
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of polysulfide dianions and radical anions in the chemical, physical and biological sciences, including sulfur-based batteries
Ralf
Steudel
 *a and
Tristram
Chivers
*b
*a and
Tristram
Chivers
*b
aInstitute of Chemistry, Technical University Berlin, D-10623 Berlin, Germany. E-mail: steudel@sulfur-research.de
bDepartment of Chemistry, The University of Calgary, Calgary, Alberta T2N 1N4, Canada. E-mail: chivers@ucalgary.ca
First published on 24th May 2019
Abstract
The well-known tendency of sulfur to catenate is exemplified by an extensive series of polysulfide dianions [Sn]2− (n = 2–9) and related radical monoanions [Sn]˙−. The dianions can be isolated as crystalline salts with appropriate cations and structurally and spectroscopically characterized. Although the smaller radical monoanions may be stabilized in zeolitic matrices, they are usually formed in solution via disproportionation or partial dissociation of the dianions as well as by electrochemical reduction of elemental sulfur. An understanding of the fundamental chemistry of these homoatomic species is key to unravelling their behaviour in a broad variety of chemical environments. This review will critically evaluate the techniques used to characterize polysulfide dianions and radical anions both in solution and in the solid state, i.e. Raman, UV-visible, EPR, NMR and X-ray absorption spectroscopy, X-ray crystallography, mass spectrometry, chromatography and high-level quantum-chemical calculations. This is followed by a discussion of recent advances in areas in which these anionic sulfur species play a crucial role, viz. alkali-metal–sulfur batteries, organic syntheses, biological chemistry, geochemical processes including metal transport, coordination complexes, atmospheric chemistry and materials science.
 Ralf Steudel | Ralf Steudel is emeritus professor of inorganic chemistry at the Technical University Berlin, Germany. He is best known for his many contributions to the chemistry of sulfur and selenium as well as for his textbooks and monographs on various aspects of inorganic chemistry. He spent a one-year sabbatical as visiting professor at the Spectroscopy Laboratory of M.I.T. (Cambridge, Mass.) and since cooperated with organic and inorganic chemists, with spectroscopists, crystallographers, theoreticians, astronomers and microbiologists around the world to explore the rich chemistry of the non-metals. As a former chairman of the 6th International Symposium on Inorganic Ring Systems held in Berlin he is a long-term member of the “IRIS family”. |
 Tristram Chivers | Tristram Chivers, a native of Bath, England, received his BSc, PhD (with Prof. R. D. Chambers FRS) and DSc degrees all from the University of Durham (UK). He joined the University of Calgary in 1969 and served as Head of the Chemistry Department from 1977 to 1982. He is currently Professor Emeritus of Chemistry. His research interests are in the area of main group element chemistry with emphasis on chalcogen compounds and inorganic ring systems. He is the author of two books: “A Guide to Chalcogen-Nitrogen Chemistry” and (with I. Manners) “Inorganic Rings and Polymers of the p-Block Elements: From Fundamentals to Applications”. |
1 Introduction
The p-block element sulfur1 is the tenth most abundant element in the universe. The crown-shaped structure of the most stable allotrope, cyclo-S8, is well-known as is the fact that elemental sulfur forms an extensive series of other cyclic allotropes, of which 16 have been structurally characterized.2 Cyclic frameworks are maintained in dications such as [S4]2+, [S8]2+ and [S19]2+, which are formed upon oxidation of elemental sulfur with strong oxidizing agents and can be isolated as crystalline salts with weakly coordinating anions.2b The radical cation [S5]˙+ produced from the dissociation of [S8]2+ in solution also has a cyclic structure, but it has not been isolated in the solid state.3,4 