development of earth-abundant transition-metal-based molecular WOCs commenced in the 2010s;4,55,244,307 this is much later than the initial development of Ru and Ir based WOCs. To obtain efficient and inexpensive metal-based WOCs for future AP application, more designs and research are required.
4.2 Overview of molecular catalysts for reduction reactions
To efficiently produce valuable fuels using the protons and electrons obtained from the oxidation of water, highly active and selective catalysts are essential for the reduction reactions (hydrogen evolution and CO2 reduction reaction).10 Molecular catalysts with the above-mentioned advantages have attracted immense attention in the field of reduction half reactions. It is necessary to note that, in the development of molecular WOCs, a number of suitable chemical oxidants allow evaluation of the catalytic activity in a simple chemical-driven water-oxidation system to thereby reveal the mechanism. On the other hand, studies on the molecular catalysis of the hydrogen evolution and CO2 reduction reactions are mainly conducted in photochemical and electrochemical systems. In this part, we present an overview of the most typical and well-studied catalysts for hydrogen evolution and CO2 reduction.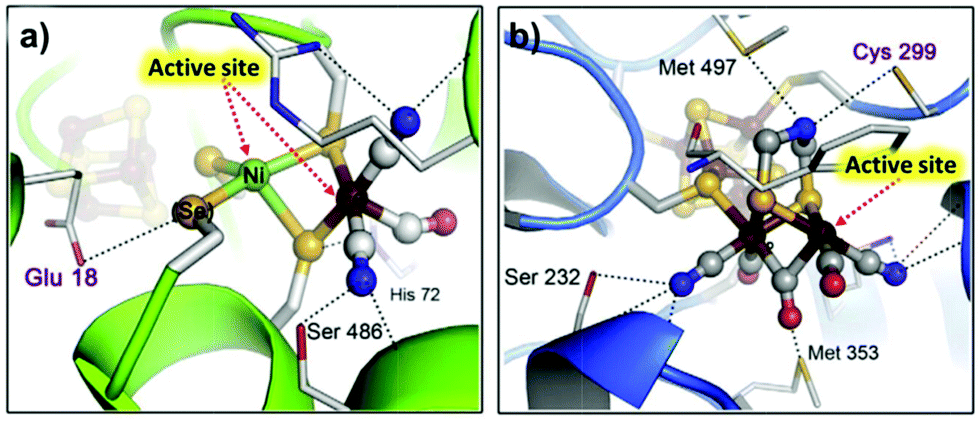 | ||
| Fig. 13 X-ray structures of (a) NiFe cluster of [NiFe(Se)]-hydrogenases; (b) 2Fe sub-cluster of the [FeFe]-hydrogenase H cluster. Adapted with permission from ref. 314. Copyright 2015 Elsevier. | ||
A reasonable design for an artificial catalyst can be facilitated by unveiling the structure and catalytic mechanism of the natural system. A general catalytic mechanism for hydrogen evolution is shown in Fig. 14.316–319 The active metal center undergoes either consecutive or coupled electron and proton transfers to generate a reactive metal-hydride intermediate, which can react in two possible ways to evolve hydrogen. In the left cycle, the metal-hydride reacts with another metal-hydride to generate H2via reductive elimination, i.e., the homolytic pathway. Alternatively, in the right cycle, the metal-hydride is further reduced and protonated to form H2, i.e., the heterolytic pathway. Taken together, to obtain a high activity for hydrogen evolution, a metal complex should have wide open coordination sites for binding the substrate and an appropriate electronic structure for generating and stabilizing a reactive metal-hydride intermediate. With these criteria in mind, hundreds of molecular HECs have been developed thus far (Fig. 15).
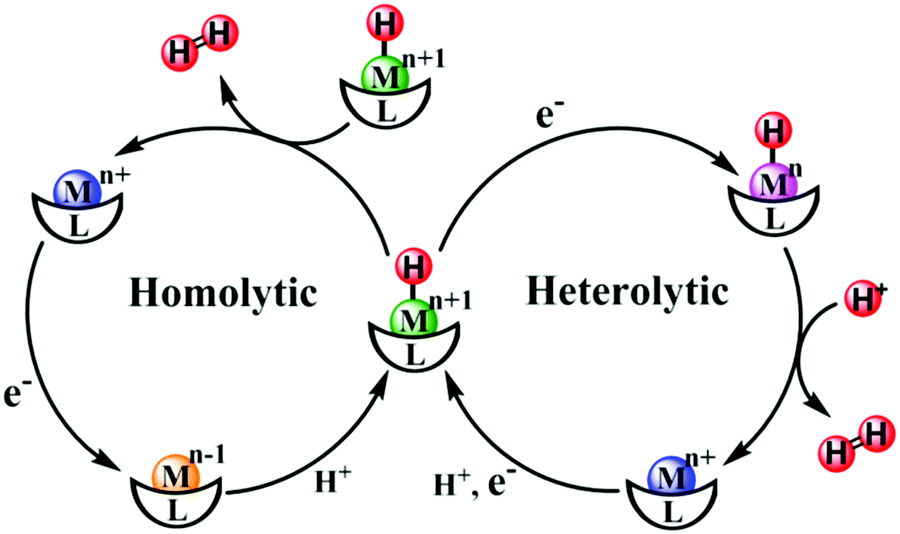 | ||
| Fig. 14 Proposed mechanistic pathways for H2 evolution by a metallic catalytic center. | ||
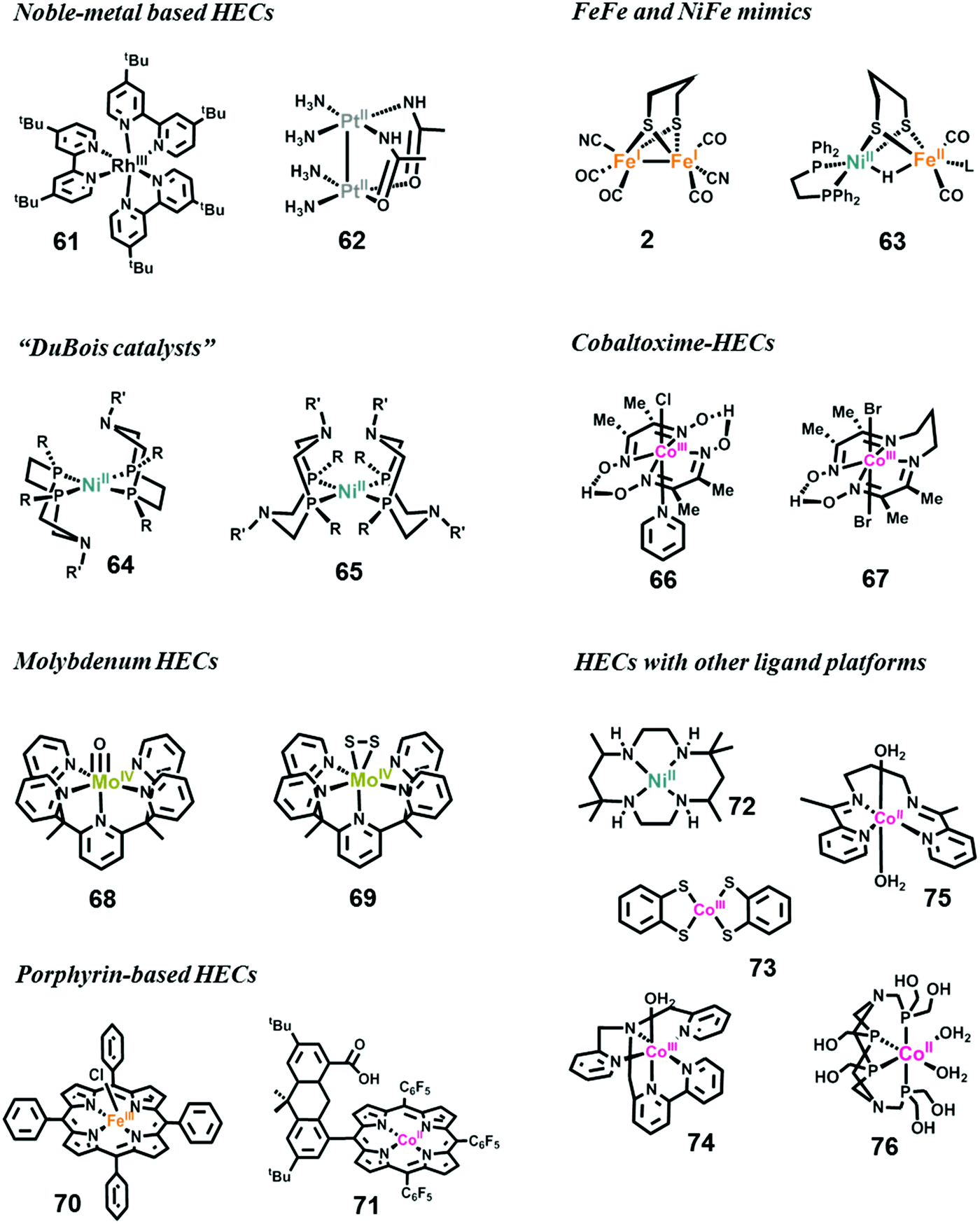 | ||
| Fig. 15 Representative structures of HECs. | ||
In the first fully molecular example of photocatalytic hydrogen evolution, the catalyst was a Rh-based complex bearing a polypyridyl ligand.320 Several Rh-based HECs have been developed for photocatalytic hydrogen evolution since then.321–323 The Bernhard group reported the most efficient Rh-based HEC by modification of the polypyridyl Rh-complex (complex 61).323 In their photocatalytic system, the catalytic TON of 61 exceeded 5000. Another large family of noble-metal-based HECs is the platinum complexes.324 Sakai et al. have developed numerous platinum complexes for photocatalytic hydrogen evolution. As a representative example, complex 62 showed outstanding catalytic performance with a TON of 100 and a high quantum efficiency of 31% in an optimized three-component photocatalytic system.
Over the last decade, studies have mainly concentrated on developing earth-abundant metal-based HECs.54,313,325–327 Chemists are engaged in the synthesis of structural and functional mimics of [FeFe]- and [NiFe]-hydrogenases to understand the mechanisms of H2 evolution by natural hydrogenases, which is key to the design of artificial HECs.39,328–332 Complexes 2 and 63 are two early available models reported by Rauchfuss et al. for [FeFe]- and [NiFe]-hydrogenase, respectively.39,330,332 More than hundred synthetic mimics have been reported thus far.333 These complexes faithfully reproduce the structure of the active center of the hydrogenases, and the related investigations provide in-depth information on the H2 evolution mechanisms. However, the complexes exhibit poor catalytic performances, i.e., low stability and larger overpotential requirements.
To obtain more advanced and cost-effective synthetic HECs, researchers have designed and synthesized various ligand platforms for earth-abundant metals, e.g., Ni, Co, Fe, and Mo.313,325,326 Thus far, the most efficient and stable molecular HECs are Ni-complexes (e.g., complex 64 and 65) bearing base-containing diphosphine ligands, which were first reported by DuBois and co-workers in 2002, and are therefore well known as “DuBois catalysts”.334–338 In an acetonitrile solution with 1.2 M H2O, the catalytic TOF of H2 production by 64 could exceed 100000 s−1 at a high overpotential.312 On the other hand, the lowest overpotential among the DuBois catalysts was exhibited by 65.339 Further studies on these catalysts proved the importance of pendant amines for proton transfer.
Another series of HECs, which are among the most efficient catalysts, is the cobaltoximes formed from the coordination of two glyoximato equatorial ligands with a cobalt ion. These complexes were originally reported in the 20th century, and their early use as a HEC was reported by Lehn in 1983340 and Espenson in 1986.341 The Peters group342 and Fontecave group343 in 2005 simultaneously revisited cobaltoximes (complex 66) and studied their electrocatalytic properties for hydrogen evolution. Following these works, cobaltoxime-based HECs have been extensively studied over the last 10 years, and have achieved the high-efficiency and low-overpotential requirement.344 Cobaltoximes can catalyze hydrogen evolution from protic solutions requiring overpotentials as low as 40 mV.325 The second generation of cobaltoxime-based HECs, cobalt diamine–dioxime complexes, was studied by the Fontecave group (complex 67).345 These diamine–dioxime complexes catalyze H2 evolution from acidic solutions with similar overpotentials to conventional cobaltoximes, but display better stability owing to the propylene bridge. The structural modification, catalytic mechanisms, and decomposition pathway of cobaltoxime catalysts have been studied in detail.346–349
Mo-Based HECs are mainly the contribution of Chang et al.350–353 The MoIV–oxo complex [Mo(PY5Me)(O)] (complex 68) is among the most efficient electrocatalysts ever reported for H2 evolution from an aqueous solution at neutral pH with good durability, and high catalytic TOF but high overpotential. After CPE on a Hg pool electrode at an overpotential of 1.0 V in a phosphate buffer at pH 7 for 71 h, 68 displayed 100% Faradaic efficiency, a TON of 6.1 × 105, and a TOF of 2.4 s−1.351 Using the same ligand framework, Chang et al. synthesized complex 69 to mimic the reactive edge site of the MoS2 material,354 which is a well-known heterogeneous HEC. Like 68, upon electrochemical reduction, 69 also showed impressive performance with 100% Faradaic efficiency, a lower-limit TON value of 3.5 × 103 calculated from the bulk solution, and an upper limit of 1.9 × 107, assuming a constant monolayer on the electrode. In subsequent work, it was proved that the PY5-based ligand also works well when cobalt is placed as the catalytic center.350 Mo complexes with Schiff base ligands have been studied recently as a catalyst for electrochemical hydrogen evolution.355,356
Porphyrin-based transition metal complexes have been widely employed for the catalysis of energy-conversion-related small-molecule activation.357 Many transition metal porphyrin complexes have been investigated as HECs, including Fe-,358,359 Co-,357,360–362 and Ni-porphyrins.363 The Fe-porphyrin complex 70 reported by Savéant and co-workers can produce H2 from an acidic DMF solution at a TOF of 105–106 s−1.358,364 The Nocera group invented a series of “hangman porphyrins”, featuring dangling proton-transfer groups, such as carboxylic acid (complex 71),359,362,363,365 sulfonic acid359 or amine,359 fixed on top of the active metal center. Due to the promoted PCET processes, these catalysts exhibited decreasing overpotential and increasing TOFs for H2 evolution.
In addition to the above main series of HECs, many other well designed ligand platforms have been applied for housing transition metals as HECs.313,326,366–369 For example, the macrocyclic Ni complex 72 and its analogues were good electrochemical HECs under organic-aqueous solutions370 and fully aqueous solutions.371 Recent studies demonstrated that dithiolene complexes 73 and the nitrogen donor ligand stabilized complex 74 could efficiently catalyze both electrochemical and photochemical H2 evolution from water reduction,372–374 and a cobalt bis(iminopyridine) complex 75 could also act as an efficient electrocatalyst for H2 production from an aqueous solution.375 Using a multihydroxy-functionalized tetraphosphine ligand, Sun and Wang obtained a self-assembled cobalt complex 76 in 2014. Although the crystal structure of 76 is not available, the complex exhibits highly-efficient activity. Electrocatalytic H2 generation from neutral water by 76 on a mercury electrode requires an overpotential of only 80 mV. The bulk electrolysis of 76 in a neutral phosphate buffer solution at −1.0 V vs. NHE generated 9.24 × 104 mol H2 per mol 76 over 20 h, with a Faradaic efficiency close to 100% and without obvious deactivation.376
| CO2 + 1e− → CO2˙− −1.90 V | (1) |
| CO2 + 2H+ + 2e− → HCOOH −0.61 V | (2) |
| CO2 + 2H+ + 2e− → CO + H2O −0.53 V | (3) |
| 2CO2 + 2H+ + 2e− → H2C2O4 −0.49 V | (4) |
| CO2 + 4H+ + 4e− → HCHO + H2O −0.48 V | (5) |
| CO2 + 6H+ + 6e− → CH3OH + H2O −0.38 V | (6) |
| CO2 + 8H+ + 8e− → CH4 + H2O −0.24 V | (7) |
Owing to the success of ReI(bpy)(CO)3X type catalysts, polypyridyl complexes of Ru,385,403–406 Os,407–409 and Ir410 have also been further developed as catalysts for CO2 reduction in recent years. As the types of Ru(tpy)(bpy)X shown in Fig. 16 (complex 78),403,404,406 Ru(bpy)2(CO)2385,411,412 and Ru(bpy)(CO)2X2405,413 have been extensively studied by different groups. For Pd-based catalysts, Dubois and co-workers reported Pd complexes using multidentate phosphine ligands (complex 79).414,415
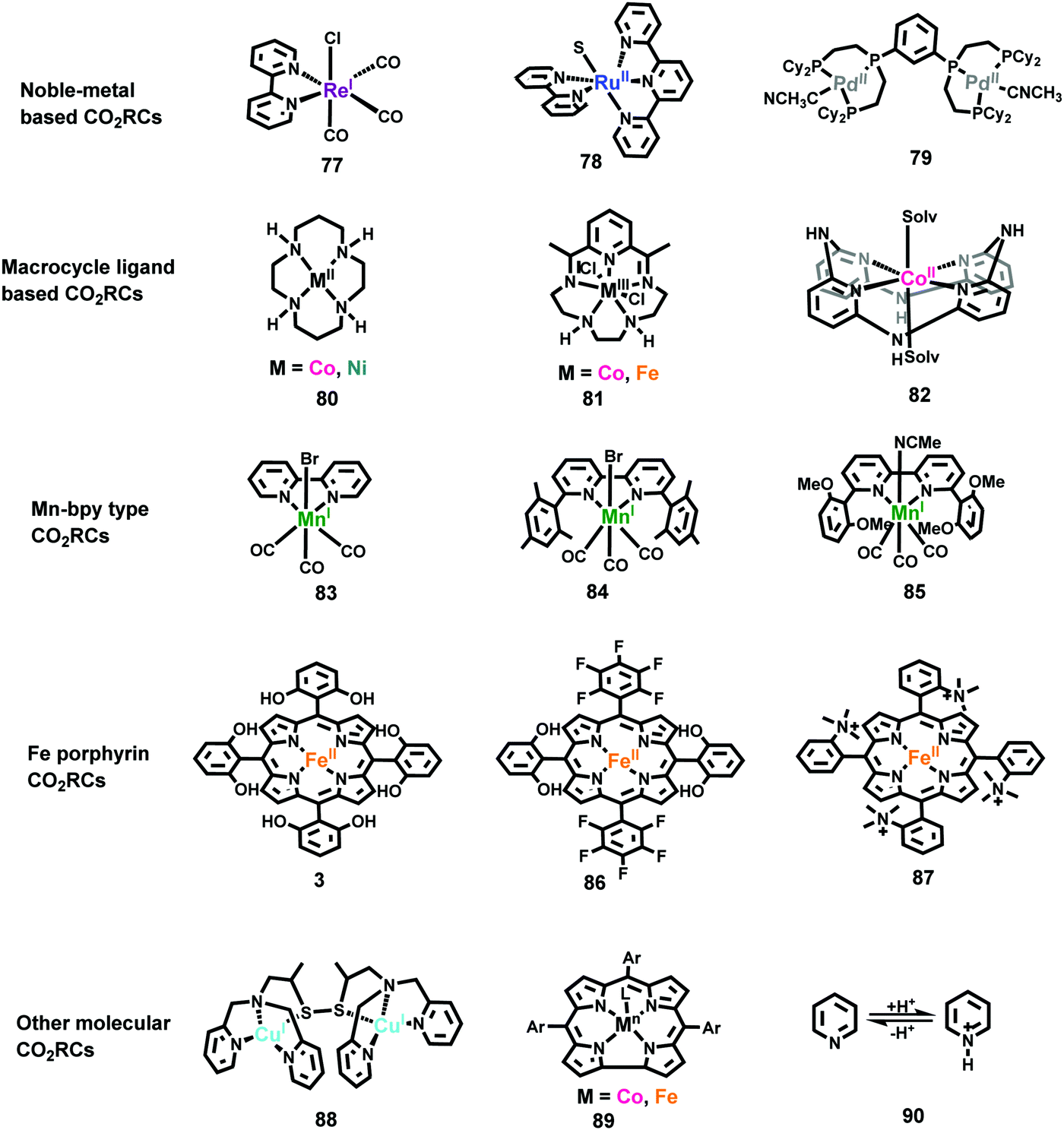 | ||
| Fig. 16 Representative structures of CO2RCs. | ||
In contrast to WOCs, the development of earth-abundant metal-based CO2 reduction molecular catalysts has been much more successful.43,377,394 For Co- and Ni-based catalysts, besides complexes with common polypyridyl ligands,416–418 tetraaza-macrocycle Co and Ni complexes, CoII(macrocycle) and NiII(macrocycle), e.g., complexes 80 and 81, have been reported as efficient catalysts for CO2 reduction.370,379,419–423 Although NiII(macrocycle) has been reported with Faradaic efficiencies over 95% in the production of CO, its good selectivity is strongly dependent on the pH of the reaction solution, and the low TOF is its drawback.421 In complex 82, the macrocycle includes four pyridines linked by pendant amine groups, which can contribute toward stabilizing the CoI-CO2 intermediate during catalysis, making 82 an efficient Co catalyst with high selectivity for CO formation.422
Replacing the Re metal in the ReI(bpy)(CO)3X catalysts with Mn metal, Deronzier and co-workers discovered a new class of highly active CO2 reduction catalysts in 2011, an example of which is complex 83, Mn(bpy)(CO)3Br.424 Subsequently, a wide variety of analogues of Mn(bpy)(CO)3Br were designed and investigated.394 To prevent deactivation due to the dimerization of Mn0 intermediates, bulky substituents with two phenyl rings were introduced into the bpy ligand (complex 84).425 By further introduction of four methoxy groups on the two phenyl rings, the proton transfer steps were promoted; consequently, the CO2 conversion overpotential was reduced by 0.55 V (complex 85).426
Another important series of noble-metal-free catalysts for CO2 conversion is iron porphyrins (e.g., complexes 3, 86, and 87), which are among the most efficient molecular catalysts for the electrochemical reduction of CO2 into CO.43,427,428 In 1991, Savéant and co-workers found that Fe0 porphyrins can reduce CO2 to CO at −1.8 V vs. SCE in DMF.429 However, the iron-porphyrin catalysts were not stable after several catalytic cycles. In further studies, they found that the addition of a hard electrophile (e.g. Mg2+) or Brønsted acids (e.g. 1-propanol, 2-pyrrolidine, and CF3CH2OH) could improve the catalytic efficiency and stability.430 In 2012, the Savéant group made a breakthrough in reducing CO2 to CO by iron porphyrins. They introduced phenolic groups in all ortho and ortho′ positions of the phenyl groups in the structure of iron porphyrin to increase the local proton effect to stabilize the Fe0 intermediate. At a low overpotential of 0.47 V, the catalyst displayed a CO Faradaic efficiency above 90% through TONs of 50 million over 4 h electrolysis without obvious degradation.41,431 Very recently, the performance of iron porphyrins for reducing CO2 to CO was dramatically boosted again by Savéant and co-workers. By introducing four charged trimethylanilinium groups in the para- or ortho-position of the phenyls in the iron porphyrin structure, complex 87 could selectively produce CO from CO2 under the test conditions with a TOF of 106 s−1 at an overpotential of only 220 mV for 80 h with no degradation observed.432
In addition to iron porphyrin complexes, porphyrin complexes with many other transition metals have been investigated for catalytic CO2 reduction.43,433–435 Interestingly, a Zn–porphyrin complex with a redox-innocent metal center was reported with a TOF as high as 14.4 s−1 and a Faradaic efficiency as high as 95% for CO2 electroreduction to CO at −1.7 V vs. NHE.433 The observation in this work that the ligand acts as the redox center during CO2 electroreduction calls for further mechanistic studies. Furthermore, Brudvig and co-workers reported a novel zinc(II)–bacteriochlorin complex to improve the stability of CO2RCs by taking advantage of the superior stability of the bioinspired bacteriochlorin ligand.436
Besides the above main class of CO2 reduction catalysts, there are many other notable reports of CO2RCs.437 For example, Bouwman and co-workers reported in 2010 a binuclear copper catalyst (complex 88) that could produce oxalate by catalyzing the one-electron reduction of CO2 at a very low overpotential.438 Metal corroles have also been reported with activity for CO2 reduction (complex 89).439 Interestingly, the pyridinium ion without the involvement of any metals has been proven to be a suitable electrocatalyst for the six-electron reduction of CO2 to methanol (complex 90).440–442 Except for pyridinium ion catalysts, molecular catalysts could only realize the one-electron and two-electron reduction of CO2, until the recent reports of a Cu-based porphyrin434 and a carbine-coordinated Ni complex,443 which are able to produce methane/ethylene and methanol, respectively. The development of efficient molecular catalysts for CO2 reduction still faces numerous challenges. Most CO2 reduction molecular catalysts still need to be able to work under organic conditions; however, the catalysts are not suitable under water oxidation conditions for future device fabrication.