However, the synthesis of the bicyclic radical cation [S8]˙+ as the blue salt Cs0.73Na0.27[S8][B12Cl12], which was characterized by X-ray diffraction, was reported in 2017.5In addition, sulfur forms a series of homoatomic polysulfide dianions [Sn]2− (n = 2–9), which have been isolated and structurally characterized as salts often in combination with solvated or chelated group 1 or 2 metal cations or large organic counter-ions, e.g. [R4N]+ (R = alkyl), [Ph4P]+, [(Ph3P)2N]+.6,7 With one exception (Section 2) these dianions form catenated, unbranched structures in the solid state. Similar to the behaviour of the dication [S8]2+, some of the long-chain dianionic species (n = 6–9) undergo an entropy-driven partial dissociation or disproportionation in solution to give polysulfide radical anions [Sn]˙− (n = 2–4), the most prevalent of which is the blue trisulfide radical anion (n = 3), which is well-known to be stabilized by entrapment in an aluminosilicate matrix, e.g. in ultramarine blue.8 A unique polysulfide radical anion is the cyclic species [S6]˙−, which forms a six-membered ring with two long central S–S bonds (263.3 pm, cf. 205 pm in cyclo-S8) in the solid state and has been isolated as a [Ph4P]+ salt.9 In 2017 a dimeric bismuth(III) complex formally containing the tetrasulfide radical anion [S4]˙− was isolated and structurally characterized (Section 5.5).10 Long-chain polysulfide radical anions [Sn]˙− (n = 5–8, 10) have been detected in the laser desorption mass spectra of cyclo-S8 in the negative ion mode (Section 3.3) and are also considered in computational studies (Section 4).11
Organic polysulfanes R–Sn–R are often called polysulfides although they are derivatives of the covalent sulfanes H–Sn–H. The same holds for the partially substituted polysulfanes R–Sn–H (“hydropolysulfides”). In aqueous solution, these sulfur-rich SH compounds (n > 1) are much stronger acids than H2S and therefore exist at pH values above 5 as mono- or di-anions, respectively.12 Their rational nomenclature, which is in line with IUPAC recommendations, can be found in a previous review.13
The broad significance of elemental sulfur and many inorganic sulfur compounds in the geo-, bio- and cosmo-spheres, has been recognized for more than thirty-five years.14 Contemporary interest has been focused on the chemical properties of polysulfide dianions and the radical anions derived from them in part because of their function in alkali metal–sulfur batteries,15 but also owing to their role in a variety of other environments. The purpose of this article is to provide a critical review of recent advances in the analytical characterization, computational investigations and applications of these catenated, anionic polysulfur species. The discussion will begin with recent developments in the synthesis and structural determinations of polysulfide salts (Section 2). Subsequently, the identification of polysulfide dianions and radical anions by various spectroscopic and chromatographic techniques (Section 3), as well as computational studies (Section 4), will be considered. Finally, the significance and applications of these species in the following areas will be evaluated (Section 5): alkali metal–sulfur batteries (Section 5.1), organic syntheses (Section 5.2), biological chemistry (Section 5.3), geochemical transformations (Section 5.4), coordination complexes (Section 5.5) and atmospheric chemistry (Section 5.6). The conjunction of dianionic and monoanionic polysulfur species in this discussion evolves naturally from the observation that the dianions are in equilibrium with the radical anions, especially in non-aqueous solvents.16 Biological and geochemical transformations involving polysulfides occur in aqueous media, whereas alkali-metal–sulfur batteries employ non-aqueous solvents or polysulfide melts. The formation of organosulfur compounds or metal complexes of polysulfide dianions and radical anions may take place in either environment.
The coverage is not intended to be comprehensive. Rather, we will attempt to provide a critical overview of the important developments that have appeared since previous reviews on these species.6–8
2 Novel syntheses and structures
The preparation and structural characterization of polysulfide dianions [Sn]2− with up to nine sulfur atoms has been described in previous reviews.6,7 Although the direct reaction of sulfur with an alkali metal in liquid ammonia is a well-established approach, the selective preparation of specific sodium oligosulfides is challenging. To this end Takata et al. reported the stoichiometric reactions of a sodium dispersion with sulfur powder in 1,2-dimethoxyethane (DME) at 70 °C to give either Na2S or Na2S2, the yields of which were determined by reaction with benzyl chloride followed by GC-MS analysis of the dibenzylsulfanes so formed (Section 3.4).17 Aromatic hydrocarbons or ketones were found to be effective catalysts to enhance both the yield and selectivity of individual salts Na2Sn (n = 1 or 2).17 In 2016, Mali and co-workers prepared a series of sodium polysulfides from the reaction of sodium metal with elemental sulfur in anhydrous methanol at 45–50 °C in order to obtain 23Na solid-state NMR spectra (Section 3.6).18 Whereas the series Na2Sn (n = 2, 4, 5) were obtained as pure compounds, X-ray diffraction patterns and solid-state 23Na NMR spectra revealed that the material with a stoichiometry of 2Na:3S crystallizes as a mixture of Na2S2 and Na2S4 rather than homogeneous Na2S3 (Section 3.6).18 In 2019 Röhr et al. obtained the high-temperature (HT) polymorph of Cs2S3 from the reaction of Cs2S2 with elemental sulfur at 800 °C followed by cooling to room temperature.19 The crystal structure of this form of Cs2S3 embodies four crystallographically independent trisulfide dianions [S3]2− of very similar molecular structures;19 such HT polymorph had been predicted earlier on the basis of Raman spectra (see Section 3.1). The solvation of the alkali-metal cation can stabilize longer chain dianions in the hexasulfide [K(THF)]2[S6] and the unique nonasulfide [K2(THF)][S9], as demonstrated in 2007 by Lerner and co-workers.20a However, a few years later the preparation of the unsolvated salt K2S6 by the reaction of potassium with stoichiometric amounts of sulfur in liquid ammonia was achieved by Getzschmann and Kaskel.20b The all-trans conformation of the S6 chain in K2S6 is in contrast with the cis,cis conformation in the solvated analogue.19,20 Larger alkali-metal cations can also stabilize longer polysulfide dianions in the solid state, e.g. the hexasulfide Cs2S6 is well-known and the structure of a new triclinic form, in which the two crystallographically independent cesium atoms adopt ten- and eleven-fold coordination, was disclosed by Pfitzner and co-workers in 2015.21 In a study of the influence of the alkali-metal cation on the stabilization of polysulfides in 2018 Lu and co-workers found that in DMSO solution the large cation Rb+ exerts a stronger stabilizing effect than the smaller cation Li+ on short-chain polysulfide dianions [Sn]2− (n = 3, 4), since the weaker solvation energy of Rb+ with respect to DMSO results in stronger cation–anion electrostatic interactions.22Lithium polysulfides Li2Sn (n = 1–8) are of particular interest in view of their role as intermediates in the operation of lithium–sulfur batteries (Section 5.1.3).15 Seminal work by Rauh and co-workers more than 40 years ago established that the stoichiometric reactions of lithium sulfide with cyclo-S8 in solvents such as DMSO, DMF or THF are the most efficient synthesis of lithium polysulfides with the nominal composition “Li2Sn” [eqn (1)],23 and that method is still used in investigations of these materials during the last few years.22,24,25 However, these products are comprised of an equilibrium mixture, both in the solid state and in solution, and there is no straightforward procedure for the separation of individual polysulfides (Section 3.4).