4.3 Promotion of intrinsic activity by precise structural control
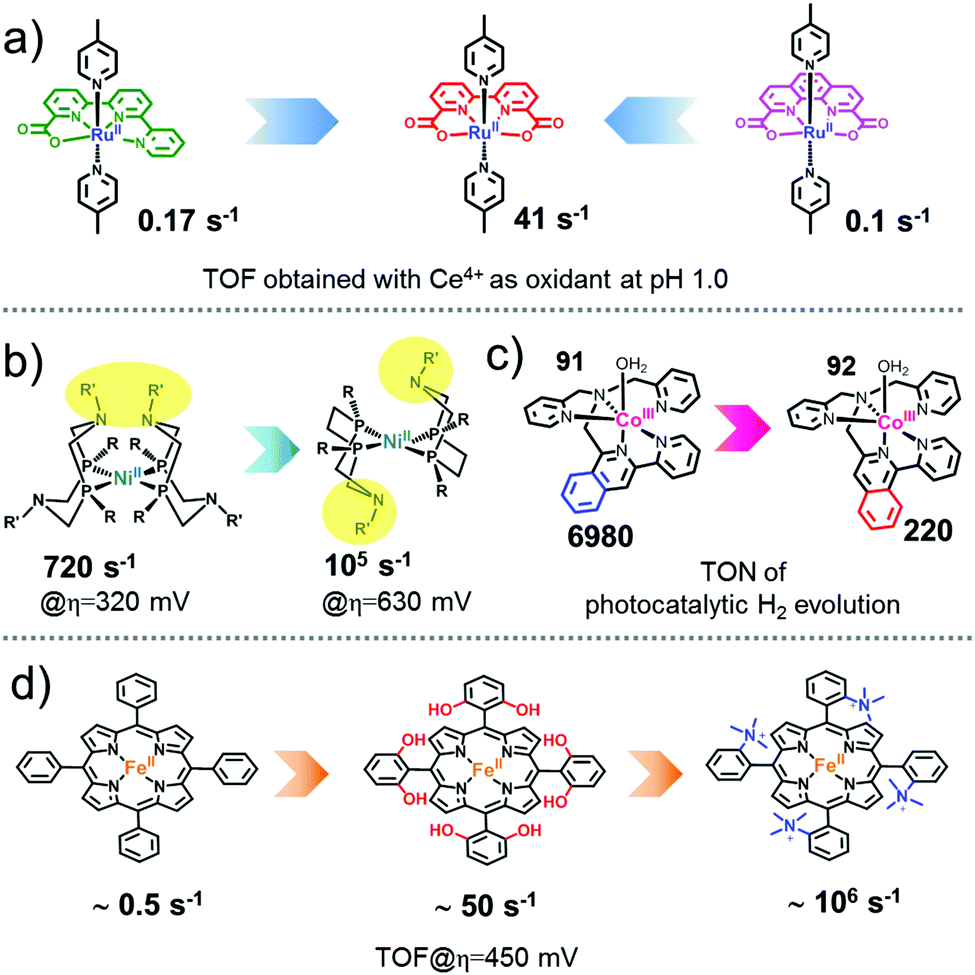
As is known, one of the basic principles of chemistry is that the structure determines the property. The activity of a catalyst closely depends on its structure. Therefore, fine adjustment of the catalyst structure is essential for the promotion of its activity. The most prominent advantage of a molecular catalyst is that it is possible to precisely regulate the electronic and steric structure to obtain extremely high catalytic performance.There are numerous examples of WOCs, HECs, and CO2RCs that highlight the importance of precise structural control for achieving new catalysts with high intrinsic activities. As shown in Fig. 17, the intrinsic activities of representative successful WOCs,174,179 HECs312,444 and CO2RCs41,432 have been improved by several orders of magnitude after precise structural adjustments. Small changes in the structure of the molecular catalyst may result in a new catalyst with higher intrinsic activity. Therefore, the development of an efficient molecular catalyst is highly feasible with a clear understanding of the catalytic mechanism and reasonable design of the structure. This is the significant advantage of developing molecular catalysts.
 | ||
| Fig. 17 Fine structural adjustments and dramatic performance (e.g. catalytic TON or TOF under comparable conditions) promotion of (a) Ru-bda WOCs; (b) Ni-diphosphine HECs; (c) pentadentate polypyridyl-amine Co HECs; and (d) Fe-porphyrin CO2RCs. | ||
5. Heterogenization of molecular catalysts – challenges of molecular catalysis
As discussed in the previous sections, there is a large gap between existing molecular catalysts and the development of effective AP devices. The heterogenization of molecular catalysts is essential to achieve simultaneous progress in the development of molecular catalysts and effective AP devices. Using the proposed model of the AP devices (Fig. 2), chemists have designed and reported many molecular catalyst-modified electrodes for AP devices during the past 10 years.445 Efficient molecular catalysts developed in the first stage, especially catalysts with low overpotential, are essential to achieve efficient AP devices. In this part, we summarize the research status of molecular-catalyst-based AP devices. These works have successfully demonstrated model design and the behavior of catalysts on the surface of electrodes. However, few studies have focused on exploring strategies and principles for the heterogenization of molecular catalysts by considering practical problems, such as the density of catalysts, exposure of the catalyst active sites, conductivity issues, enhancement of mass transport, and improvement in the stability of the molecule-catalyst-derived material or electrode. Setting aside the research on the development of more efficient and cheaper molecular catalysts, the heterogenization of molecular catalysts constitutes the main challenge toward their application in AP devices.5.1 Molecular-catalyst-modified dye-sensitized PEC cells (DSPEC cells)
The concept of a DSPEC cell has evolved from the dye-sensitized solar cell (DSSC), which was first proposed by O’Regan and Grätzel in 1991 (Fig. 18a).446 A new type of solar cell was invented by sensitizing nanocrystalline films of TiO2 with RuII dyes for molecular-level light absorption. An essential feature in these devices is that photo-excited electrons of RuII are irreversibly injected into the conduction band of TiO2, yielding the oxidized dye RuIII. RuIII is reduced to regenerate RuII dye by electron transfer from a redox species in the electrolyte solution, which is then reduced at the counter electrode.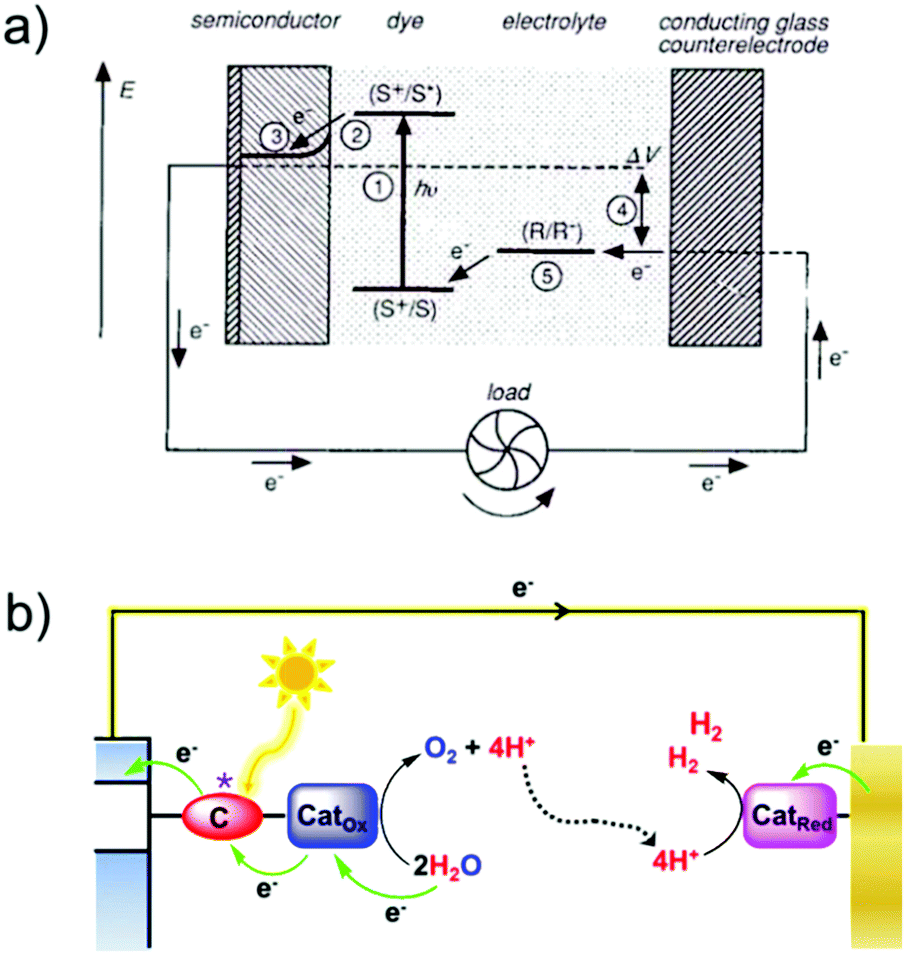 | ||
| Fig. 18 Schematic representation of (a) principle of dye-sensitized solar cells. Reprinted with permission from ref. 446. Copyright 1991 Nature publications. (b) Photoelectrochemical cells for water splitting. | ||
Instead of simply generating electricity, the cell can be modified with a molecular assembly of chromophore-catalysts (e.g., WOC, HER or CO2RR) to use the electron flow to drive reactions that produce chemical fuels.79 As shown in Fig. 18b, the light excitation of chromophore (C) leads to the generation of its excited state C*, which subsequently transfers electrons to the conduction band of TiO2 to form the oxidative state C+. C+ is reduced by the electrons from the catalytic oxidation of H2O to O2. Electrons diffuse through the TiO2 film to the conductive contactor, and then to the cathode, where the electrons are finally used to catalytically reduce protons into H2. This provides a straightforward theoretical basis for water splitting and CO2 reduction based on the molecular chromophore and molecular catalyst.
 | ||
| Fig. 19 Schematic of the water splitting dye sensitized solar cell, with (a) Ru–Ru chromophore-catalyst molecular assembly; (b) Ru-based molecular chromophore and IrO2 WOC. Reprinted with permission from ref. 449. Copyright 2013 American Chemical Society. | ||
In 2009, Mallouk and co-workers rekindled research on the development of DSPEC cells by reporting the first example of a DSPEC cell with a photoanode for visible-light-driven water oxidation.449–451 They reported a DSPEC cell with a photoanode consisting of a Ru-based molecular chromophore and the state-of-the-art WOC IrO2 (Fig. 19b). A photocurrent of 30 μA for water oxidation was obtained by this DSPEC cell. The low photocurrent indicates that the electron transfer from IrO2 to the Ru chromophore is poor. To overcome this inefficient electron transfer, the photoanode was improved by introducing an electron-transfer mediator.450 This strategy enhanced the photocurrent three-fold. The works of the Mallouk group encouraged many following reports of DSPEC cells.
Because of the challenge in synthesizing a chromophore-catalyst molecular assembly, molecules entrapped in a Nafion polymer layer have been used to modify molecular catalysts onto the electrode in a simple method to build DSPEC cells (Fig. 20).452,453 A bioinspired manganese molecular catalyst452 and the state-of-the-art molecular WOC Ru-bda453 were immobilized on a TiO2 film sensitized by RuII molecular chromophores to fabricate DSPEC cells. The achieved photocurrents of water splitting by these DSPEC cells were still only tens of μA. The reason for this low efficiency may be the inefficient electron transfer from the polymer coated catalysts to the excited Ru dye adsorbed on the surface of TiO2. Nevertheless, these works further demonstrated the success of water splitting by molecular catalyst-modified DSPEC cells.
 | ||
| Fig. 20 Depiction of PEC devices consisting of photoanodes immobilized by molecular WOCs embedded in a Nafion film. (a) Ru–sensitized TiO2 film coated with the “Mn4O4” WOC embedded in a Nafion film. Reprinted with permission from ref. 452. Copyright 2013 American Chemical Society. (b) Ru sensitized TiO2 film coated with the Ru-bda WOC embedded in a Nafion film. Reproduced by permission of The Royal Society of Chemistry. | ||
In 2011, a seminal work reported the co-adsorption of a photoanode by a high-potential Zn-based porphyrin and a Cp*–iridium molecular WOC.454 As shown in Fig. 21a, the photoanode provided a photocurrent density of 30 μA cm−2 for water oxidation. This work is the first example to prove that the self-assembled co-adsorption of a molecular chromophore and molecular catalyst on a TiO2 film can also result in the formation of a photoanode capable of visible-light-driven water oxidation. Employing the same strategy, the Sun group in 2013 made a breakthrough in improving the performance of this family of DSPEC cells by employing the highly efficient Ru-bda WOC.163 Under an external bias of 0.2 V vs. NHE, the DSPEC cell shown in Fig. 21b displayed a high initial photocurrent density of more than 1.7 mA cm−2 and a steady photocurrent density of 0.8 mA cm−2. Oxygen evolution on the photoanode was distinct, and the determined Faradaic efficiency was also high. Apparently, the dramatically improved performance of the DSPEC cell could be attributed to the excellent catalytic activity of the Ru-bda WOC with a low overpotential and high catalytic rate. The simple but advanced method of self-assembled co-adsorption is also essential, if we consider the above photoanode with the Ru-bda WOC entrapped in the Nafion polymer layer.
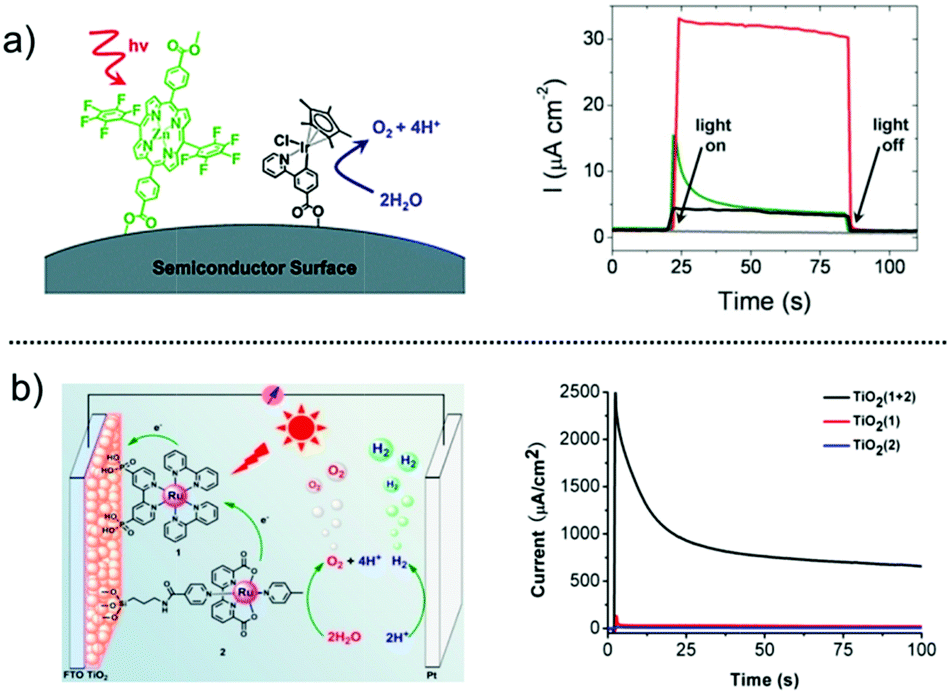 | ||
| Fig. 21 Schematic of PEC devices consisting of photoanodes coadsorbed with molecular photosensitizers and WOCs. (a) Photoanodes immobilized with a zinc porphyrin photosensitizer and Cp*–Ir WOC. Adapted from ref. 454 with permission from The Royal Society of Chemistry. (b) Photoanodes immobilized with a Ru photosensitizer and Ru-bda WOC. Adapted with permission from ref. 163. Copyright 2013 American Chemical Society. | ||
On the basis of these proof-of-concept examples and the breakthrough work, an increasing number of photoanodes have been developed for efficient and stable DSPEC cells.18,20,455,456 Considering the benefits of intra-assembly electron transfer, various strategies have been investigated to produce chromophore-catalyst molecular assemblies for DSPEC cells, including bpm ligand-linked assembly,457 covalent bond linked assembly,458–461 self-assembly via Zr “chemical glue”462–464 and self-assembly via electro-polymerization.465–467 For several of these DSPEC cells, the working mechanisms and dynamics have been elucidated.78,461,468 As an example, a summary of the relevant time scales for light absorption, electron injection, intra-assembly electron transfer and other steps is shown in Fig. 22 for the related chromophore-catalyst molecular assembly-modified TiO2 photoanode.20,461,468 It is clear that the intra-molecule electron transfer is ultrafast, whereas the back electron transfer from TiO2 to the Ru catalyst core is rather slow.
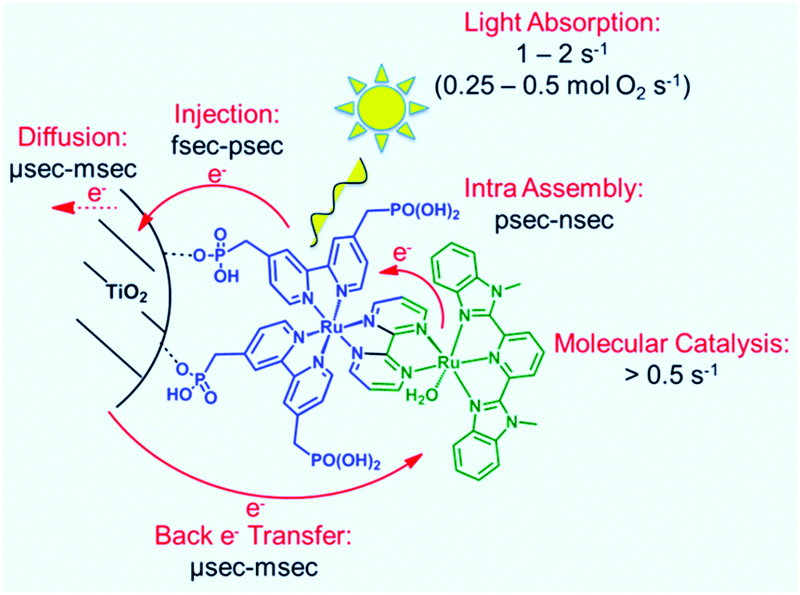 | ||
| Fig. 22 Summary of relevant time scales for light absorption, electron injection, and intra-assembly electron transfer of a photoanode modified with Ru–Ru chromophore-catalyst molecular assembly. Adapted with permission from ref. 20. Copyright 2015 American Chemical Society. | ||
The stability of the adsorption of molecules on the metal oxide surface is always a challenge. To stabilize the attachment of functional molecules to the surface of the metal-oxide substrate, various methods have been attempted, such as electro-assembly,465–467 the use of hydrophobic polymer overlayers,471 silatrane surface bindings,472–474 siloxane surface bindings475,476 and hydrophobic environments.477 As shown in Fig. 23a, the most effective stabilization was recently achieved by Meyer and co-workers via embedding the functional molecules into thin overlayers of Al2O3 or TiO2 formed by atomic layer deposition (ALD). After the deposition of a protective layer, the long-term stability of the Ru molecular WOC-modified ITO electrodes for water oxidation was dramatically enhanced (Fig. 23b).20,469,470,478,479
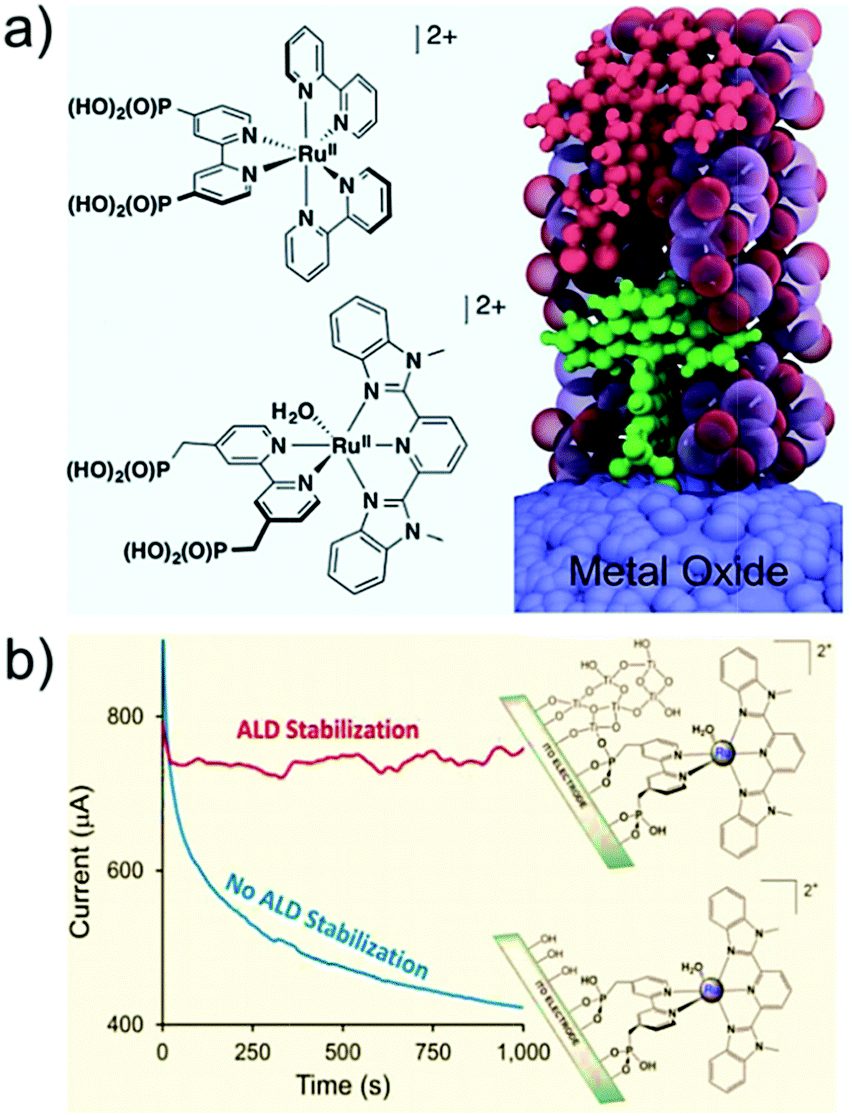 | ||
| Fig. 23 (a) Depiction of the ALD layer-protected surface assembly with a Ru photosensitizer molecule (green molecule) and a Ru WOC molecule (red molecule) embedded in 3 nm of Al2O3. Adapted from ref. 469 with permission from The Royal Society of Chemistry. (b) Electrolysis curves for water oxidation catalysis by a Ru WOC surface bound to ITO with and without a 1 nm TiO2 protective layer at pH 11 at 1.40 V vs. NHE. Adapted with permission from ref. 470. Copyright 2013 National Academy of Sciences. | ||
A high catalytic rate for water oxidation is essential to prevent electron recombination and consequently achieve high photocatalytic performance. Owing to the remarkable activity, Ru-bda type WOCs have been extensively employed for the development of DSPEC cells in the last three years by not only the Sun group but also many other scientists.161,458,480–486 A photocurrent density of approximately 1.0 mA cm−2 can be achieved for DSPEC cells with a Ru photosensitizer and Ru-bda type WOCs, with significantly improved stability.165
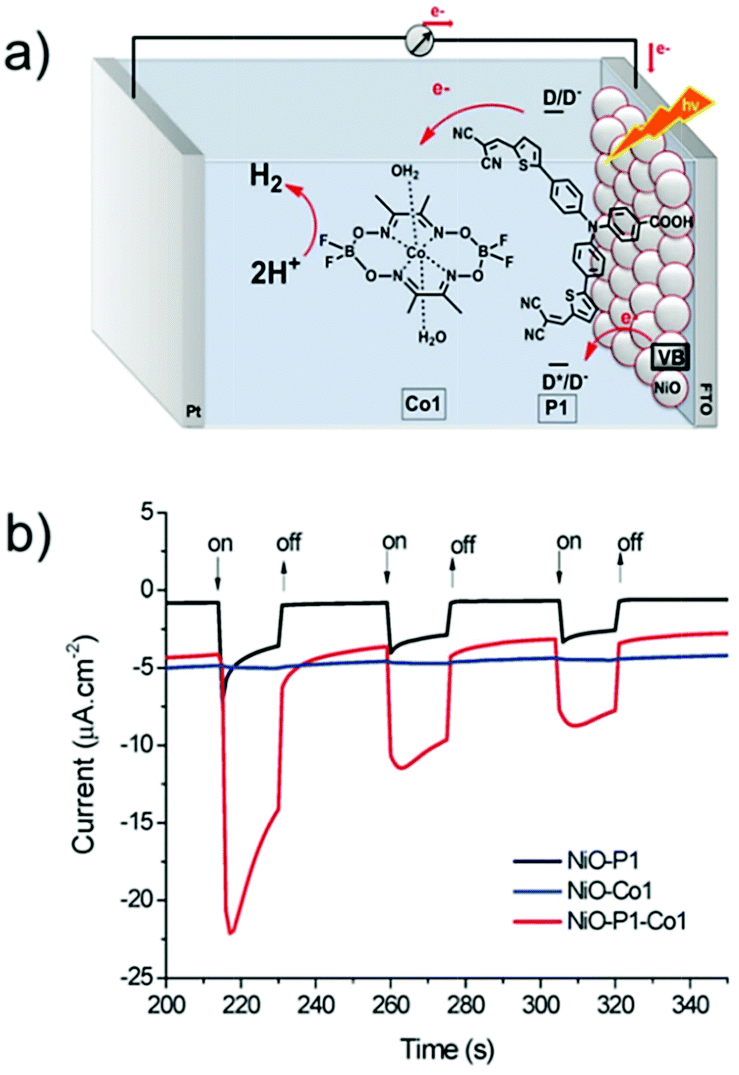 | ||
| Fig. 24 (a) Depiction of PEC devices consisting of a photocathode based on an organic dye sensitized nanostructured NiO film coated with cobaltoxime HEC. (b) The photoelectrochemical performance. Adapted with permission of The Royal Society of Chemistry. | ||
Several strategies as shown above for fabricating molecular-catalyst-modified dye-sensitized photoanodes, including chromophore catalyst self-assembled co-adsorption,491–494 adsorption of chromophore-catalyst assembly495–497 and self-assembly of a system via Zr “chemical glue”,498,499 have been duplicated for the development of molecular-catalyst-modified dye-sensitized photocathodes. For example, Ott, Hammarström and their co-workers researched the light-driven electron transfer dynamics of a photocathode with the co-adsorption of a Coumarin-343 sensitizer and iron-dithiolate HECs (Fig. 25a).491,494,500,501 By coordinating a cobaltoxime catalyst to the RuII dye molecule with a free pyridyl ligand already on a NiO electrode, Wu and co-workers obtained a chromophore-catalyst assembly-immobilized photocathode (Fig. 25b). One important benefit of this approach is that it does not require a readily synthesized chromophore-catalyst assembly.496
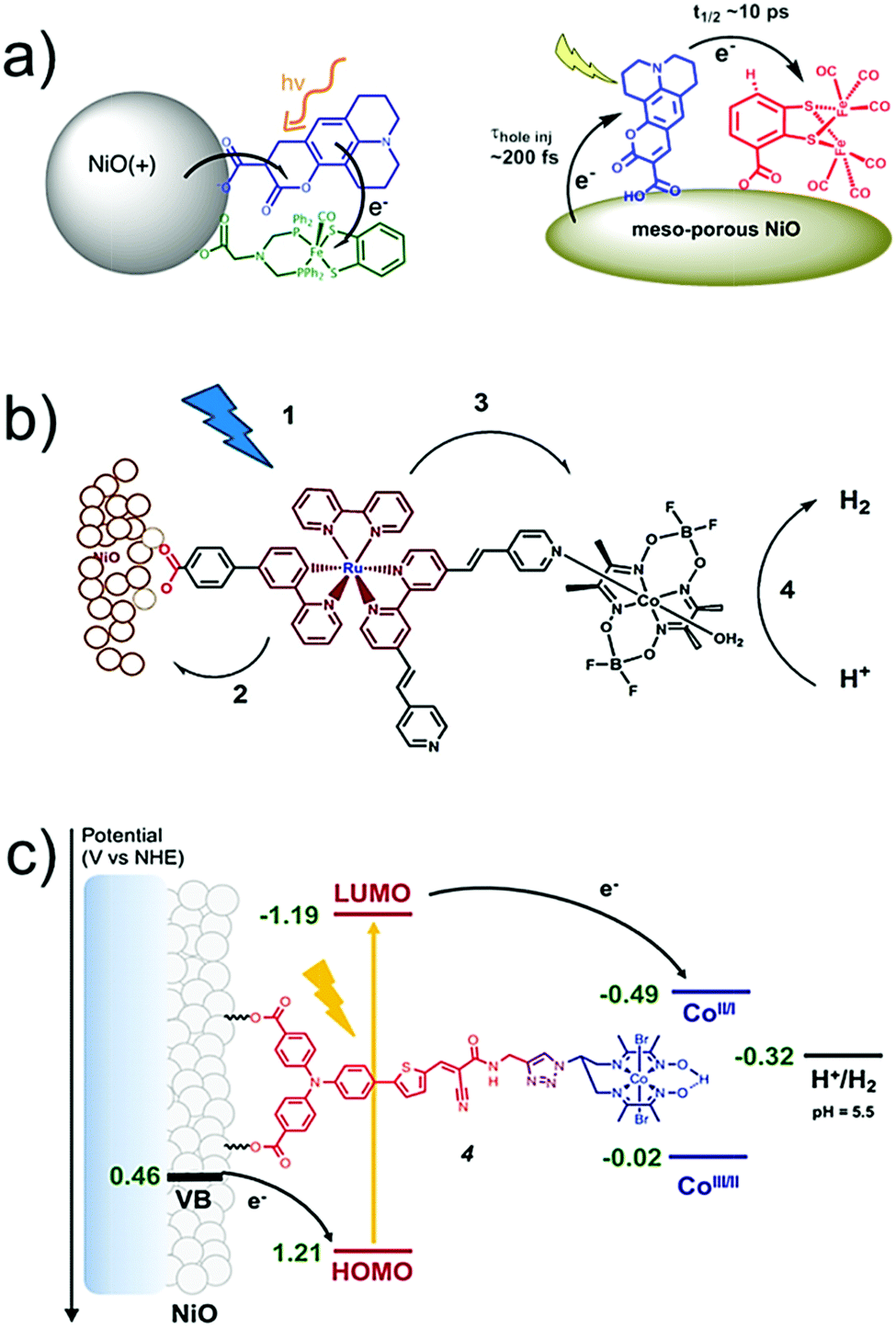 | ||
| Fig. 25 Schematic representation of (a) the sequence of photoinduced electron transfer processes leading to iron catalyst reduction in coumarin dye and an iron catalyst co-sensitized NiO photocathode. (b) RuII dye cobaltoxime catalyst assembly-immobilized photocathode. Adapted with permission from ref. 496. Copyright 2013 American Chemical Society. (c) Organic dye cobaltoxime catalyst assembly-immobilized photocathode. Adapted with permission from ref. 497. Copyright 2016 American Chemical Society. | ||
In very recent years, two covalently linked organic dye–cobaltoxime catalyst assemblies were separately reported by the Artero group497 and Tian group (Fig. 25c).495 Tian and co-workers deeply investigated the mechanism of their system to facilitate the better design and further promotion of more efficient organic dye–molecular catalyst assemblies. During the preparation of this review, Artero and co-workers reported insights into the mechanism and aging of a photocathode with the co-adsorption of an organic push–pull dye and a cobaltoxime catalyst.493 They suggested decomposition pathways of the dye and a triazole linkage to immobilize the catalyst onto NiO, providing grounds for the development of more advanced molecular DS-PEC components. Instead of NiO, Reisner and co-workers recently employed a p-type semiconductor CuCrO2 as a novel hole-collector substrate for fabricating a DSPEC cell upon the co-adsorption of a phosphonated diketopyrrolopyrrole dye with a Ni-bis(diphosphine) catalyst (Fig. 26a).492 This work demonstrated the importance of developing new delafossite structures as p-type semiconductors for the production of solar fuels.