| Li2S + (n − 1)/8S8 → Li2Sn (n = 3–8) | (1) |
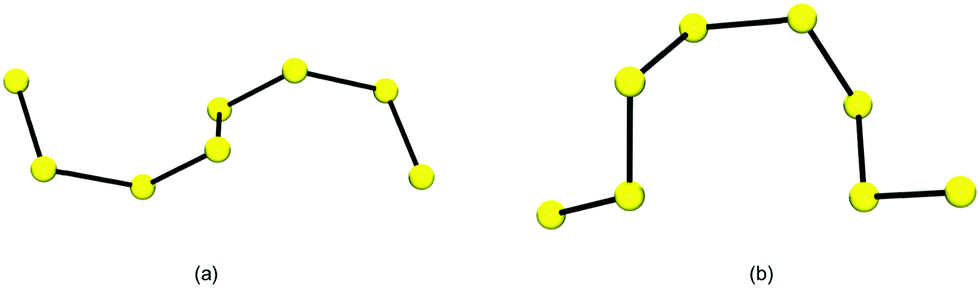 | ||
| Fig. 1 Conformation of the [S8]2− chain in (a) [Ni(N-MeIm)6][S8] and (b) [Et3NH]2[S8] (adapted from Fig. 3 in ref. 26a). | ||
In 2017, prompted by the interest in magnesium–sulfur batteries, Bieker and co-workers28 extended the approach of Rauchfuss et al.26a to the synthesis of the DMSO analogue [Mg(DMSO)6][S8] (DMSO = dimethyl sulfoxide) [eqn (2)]. They found that solutions of magnesium polysulfides undergo disproportionation and dissociation equilibria in a similar manner to the well-established behaviour of lithium polysulfides.28
| Mg + S8 + 6L → [Mg(L)6][S8] (L = N-methylimidazole or DMSO) | (2) |
| [Cat]+[MeOCO2]− + H2S → [Cat]+[HS]− + MeOH + CO2 | (3) |
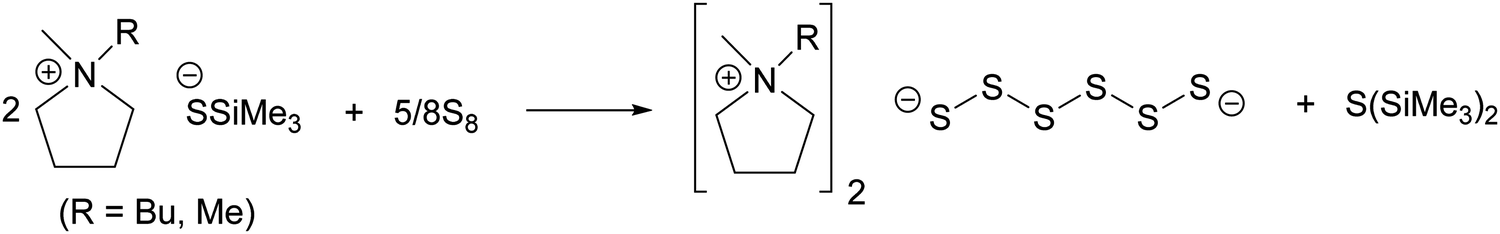 | ||
| Scheme 1 Synthesis of pyrrolidinium hexasulfides. | ||
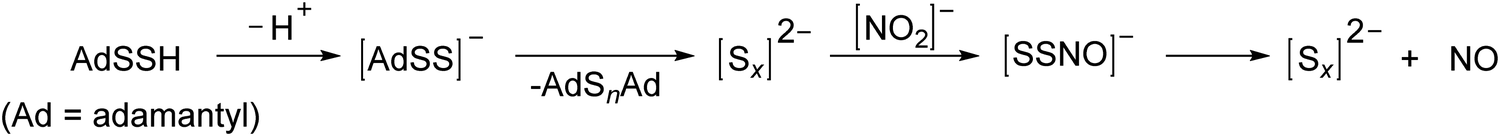 | ||
| Scheme 2 Involvement of polysulfides in the formation of NO from persulfides and nitrite. | ||
In 2018 the first structurally characterized example of a branched polysulfide [SS3]2−, the perthio form of the sulfite ion [SO3]2−, was reported by Seff and co-workers.32a This unique polysulfide was formed via disproportionation of sulfur vapour at 623 K in a zeolite matrix with the equally unusual trisulfur dication [S3]2+ as the counter-ion. The [SS3]2− and [S3]2+ ions are sequestered in cavities of the matrix with S–S bond lengths of approximately 216(5) and 211(8) pm, respectively, as determined by X-ray diffraction (Fig. 2); the high values of the estimated standard deviations are the result of disorder problems.32a
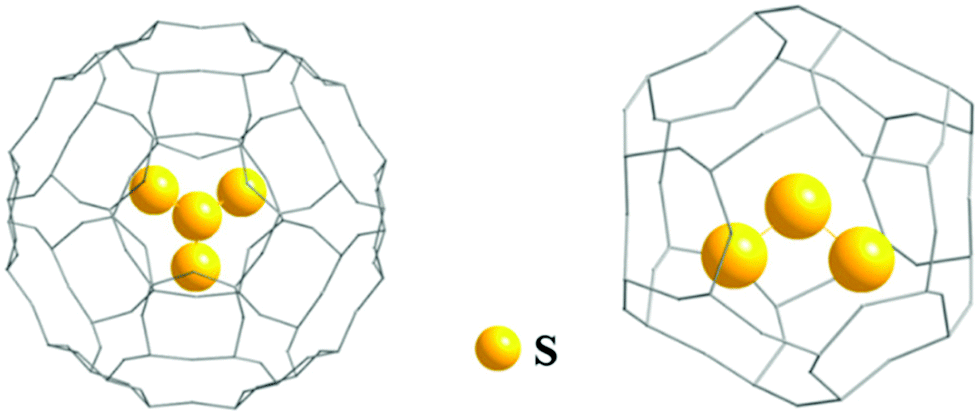 | ||
| Fig. 2 Schematic representation of entrapment of [SS3]2− and [S3]2+ ions in a zeolite matrix (adapted with permission from the graphical abstract in ref. 32a). | ||
Strong interactions with the surrounding sodium ([SS3]2−) and oxygen atoms ([S3]2+) of the zeolite matrix are thought to stabilize these unusual ions. The trapping of [S3]2+ in this way is reminiscent of the well-known encapsulation of the [S3]˙− radical ion in sodalite (Section 3.5).8 In addition, it is noted that in 2013 the neutral trisulfur molecule S3 has been selectively sequestered in the pores of a coordination network formed by the polymeric complex [(TPT)3ZnI2]n {TPT = 2,4,6-tris(4-pyridyl)triazine}.33
3 Characterisation of polysulfide dianions and radical anions
The broad variety of spectroscopic, as well as chromatographic and computational techniques, that are available for the characterization of polysulfide dianions and radical anions in solution or the solid state are discussed in the following sections. In each case the capabilities as well as the limitations of the method are considered. Indeed, as emphasized in a recent book chapter by Prendergast et al.,15a it is essential to employ several independent techniques before firm conclusions about the identity of a particular species are made. The use of a combination of UV-visible, IR/Raman and EPR spectra to identify the blue species formed in solutions of polysulfide dianions in electron-pair donor solvents as the trisulfide radical anion [S3]˙− is a cogent example of this dictum.34 Similarly, UV-visible and EPR spectra have been employed to confirm the presence of [S3]˙− in the recent applications of this species as a reagent in organic synthesis (Section 5.2). The singular ability of Raman spectroscopy for the identification of [S3]˙− is impressively demonstrated by the use of this technique to establish that the blue mineral recently found in a monastery at Dalheim, Germany, in the dental calculus of a medieval (11th or early 12th century by radiocarbon dating) religious woman is lapis lazuli.35 The characteristic symmetric stretching vibration of [S3]˙− was observed at 548 cm−1 with an overtone at 1096 cm−1 (Fig. 3) (Section 3.1).8 This significant anthropological finding casts doubt on the widely held assumption concerning the limited availability of lapis lazuli in medieval Europe and suggests that the woman was working on valuable illuminated manuscripts.35