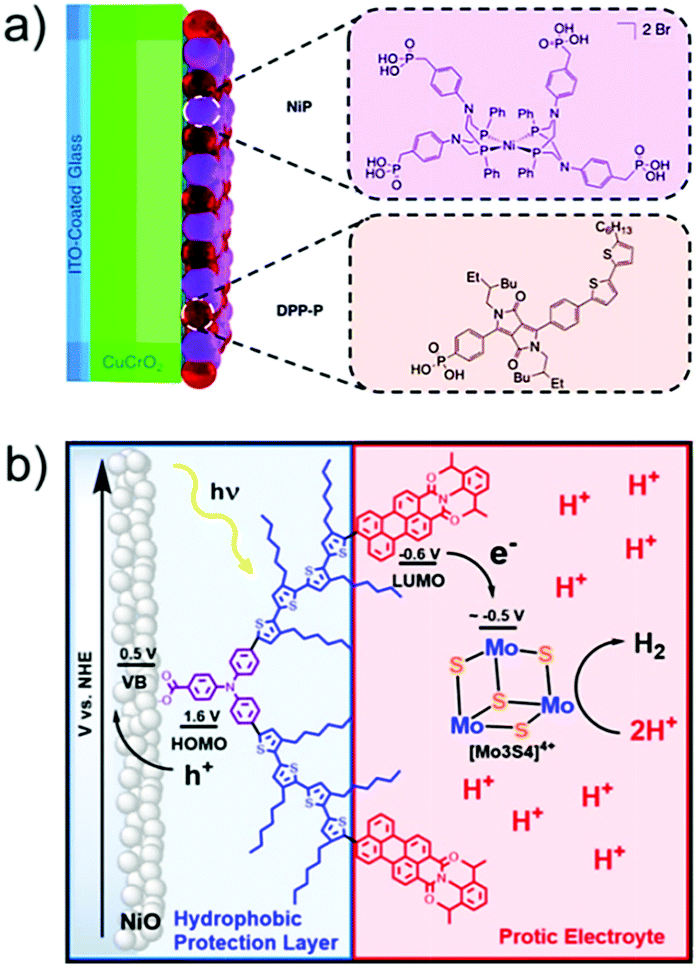 | ||
| Fig. 26 Schematic representation of H2-evolving photocathodes (a) co-immobilized by an organic dye (DPP-P) and Ni-bis(diphosphine) HEC on the CuCrO2 electrode. Reproduced from ref. 492 with permission from The Royal Society of Chemistry. (b) Modified by a membrane mimicking organic dye working in an electrolyte solution with [Mo3S4]4+ HEC. Reprinted with permission from ref. 502. Copyright 2016 American Chemical Society. | ||
As the development of photocathode-based DSPEC cells is a relatively recent research area, in most of the above reported cases, the photocurrent densities were of the order of tens of microamperes per square centimeter. Nevertheless, the strategies aimed at building a device and the subsequent revealed mechanisms are promising for the future development of more efficient DSPEC cells. Recent work from the Wu group shows promise. Using an organic push-double-pull dye BH4-sensitized NiO photocathode and a [Mo3S4]4+ cluster as a catalyst in an electrolyte solution (Fig. 26b), they achieved a proton-reducing photocurrent of approximately 183 μA cm−2 (0 V vs. NHE with 300 W Xe lamp) for an unprecedented 16 h without degradation.502 Further improvement in the catalytic photocurrent is expected if the catalyst is modified on the electrode, as it will facilitate electron transfer between the dye and catalyst and also increase the local concentration of catalysts.
Encouraged by the development of DSPEC cells for water splitting, CO2RC molecular-catalyst-modified DSPEC cells have also been reported in recent years. The first example of a DSPEC cell for the reduction of CO2 was reported by Inoue and co-workers with a photocathode immobilized by a chromophore-catalyst assembly of a zinc porphyrin photosensitizer and a bipyridyl rhenium catalyst (Fig. 27a).504 Due to the fast intramolecular electron transfer from zinc porphyrin to the rhenium catalyst, the hole-injection efficiency of this system was as high as 98.2%. A catalytic photocurrent of 20 μA was obtained with a low Faradaic efficiency of 6.2% for CO2 reduction to CO, and the TON was determined to be 10. Decomposition of the sensitizer part is one reason for the moderate performance, as a TON of 122 was achieved when more separate zinc porphyrin was adsorbed onto the photocathode. This was further proved by a subsequent work from the group of Ishitani et al. The authors synthesized a Ru photosensitizer–Re catalyst assembly to sensitize a NiO film to form a photocathode for light-driven CO2 reduction (Fig. 27b). With a bias potential of −1.25 V vs. Ag/AgNO3 under light (λ > 460) illumination, a TON of 32 and a Faradaic efficiency of 71% were achieved for the reduction of CO2 into CO by this DSPEC cell.503 Furthermore, they reported a new hybrid photocathode, a Ru(II)–Re(I) supramolecular photocatalyst-modified CuGaO2 p-type semiconductor (RuRe/CuGaO2), instead of the NiO. By the RuRe/CuGaO2 hybrid photocathode, a 400 mV more positive onset potential was achieved for the conversion of CO2 to CO in an aqueous electrolyte solution.505
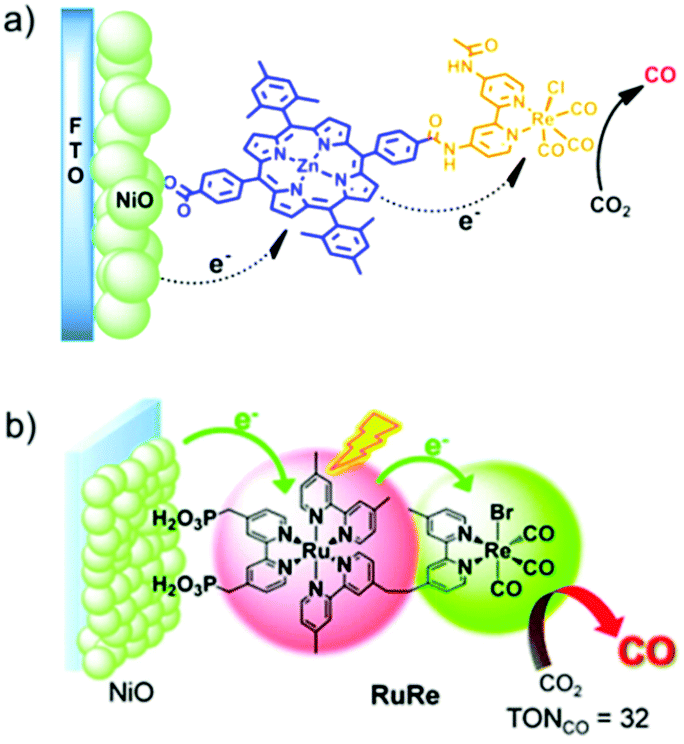 | ||
| Fig. 27 Schematic representation of CO2-reduction photocathodes (a) immobilized by a chromophore-catalyst assembly of a zinc porphyrin photosensitizer and Re-bpy CO2RC; (b) modified with a Ru photosensitizer–Re catalyst assembly. Reproduced from ref. 503 with permission from The Royal Society of Chemistry. | ||
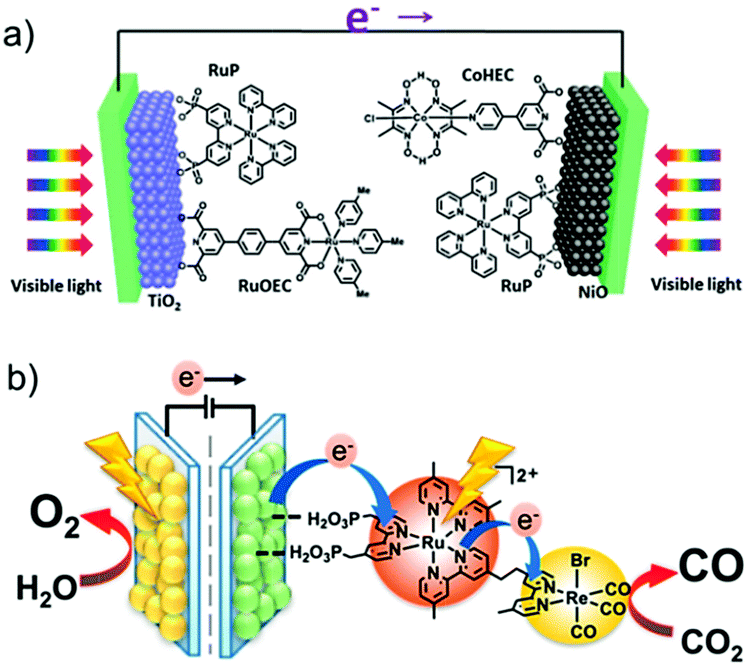 | ||
| Fig. 28 Schematic representation of (a) a tandem DSPEC cell for water splitting based on molecular-WOC-modified TiO2 as the photoanode and molecular-HEC-modified NiO as the photocathode. Reproduced by permission of The Royal Society of Chemistry. (b) A hybrid photoelectrochemical cell with TaON as the photoanode and chromophore-catalyst-assembly-sensitized NiO as the photocathode. Adapted with permission from ref. 506. Copyright 2018 American Chemical Society. | ||
5.2 PEC cells with molecular catalyst modified VLA-SC photoelectrodes
In the DSPEC cells, dyes are used to absorb light to generate charge separation, and transparent semiconductors, such as TiO2 and NiO, are used to transport charge. Indeed, the earliest PEC cell was reported with a TiO2 film under UV light in the 1972 paper of Fujishima and Honda (Fig. 29).510 In contrast to the dye sensitized TiO2 film working under visible light, the PEC cell with a bare TiO2 film under UV-light illumination works according to a different mechanism. UV light is absorbed by TiO2, and it consequently generates charge separation in the energy band of TiO2. The electrons flow via the conduction band to the counter electrode for reduction reactions, and the holes diffuse to the TiO2 film surface to oxidize water (Fig. 29).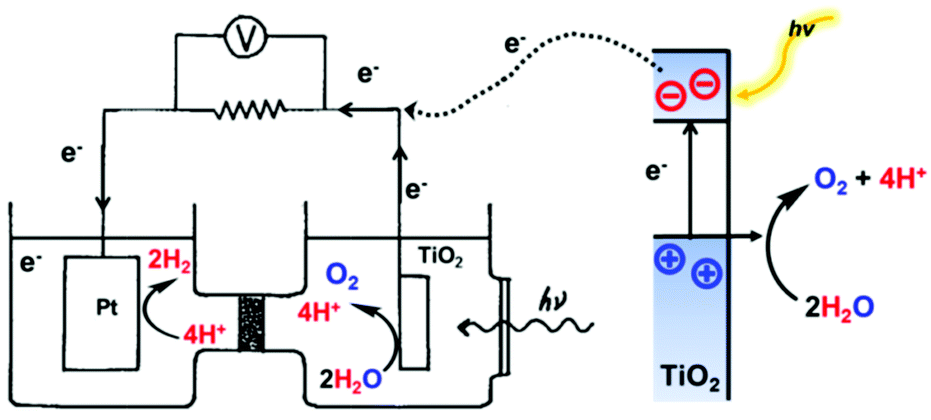 | ||
| Fig. 29 Simple PEC cell reported by Fujishima and Honda in 1972 and the working mechanism. Adapted with permission from ref. 510. Copyright 1972 Nature publications. | ||
However, bare TiO2 cannot be used to produce an applicable PEC cell because a large band gap of 3.1 eV makes it unable to absorb visible light, which accounts for over 95% irradiation of the solar spectrum. In contrast, visible-light-absorbing semiconductors (VLA-SCs) with narrow band gaps and a suitable energy band for driving the reactions involved in AP can be directly employed to build PEC cells without the sensitization of dyes.26 For example, a PEC cell with a p-GaAs–n-GaAs–p-GaInP2 multijunction photoelectrode was reported in 1998 by Turner and co-workers for hydrogen evolution.511 Under visible-light illumination, electrons flow toward the illuminated surface and holes flow toward the ohmic contact. The hydrogen production efficiency of this system is 12.4%.
In recent years, n-type VLA-SCs, e.g., Fe2O3, WO3, and BiVO4, have been extensively studied as a photoanode for photoelectrochemical water oxidation, and p-type VLA-SCs, such as p-Si, InP, GaP, and InGaP, have been employed as a photocathode for proton and CO2 reductions (Fig. 30).34,512 Three basic properties of a VLA-SC for efficient photosynthesis are good light absorption ability, high efficiency of charge separation and migration, and a powerful energy band to drive the photosynthetic reaction, which can be dominated by adjusting the band gap and by tuning its physical features, such as crystal structure, crystallinity, and particle size.
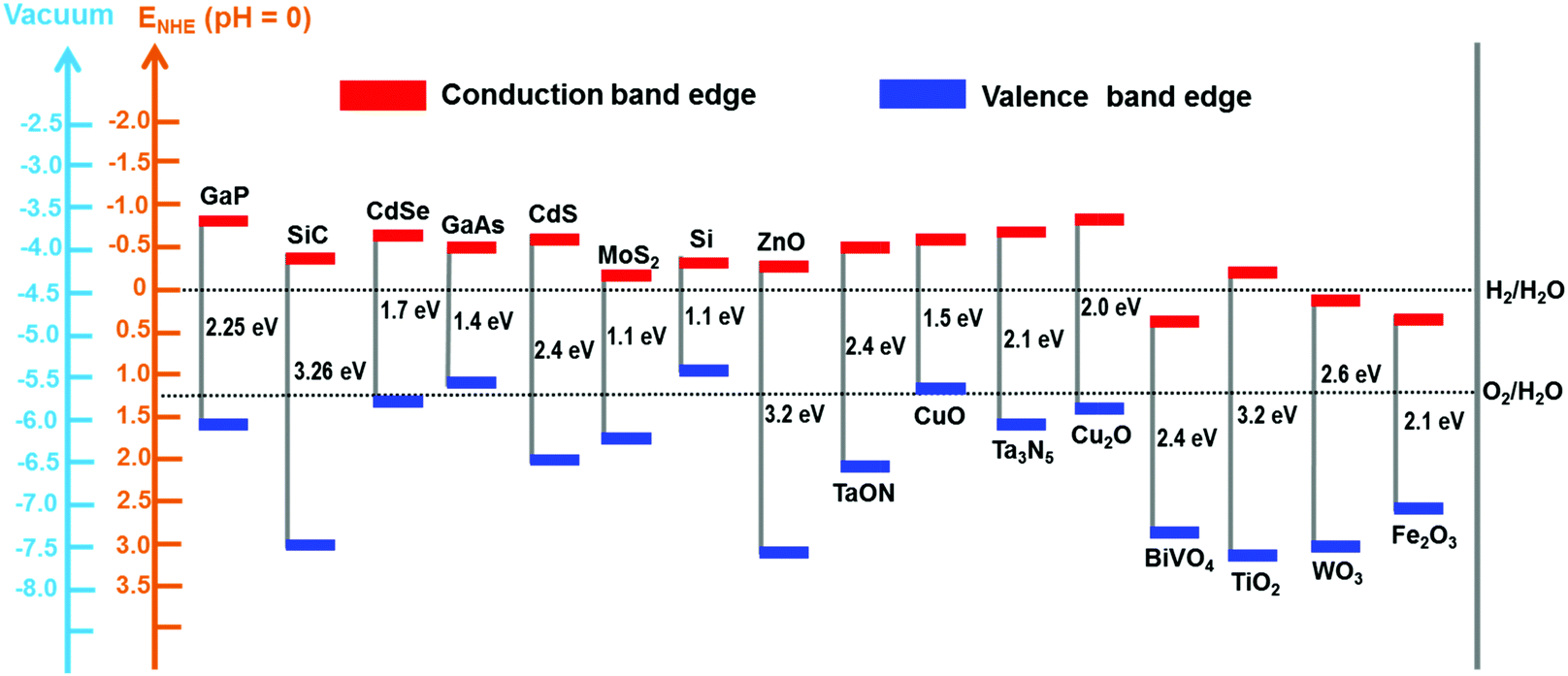 | ||
| Fig. 30 Band gaps and band positions for common semiconductors. | ||
However, for a bare VLA-SC, even if it possesses the above-mentioned basic properties, in most cases, it may show a very low catalytic photocurrent because of the recombination of a vast majority of photo-generated electrons and holes on the surface before being used to drive reactions, which are uphill reactions with a slow reaction rate (Fig. 31a). Therefore, cocatalysts modified on the surface of the semiconductors to accelerate the reaction rates are considered to be the other essential components to fabricate efficient VLA-SC-based PEC cells.513,514 The use of a molecular catalyst is one of the best approaches for the modification of photoelectrodes (Fig. 31b). With high TOFs, molecular catalysts can effectively restrain the combination of electrons and holes on the surface of photoelectrodes. The modification by molecular catalysts is homogeneous and at a molecular level, thereby facilitating direct transfer of charge to every catalytic site. In general, the cover of a molecular layer will not affect the light absorption of the semiconductor and the mass transfer.
 | ||
| Fig. 31 Schematic representation of (a) catalytic reactions on the surface of VLA-SCs and pathway of charge recombination. Reprinted with permission from ref. 513. Copyright 2013 American Chemical Society. (b) Molecular-catalyst-modified semiconductor. | ||
The earliest example of molecular-catalyst-based VLA-SC hybrid devices was reported by Mueller-Westerhoff in 1984. Proton reduction catalyst 1-methyl-[1.1]ferrocenophane was immobilized onto a p-Si photocathode via the formation of a co-polymer film with poly(chloromethyl)styrene.515 By this hybrid device, a photocurrent density of 194 mA cm−2 was achieved at −0.54VSCE in the HBF3OH electrolyte under 870 mW cm−2. Moreover, it maintained high performance for 5 days. However, the continuous reports of molecular-catalyst-based VLA-SC hybrid devices did not appear until the 2010s. Development of molecular-catalyst-based VLA-SC hybrid devices for artificial photosynthesis has attracted considerable research attention since the last 5 years.26
VLA-SC hybrid photoanodes modified by two pioneer noble-metal-free molecular catalysts were developed by the Bartlett group and the Sun group in 2014.235,518 Bartlett and co-workers synthesized non-heme-iron WOC Fe(mcp)(Cl)2 with two phosphate groups to modify the WO3 electrode (Fig. 32a).518 After modification of the iron catalyst, the catalytic photocurrent density of WO3 under 100 mW cm−2 illumination increased from 0.7 mA cm−2 to 1.3 mA cm−2. The combination of non-heme-iron WOC and WO3 photoanode was further studied by the Reisner group.519 The Sun group for the first time modified VLA-SCs α-Fe2O3 with the OEC mimic “Co4O4 cubane” catalyst (Fig. 32b).235 Under the same conditions, α-Fe2O3 with the modification of the “Co4O4 cubane” catalyst exhibited a transient photocurrent that was eight times higher than that of the control α-Fe2O3 electrode, indicating that “Co4O4 cubane” is effective at accelerating the water-oxidation rate on the surface of the VLA-SC. On the basis of this work, “Co4O4 cubane” was employed as cocatalysts on other VLA-SCs, such as BiVO4237,238 and Ta3N5.520 A stable photocurrent density of 5 mA cm−2 was obtained by the “Co4O4 cubane” modified BiVO4 photoanode at 1.23 VRHE in a pH 9.3 solution.238 Ta3N5 integrated with hole storage layers of Ni(OH)x/ferrihydrite and a layer of “Co4O4 cubane” and Ir-based catalysts displayed a high photocurrent density of 12.1 mA cm−2 at 1.23 VRHE under 100 mW cm−2 illumination.520
 | ||
| Fig. 32 Schematic representation and performances of PEC cells based on molecular-WOC-modified n-type VLA-SC photoanodes. (a) Fe-mcp-WOC-modified WO3 photoanode. Adapted with permission from ref. 518. Copyright 2014 American Chemical Society. (b) “Co4O4 cubane”-WOC-immobilized α-Fe2O3 photoanode. Adapted with permission from ref. 235. Copyright 2014 American Chemical Society. | ||
The effect of modification of molecular catalysts on VLA-SCs has been further proved by more examples in recent years, including α-Fe2O3 modified with a Ru-pdc complex,521 binuclear Ir complex,183,522–524 and Co-phthalocyanines,525 and BiVO4 modified with Co-porphyrin,526 Co-salophen527 and Cu-porphyrin.528
The group of Moore reported a family of cobaltoxime/GaP hybrid photocathodes by coordinating cobaltoxime to the pyridyl or imidazolyl groups in the polymer coated on the GaP electrode (Fig. 33a). This is a simple but effective method to modify an electrode with a large amount of the molecular catalyst.542–546 At the same time, the polymer coating can also be a protective layer for the VLA-SC film. The photocurrent density of this series of hybrid photocathodes for hydrogen evolution is around 1–2 mA cm−2. For the example shown in Fig. 33b, the photocathode displayed a photocurrent density of 2.4 mA cm−2 at 0.31 V in 1 M phosphate buffer solution under 100 mW cm−2 illumination.542 Comparable photocatalytic performance has also been achieved for the GaP electrode modified with metalloporphyrins by a UV-induced covalent grafting approach.547 Obviously, the modification of molecular HER catalysts on p-semiconductors can dramatically increase the photocatalytic current density and positively shift the onset potential for hydrogen evolution.
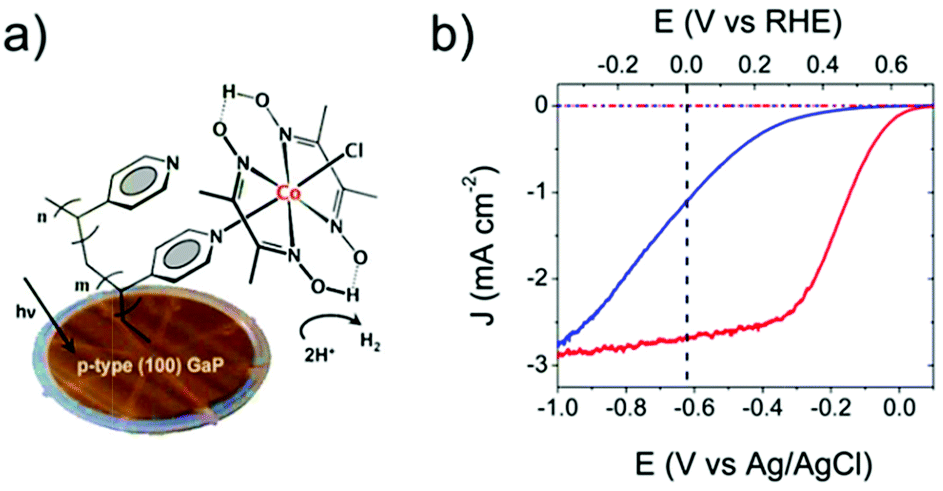 | ||
| Fig. 33 (a) Schematic representation and (b) linear sweep voltammograms (LSVs) of a cobaltoxime-modified GaP photocathode for H2 evolution. Adapted with permission from ref. 542. Copyright 2013 American Chemical Society. | ||
A breakthrough in this field was made by Turner and co-workers. The authors prepared a highly efficient p-GaInP2 photocathode protected by a 35-nm TiO2 layer immobilized with a cobaltoxime molecular catalyst (GaInP2–TiO2–cobaltoxime) (Fig. 34).548 Atomic layer-deposited TiO2 on top of the cobaltoxime catalyst layer makes the final GaInP2–TiO2–cobaltoxime–TiO2 electrode corrosion resistant in a basic solution. In 0.1 M NaOH solution, the GaInP2–TiO2–cobaltoxime–TiO2 electrode under 1 sun light illumination showed a photocurrent density of 11 mA cm−2 at 0 VRHE and an onset potential at 0.75 VRHE. Moreover, a TON of 1.39 × 105 was attained after 16 h of photoelectrolysis. The performance of the GaInP2–TiO2–cobaltoxime–TiO2 electrode is comparable with that of Pt-modified GaInP2. The most active and robust molecular catalysts for HER, Ni-based “DuBois catalysts”, have also been investigated as a cocatalyst on the surface of VLA-SCs for photoelectrochemical hydrogen evolution.549–552
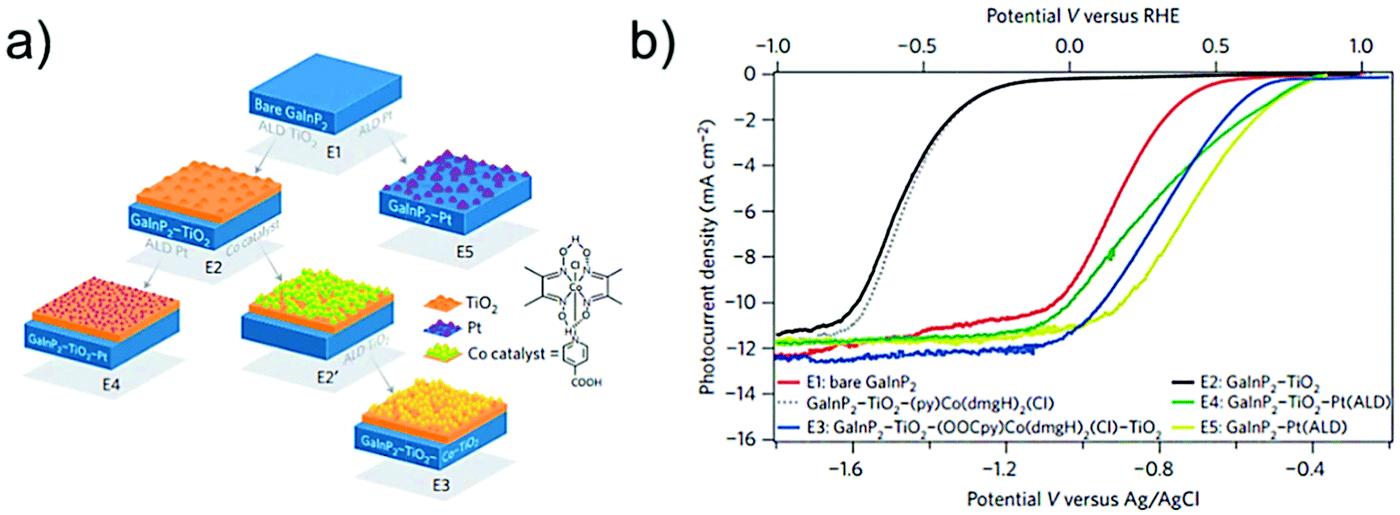 | ||
| Fig. 34 (a) Schematic representation of surface modification for the p-GaInP2 photoelectrodes; (b) LSVs of the cobaltoxime-modified p-GaInP2 photoelectrodes and the control photoelectrodes. Adapted with permission from ref. 548. Copyright 2016 Nature publications. | ||
The modification of the CO2 reduction catalyst enables p-type semiconductors to perform the photochemical reduction of CO2 to form chemical fuels. Sato and co-workers reported the first example of molecular-catalyst-modified VLA-SC-based photocathodes with Zn-doped p-InP as a light absorber immobilized by a ruthenium complex polymer (RuCP) as a CO2 reduction catalyst (Fig. 35).553 CO2 was successfully reduced into HCOO− by this device. This RuCP/p-InP photocathode was further investigated by Arai and co-workers for CO2 reduction.554–556
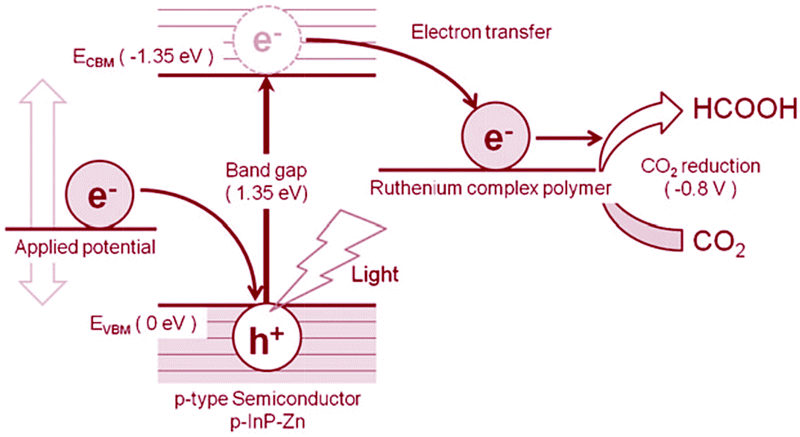 | ||
| Fig. 35 Schematic energy diagram of the RCP/p-InP-Zn photocathode producing HCOOH under visible-light irradiation. Reproduced from ref. 553 with permission from The Royal Society of Chemistry. | ||
The n-type VLA-SC photoanode and the p-type VLA-SC photocathode can be coupled by a wire. As the example shown in Fig. 36a, a BiVO4/Cu2O photoanode/photocathode tandem cell was reported for overall unassisted solar water splitting.557 Another strategy is to fabricate a multijunction photoelectrode. The multijunction photoelectrode can be coupled with the counter electrode by a wire (Fig. 36b) or integrated with the counter electrode into a chip to form a monolithic PEC device (Fig. 36c). These spontaneous PEC devices have attracted attention since 1970s.17 Efficiencies of a number of devices are high over 10%, and the highest reported efficiency is 18%.511,559–562 However, the catalysts used in these devices still depended on precious-metal-based inorganic materials, which are rare and not atom-economic. One promising approach to change this situation is to employ molecular catalysts.
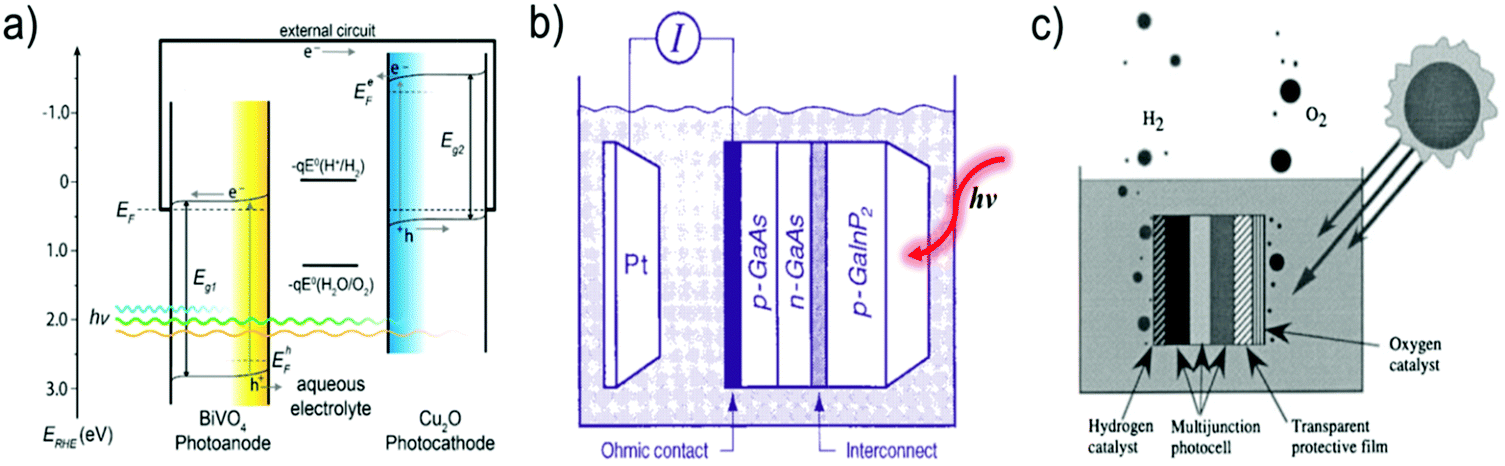 | ||
| Fig. 36 Schematic representation of (a) PEC tandem cells based on wire-linked n-type VLA-SC photoanode and p-type VLA-SC photocathode. Reprinted with permission from ref. 557. Copyright 2014 American Chemical Society. (b) PEC cell with a p-GaAs–n-GaAs–p-GaInP2 multijunction photoelectrode coupled with the counter electrode by a wire. Adapted with permission from ref. 511. Copyright 1998 American Association for the Advancement of Science (AAAS). (c) Monolithic PEC device integrated with a multijunction of different VLA-SCs and counter electrode for water splitting. Adapted with permission from ref. 558. Copyright 1998 American Chemical Society. | ||
So far VLA-SC based PEC tandem cells modified by a molecular catalyst have rarely been reported. By coupling the RuCP/p-InP photocathode with a reduced SrTiO3 (r-STO) photoanode, Arai and co-workers reported a full device for solar formate production from CO2 and H2O with no external electrical bias (Fig. 37).554 A stable photocurrent of 140 μA cm−2 was achieved during 3 h of illumination for the r-STO//p-InP/RuCP system, which was seven times higher than that of the previous TiO2(P25)//p-InP/RuCP system.556 The conversion efficiency from solar to chemical energy was increased from 0.03% to 0.14%. A monolithic PEC device has also been fabricated with this r-STO//p-InP/RuCP system.
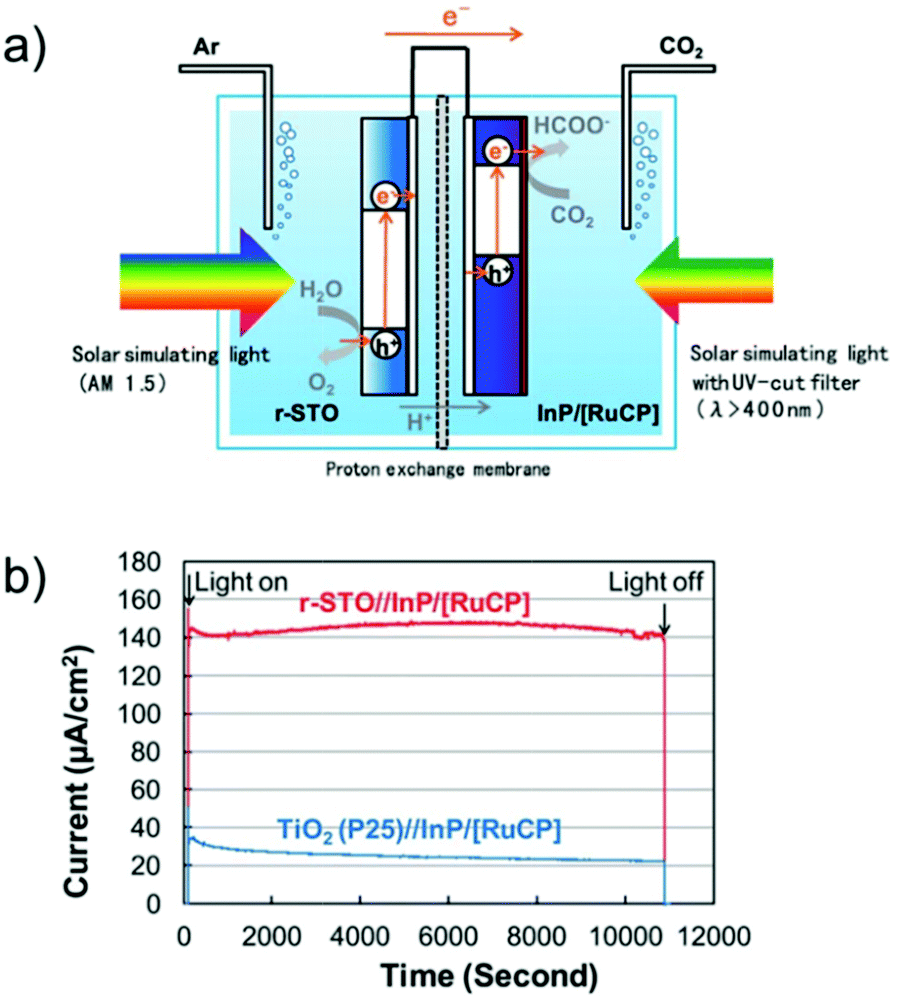 | ||
| Fig. 37 (a) Schematic representation and (b) long-term photocurrent of the PEC tandem cells for reduction of CO2 integrated with a reduced SrTiO3 photoanode and a RuCP molecular CO2RC decorated InP photocathode. Reproduced from ref. 554 with permission from The Royal Society of Chemistry. | ||
The same group further improved the solar-to-chemical-energy conversion efficiency to 4.6% by engineering a monolithic tablet-shaped device (IrOx/SiGe-jn/CC/p-RuCP), which is composed of a porous RuCP as a CO2 reduction catalyst, IrOx as a WOC, and a triple-junction of amorphous silicon–germanium (Fig. 38).555 In this efficient device, the first key technology is the selective CO2 photoreduction in aqueous media, which was realized by the employment of the molecular CO2 reduction catalyst. Therefore, the semiconductor–molecular catalyst hybrid system is a promising approach for the AP.
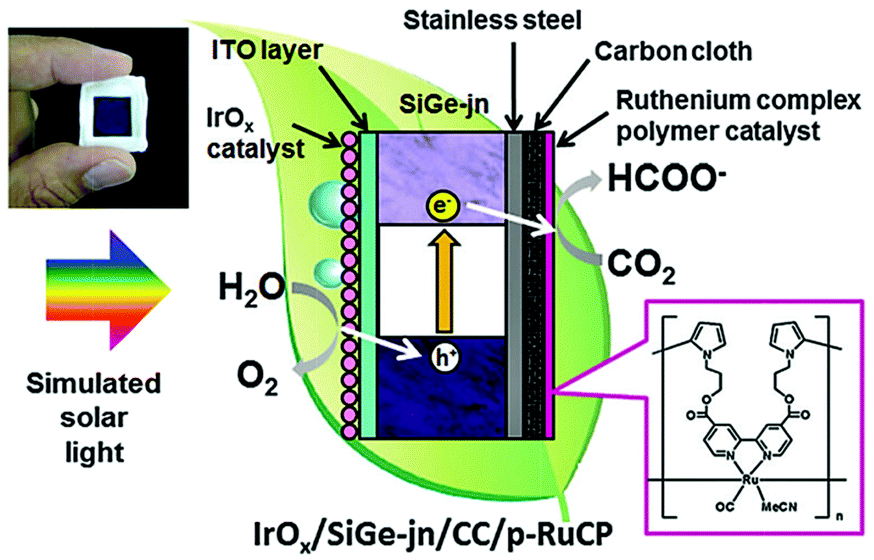 | ||
| Fig. 38 Schematic representation of the IrOx/SiGe-jn/CC/p-RuCP monolithic one-chip device for CO2 photoreduction developed by Arai and co-workers. Reproduced from ref. 555 with permission from The Royal Society of Chemistry. | ||
5.3 Application of molecular catalysts in PVE devices – a promising strategy
As an alternative of the PEC cell, the PVE device shown in Fig. 2 has become attractive in the past 5 years. Indeed, as the most direct and “brute force” approach, hydrogen production from water electrolysis driven by solar cells was proposed decades ago.563 It consists of separated traditional photovoltaic solar cells wired to an electrolyzer for water splitting or water-oxidation-coupled CO2 reduction. The separation of the light absorber part from an electrolyte offers several advantages: (1) two components, i.e., solar cell and electrolyzer, can be investigated and developed separately; (2) light absorber can be protected from the damage of the electrochemical process.In the past seven years, the prices of crystalline silicon (c-Si) solar cells and PV modules have dramatically decreased by 86% and 77%, respectively. In the meantime, average efficiencies of commercial c-Si solar cells have been improved to 17.5% for multicrystalline silicon and 19.5% for monocrystalline silicon.564,565 Moreover, new generations of solar cells have been extensively developed, particularly perovskite solar cells with a simple preparation process, low-cost, and high laboratorial efficiency over 22%.42,566
On the other hand, there are commercially available technologies to produce hydrogen through water electrolysis on a large scale. Before the realization of hydrogen production via coal gasification and natural gas steam reforming in the 1950s, hydrogen was mainly produced by water electrolysis.568 Two main commercial water-electrolysis technologies are the alkaline water electrolysis and proton exchange membrane (PEM) water electrolysis, as shown in Fig. 39a and b. Since the discovery of the electrolysis phenomenon by Troostwijk and Diemann in 1789,569,570 alkaline electrolysis has become a matured technology for the production of H2 up to the megawatt scale.567 The most obvious advantage of alkaline water electrolysis is that both the anode and the cathode are noble-metal free materials, the anode is based on Ni–Co–Fe and the cathode is based on Ni–C. This is one of the important reasons that alkaline water electrolysis has been utilized on a commercial level worldwide for many years. However, for over a century, the employment of a strong alkaline electrolyte (30% KOH) and a low maximum achievable current density (0.2–0.4 A cm−2) are the two major issues occurring in alkaline water electrolysis. To overcome these problems, PEM water electrolysis has been developed since the 1960s.567 A typical PEM device requires only pure water as the reactant, and it is capable of obtaining a considerably higher current density (0.6–2 A cm−2). In addition, the PEM device has a compact system design, which is suitable for domestic application. Like the commercialized PEC water electrolyzer displayed in Fig. 39c (size of the inside electrode is 10 cm diameter), it can produce H2 at a rate of 18 L h−1. PEM water electrolysis is an advanced and promising technology that can be combined with solar cells for practical application of AP. However, the broad application of PEM technology for water electrolysis is limited by its high cost arising from the usage of valuable materials. One of the major issues for this high cost is that only noble metal-based catalysts, such as RuO2, IrO2, and black Pt, can work well in the PEM cells because of their locally strong acidic corrosive environment. The development of efficient and stable catalysts that can function under acidic conditions is urgently required for PEM water electrolysis.
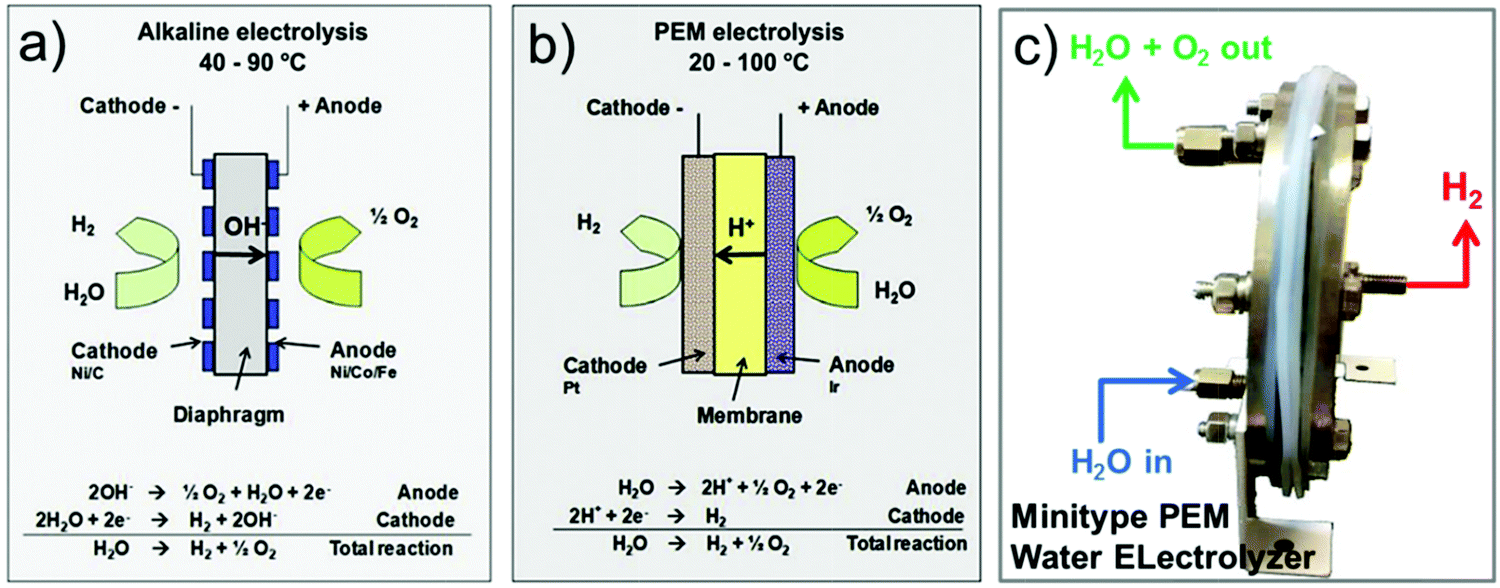 | ||
| Fig. 39 Schematic representation of the operating principle of (a) an alkaline and (b) PEM water electrolysis cell. Reprinted with permission from ref. 567. Copyright 2013 Elsevier. (c) Photograph of a commercialized mini-type PEM electrolyzer for H2 production. | ||
The development of applicable PVE devices for H2 production from H2O based on the available technologies is the best approach. To date, PVE devices, compared with the PEC cells, have been considered to be closer to their practical application. However, concerning the high cost and the huge demand, the replacement of noble-metal-based catalysts must be addressed. Molecular chemistry provides good opportunities for developing alternative cost-effective materials, sufficiently stable in strong acidic media. In this context, as discussed previously, the first challenge is to develop a low-cost and efficient molecular catalyst. The second challenge is to explore suitable electronic carriers for catalyst loading. The third challenge is to develop strategies for stable coating of molecular catalysts on the carrier (Fig. 40).
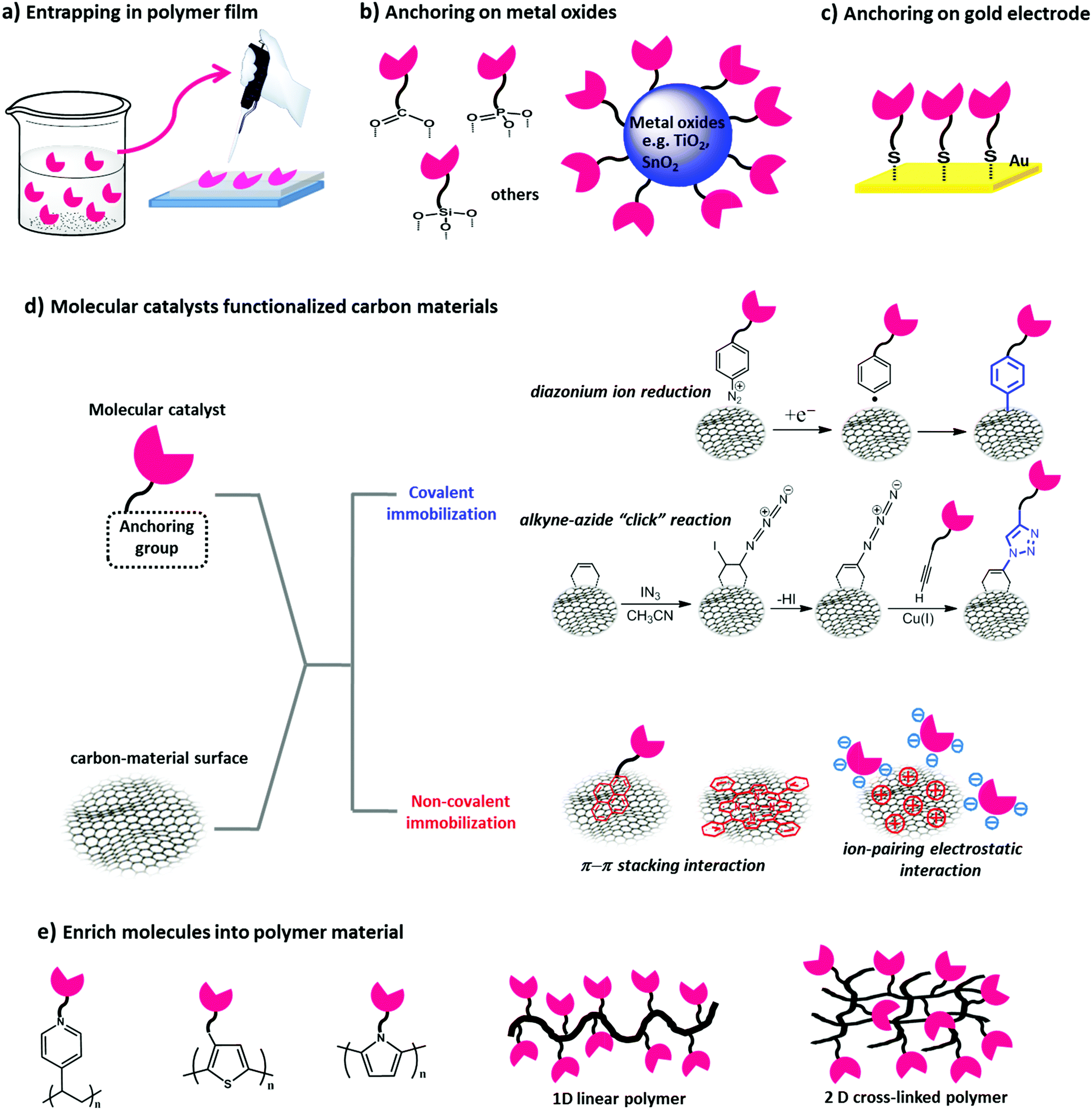 | ||
| Fig. 40 Schematic representation of various strategies for heterogenizing molecular catalysts into materials. | ||
Many strategies have been developed to modify molecular catalysts on various conductive substrates in order to investigate their electrocatalytic properties. A direct and simple approach to place molecular catalysts onto the surface of solid electrodes is entrapping the catalysts within a film generated from polymeric materials, such as polyvinyl, hydrophilic polystyrene derivatives and Nafion™ (Fig. 39a).222,235,361,571 This method is suitable for preliminary evaluation of electrocatalytic properties; however, it is not promising for the applicable design because of its obvious drawbacks, such as catalyst leaking and limited mass and charge transfer. Based on the many examples of PEC devices,20,25 molecular catalysts can be immobilized on the surface of metal oxides, e.g., TiO2 and SnO2, via acid groups such as carboxylic, phosphate and silicate groups (Fig. 39b).78,457,572,573 The drawbacks of this methodology for catalyst immobilization are low loading amount and instability in aqueous media. A molecular catalyst functionalized with sulfur groups can be easily grafted on an Au electrode (Fig. 39c),574,575 which, like the glass carbon (GC) electrode, is a classic planar substrate electrode for investigating the monomolecular layer. There are several conventional strategies for the modification of the GC electrode via covalent attachment, i.e., the “click” reaction route,576,577 diazonium reduction route,376,578–580 and methods designed by combining these two routes (Fig. 39d).581,582 These covalent anchoring methods can also be employed to immobilize other advanced carbon materials, such as carbon nanotubes and graphene.583–585 In addition, non-covalent immobilization of molecular catalysts on carbon nanotubes and graphene has also been realized by the introduction of pyrene substituents on target molecules to form π–π stacking interactions with the surface of carbon materials (Fig. 39d).166,586–594 Because carbon nanotubes and graphene have good electro-oxidation stability, excellent charge transfer ability, high surface area and feasibility of modification, hybrid materials/electrodes made of carbon nanotube/graphene and molecular catalysts are promising to be employed by applicable devices for AP.
The strategy of involving the modification of a monomolecular layer on the porous carbon nanomaterial can provide sufficient exposure of active sites and increase the loading amount. However, in the view of application, the loading amount of the molecular catalyst should be further enhanced to a level higher than that of the monomolecular layer. The final solution is heterogenization of molecular catalysts into functional materials. Polymerization is an easy, tunable, and effective route to enrich molecules into polymer materials, which can be conductive and processable. There are several reports on electrodes generated from polymerization of molecular catalysts.482,595–598 For example, the Ru-bda WOC functionalized with a thiophene group is immobilized to the surface of electrodes by electropolymerization.599 The polymer film of Ru-bda molecular WOC can electrochemically catalyze water oxidation under an overpotential of ∼500 mV to reach a current density of 5 mA cm−2. The first electropolymeric material incorporating the Fe–Fe hydrogenase mimic was reported by Pickett and co-workers although a high overpotential was required for this polymeric material; this work has taken several early steps toward a functional material containing a molecular catalyst.595
Metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) are advanced materials formed by orderly accumulation of small-molecule units. The development of various structures of MOFs and COFs has attracted considerable attention in the material field, and significant improvement has been achieved on the design and synthesis of MOFs601–603 and COFs.604,605 MOFs and COFs have been widely investigated as catalysts because of their large porosities, uniform pore sizes, redox properties, diversified/tunable pore surfaces, and some other unique structural features.606 Therefore, it is one of the promising approaches to heterogenize molecular catalysts into MOF or COF materials for AP.164,435,600,607–613 As the examples shown in Fig. 41, MOFs and COFs generated from functionalized cobalt-porphyrins have been reported with a prominent performance of CO2 electroreduction.435,600 The latter displayed high Faradaic efficiency of 90% and TONs up to 2.9 × 105 (with initial TOF of 9400 h−1) at pH 7 with an overpotential of −0.55 V with no degradation over 24 h, indicating the high potential of these structures.435
 | ||
| Fig. 41 Schematic representation of (a) MOFs generated from functionalized cobalt-porphyrins for efficient CO2 electroreduction. Adapted with permission from ref. 600. Copyright 2015 American Chemical Society. (b) COFs generated from functionalized cobalt-porphyrins for efficient CO2 electroreduction. Adapted with permission from ref. 435. Copyright 2015 American Association for the Advancement of Science (AAAS). | ||
However, most polymers, MOFs and COFs have low stability and conductivity in aqueous solutions under electrochemical conditions.601,614 In addition to promoting the intrinsic activity of molecular catalysts, the improvement of stability and electron conductivity of the materials heterogenized from molecular catalysts is another challenge to produce applicable AP devices based on molecular catalysts.
6. Underestimated power of molecular catalysts for AP
6.1 Intrinsic activity of molecular catalyst ≠ performance of AP device
The performance of an AP device is depended on many factors. However, an efficient catalyst is one of the most essential elements. This is also the reason why the development of catalysts with high intrinsic activity has become extensively attractive. A suitable system for the evaluation and comparison of a new catalyst is important for the development of the field. The most reliable parameter to judge a good catalyst is low overpotential, because the role of a catalyst in a certain reaction is to lower the activation energy, i.e., facilitating the reaction under a lower overpotential. TONs and TOFs are also important direct parameters to evaluate a catalyst. However, TONs and TOFs are closely related to the testing systems and conditions, which need to be carefully considered when catalysts are compared based on TONs and TOFs.Nevertheless, efficient catalysts with a high intrinsic activity are necessary but not the only factors attributable to the good performance of a device or an electrode.30,615 At the initial stage in the development of molecular catalysts, the activity is evaluated by employing sacrificial reagents (e.g., Ce4+ oxidant for water oxidation) and/or under homogeneous electrocatalytic experiments, because these conditions can provide an adequate driving force and simple investigation systems. In many cases, when an efficient molecular catalyst is employed to construct a PEC cell or to modify an electrode, the device exhibits poor performance. However, this cannot indicate that the catalyst is not excellent or not promising for an applicable AP device, because the performance of a PEC cell or an electrolyzer is determined by a number of factors, such as catalyst loading amount, loading method, electrolyte, device design for mass transfer and proton transfer.
For example, the Ru-bda WOC exhibited an activity similar to the Nature's OEC when it is evaluated with Ce4+ as the oxidant under acidic conditions;146–149 however, it exhibited very moderate performance under the three-component photocatalytic system.145,616,617 In particular, when the Ru-bda WOC was employed for the first time to construct a DSPEC cell as shown in Fig. 20b, it yielded a photocurrent density of only 40 μA cm−2.453 However, after the photoanode was improved by the co-absorption strategy, a photocurrent density of 1.7 mA cm−2 was obtained.163 A 40-fold enhancement of device performance was achieved with the same catalyst but different device design. Without Ru-bda as the catalyst, no other DSPEC cells have been reported with comparable performance. The super-low overpotential and super-high TOF guarantee the good performance of the DSPEC cell. As another example, the performance of photocatalytic H2 evolution of the same [FeFe]-hydrogenase HECs has been investigated under systems using quantum dots (QDs) and the molecular photosensitizer [Ru(bpy)3]2+ in water. Under the same conditions, TONs of the [FeFe]-hydrogenase HECs working under the system with CdSe QDs are two orders of magnitude higher than those of the system with [Ru(bpy)3]2+.618,619 The above examples clearly show that the activity of a catalyst cannot completely determine the efficiency of an AP device or a photocatalytic system, although an efficient catalyst is necessary for developing applicable AP devices in future.
6.2 Efficient molecular catalyst ≠ high catalytic current density of an electrode
Electrocatalytic water splitting has been a widely discussed topic since the past five years because it is believed that it can realize an applicable PVE device in the near future. Nowadays, electrocatalysts for AP are ranked on the basis of the current density (per geometric area of an electrode, e.g. 10 mA cm−2) at a certain overpotential.30 Although several super-efficient catalysts for reactions involved in AP, including water oxidation, proton reduction, and CO2 reduction, have been developed, the current density of electrodes modified by molecular catalysts is still significantly lower than that of inorganic-material-based electrodes. This is one of the main reasons that make the development of molecular catalyst getting less attractive for applicable AP devices in recent years.The unsatisfactory performance of molecular-catalyst-based electrodes can be attributed to the following two reasons: the challenge of heterogenization of molecular catalysts has not yet been overcome. Consequently, the low loading amount and the stability of immobilization dramatically limit the performance of molecular-catalyst-based electrodes. Secondly, the electrochemical testing system employed in the laboratory is not beneficial for exhibiting the activity of molecular-catalyst-based electrodes. Because water oxidation is the bottleneck of AP, we consider it as an example. Water oxidation involves the removal of four electrons and four protons. The electrons can be easily transported to a counter electrode, whereas the protons will accumulate in the diffuse double layer, instantly leading to a local pH drop, which will significantly increase the thermodynamic barrier and decrease the catalytic current. To date, the electrochemical water-oxidation performance of an electrode has been evaluated in a simple three-electrode electrochemical cell, i.e., a beaker-like cell fixed with working, reference, and counter electrodes. However, in this kind of test system, the catalytic performance will be strongly limited by the proton and mass transfers.
To obtain high electrocatalytic efficiency, i.e., high catalytic current density, the released protons must be fleetly transported to the bulk solution. For this, one possible solution is the employment of a highly concentrated strong alkaline electrolyte solution, such as 6 M KOH electrolyte solution used in the industrial alkaline water electrolysis620 or 1 M KOH electrolyte solution widely employed in the laboratory beaker-like electrolyzer.621 By using this strategy, the current density of the order of several hundreds of mA cm−2 (less than 500 mA cm−2) can be achieved.620,621 However, for most molecular WOCs based on organometallic compounds, maintaining the stability of compounds under electrooxidation conditions in a strong alkaline solution is already a considerable challenge, because OH− is a strong σ-donating ligand, which may replace the ligand of the molecular catalyst when it is oxidized to high valence. For this reason, many molecular catalysts catalyzing under alkaline conditions are found transformed into metal oxide/hydroxide, serving as true catalysts, thereby imparting good stability and high catalytic activity because of the low thermodynamic requirement of water oxidation under high pH conditions.308,622
Nevertheless, without the alkaline condition, the performance of any type of water-oxidation electrodes is limited in a range of several mA cm−2 of current density under a moderate overpotential. Even with the state-of-the-art Ru-bda WOC, only 0.22 mA cm−2 has been obtained for the Ru-bda-modified carbon nanotube electrode at 0.58 V overpotential in a neutral Na2SO4 solution.166 When the polymerization method was conducted to improve the catalyst loading amount, current density above 4.0 mA cm−2 at 0.48 V overpotential can be achieved in a phosphate buffer solution, where phosphate can functionalize as a proton shuttle.599 The so called “Co-Pi” is an efficient inorganic WOC based on cobalt oxide. Although the “Co-Pi” is highly active under neutral conditions, even with a considerably higher loading amount, the catalytic current density of the Co-Pi electrode is also of the order of several mA cm−2.623
Another methodology to solve the problem of proton transfer is the PEM electrolyzer. For the PEM electrolyzer, an electrolyte is avoided. With this technology, current density between 0.6 and 2 A cm−2 can be reached. However, because of its complicated assembly process, the performance of the molecular-catalyst-based electrode has not been widely reported. The only available example was provided by Millet and co-workers demonstrating the incorporation of the Ru(tpy)(bpy) type WOC into a PEM cell.624 A Ru(tpy)(bpy)-type WOC is far less efficient compared to a Ru-bda-type WOC. However, when it is incorporated into a PEM cell, a current density of 1 A cm−2 has been reached at 1.9 V, although this performance is not as good as that exhibited by the IrO2 catalyst under the same testing conditions. It should be noted that the intrinsic activity of the Ru(tpy)(bpy)-type WOC is far lower than that of most of the efficient molecular WOCs. This report proves that high catalytic current can be anticipated by the molecular-catalyst-modified electrode.
Apparently, the power of molecular catalysts for AP is significantly underestimated because of the non-standard comparison. The current density normalized to the geometric area of an electrode is closely dependent on the loading amount of catalysts, the testing systems, and the actual electrochemically active surface area of the substrate electrode. For example, the actual surface area of the Ni foam electrode substrate is far higher than the value of its geometric area measured by a ruler. Therefore, it is not a valid parameter to indicate the intrinsic activity of a given electrocatalyst. Finally, the performance of an electrode is dominated by the intrinsic activity of the modified catalyst. Moreover, the application of an efficient molecular catalyst in an appropriate manner by careful design of the modification methods and the structure of the electrolyzer is also a great challenge.
6.3 The stability of molecular catalyst engineered devices
Low long-term-stability is one major limitation of AP devices based on molecular-catalyst-engineered electrodes. Many efficient molecular catalysts, including water oxidation catalysts, proton reduction catalysts, and CO2 reduction catalysts, have been clearly reported to exhibit much lower overpotentials, extremely higher TOFs and TONs as compared to the conventional inorganic-material-based catalysts, such as metal alloys, oxides, and hydroxides.4,43,55,313 In spite of this, there arises a question of why the stabilities of the electrodes engineered from these much more efficient molecular catalysts are not comparable with the electrodes generated from inorganic-material-based catalysts. To address this question, we propose here several reasons and possible solutions.First, the stability of molecule-integrated electrodes should be distinguished from that of molecular catalysts. A molecular catalyst with a high TOF and TON certainly is a stable catalyst; however, it is very normal that the functional electrode fabricated from the molecular catalyst has very poor stability. Besides the intrinsic stability of the modified catalyst, many factors can impart low stability to an electrode, e.g., detachment of an electrode film from the substrate, desorption of molecules, breaking of the linking bridge groups, and alteration of the catalytic mechanism on an electrode surface.
Second, catalytic kinetics, conditions, and local environments of molecular catalysts operating on an electrode surface are significantly different from those in homogeneous solutions. The performance of an electrode is dominated not only by the intrinsic activity of the molecular catalyst but also by the electron transfer rate between an active site and a supporting electrode, the reactant substrate transformation and binding to an active site, proton transfer between a catalyst and a solution phase, local pH change, and product removal from the electrode. If any of these steps is inefficient, the performance of the modified electrode will be dramatically limited, and the decomposition of catalysts will occur because of the accumulation of intermediates or the changes in local conditions. These types of decomposition occur in laboratory experiments, but they may be effectively prevented in practical industrial tests by the use of well-designed cells and technologies.
Third, the comparison of the stabilities of molecular-catalyst-engineered electrodes with those of inorganic-material-based electrodes is unequal if the loading amounts of catalysts are considered. The loading amounts of a molecular catalyst on an electrode are of the order of nmol cm−2,166,298,592 in contrast, it is usually more than an order of μmol cm−2 for an inorganic-material-based catalyst.621,625,626 It is noticed that the catalytic current is dramatically influenced by the loading amount of catalysts; therefore there is a factor called mass activity (A g−1). Indeed, the stability of an electrode can also be dependent on the loading amounts of catalysts. The decomposition of 1 nmol catalyst is definitely much faster than that of 1 μmol catalyst. Therefore, if the loading amounts of catalysts are also considered and discussed during the stability comparison, the stability of molecular-catalyst-based electrodes may not be a real drawback and has a high possibility to be remarkably upgraded in future research.
In the present context, it is obvious that the power of molecular catalysts for AP has been under estimated, and the molecular catalyst is very promising for the realization of applicable AP devices. Because the mechanistic study of a molecular catalyst is relatively easier than material-based catalysis, it is more possible to protect the catalyst via blocking the decomposition pathways. Under suitable testing conditions, molecular catalysts will display not only superior intrinsic activity but also high working catalytic current density and stability.
7. Concluding remarks and outlook
Promising catalysts with high intrinsic activities are still the basis for producing chemical fuels from solar energy via AP. The development of molecular catalysts with plenty of specific advantages, as summarized above in this review, is the most effective way to understand catalytic mechanisms in detail and to obtain high intrinsic catalytic activities. The field of molecular AP, including the development of molecular catalysts (e.g., WOCs, HECs, and CO2RCs) and the assembly of molecular-catalyst-based AP devices, has thrived during the past 15 years. In this review, the history and the present developing state of AP based on molecules have been summarized to show the advantages and challenges of molecular catalysts, as well as the future of molecular-catalyst-based AP for renewable energy conversion.7.1 Present achievements in molecular-catalyst-based AP
In recent decades, abundant molecular catalysts for the reactions involved in AP, i.e., water oxidation, hydrogen evolution, and CO2 reduction, have been designed and investigated, and several successful catalysts with outstanding catalytic performances have been obtained. For the WOCs, the Ru-bda series can reach a catalytic TOF of 1000 s−1 and a TON of 100000.80,142,143 WOCs based on iron, which is the most cheap and abundant metal, can also achieve a considerable catalytic performance, with TOFs of 2.2 s−1 and TONs of 1000.278 The structures of NiFe- and FeFe-hydrogenases have been successfully mimicked.39,330,332 Although most of the mimics showed low activity and high overpotentials for hydrogen evolution, detailed studies of these molecular models have provided useful information for understanding the mechanism of hydrogen evolution via metal catalytic sites. Fortunately, several noble-metal-free HECs with excellent catalytic properties have been explored. Cobaltoxime HECs have overpotentials less than 100 mV towards hydrogen evolution.325 With a high overpotential, Ni-diphosphine HECs can display catalytic TOFs over 100000 s−1.312 Fe porphyrins have been, so far, the most efficient molecular catalysts for the reduction of CO2 into CO, showing high catalytic rate, high selectivity, and high robustness. With an overpotential of only 220 mV, Fe porphyrin CO2RCs can convert CO2 into CO at a TOF of 106 s−1 for 80 h without obvious degradation.432 On the other hand, studies to understand the catalytic mechanisms of these catalysts have also been fruitful. A toolbox is now available to allow rational design of more promising molecular catalysts for AP.
For an AP device based on molecular catalysis, a high intrinsic activity of the molecular catalyst, especially low overpotential, is an essential basis for obtaining high efficiency. Facilitated by the presence of plenty of efficient molecular catalysts, many design concepts for AP devices with molecular catalysts have been proven in the last five years, including DSPEC cells with photoanodes modified by molecules, DSPEC cells with photocathodes modified by molecules, DSPEC tandem cells based on molecule-modified photoelectrodes, molecular-catalyst–VLA-SC hybrid devices, and molecular-catalyst engineered materials as electrodes for PVE devices. The design and performance of AP devices with molecular catalysts have been much improved. For example, the successful development of the Ru-bda WOC with an extremely low potential allowed DSPEC cells based on molecular-WOC-modified photoanodes to flourish. The photocurrents generated by molecular DSPEC cells have been improved by about two orders of magnitude to 1.7 mA cm−2.163 With the modification of cobaltoxime HECs, a composite p-GaInP2 photocathode mediates H2 production with a current density of 11 mA cm−2, which is close to the light-limited current of 12.5 mA cm−2.548 Several successful molecular-catalyst engineered electrode materials are readily available with very low overpotentials and good stabilities for PVE devices, for example, Ru-bda WOC functionalized MWCNTs,166 cobaltoxime HECs/Ni-diphosphine functionalized MWCNTs,579,581,586 and Fe-porphyrin CO2RC constructed MOF materials.435,600 In addition, the strategies advanced for the immobilization of molecular catalysts on electrodes, the mechanistic understanding of catalysis on the surface of electrodes, and the determination of the kinetics of electron transfer on photoelectrodes have also been gradually developed. This is extremely valuable for further development of AP devices based on molecular catalysts toward practical applications.
7.2 Challenges for the future of molecular-catalyst-based AP
Although the concept of AP was first proposed in 1912 by Giacomo Ciamician,627 investigations into AP based on molecular catalysis were triggered much later, and intensive interest has only been aroused in recent 20 years. Moreover, most of the efforts of the community during this recent period have contributed to research for the development of efficient molecular catalysts. Work on assembling AP devices with molecular catalysts has been widely conducted in the last five to ten years. Most of these studies are only at the level of proof of concept. Because of the complexity of complete molecular-catalyst-based AP devices, the performance can be dominated by many different factors, as discussed in previous sections. Indeed, so far, there has not been even a set of standard cell designs or operating conditions for AP devices to be based on, which in turn is adverse to the development of molecular AP devices.Apparently, the present research is largely insufficient to realize a practically applicable device because an ideal AP device for solar fuels is highly challenging involving not only the fundamentals of light absorption, charge transfer, and difficult catalysis but also engineering problems, such as device design and mass transfer. It is entirely understandable that, although some encouraging progress has been made in the past ten years in the development of molecular AP devices, none of these devices really satisfy the requirements for practical application or solve the issues of low stabilities of AP devices based on molecular catalysts. Therefore, it is too early to judge that AP devices based on molecular catalysts are not promising. Considering the prominent advantages of molecular catalysts, the construction of practically applicable AP devices based on molecular catalysts is still highly promising once the challenges are overcome. The development of molecular catalysts may still be the most effective way to solve the catalysis issues of AP.
In light of the above analysis and the present state of the field, the following challenges need to be addressed for the future of developing AP devices based on molecular catalysts:
(1) To develop earth-abundant metal based efficient catalysts working under aqueous conditions, especially under acidic conditions. WOCs with low overpotential and high TOF and TON are available. However, they are based on the noble metal ruthenium. Earth-abundant metal based WOCs are urgently needed now. For example, iron-based WOCs are promising candidates. To face the extremely low local pH problem, the development of WOCs that are efficient and stable under acidic conditions is necessary. For HECs, earth-abundant metal based catalysts with low overpotential and high TOF have been obtained, but the big issue is that their best performances are highly dependent on organic solvent systems. The same problem also exists for the development of CO2RCs. Moreover, the design of CO2RCs to break through the limit of two-electron reduction to produce liquid fuels is another challenge.
(2) To enhance the development of molecular electrocatalysts for water oxidation. Over the last 30 years, the development of molecular catalyst has mainly been carried out under homogeneous catalytic conditions with sacrificial reagents, such as Ce4+ and Ru3+ complexes, sodium peroxodisulfate, oxone, or sodium periodate.628 However, WOCs will finally be immobilized on electrodes for electrochemical water oxidation. Considering this point, the development of molecular electrocatalysts is closer to the aim of using molecular WOCs for AP devices. On the other hand, the investigation of catalysts is limited to the driving force of the sacrificial reagent and is also influenced by the instability of the sacrificial reagent, the involvement of active oxygen atom in the sacrificial reagent, and the special pH requirements of the sacrificial reagent. Harsh oxidative conditions may also lead to decomposition of the targeted catalysts. In contrast to the investigations with sacrificial reagents, the development of molecular electrocatalysts is much easier, milder and more adjustable.629,630
(3) To expand light absorbers, explore new transparent p-type semiconductors and upgrade the anchoring groups of functional molecules for PEC cells. In previous reports, Ru-based dyes have been the most used light absorber to build molecular-catalyst-modified PEC cells. Low stability, moderate light-absorption ability, and high cost mean they are not a good choice for practical applications. Inorganic semiconductors are a good replacement. The development of molecular catalyst-VLA-SC hybrid devices should be continued. With plenty of advantages, pure organic dyes can be more widely applied to PEC cells. In the reported PEC cells, the common anchoring groups of molecules are carboxylic or phosphate groups. However, in contrast to their utilization in dye-sensitized solar cells, these anchoring groups are not perfectly suited for use in PEC cells because the adsorption through carboxylic or phosphate groups cannot tolerate the aqueous electrolyte environment for long periods. Stronger binding groups should be designed and investigated for long-term stable PEC cells. Hydroxamate, acetylacetonate and 2,6-pyridinedicarboxylate groups are possible anchoring groups for stable PEC cells.
(4) To heterogenize molecular catalysts into materials. Heterogenization of molecular catalysts is an essential stage to bridge the gap between molecular catalysts and applicable AP devices. It could well remove the disadvantages of molecular catalysts, e.g., low loading amounts and low stability, by merging homogeneous and heterogeneous catalysis. PVE devices can indirectly convert solar energy into fuels. General investigation into PVE devices began much earlier than the study of PEC cells, and the development of PVE devices is now closer to practical application. Alkaline water electrolysis has been a mature industrial technology for decades. Moreover, plenty of inorganic materials have been reported in the last five years as efficient catalysts for water splitting under alkaline conditions. On the other hand, the practical utilization of PEM electrolyzers, which are ideal for domestic applications, will be limited because of their dependency on noble-metal-based catalysts. Molecular chemistry offers new possibilities for this technology. For PEM water electrolyzers, inexpensive materials that can work in acidic environments are urgently required. Developing molecular-catalyst engineered materials will facilitate the advance of PEM technology. PEM technology can probably be further applied to convert CO2 and N2 into chemicals.
(5) To reveal the reasons for deactivation of molecular catalyst based AP devices, including the reasons for the decomposition of modified catalysts. The performance of a complete molecular-catalyst-based AP device is affected by many factors. Relative to homogeneous catalysis, more complicated reasons can lead to the decomposition of catalysts modified on electrodes. A deep understanding of the reasons for the deactivation of molecular-catalyst-based AP devices is essential to improve the stability and activity of AP devices for practical applications.
(6) To set the standards for comprehensive evaluation of molecular catalysts, molecular-catalyst engineered materials and molecular-catalyst-based devices. The performance of a catalyst can be completely different under different test systems, such as chemical catalysis, photochemical catalysis, electrochemical catalysis, or photoelectrochemical catalysis. For example, the TOF of a molecular catalyst evaluated by homogeneous electrochemical catalysis is normally several orders of magnitude higher than that obtained from homogeneous chemical catalysis. Comparisons should be made under the same test system with similar detailed parameters, such as oxidant/reductant, solvent, pH value and light intensity. The evaluation of the performance of electrodes needs to be conducted with comparable conductive substrates. The geometric area, electrochemically active surface area, and loading amount of catalysts must also be comparable and clearly stated; otherwise, any discussions based on the obtained current densities, e.g., 10 mA cm−2@η mV, are meaningless.
It is certain that AP based on molecular catalysts is promising. Much has been attained, but more challenges remain.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
We acknowledge financial support for this work from the Swedish Research Council (2017-00935), Swedish Energy Agency, Knut and Alice Wallenberg Foundation, and National Basic Research Program of China (973 program, 2014CB239402).References
- R. L. Purchase and H. J. de Groot, Interface Focus, 2015, 5, 20150014 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed.
- H. Dau, E. Fujita and L. Sun, ChemSusChem, 2017, 10, 4228–4235 CrossRef CAS PubMed.
- M. D. Kärkas, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4493 CrossRef CAS PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- S. A. Crowe, L. N. Dossing, N. J. Beukes, M. Bau, S. J. Kruger, R. Frei and D. E. Canfield, Nature, 2013, 501, 535–538 CrossRef CAS PubMed.
- A. Amunts, O. Drory and N. Nelson, Nature, 2007, 447, 58–63 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- W. M. Singh, D. Pegram, H. Duan, D. Kalita, P. Simone, G. L. Emmert and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 1653–1656 CrossRef CAS PubMed.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CAS.
- T. Shimada, A. Kumagai, S. Funyu, S. Takagi, D. Masui, Y. Nabetani, H. Tachibana, D. A. Tryk and H. Inoue, Faraday Discuss., 2012, 155, 145–163 RSC.
- S. Fukuzumi, Y. M. Lee and W. Nam, Chem. – Eur. J., 2018, 24, 5016–5031 CrossRef CAS PubMed.
- K. Li and Y. Sun, Chem. – Eur. J., 2018, 24, 18258–18270 CrossRef CAS PubMed.
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- X. Ding, L. Zhang, Y. Wang, A. Liu and Y. Gao, Coord. Chem. Rev., 2018, 357, 130–143 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- J. Seo, H. Nishiyama, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2018, 57, 8396–8415 CrossRef CAS PubMed.
- K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- S. S. Kalanur, L. T. Duy and H. Seo, Top. Catal., 2018, 61, 1043–1076 CrossRef CAS.
- Y. Park, K. J. McDonald and K. S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- E. A. Gibson, Chem. Soc. Rev., 2017, 46, 6194–6209 RSC.
- M. Wang, Y. Yang, J. Shen, J. Jiang and L. Sun, Sustainable Energy Fuels, 2017, 1, 1641–1663 RSC.
- T. Nann, S. K. Ibrahim, P. M. Woi, S. Xu, J. Ziegler and C. J. Pickett, Angew. Chem., Int. Ed., 2010, 49, 1574–1577 CrossRef CAS PubMed.
- R. Wick and S. D. Tilley, J. Phys. Chem. C, 2015, 119, 26243–26257 CrossRef CAS.
- B. Zhang, Q. Daniel, M. Cheng, L. Fan and L. Sun, Faraday Discuss., 2017, 198, 169–179 RSC.
- C. Wei and Z. J. Xu, Small Methods, 2018, 2, 1800168 CrossRef.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- R. Argazzi, N. Y. Murakami Iha, H. Zabri, F. Odobel and C. A. Bignozzi, Coord. Chem. Rev., 2004, 248, 1299–1316 CrossRef CAS.
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC.
- T. Yao, X. An, H. Han, J. Q. Chen and C. Li, Adv. Energy Mater., 2018, 8, 1800210 CrossRef.
- H. L. Wu, X. B. Li, C. H. Tung and L. Z. Wu, Adv. Sci., 2018, 5, 1700684 CrossRef PubMed.
- Y. Xu, X. Wang, W. L. Zhang, F. Lv and S. Guo, Chem. Soc. Rev., 2018, 47, 586–625 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 9476–9477 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codola, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- A. Polman, M. Knight, E. C. Garnett, B. Ehrler and W. C. Sinke, Science, 2016, 352, aad4424 CrossRef PubMed.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2016, 7, 70–88 CrossRef.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- B. Reuillard, K. H. Ly, T. E. Rosser, M. F. Kuehnel, I. Zebger and E. Reisner, J. Am. Chem. Soc., 2017, 139, 14425–14435 CrossRef CAS PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed.
- Y. Zhao, X. Yan, K. R. Yang, S. Cao, Q. Dong, J. E. Thorne, K. L. Materna, S. Zhu, X. Pan, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, ACS Cent. Sci., 2018, 4, 1166–1172 CrossRef CAS PubMed.
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Yei, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pang, J. Guo, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902–2907 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- A. Dhingra, A. R. Portis and H. Daniell, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6315–6320 CrossRef CAS PubMed.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS PubMed.
- M. M. Najafpour, G. Renger, M. Holynska, A. N. Moghaddam, E. M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J. R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- J. R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- T. Lohmiller, V. Krewald, A. Sedoud, A. W. Rutherford, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, J. Am. Chem. Soc., 2017, 139, 14412–14424 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- M. Perez-Navarro, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Curr. Opin. Chem. Biol., 2016, 31, 113–119 CrossRef CAS PubMed.
- D. J. Vinyard, S. Khan and G. W. Brudvig, Faraday Discuss., 2015, 185, 37–50 RSC.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- J. Barber, Nat. Plants, 2017, 3, 17041 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Dalton Trans., 2018, 47, 14381–14387 RSC.
- B. Zhang, Q. Daniel, L. Fan, T. Liu, Q. Meng and L. Sun, iScience, 2018, 4, 144–152 CrossRef CAS PubMed.
- B. Zhang, H. Chen, Q. Daniel, B. Philippe, F. Yu, M. Valvo, Y. Li, R. B. Ambre, P. Zhang, F. Li, H. Rensmo and L. Sun, ACS Catal., 2017, 7, 6311–6322 CrossRef CAS.
- P. Garrido-Barros, C. Gimbert-Surinach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088–6098 RSC.
- J. Hessels, R. J. Detz, M. T. M. Koper and J. N. H. Reek, Chem. – Eur. J., 2017, 23, 16413–16418 CrossRef CAS PubMed.
- S. Romanin, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef PubMed.
- L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296–3313 RSC.
- T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148–6169 RSC.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. M. Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- J. Nyhlen, L. Duan, B. Akermark, L. Sun and T. Privalov, Angew. Chem., Int. Ed., 2010, 49, 1773–1777 CrossRef CAS PubMed.
- M. T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- K. Nagoshi, S. Yamashita, M. Yagi and M. Kaneko, J. Mol. Catal. A: Chem., 1999, 144, 71–76 CrossRef CAS.
- J. P. Collin and J. P. Sauvage, Inorg. Chem., 1986, 25, 135–141 CrossRef CAS.
- L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS PubMed.
- D. Moonshiram, I. Alperovich, J. J. Concepcion, T. J. Meyer and Y. Pushkar, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3765–3770 CrossRef CAS PubMed.
- J. A. Gilbert, D. S. Eggleston, W. R. Murphy Jr., D. A. Geselowitz, S. W. Gersten, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 3855–3864 CrossRef CAS.
- J. A. Stull, T. A. Stich, J. K. Hurst and R. D. Britt, Inorg. Chem., 2013, 52, 4578–4586 CrossRef CAS PubMed.
- D. Moonshiram, J. W. Jurss, J. J. Concepcion, T. Zakharova, I. Alperovich, T. J. Meyer and Y. Pushkar, J. Am. Chem. Soc., 2012, 134, 4625–4636 CrossRef CAS PubMed.
- H. Yamada and J. K. Hurst, J. Am. Chem. Soc., 2000, 122, 5303–5311 CrossRef CAS.
- H. Yamada, W. F. Siems, T. Koike and J. K. Hurst, J. Am. Chem. Soc., 2004, 126, 9786–9795 CrossRef CAS PubMed.
- J. L. Cape and J. K. Hurst, J. Am. Chem. Soc., 2008, 130, 827–829 CrossRef CAS PubMed.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727–1752 CrossRef CAS PubMed.
- R. A. Binstead, C. W. Chronister, J. Ni, C. M. Hartshorn and T. J. Meyer, J. Am. Chem. Soc., 2000, 122, 8464–8473 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17632–17635 CrossRef CAS PubMed.
- X. Yang and M.-H. Baik, J. Am. Chem. Soc., 2006, 128, 7476–7485 CrossRef CAS PubMed.
- H. Seo, K. H. Cho, H. Ha, S. Park, J. S. Hong, K. Jin and K. T. Nam, J. Korean Ceram. Soc., 2017, 54, 1–8 CrossRef CAS.
- B. M. Hunter, H. B. Gray and A. M. Muller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- B. A. Moyer and T. J. Meyer, Inorg. Chem., 1981, 20, 436–444 CrossRef CAS.
- B. A. Moyer and T. J. Meyer, J. Am. Chem. Soc., 1978, 100, 3601–3603 CrossRef CAS.
- L. Tong and R. P. Thummel, Chem. Sci., 2016, 7, 6591–6603 RSC.
- C. J. Gagliardi, A. K. Vannucci, J. J. Concepcion, Z. Chen and T. J. Meyer, Energy Environ. Sci., 2012, 5, 7704–7717 RSC.
- C. Sens, I. Romero, M. Rodriguez, A. Llobet, T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798–7799 CrossRef CAS PubMed.
- S. Romain, F. Bozoglian, X. Sala and A. Llobet, J. Am. Chem. Soc., 2009, 131, 2768–2769 CrossRef CAS.
- X. Sala, S. Maji, R. Bofill, J. García-Antón, L. Escriche and A. Llobet, Acc. Chem. Res., 2014, 47, 504–516 CrossRef CAS.
- C. Sens, M. Rodríguez, I. Romero and A. Llobet, Inorg. Chem., 2003, 42, 2040–2048 CrossRef CAS.
- L. Francas, X. Sala, E. Escudero-Adan, J. Benet-Buchholz, L. Escriche and A. Llobet, Inorg. Chem., 2011, 50, 2771–2781 CrossRef CAS PubMed.
- J. Mola, C. Dinoi, X. Sala, M. Rodriguez, I. Romero, T. Parella, X. Fontrodona and A. Llobet, Dalton Trans., 2011, 40, 3640–3646 RSC.
- C. Sens, M. Rodríguez, I. Romero and A. Llobet, Inorg. Chem., 2003, 42, 8385–8394 CrossRef CAS PubMed.
- J. Aguiló, L. Francàs, H. J. Liu, R. Bofill, J. García-Antón, J. Benet-Buchholz, A. Llobet, L. Escriche and X. Sala, Catal. Sci. Technol., 2014, 4, 190–199 RSC.
- L. Francas, X. Sala, J. Benet-Buchholz, L. Escriche and A. Llobet, ChemSusChem, 2009, 2, 321–329 CrossRef CAS PubMed.
- Z. Deng, H.-W. Tseng, R. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835–1848 CrossRef CAS.
- R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS.
- T. Wada, K. Tsuge and K. Tanaka, Angew. Chem., Int. Ed., 2000, 39, 1479–1482 CrossRef CAS PubMed.
- T. A. Betley, Q. Wu, T. V. Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849–1861 CrossRef CAS PubMed.
- L. Chen, M. Wang, K. Han, P. Zhang, F. Gloaguen and L. Sun, Energy Environ. Sci., 2014, 7, 329–334 RSC.
- T. Wada, J. T. Muckerman, E. Fujitab and K. Tanaka, Dalton Trans., 2011, 40, 2225–2233 RSC.
- T. Wada, H. Ohtsu and K. Tanaka, Chem. – Eur. J., 2012, 18, 2374–2381 CrossRef CAS PubMed.
- H. Y. Du, S. C. Chen, X. J. Su, L. Jiao and M. T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS PubMed.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto and C. P. Berlinguette, Eur. J. Inorg. Chem., 2010, 3135–3142 CrossRef CAS.
- J. J. Concepcion, M.-K. Tsai, J. T. Muckerman and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 1545–1557 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS PubMed.
- H.-W. Tseng, R. Zong, J. T. Muckerman and R. Thummel, Inorg. Chem., 2008, 47, 11763–11773 CrossRef CAS.
- N. Kaveevivitchai, R. Zong, H. W. Tseng, R. Chitta and R. P. Thummel, Inorg. Chem., 2012, 51, 2930–2939 CrossRef CAS PubMed.
- Y. M. Badiei, D. E. Polyansky, J. T. Muckerman, D. J. Szalda, R. Haberdar, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2013, 52, 8845–8850 CrossRef CAS PubMed.
- D. E. Polyansky, J. T. Muckerman, J. Rochford, R. Zong, R. P. Thummel and E. Fujita, J. Am. Chem. Soc., 2011, 133, 14649–14665 CrossRef CAS PubMed.
- S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182–183 CrossRef CAS.
- A. Kimoto, K. Yamauchi, M. Yoshida, S. Masaoka and K. Sakai, Chem. Commun., 2012, 48, 239–241 RSC.
- J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277–1279 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2011, 50, 3662–3672 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson, B. D. Koivisto, H. D. Osthoff and C. P. Berlinguette, J. Am. Chem. Soc., 2010, 132, 16094–16106 CrossRef CAS PubMed.
- M. Yagi, S. Tajima, M. Komia and H. Yamazaki, Dalton Trans., 2011, 40, 3802–3804 RSC.
- Y. Tamaki, A. K. Vannucci, C. J. Dares, R. A. Binstead and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 6854–6857 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion, X. Hu, W. Yang, P. G. Hoertz and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7225–7229 CrossRef CAS PubMed.
- Y. Xu, T. Åkermark, V. Gyollai, D. Zou, L. Eriksson, L. Duan, R. Zhang, B. Åkermark and L. Sun, Inorg. Chem., 2009, 48, 2717–2719 CrossRef CAS PubMed.
- Y. Xu, L. Duan, L. Tong, B. Akermark and L. Sun, Chem. Commun., 2010, 46, 6506–6508 RSC.
- Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 8934–8937 CrossRef CAS PubMed.
- L. Wang, L. Duan, Y. Wang, M. S. Ahlquist and L. Sun, Chem. Commun., 2014, 50, 12947–12950 RSC.
- L. Duan, C. M. Araujob, M. S. G. Ahlquist and L. Sun, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15584–15588 CrossRef CAS PubMed.
- L. Wang, L. Duan, B. Stewart, M. Pu, J. Liu, T. Privalov and L. Sun, J. Am. Chem. Soc., 2012, 134, 18868–18880 CrossRef CAS PubMed.
- L. Duan, Y. Xu, P. Zhang, M. Wang and L. Sun, Inorg. Chem., 2010, 49, 209–215 CrossRef CAS PubMed.
- Y. Jiang, F. Li, F. Huang, B. Zhang and L. Sun, Chin. J. Catal., 2013, 34, 1489–1495 CrossRef CAS.
- Y. Sato, S.-y. Takizawa and S. Murata, Eur. J. Inorg. Chem., 2015, 5495–5502 CrossRef CAS.
- L. Duan, L. Wang, A. K. Inge, A. Fischer, X. Zou and L. Sun, Inorg. Chem., 2013, 52, 7844–7852 CrossRef CAS PubMed.
- Y. Xie, D. W. Shaffer and J. J. Concepcion, Inorg. Chem., 2018, 57, 10533–10542 CrossRef CAS PubMed.
- Q. Daniel, P. Huang, T. Fan, Y. Wang, L. Duan, L. Wang, F. Li, Z. Rinkevicius, F. Mamedov, M. S. G. Ahlquist, S. Styring and L. Sun, Coord. Chem. Rev., 2017, 346, 206–215 CrossRef CAS.
- D. Lebedev, Y. Pineda-Galvan, Y. Tokimaru, A. Fedorov, N. Kaeffer, C. Coperet and Y. Pushkar, J. Am. Chem. Soc., 2018, 140, 451–458 CrossRef CAS PubMed.
- N. Song, J. J. Concepcion, R. A. Binstead, J. A. Rudd, A. K. Vannucci, C. J. Dares, M. K. Coggins and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4935–4940 CrossRef CAS PubMed.
- S. Zhan, D. Martensson, M. Purg, S. C. L. Kamerlin and M. S. G. Ahlquist, Angew. Chem., Int. Ed., 2017, 56, 6962–6965 CrossRef CAS PubMed.
- T. Fan, S. Zhan and M. S. G. Ahlquist, ACS Catal., 2016, 6, 8308–8312 CrossRef CAS.
- B. Zhang, F. Li, R. Zhang, C. Ma, L. Chen and L. Sun, Chem. Commun., 2016, 52, 8619–8622 RSC.
- R. Matheu, A. Ghaderian, L. Francàs, P. Chernev, M. Z. Ertem, J. Benet-Buchholz, V. S. Batista, M. Haumann, C. Gimbert-Suriñach, X. Sala and A. Llobet, Chem. – Eur. J., 2018, 24, 12838–12847 CrossRef CAS PubMed.
- M. Schulze, V. Kunz, P. D. Frischmann and F. Wurthner, Nat. Chem., 2016, 8, 576–583 CrossRef CAS PubMed.
- Y. Xie, D. W. Shaffer, A. Lewandowska-Andralojc, D. J. Szalda and J. J. Concepcion, Angew. Chem., Int. Ed., 2016, 55, 8067–8071 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, J. Am. Chem. Soc., 2017, 139, 15347–15355 CrossRef CAS PubMed.
- D. Wang, S. L. Marquard, L. Troian-Gautier, M. V. Sheridan, B. D. Sherman, Y. Wang, M. S. Eberhart, B. H. Farnum, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 719–726 CrossRef CAS PubMed.
- Y. K. Eom, L. Nhon, G. Leem, B. D. Sherman, D. Wang, L. Troian-Gautier, S. Kim, J. Kim, T. J. Meyer, J. R. Reynolds and K. S. Schanze, ACS Energy Lett., 2018, 3, 2114–2119 CrossRef CAS.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- A. Bhunia, B. A. Johnson, J. Czapla-Masztafiak, J. Sa and S. Ott, Chem. Commun., 2018, 54, 7770–7773 RSC.
- D. Wang, M. S. Eberhart, M. V. Sheridan, K. Hu, B. D. Sherman, A. Nayak, Y. Wang, S. L. Marquard, C. J. Dares and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8523–8528 CrossRef CAS PubMed.
- F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 12276–12279 CrossRef CAS PubMed.
- B. Yang, X. Jiang, Q. Guo, T. Lei, L. P. Zhang, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 6229–6234 CrossRef CAS PubMed.
- B. Li, F. Li, S. Bai, Z. Wang, L. Sun, Q. Yang and C. Li, Energy Environ. Sci., 2012, 5, 8229–8233 RSC.
- F. Yu, D. Poole, 3rd, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanovic-Burmazovic and J. N. H. Reek, Angew. Chem., Int. Ed., 2018, 57, 11247–11251 CrossRef CAS PubMed.
- C. J. Richmond, R. Matheu, A. Poater, L. Falivene, J. Benet-Buchholz, X. Sala, L. Cavallo and A. Llobet, Chem. – Eur. J., 2014, 20, 17282–17286 CrossRef CAS PubMed.
- Y. Jiang, F. Li, B. Zhang, X. Li, X. Wang, F. Huang and L. Sun, Angew. Chem., Int. Ed., 2013, 52, 3398–3401 CrossRef CAS PubMed.
- L. L. Zhang, Y. Gao, Z. Liu, X. Ding, Z. Yu and L. C. Sun, Dalton Trans., 2016, 45, 3814–3819 RSC.
- S. Zhan, R. Zou and M. S. G. Ahlquist, ACS Catal., 2018, 8, 8642–8648 CrossRef CAS.
- T. Fan, L. Duan, P. Huang, H. Chen, Q. Daniel, M. S. G. Ahlquist and L. Sun, ACS Catal., 2017, 7, 2956–2966 CrossRef CAS.
- L. Duan, Y. Xu, M. Gorlov, L. Tong, S. Andersson and L. Sun, Chem. – Eur. J., 2010, 16, 4659–4668 CrossRef CAS PubMed.
- Q. Daniel, L. Duan, B. J. J. Timmer, H. Chen, X. Luo, R. Ambre, Y. Wang, B. Zhang, P. Zhang, L. Wang, F. Li, J. Sun, M. Ahlquist and L. Sun, ACS Catal., 2018, 8, 4375–4382 CrossRef CAS.
- R. Matheu, M. Z. Ertem, M. Pipelier, J. Lebreton, D. Dubreuil, J. Benet-Buchholz, X. Sala, A. Tessier and A. Llobet, ACS Catal., 2018, 8, 2039–2048 CrossRef CAS.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525–6532 CrossRef CAS.
- L. Tong, L. Duan, Y. Xu, T. Privalov and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 445–449 CrossRef CAS PubMed.
- V. Kunz, M. Schulze, D. Schmidt and F. Würthner, ACS Energy Lett., 2017, 2, 288–293 CrossRef CAS.
- V. Kunz, J. O. Lindner, M. Schulze, M. I. S. Röhr, D. Schmidt, R. Mitrić and F. Würthner, Energy Environ. Sci., 2017, 10, 2137–2153 RSC.
- V. Kunz, D. Schmidt, M. I. S. Röhr, R. Mitrić and F. Würthner, Adv. Energy Mater., 2017, 7, 1602939 CrossRef.
- J. M. Thomsen, D. L. Huang, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2015, 44, 12452–12472 RSC.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210–217 CrossRef CAS PubMed.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730–8731 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS PubMed.
- T. P. Brewster, J. D. Blakemore, N. D. Schley, C. D. Incarvito, N. Hazari, G. W. Brudvig and R. H. Crabtree, Organometallics, 2011, 30, 965–973 CrossRef CAS.
- M. Navarro, C. A. Smith, M. Li, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2018, 24, 6386–6398 CrossRef CAS PubMed.
- R. Lalrempuia, N. D. McDaniel, H. Muller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765–9768 CrossRef CAS PubMed.
- A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218–9219 RSC.
- D. G. Hetterscheid and J. N. Reek, Chem. Commun., 2011, 47, 2712–2714 RSC.
- S. B. Sinha, D. Y. Shopov, L. S. Sharninghausen, C. J. Stein, B. Q. Mercado, D. Balcells, T. B. Pedersen, M. Reiher, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2017, 139, 9672–9683 CrossRef CAS PubMed.
- L. S. Sharninghausen, S. B. Sinha, D. Y. Shopov, B. Choi, B. Q. Mercado, X. Roy, D. Balcells, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2016, 138, 15917–15926 CrossRef CAS PubMed.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS.
- Y. Naruta, M. Sasayama and T. Sasaki, Angew. Chem., Int. Ed., 1994, 33, 1839–1841 CrossRef.
- Y. Shimazaki, T. Nagano, H. Takesue, B. H. Ye, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2004, 43, 98–100 CrossRef PubMed.
- J. Limburg, J. S. Vrettos, H. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423–430 CrossRef CAS PubMed.
- J. Limburg, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2761–2762 CrossRef CAS.
- C. Baffert, S. Romain, A. Richardot, J.-C. Leprêtre, B. Lefebvre, A. Deronzier and M.-N. Collomb, J. Am. Chem. Soc., 2005, 127, 13694–13704 CrossRef CAS PubMed.
- M. Hirahara, H. Yamazaki, S. Yamada, K. Matsubara, K. Saito, T. Yui and M. Yagi, Catal. Sci. Technol., 2013, 3, 1776–1781 RSC.
- M. Yagi and K. Narita, J. Am. Chem. Soc., 2004, 126, 8084–8085 CrossRef CAS PubMed.
- M. Yagi, M. Toda, S. Yamada and H. Yamazaki, Chem. Commun., 2010, 46, 8594–8596 RSC.
- G. Li, E. M. Sproviero, R. C. Snoeberger Iii, N. Iguchi, J. D. Blakemore, R. H. Crabtree, G. W. Brudvig and V. S. Batista, Energy Environ. Sci., 2009, 2, 230–238 RSC.
- G. Li, E. M. Sproviero, W. R. McNamara, R. C. Snoeberger III, R. H. Crabtree, G. W. Brudvig and V. S. Batista, J. Phys. Chem. B, 2010, 114, 14214–14222 CrossRef CAS PubMed.
- B. Nepal and S. Das, Angew. Chem., Int. Ed., 2013, 52, 7224–7227 CrossRef CAS PubMed.
- C. Baffert, J.-M. Latour, L. P. Nielsenb, M.-N. Collomb, K. H. Lund and N. Thorup, Dalton Trans., 2003, 1765–1772 RSC.
- Y. Gao, T. Åkermark, J. Liu, L. Sun and B. Åkermark, J. Am. Chem. Soc., 2009, 131, 8726–8727 CrossRef CAS PubMed.
- E. A. Karlsson, B. L. Lee, T. Akermark, E. V. Johnston, M. D. Karkas, J. Sun, O. Hansson, J. E. Backvall and B. Akermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- L. Ma, Q. Wang, W. L. Man, H. K. Kwong, C. C. Ko and T. C. Lau, Angew. Chem., Int. Ed., 2015, 54, 5246–5249 CrossRef CAS PubMed.
- M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed.
- C. Chen, Y. Li, G. Zhao, R. Yao and C. Zhang, ChemSusChem, 2017, 10, 4403–4408 CrossRef CAS PubMed.
- P. Huang, A. Magnuson, R. Lomoth, M. Abrahamsson, M. Tamm, L. Sun, B. van Rotterdam, J. Park, L. Hammarström, B. Åkermark and S. Styring, J. Inorg. Biochem., 2002, 91, 159–172 CrossRef CAS PubMed.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- L. Sun, H. Berglund, R. Davydov, T. Norrby, L. Hammarström, P. Korall, A. Börje, C. Philouze, K. Berg, A. Tran, M. Andersson, G. Stenhagen, J. Mårtensson, M. Almgren, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1997, 119, 6996–7004 CrossRef CAS.
- A. Johansson, M. Abrahamsson, A. Magnuson, P. Huang, J. Mårtensson, S. Styring, L. Hammarström, L. Sun and B. Åkermark, Inorg. Chem., 2003, 42, 7502–7511 CrossRef CAS PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- J. S. Kanady, P. H. Lin, K. M. Carsch, R. J. Nielsen, M. K. Takase, W. A. Goddard, 3rd and T. Agapie, J. Am. Chem. Soc., 2014, 136, 14373–14376 CrossRef CAS PubMed.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- G. Maayan, N. Gluz and G. Christou, Nat. Catal., 2017, 1, 48–54 CrossRef.
- M. Anbar and I. Pecht, J. Am. Chem. Soc., 1967, 89, 2553–2556 CrossRef CAS.
- D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, Chem. Commun., 2012, 48, 2107–2109 RSC.
- D. K. Dogutan, R. McGuire, Jr. and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- H. Lei, A. Han, F. Li, M. Zhang, Y. Han, P. Du, W. Lai and R. Cao, Phys. Chem. Chem. Phys., 2014, 16, 1883–1893 RSC.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606–7616 RSC.
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903–7907 RSC.
- H. Chen, Z. Sun, X. Liu, A. Han and P. Du, J. Phys. Chem. C, 2015, 119, 8998–9004 CrossRef CAS.
- E. Pizzolato, M. Natali, B. Posocco, A. Montellano Lopez, I. Bazzan, M. Di Valentin, P. Galloni, V. Conte, M. Bonchio, F. Scandola and A. Sartorel, Chem. Commun., 2013, 49, 9941–9943 RSC.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS PubMed.
- G. La Ganga, F. Puntoriero, S. Campagna, I. Bazzan, S. Berardi, M. Bonchio, A. Sartorel, M. Natali and F. Scandola, Faraday Discuss., 2012, 155, 177–190 RSC.
- S. Berardi, G. La Ganga, M. Natali, I. Bazzan, F. Puntoriero, A. Sartorel, F. Scandola, S. Campagna and M. Bonchio, J. Am. Chem. Soc., 2012, 134, 11104–11107 CrossRef CAS PubMed.
- M. D. Symes, D. A. Lutterman, T. S. Teets, B. L. Anderson, J. J. Breen and D. G. Nocera, ChemSusChem, 2013, 6, 65–69 CrossRef CAS PubMed.
- X. Zhou, F. Li, H. Li, B. Zhang, F. Yu and L. Sun, ChemSusChem, 2014, 7, 2453–2456 CrossRef CAS PubMed.
- B. Zhang, F. Li, F. Yu, X. Wang, X. Zhou, H. Li, Y. Jiang and L. Sun, ACS Catal., 2014, 4, 804–809 CrossRef CAS.
- Y. Wang, F. Li, H. Li, L. Bai and L. Sun, Chem. Commun., 2016, 52, 3050–3053 RSC.
- S. Ye, C. Ding, R. Chen, F. Fan, P. Fu, H. Yin, X. Wang, Z. Wang, P. Du and C. Li, J. Am. Chem. Soc., 2018, 140, 3250–3256 CrossRef CAS PubMed.
- Y. Wang, F. Li, X. Zhou, F. Yu, J. Du, L. Bai and L. Sun, Angew. Chem., Int. Ed., 2017, 56, 6911–6915 CrossRef CAS PubMed.
- P. F. Smith, L. Hunt, A. B. Laursen, V. Sagar, S. Kaushik, K. U. D. Calvinho, G. Marotta, E. Mosconi, F. De Angelis and G. C. Dismukes, J. Am. Chem. Soc., 2015, 137, 15460–15468 CrossRef CAS PubMed.
- A. I. Nguyen, M. S. Ziegler, P. Ona-Burgos, M. Sturzbecher-Hohne, W. Kim, D. E. Bellone and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 12865–12872 CrossRef CAS PubMed.
- C. N. Brodsky, R. G. Hadt, D. Hayes, B. J. Reinhart, N. Li, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3855–3860 CrossRef CAS PubMed.
- R. G. Hadt, D. Hayes, C. N. Brodsky, A. M. Ullman, D. M. Casa, M. H. Upton, D. G. Nocera and L. X. Chen, J. Am. Chem. Soc., 2016, 138, 11017–11030 CrossRef CAS PubMed.
- F. Evangelisti, R. More, F. Hodel, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2015, 137, 11076–11084 CrossRef CAS PubMed.
- M. Kondo and S. Masaoka, Chem. Lett., 2016, 45, 1220–1231 CrossRef CAS.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- T. Zhang, C. Wang, S. Liu, J. L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689–12698 CrossRef CAS PubMed.
- J. S. Pap, L. Szyrwiel, D. Sranko, Z. Kerner, B. Setner, Z. Szewczuk and W. Malinka, Chem. Commun., 2015, 51, 6322–6324 RSC.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384–3387 CrossRef CAS.
- B. Rudshteyn, K. J. Fisher, H. M. C. Lant, K. R. Yang, B. Q. Mercado, G. W. Brudvig, R. H. Crabtree and V. S. Batista, ACS Catal., 2018, 8, 7952–7960 CrossRef CAS.
- X. J. Su, M. Gao, L. Jiao, R. Z. Liao, P. E. Siegbahn, J. P. Cheng and M. T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- J.-W. Wang, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Coord. Chem. Rev., 2019, 378, 237–261 CrossRef CAS.
- M. Zhang, M. T. Zhang, C. Hou, Z. F. Ke and T. B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042–13048 CrossRef CAS PubMed.
- J.-W. Wang, C. Hou, H.-H. Huang, W.-J. Liu, Z.-F. Ke and T.-B. Lu, Catal. Sci. Technol., 2017, 7, 5585–5593 RSC.
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed.
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das, A. Thapper, J. Uhlig, P. Dinér and L. Sun, J. Catal., 2016, 335, 72–78 CrossRef CAS.
- J. Lin, P. Kang, X. Liang, B. Ma and Y. Ding, Electrochim. Acta, 2017, 258, 353–359 CrossRef CAS.
- G. Y. Luo, H. H. Huang, J. W. Wang and T. B. Lu, ChemSusChem, 2016, 9, 485–491 CrossRef CAS PubMed.
- D. Wang and C. O. Bruner, Inorg. Chem., 2017, 56, 13638–13641 CrossRef CAS PubMed.
- A. Singh, S. L. Y. Chang, R. K. Hocking, U. Bach and L. Spiccia, Energy Environ. Sci., 2013, 6, 579–586 RSC.
- A. Singh, S. L. Y. Chang, R. K. Hocking, U. Bach and L. Spiccia, Catal. Sci. Technol., 2013, 3, 1725–1732 RSC.
- D. Wang, G. Ghirlanda and J. P. Allen, J. Am. Chem. Soc., 2014, 136, 10198–10201 CrossRef CAS PubMed.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- M. Costas, K. Chen and L. Que Jr., Coord. Chem. Rev., 2000, 200–202, 517–544 CrossRef CAS.
- T. Liu, B. Zhang and L. Sun, Chem. – Asian J., 2019, 14, 31–43 CrossRef CAS PubMed.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS PubMed.
- E. L. Demeter, S. L. Hilburg, N. R. Washburn, T. J. Collins and J. R. Kitchin, J. Am. Chem. Soc., 2014, 136, 5603–5606 CrossRef CAS PubMed.
- R.-Z. Liao, X.-C. Li and P. E. M. Siegbahn, Eur. J. Inorg. Chem., 2014, 728–741 CrossRef CAS.
- M. Z. Ertem, L. Gagliardi and C. J. Cramer, Chem. Sci., 2012, 3, 1293–1299 RSC.
- G. Panchbhai, W. M. Singh, B. Das, R. T. Jane and A. Thapper, Eur. J. Inorg. Chem., 2016, 3262–3268 CrossRef CAS.
- W. A. Hoffert, M. T. Mock, A. M. Appel and J. Y. Yang, Eur. J. Inorg. Chem., 2013, 3846–3857 CrossRef CAS.
- Z. Codola, I. Garcia-Bosch, F. Acuna-Pares, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 8042–8047 CrossRef CAS PubMed.
- F. Acuna-Pares, M. Costas, J. M. Luis and J. Lloret-Fillol, Inorg. Chem., 2014, 53, 5474–5485 CrossRef CAS PubMed.
- F. Acuna-Pares, Z. Codola, M. Costas, J. M. Luis and J. Lloret-Fillol, Chem. – Eur. J., 2014, 20, 5696–5707 CrossRef CAS PubMed.
- Z. Codola, L. Gomez, S. T. Kleespies, L. Que, Jr., M. Costas and J. Lloret-Fillol, Nat. Commun., 2015, 6, 5865 CrossRef CAS PubMed.
- E. E. Kasapbasi and M. H. Whangbo, Inorg. Chem., 2012, 51, 10850–10855 CrossRef CAS PubMed.
- L. D. Wickramasinghe, R. Zhou, R. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260–13263 CrossRef CAS PubMed.
- M. M. Najafpour, A. N. Moghaddam, D. J. Sedigh and M. Hołyńska, Catal. Sci. Technol., 2014, 4, 30–33 RSC.
- A. R. Parent, T. Nakazono, S. Lin, S. Utsunomiya and K. Sakai, Dalton Trans., 2014, 43, 12501–12513 RSC.
- Y. Liu, R. Xiang, X. Du, Y. Ding and B. Ma, Chem. Commun., 2014, 50, 12779–12782 RSC.
- B. Zhang, F. Li, F. Yu, H. Cui, X. Zhou, H. Li, Y. Wang and L. Sun, Chem. – Asian J., 2014, 9, 1515–1518 CrossRef CAS PubMed.
- W. P. To, T. Wai-Shan Chow, C. W. Tse, X. Guan, J. S. Huang and C. M. Che, Chem. Sci., 2015, 6, 5891–5903 RSC.
- M. K. Coggins, M. T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- Y. V. Geletii, B. Botar, P. Kogerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- C. Besson, Z. Huang, Y. V. Geletii, S. Lense, K. I. Hardcastle, D. G. Musaev, T. Lian, A. Proust and C. L. Hill, Chem. Commun., 2010, 46, 2784–2786 RSC.
- M. Murakami, D. Hong, T. Suenobu, S. Yamaguchi, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11605–11613 CrossRef CAS PubMed.
- P.-E. Car, M. Guttentag, K. K. Baldridge, R. Alberto and G. R. Patzke, Green Chem., 2012, 14, 1680–1688 RSC.
- R. Cao, H. Ma, Y. V. Geletii, K. I. Hardcastle and C. L. Hill, Inorg. Chem., 2009, 48, 5596–5598 CrossRef CAS PubMed.
- G. Zhu, E. N. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Song and C. L. Hill, Dalton Trans., 2012, 41, 13043–13049 RSC.
- A. E. Kuznetsov, Y. V. Geletii, C. L. Hill, K. Morokuma and D. G. Musaev, J. Am. Chem. Soc., 2009, 131, 6844–6854 CrossRef CAS PubMed.
- A. Sartorel, P. Miró, E. Salvadori, S. Romain, M. Carraro, G. Scorrano, M. D. Valentin, A. Llobet, C. Bo and M. Bonchio, J. Am. Chem. Soc., 2009, 131, 16051–16053 CrossRef CAS PubMed.
- Y. V. Geletii, C. Besson, Y. Hou, Q. Yin, D. G. Musaev, D. Quiñonero, R. Cao, K. I. Hardcastle, A. Proust, P. Kögerler and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 17360–17370 CrossRef CAS PubMed.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, S. Rapino, L. Hoober-Burkhardt, T. Da Ros, M. Marcaccio, G. Scorrano, F. Paolucci, M. Bonchio and M. Prato, ChemSusChem, 2011, 4, 1447–1451 CrossRef CAS PubMed.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, F. M. Toma, M. Prato and M. Bonchio, Energy Environ. Sci., 2012, 5, 5592–5603 RSC.
- E. Anxolabehere-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2012, 134, 6104–6107 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, Y. S. Chaudhary and F. A. Armstrong, Energy Environ. Sci., 2012, 5, 7470–7490 RSC.
- K. S. Joya and H. J. M. de Groot, Int. J. Hydrogen Energy, 2012, 37, 8787–8799 CrossRef CAS.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS.
- L. Yu, Y. Ding and M. Zheng, Appl. Catal., B, 2017, 209, 45–52 CrossRef CAS.
- R. Al-Oweini, A. Sartorel, B. S. Bassil, M. Natali, S. Berardi, F. Scandola, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2014, 53, 11182–11185 CrossRef CAS PubMed.
- M. Blasco-Ahicart, J. Soriano-Lopez, J. J. Carbo, J. M. Poblet and J. R. Galan-Mascaros, Nat. Chem., 2018, 10, 24–30 CrossRef CAS PubMed.
- M. D. Karkas and B. Akermark, Dalton Trans., 2016, 45, 14421–14461 RSC.
- X. Wu, F. Li, B. Zhang and L. Sun, J. Photochem. Photobiol., C, 2015, 25, 71–89 CrossRef CAS.
- Q. Daniel, R. B. Ambre, B. Zhang, B. Philippe, H. Chen, F. Li, K. Fan, S. Ahmadi, H. Rensmo and L. Sun, ACS Catal., 2017, 7, 1143–1149 CrossRef CAS.
- J. Li, R. Guttinger, R. More, F. Song, W. Wan and G. R. Patzke, Chem. Soc. Rev., 2017, 46, 6124–6147 RSC.
- M. Frey, ChemBioChem, 2002, 3, 153–160 CrossRef CAS.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 RSC.
- J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. Adams, Biochim. Biophys. Acta, 2015, 1853, 1350–1369 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817–1824 CrossRef CAS PubMed.
- J. T. Muckerman and E. Fujita, Chem. Commun., 2011, 47, 12456–12458 RSC.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 16774–16776 CrossRef CAS PubMed.
- B. H. Solis and S. Hammes-Schiffer, J. Am. Chem. Soc., 2011, 133, 19036–19039 CrossRef CAS PubMed.
- M. Kirch, J.-M. Lehn and J.-P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, Angew. Chem., Int. Ed., 2011, 50, 728–731 CrossRef CAS PubMed.
- T. Stoll, M. Gennari, I. Serrano, J. Fortage, J. Chauvin, F. Odobel, M. Rebarz, O. Poizat, M. Sliwa, A. Deronzier and M. N. Collomb, Chem. – Eur. J., 2013, 19, 782–792 CrossRef CAS PubMed.
- E. D. Cline, S. E. Adamson and S. Bernhard, Inorg. Chem., 2008, 47, 10378–10388 CrossRef CAS PubMed.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753–2766 CrossRef CAS.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- N. Coutard, N. Kaeffer and V. Artero, Chem. Commun., 2016, 52, 13728–13748 RSC.
- V. Fourmond, S. Canaguier, B. Golly, M. J. Field, M. Fontecave and V. Artero, Energy Environ. Sci., 2011, 4, 2417–2427 RSC.
- S. Canaguier, M. Field, Y. Oudart, J. Pecaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876–5878 RSC.
- B. E. Barton, C. M. Whaley, T. B. Rauchfuss and D. L. Gray, J. Am. Chem. Soc., 2009, 131, 6942–6943 CrossRef CAS PubMed.
- D. Sil, Z. Martinez, S. Ding, N. Bhuvanesh, D. J. Darensbourg, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2018, 140, 9904–9911 CrossRef CAS PubMed.
- B. E. Barton and T. B. Rauchfuss, J. Am. Chem. Soc., 2010, 132, 14877–14885 CrossRef CAS PubMed.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- C. J. Curtis, A. Miedaner, W. W. Ellis and D. L. DuBois, J. Am. Chem. Soc., 2002, 124, 1918–1925 CrossRef CAS.
- M. R. Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef PubMed.
- D. H. Pool and D. L. DuBois, J. Organomet. Chem., 2009, 694, 2858–2865 CrossRef CAS.
- U. J. Kilgore, J. A. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. R. DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2011, 133, 5861–5872 CrossRef CAS PubMed.
- J. Hawecker, J. M. Lehn and R. Ziessel, Nouv. J. Chim., 1983, 7, 271–277 CAS.
- P. Connolly and J. H. Espenson, Inorg. Chem., 1986, 25, 2684–2688 CrossRef CAS.
- X. Hu, B. M. Cossairt, B. S. Brunschwig, N. S. Lewis and J. C. Peters, Chem. Commun., 2005, 4723–4725 RSC.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- P. A. Jacques, V. Artero, J. Pecaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS PubMed.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS PubMed.
- C. C. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS PubMed.
- B. H. Solis, Y. Yu and S. Hammes-Schiffer, Inorg. Chem., 2013, 52, 6994–6999 CrossRef CAS PubMed.
- A. Bhattacharjee, E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave, M. J. Field and V. Artero, Chem. – Eur. J., 2013, 19, 15166–15174 CrossRef CAS PubMed.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- E. J. Sundstrom, X. Yang, V. S. Thoi, H. I. Karunadasa, C. J. Chang, J. R. Long and M. Head-Gordon, J. Am. Chem. Soc., 2012, 134, 5233–5242 CrossRef CAS PubMed.
- V. S. Thoi, H. I. Karunadasa, Y. Surendranath, J. R. Long and C. J. Chang, Energy Environ. Sci., 2012, 5, 7762–7770 RSC.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- J.-P. Cao, T. Fang, L.-L. Zhou, L.-Z. Fu and S. Zhan, Electrochim. Acta, 2014, 147, 129–135 CrossRef CAS.
- J.-P. Cao, L.-L. Zhou, L.-Z. Fu and S. Zhan, J. Power Sources, 2014, 272, 169–175 CrossRef CAS.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 3982–3983 CrossRef CAS.
- D. J. Graham and D. G. Nocera, Organometallics, 2014, 33, 4994–5001 CrossRef CAS.
- R. M. Kellett and T. G. Spiro, Inorg. Chem., 1985, 24, 2373–2377 CrossRef CAS.
- R. M. Kellett and T. G. Spiro, Inorg. Chem., 1985, 24, 2378–2382 CrossRef CAS.
- C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef CAS PubMed.
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001–15006 CrossRef CAS PubMed.
- C. Costentin, H. Dridi and J. M. Saveant, J. Am. Chem. Soc., 2014, 136, 13727–13734 CrossRef CAS PubMed.
- M. M. Roubelakis, D. K. Bediako, D. K. Dogutan and D. G. Nocera, Energy Environ. Sci., 2012, 5, 7737–7740 RSC.
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304-305, 3–19 CrossRef CAS PubMed.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pecaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
- T. Straistari, R. Hardre, J. Fize, S. Shova, M. Giorgi, M. Reglier, V. Artero and M. Orio, Chem. – Eur. J., 2018, 24, 8779–8786 CrossRef CAS PubMed.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- J.-P. Collin, A. Jouaiti and J.-P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed.
- W. R. McNamara, Z. Han, C.-J. M. Yin, W. W. Brennessel, P. L. Holland and R. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15594–15599 CrossRef CAS PubMed.
- B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2011, 133, 18070–18073 CrossRef CAS PubMed.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- F. Wang, ChemSusChem, 2017, 10, 4393–4402 CrossRef CAS PubMed.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159 RSC.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, J. Photochem. Photobiol., A, 1996, 96, 171–174 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- B. P. Sullivan, C. M. Bolinger, D. Conrad, W. J. Vining and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 1414–1416 RSC.
- J. R. Pugh, M. R. M. Bruce, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1991, 30, 86–91 CrossRef CAS.
- H. Ishida, H. Tanaka, K. Tanaka and T. Tanaka, J. Chem. Soc., Chem. Commun., 1987, 131–132 RSC.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- H. Ishida, T. Terada, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 29, 905–911 CrossRef CAS.
- S. Chardon-Noblat, M.-N. Collomb-Dunand-Sauthier, A. Deronzier, R. Ziessel and D. Zsoldos, Inorg. Chem., 1994, 33, 4410–4412 CrossRef CAS.
- M.-N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, J. Phys. Chem., 1993, 97, 5973–5979 CrossRef CAS.
- C. Caix, S. Chardon-Noblat and A. Deronzier, J. Electroanal. Chem., 1997, 434, 163–170 CrossRef CAS.
- C. M. Bolinger, B. P. Sullivan, D. Conrad, J. A. Gilbert, N. Story and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 796–797 RSC.
- L. Duan, G. F. Manbeck, M. Kowalczyk, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2016, 55, 4582–4594 CrossRef CAS PubMed.
- P. Paul, B. Tyagi, A. K. Bilakhiya, M. M. Bhadbhade, E. Suresh and G. Ramachandraiah, Inorg. Chem., 1998, 37, 5733–5742 CrossRef CAS.
- A. Sinopoli, N. T. La Porte, J. F. Martinez, M. R. Wasielewski and M. Sohail, Coord. Chem. Rev., 2018, 365, 60–74 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- F. P. A. Johnson, M. W. George, F. Hartl and J. J. Turner, Organometallics, 1996, 15, 3374–3387 CrossRef CAS.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- S. A. Chabolla, E. A. Dellamary, C. W. Machan, F. A. Tezcan and C. P. Kubiak, Inorg. Chim. Acta, 2014, 422, 109–113 CrossRef CAS.
- E. E. Benson and C. P. Kubiak, Chem. Commun., 2012, 48, 7374–7376 RSC.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef PubMed.
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 CrossRef CAS PubMed.
- C. M. Bolinger, N. Story, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4582–4587 CrossRef CAS.
- C. W. Machan, M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 8564–8571 CrossRef CAS PubMed.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC.
- M. R. M. Bruce, E. Megehee, B. P. Sullivan, H. Thorp, T. R. O’Toole, A. Downard and T. J. Meyer, Organometallics, 1988, 7, 238–240 CrossRef CAS.
- J. Chauvin, F. Lafolet, S. Chardon-Noblat, A. Deronzier, M. Jakonen and M. Haukka, Chem. – Eur. J., 2011, 17, 4313–4322 CrossRef CAS PubMed.
- M. R. M. Bruce, E. Megehee, B. P. Sullivan, H. H. Thorp, T. R. O'Toole, A. Downard, J. R. Pugh and T. J. Meyer, Inorg. Chem., 1992, 31, 4864–4873 CrossRef CAS.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed.
- P. Voyame, K. E. Toghill, M. A. Mendez and H. H. Girault, Inorg. Chem., 2013, 52, 10949–10957 CrossRef CAS PubMed.
- Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6, 3063–3074 RSC.
- J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. DuBois, Organometallics, 2006, 25, 3345–3351 CrossRef CAS.
- D. L. DuBois, A. Miedaner and R. C. Haltiwanger, J. Am. Chem. Soc., 1991, 113, 8753–8764 CrossRef CAS.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef CAS.
- T. Hirose, S. Shigaki, M. Hirose and A. Fushimi, J. Fluorine Chem., 2010, 131, 915–921 CrossRef CAS.
- A. G. M. M. Hossain, T. Nagaoka and K. Ogura, Electrochim. Acta, 1997, 42, 2577–2585 CrossRef.
- S. Matsuoka, K. Yamamoto, T. Ogata, M. Kusaba, N. Nakashima, E. Fujita and S. Yanagida, J. Am. Chem. Soc., 1993, 115, 601–609 CrossRef CAS.
- C.-M. Che, S.-T. Mak, W.-O. Lee and K.-W. Fung, J. Chem. Soc., Dalton Trans., 1988, 2153–2159 RSC.
- K. Mochizuki, S. Manaka, I. Takeda and T. Kondo, Inorg. Chem., 1996, 35, 5132–5136 CrossRef CAS.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS PubMed.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- K. T. Ngo, M. McKinnon, B. Mahanti, R. Narayanan, D. C. Grills, M. Z. Ertem and J. Rochford, J. Am. Chem. Soc., 2017, 139, 2604–2618 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Saveant, Acc. Chem. Res., 2015, 48, 2996–3006 CrossRef CAS PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- M. Hammouche, D. Lexa, M. Momenteau and J. M. Saveant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- Y. Wu, J. Jiang, Z. Weng, M. Wang, D. L. J. Broere, Y. Zhong, G. W. Brudvig, Z. Feng and H. Wang, ACS Cent. Sci., 2017, 3, 847–852 CrossRef CAS PubMed.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- J. Jiang, A. J. Matula, J. R. Swierk, N. Romano, Y. Wu, V. S. Batista, R. H. Crabtree, J. S. Lindsey, H. Wang and G. W. Brudvig, ACS Catal., 2018, 8, 10131–10136 CrossRef CAS.
- K. A. Grice, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313–315 CrossRef CAS PubMed.
- J. Grodkowski, P. Neta, E. Fujita, A. Mahammed, L. Simkhovich and Z. Gross, J. Phys. Chem. A, 2002, 106, 4772–4778 CrossRef CAS.
- G. Seshadri, C. Lin and A. B. Bocarsly, J. Electroanal. Chem., 1994, 372, 145–150 CrossRef CAS.
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS PubMed.
- E. B. Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Morris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539–11551 CrossRef PubMed.
- Z. Lu and T. J. Williams, ACS Catal., 2016, 6, 6670–6673 CrossRef CAS.
- P. Wang, G. Liang, M. R. Reddy, M. Long, K. Driskill, C. Lyons, B. Donnadieu, J. C. Bollinger, C. E. Webster and X. Zhao, J. Am. Chem. Soc., 2018, 140, 9219–9229 CrossRef CAS PubMed.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- J. A. Treadway, J. A. Moss and T. J. Meyer, Inorg. Chem., 1999, 38, 4386–4387 CrossRef CAS PubMed.
- B. C. O'Regan and J. R. Durrant, Acc. Chem. Res., 2009, 42, 1799–1808 CrossRef PubMed.
- W. J. Youngblood, S. H. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- Y. Zhao, J. R. Swierk, J. D. Megiatto Jr, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS PubMed.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS PubMed.
- L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldt and L. Sun, Chem. Commun., 2010, 46, 7307–7309 RSC.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H.-E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389–2392 RSC.
- F. Li, H. Yang, W. Li and L. Sun, Joule, 2018, 2, 36–60 CrossRef CAS.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 RSC.
- J. J. Concepcion, J. W. Jurss, P. G. Hoertz and T. J. Meyer, Angew. Chem., Int. Ed., 2009, 48, 9473–9476 CrossRef CAS PubMed.
- B. D. Sherman, Y. Xie, M. V. Sheridan, D. Wang, D. W. Shaffer, T. J. Meyer and J. J. Concepcion, ACS Energy Lett., 2016, 2, 124–128 CrossRef.
- W. Song, C. R. K. Glasson, H. Luo, K. Hanson, M. K. Brennaman, J. J. Concepcion and T. J. Meyer, J. Phys. Chem. Lett., 2011, 2, 1808–1813 CrossRef CAS.
- M. R. Norris, J. J. Concepcion, Z. Fang, J. L. Templeton and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 13580–13583 CrossRef CAS PubMed.
- D. L. Ashford, W. Song, J. J. Concepcion, C. R. Glasson, M. K. Brennaman, M. R. Norris, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 19189–19198 CrossRef CAS PubMed.
- M. V. Sheridan, B. D. Sherman, R. L. Coppo, D. Wang, S. L. Marquard, K.-R. Wee, N. Y. Murakami Iha and T. J. Meyer, ACS Energy Lett., 2016, 1, 231–236 CrossRef CAS.
- X. Ding, Y. Gao, L. Zhang, Z. Yu, J. Liu and L. Sun, ACS Catal., 2014, 4, 2347–2350 CrossRef CAS.
- K. Hanson, D. A. Torelli, A. K. Vannucci, M. K. Brennaman, H. Luo, L. Alibabaei, W. Song, D. L. Ashford, M. R. Norris, C. R. K. Glasson, J. J. Concepcion and T. J. Meyer, Angew. Chem., Int. Ed., 2012, 51, 12782–12785 CrossRef CAS PubMed.
- D. L. Ashford, B. D. Sherman, R. A. Binstead, J. L. Templeton and T. J. Meyer, Angew. Chem., Int. Ed., 2015, 54, 4778–4781 CrossRef CAS PubMed.
- D. L. Ashford, A. M. Lapides, A. K. Vannucci, K. Hanson, D. A. Torelli, D. P. Harrison, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 6578–6581 CrossRef CAS PubMed.
- A. M. Lapides, D. L. Ashford, K. Hanson, D. A. Torelli, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 15450–15458 CrossRef CAS PubMed.
- L. Wang, D. L. Ashford, D. W. Thompson, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. C, 2013, 117, 24250–24258 CrossRef CAS.
- A. M. Lapides, B. D. Sherman, M. K. Brennaman, C. J. Dares, K. R. Skinner, J. L. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- A. K. Vannuccia, L. Alibabaeia, M. D. Losegob, J. J. Concepciona, B. Kalanyanb, G. N. Parsonsb and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef PubMed.
- K. R. Wee, M. K. Brennaman, L. Alibabaei, B. H. Farnum, B. Sherman, A. M. Lapides and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 13514–13517 CrossRef CAS PubMed.
- K. L. Materna, B. Rudshteyn, B. J. Brennan, M. H. Kane, A. J. Bloomfield, D. L. Huang, D. Y. Shopov, V. S. Batista, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2016, 6, 5371–5377 CrossRef CAS.
- B. J. Brennan, M. J. Llansola Portoles, P. A. Liddell, T. A. Moore, A. L. Moore and D. Gust, Phys. Chem. Chem. Phys., 2013, 15, 16605–16614 RSC.
- K. L. Materna, B. J. Brennan and G. W. Brudvig, Dalton Trans., 2015, 44, 20312–20315 RSC.
- K. Szpakolski, K. Latham, C. Rix, R. A. Rani and K. Kalantar-zadeh, Polyhedron, 2013, 52, 719–732 CrossRef CAS.
- D. G. Brown, P. A. Schauer, J. Borau-Garcia, B. R. Fancy and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1692–1695 CrossRef CAS PubMed.
- S. M. Zakeeruddin, M. K. Nazeeruddin, R. Humphry-Baker, P. Péchy, P. Quagliotto, C. Barolo, G. Viscardi and M. Grätzel, Langmuir, 2002, 18, 952–954 CrossRef CAS.
- K. Hanson, M. D. Losego, B. Kalanyan, D. L. Ashford, G. N. Parsons and T. J. Meyer, Chem. Mater., 2012, 25, 3–5 CrossRef.
- K. Hanson, M. D. Losego, B. Kalanyan, G. N. Parsons and T. J. Meyer, Nano Lett., 2013, 13, 4802–4809 CrossRef CAS PubMed.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430–1439 RSC.
- L. Zhang, Y. Gao, X. Ding, Z. Yu and L. Sun, ChemSusChem, 2014, 7, 2801–2804 CrossRef CAS PubMed.
- F. Li, K. Fan, L. Wang, Q. Daniel, L. Duan and L. Sun, ACS Catal., 2015, 5, 3786–3790 CrossRef CAS.
- X. Ding, Y. Gao, L. Ye, L. Zhang and L. Sun, ChemSusChem, 2015, 8, 3992–3995 CrossRef CAS PubMed.
- Y. Gao, L. Zhang, X. Ding and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 12008–12013 RSC.
- D. Wang, M. V. Sheridan, B. Shan, B. H. Farnum, S. L. Marquard, B. D. Sherman, M. S. Eberhart, A. Nayak, C. J. Dares, A. K. Das, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 14518–14525 CrossRef CAS PubMed.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 16248–16255 CrossRef CAS PubMed.
- H. Tian, ChemSusChem, 2015, 8, 3746–3759 CrossRef CAS PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- L. J. Antila, P. Ghamgosar, S. Maji, H. Tian, S. Ott and L. Hammarström, ACS Energy Lett., 2016, 1, 1106–1111 CrossRef CAS.
- C. E. Creissen, J. Warnan and E. Reisner, Chem. Sci., 2018, 9, 1439–1447 RSC.
- N. Kaeffer, C. D. Windle, R. Brisse, C. Gablin, D. Leonard, B. Jousselme, M. Chavarot-Kerlidou and V. Artero, Chem. Sci., 2018, 9, 6721–6738 RSC.
- J. M. Gardner, M. Beyler, M. Karnahl, S. Tschierlei, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 19322–19325 CrossRef CAS PubMed.
- P. B. Pati, L. Zhang, B. Philippe, R. Fernández-Terán, S. Ahmadi, L. Tian, H. Rensmo, L. Hammarström and H. Tian, ChemSusChem, 2017, 10, 2480–2495 CrossRef CAS PubMed.
- Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- B. Shan, A. K. Das, S. Marquard, B. H. Farnum, D. Wang, R. M. Bullock and T. J. Meyer, Energy Environ. Sci., 2016, 9, 3693–3697 RSC.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- M. G. Gatty, S. Pullen, E. Sheibani, H. Tian, S. Ott and L. Hammarström, Chem. Sci., 2018, 9, 4983–4991 RSC.
- A. M. Brown, L. J. Antila, M. Mirmohades, S. Pullen, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 8060–8063 CrossRef CAS PubMed.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef CAS.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS PubMed.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- A. G. Tamirat, J. Rick, A. A. Dubale, W.-N. Su and B.-J. Hwang, Nanoscale Horiz., 2016, 1, 243–267 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
- U. T. Mueller-Westerhoff and A. Nazzal, J. Am. Chem. Soc., 1984, 106, 5381–5382 CrossRef CAS.
- D. K. Zhong, S. Zhao, D. E. Polyansky and E. Fujita, J. Catal., 2013, 307, 140–147 CrossRef CAS.
- X. Chen, X. Ren, Z. Liu, L. Zhuang and J. Lu, Electrochem. Commun., 2013, 27, 148–151 CrossRef CAS.
- B. M. Klepser and B. M. Bartlett, J. Am. Chem. Soc., 2014, 136, 1694–1697 CrossRef CAS PubMed.
- T. E. Rosser, M. A. Gross, Y. H. Lai and E. Reisner, Chem. Sci., 2016, 7, 4024–4035 RSC.
- G. Liu, S. Ye, P. Yan, F. Xiong, P. Fu, Z. Wang, Z. Chen, J. Shi and C. Li, Energy Environ. Sci., 2016, 9, 1327–1334 RSC.
- K. Fan, F. Li, L. Wang, Q. Daniel, H. Chen, E. Gabrielsson, J. Sun and L. Sun, ChemSusChem, 2015, 8, 3242–3247 CrossRef CAS PubMed.
- W. Li, D. He, S. W. Sheehan, Y. He, J. E. Thorne, X. Yao, G. W. Brudvig and D. Wang, Energy Environ. Sci., 2016, 9, 1794–1802 RSC.
- L. Badia-Bou, E. Mas-Marza, P. Rodenas, E. M. Barea, F. Fabregat-Santiago, S. Gimenez, E. Peris and J. Bisquert, J. Phys. Chem. C, 2013, 117, 3826–3833 CrossRef CAS.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef CAS PubMed.
- K. S. Joya, N. Morlanes, E. Maloney, V. Rodionov and K. Takanabe, Chem. Commun., 2015, 51, 13481–13484 RSC.
- B. Liu, J. Li, H. L. Wu, W. Q. Liu, X. Jiang, Z. J. Li, B. Chen, C. H. Tung and L. Z. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 18577–18583 CrossRef CAS PubMed.
- Y. Liu, Y. Jiang, F. Li, F. Yu, W. Jiang and L. Xia, J. Mater. Chem. A, 2018, 6, 10761–10768 RSC.
- S. Nakashima, R. Negishi and H. Tada, Chem. Commun., 2016, 52, 3665–3668 RSC.
- Y. Hou, B. L. Abrams, P. C. Vesborg, M. E. Bjorketun, K. Herbst, L. Bech, A. M. Setti, C. D. Damsgaard, T. Pedersen, O. Hansen, J. Rossmeisl, S. Dahl, J. K. Norskov and I. Chorkendorff, Nat. Mater., 2011, 10, 434–438 CrossRef CAS PubMed.
- B. Seger, K. Herbst, T. Pedersen, B. Abrams, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Electrochem. Soc., 2014, 161, H722–H724 CrossRef.
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 8, 5172–5180 RSC.
- Y. H. Lai, H. S. Park, J. Z. Zhang, P. D. Matthews, D. S. Wright and E. Reisner, Chem. – Eur. J., 2015, 21, 3919–3923 CrossRef CAS PubMed.
- C.-Y. Lee, H. S. Park, J. C. Fontecilla-Camps and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 5971–5974 CrossRef CAS PubMed.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- M. Kato, J. Z. Zhang, N. Paul and E. Reisner, Chem. Soc. Rev., 2014, 43, 6485–6497 RSC.
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS PubMed.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- J. Z. Zhang, K. P. Sokol, N. Paul, E. Romero, R. van Grondelle and E. Reisner, Nat. Chem. Biol., 2016, 12, 1046–1052 CrossRef CAS PubMed.
- D. Mersch, C. Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- D. H. Nam, J. Z. Zhang, V. Andrei, N. Kornienko, N. Heidary, A. Wagner, K. Nakanishi, K. P. Sokol, B. Slater, I. Zebger, S. Hofmann, J. C. Fontecilla-Camps, C. B. Park and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 10595–10599 CrossRef CAS PubMed.
- K. P. Sokol, W. E. Robinson, A. R. Oliveira, J. Warnan, M. M. Nowaczyk, A. Ruff, I. A. C. Pereira and E. Reisner, J. Am. Chem. Soc., 2018, 140, 16418–16422 CrossRef CAS PubMed.
- A. Krawicz, J. Yang, E. Anzenberg, J. Yano, I. D. Sharp and G. F. Moore, J. Am. Chem. Soc., 2013, 135, 11861–11868 CrossRef CAS PubMed.
- D. Cedeno, A. Krawicz, P. Doak, M. Yu, J. B. Neaton and G. F. Moore, J. Phys. Chem. Lett., 2014, 5, 3222–3226 CrossRef CAS PubMed.
- A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, Ind. Eng. Chem. Res., 2016, 55, 5306–5314 CrossRef CAS.
- A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, ACS Appl. Mater. Interfaces, 2016, 8, 10038–10047 CrossRef CAS PubMed.
- A. Krawicz, D. Cedeno and G. F. Moore, Phys. Chem. Chem. Phys., 2014, 16, 15818–15824 RSC.
- D. Khusnutdinova, A. M. Beiler, B. L. Wadsworth, S. I. Jacob and G. F. Moore, Chem. Sci., 2017, 8, 253–259 RSC.
- J. Gu, Y. Yan, J. L. Young, K. X. Steirer, N. R. Neale and J. A. Turner, Nat. Mater., 2016, 15, 456–460 CrossRef CAS PubMed.
- G. F. Moore and I. D. Sharp, J. Phys. Chem. Lett., 2013, 4, 568–572 CrossRef CAS PubMed.
- H. J. Kim, J. Seo and M. J. Rose, ACS Appl. Mater. Interfaces, 2016, 8, 1061–1066 CrossRef CAS PubMed.
- J. Seo, R. T. Pekarek and M. J. Rose, Chem. Commun., 2015, 51, 13264–13267 RSC.
- D. Cedeno, A. Krawicz and G. F. Moore, Interface Focus, 2015, 5, 20140085 CrossRef PubMed.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 RSC.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- P. Bornoz, F. F. Abdi, S. D. Tilley, B. Dam, R. van de Krol, M. Graetzel and K. Sivula, J. Phys. Chem. C, 2014, 118, 16959–16966 CrossRef CAS.
- R. E. Rocheleau, E. L. Miller and A. Misra, Energy Fuels, 1998, 12, 3–10 CrossRef CAS.
- O. Khaselev, A. Bansal and J. A. Turner, Int. J. Hydrogen Energy, 2001, 26, 127–132 CrossRef CAS.
- R. C. Kainthla, B. Zelenay and J. O. M. Bockris, J. Electrochem. Soc., 1987, 134, 841–845 CrossRef CAS.
- G. Peharz, F. Dimroth and U. Wittstadt, Int. J. Hydrogen Energy, 2007, 32, 3248–3252 CrossRef CAS.
- S. Licht, B. Wang, S. Mukerji, T. Soga, M. Umeno and H. Tributsch, J. Phys. Chem. B, 2000, 104, 8920–8924 CrossRef CAS.
- A. Heller, Science, 1984, 223, 1141–1148 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- D. M. Powell, M. T. Winkler, H. J. Choi, C. B. Simmons, D. B. Needleman and T. Buonassisi, Energy Environ. Sci., 2012, 5, 5874–5883 RSC.
- F. Sahli, J. Werner, B. A. Kamino, M. Brauninger, R. Monnard, B. Paviet-Salomon, L. Barraud, L. Ding, J. J. Diaz Leon, D. Sacchetto, G. Cattaneo, M. Despeisse, M. Boccard, S. Nicolay, Q. Jeangros, B. Niesen and C. Ballif, Nat. Mater., 2018, 17, 820–826 CrossRef CAS PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- W. Kreuter and H. Hofmann, Int. J. Hydrogen Energy, 1998, 23, 661–666 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1999, 476, 90–91 CrossRef CAS.
- A. Ursua, L. M. Gandia and P. Sanchis, Proc. IEEE, 2012, 100, 410–426 CAS.
- F. Zhao, J. Zhang, T. Abe, D. Wohrle and M. Kaneko, J. Mol. Catal. A: Chem., 1999, 145, 245–256 CrossRef CAS.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- A. Reynal, J. Willkomm, N. M. Muresan, F. Lakadamyali, M. Planells, E. Reisner and J. R. Durrant, Chem. Commun., 2014, 50, 12768–12771 RSC.
- B. R. Lydon, A. Germann and J. Y. Yang, Inorg. Chem. Front., 2016, 3, 836–841 RSC.
- S. Mukherjee, A. Mukherjee, A. Bhagi-Damodaran, M. Mukherjee, Y. Lu and A. Dey, Nat. Commun., 2015, 6, 8467 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- A. K. Das, M. H. Engelhard, S. Lense, J. A. Roberts and R. M. Bullock, Dalton Trans., 2015, 44, 12225–12233 RSC.
- P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J.-M. Savéant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Metaye, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- M. A. Alonso-Lomillo, O. Rüdiger, A. Maroto-Valiente, M. Velez, I. Rodríguez-Ramos, F. J. Muñoz, V. M. Fernández and A. L. De Lacey, Nano Lett., 2007, 7, 1603–1608 CrossRef CAS PubMed.
- E. S. Andreiadis, P. A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pecaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed.
- L. Tong, M. Gothelid and L. Sun, Chem. Commun., 2012, 48, 10025–10027 RSC.
- K. E. deKrafft, C. Wang, Z. Xie, X. Su, B. J. Hinds and W. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 608–613 CrossRef CAS PubMed.
- M. Tavakkoli, M. Nosek, J. Sainio, F. Davodi, T. Kallio, P. M. Joensuu and K. Laasonen, ACS Catal., 2017, 7, 8033–8041 CrossRef CAS.
- X. Zhou, T. Zhang, C. W. Abney, Z. Li and W. Lin, ACS Appl. Mater. Interfaces, 2014, 6, 18475–18479 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- B. Reuillard, J. Warnan, J. J. Leung, D. W. Wakerley and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 3952–3957 CrossRef CAS PubMed.
- H. Lei, C. Liu, Z. Wang, Z. Zhang, M. Zhang, X. Chang, W. Zhang and R. Cao, ACS Catal., 2016, 6, 6429–6437 CrossRef CAS.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- R. Matheu, I. A. Moreno-Hernandez, X. Sala, H. B. Gray, B. S. Brunschwig, A. Llobet and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 11345–11348 CrossRef CAS PubMed.
- P. Garrido-Barros, C. Gimbert-Surinach, D. Moonshiram, A. Picon, P. Monge, V. S. Batista and A. Llobet, J. Am. Chem. Soc., 2017, 139, 12907–12910 CrossRef CAS PubMed.
- J. Creus, R. Matheu, I. Peñafiel, D. Moonshiram, P. Blondeau, J. Benet-Buchholz, J. García-Antón, X. Sala, C. Godard and A. Llobet, Angew. Chem., Int. Ed., 2016, 55, 15382–15386 CrossRef CAS PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- X. Li, H. Lei, X. Guo, X. Zhao, S. Ding, X. Gao, W. Zhang and R. Cao, ChemSusChem, 2017, 10, 4632–4641 CrossRef CAS PubMed.
- S. K. Ibrahim, X. Liu, C. Tard and C. J. Pickett, Chem. Commun., 2007, 1535–1537 RSC.
- W. W. Kramer and C. C. L. McCrory, Chem. Sci., 2016, 7, 2506–2515 RSC.
- X. M. Hu, Z. Salmi, M. Lillethorup, E. B. Pedersen, M. Robert, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Chem. Commun., 2016, 52, 5864–5867 RSC.
- L. Wang, K. Fan, Q. Daniel, L. Duan, F. Li, B. Philippe, H. Rensmo, H. Chen, J. Sun and L. Sun, Chem. Commun., 2015, 51, 7883–7886 RSC.
- L. Wang, K. Fan, H. Chen, Q. Daniel, B. Philippe, H. Rensmo and L. Sun, Catal. Today, 2017, 290, 73–77 CrossRef CAS.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- B. Zhu, R. Zou and Q. Xu, Adv. Energy Mater., 2018, 8, 1801193 CrossRef.
- C.-C. Hou and Q. Xu, Adv. Energy Mater., 2018, 1801307 CrossRef.
- C. Y. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS.
- M. B. Chambers, X. Wang, N. Elgrishi, C. H. Hendon, A. Walsh, J. Bonnefoy, J. Canivet, E. A. Quadrelli, D. Farrusseng, C. Mellot-Draznieks and M. Fontecave, ChemSusChem, 2015, 8, 603–608 CrossRef CAS PubMed.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- T. Zheng and L. Li, New J. Chem., 2018, 42, 2526–2536 RSC.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- S. Cui, M. Qian, X. Liu, Z. Sun and P. Du, ChemSusChem, 2016, 9, 2365–2373 CrossRef CAS PubMed.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- M. Wang, J. Liu, C. Guo, X. Gao, C. Gong, Y. Wang, B. Liu, X. Li, G. G. Gurzadyan and L. Sun, J. Mater. Chem. A, 2018, 6, 4768–4775 RSC.
- N. Li, J. Xu, R. Feng, T. L. Hu and X. H. Bu, Chem. Commun., 2016, 52, 8501–8513 RSC.
- S. Sun, H. Li and Z. J. Xu, Joule, 2018, 2, 1024–1027 CrossRef.
- H. Li, F. Li, B. Zhang, X. Zhou, F. Yu and L. Sun, J. Am. Chem. Soc., 2015, 137, 4332–4335 CrossRef CAS PubMed.
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417–2420 CrossRef CAS PubMed.
- J.-X. Jian, C. Ye, X.-Z. Wang, M. Wen, Z.-J. Li, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2016, 9, 2083–2089 RSC.
- L. Z. Wu, B. Chen, Z. J. Li and C. H. Tung, Acc. Chem. Res., 2014, 47, 2177–2185 CrossRef CAS PubMed.
- S. Marini, P. Salvi, P. Nelli, R. Pesenti, M. Villa, M. Berrettoni, G. Zangari and Y. Kiros, Electrochim. Acta, 2012, 82, 384–391 CrossRef CAS.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- P. Zhang, L. Li, D. Nordlund, H. Chen, L. Fan, B. Zhang, X. Sheng, Q. Daniel and L. Sun, Nat. Commun., 2018, 9, 381 CrossRef PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- N. Mbemba Kiele, C. Herrero, A. Ranjbari, A. Aukauloo, S. A. Grigoriev, A. Villagra and P. Millet, Int. J. Hydrogen Energy, 2013, 38, 8590–8596 CrossRef CAS.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- F. Dionigi and P. Strasser, Adv. Eng. Mater., 2016, 6, 1600621 Search PubMed.
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247–2252 RSC.
- C. Costentin, S. Drouet, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |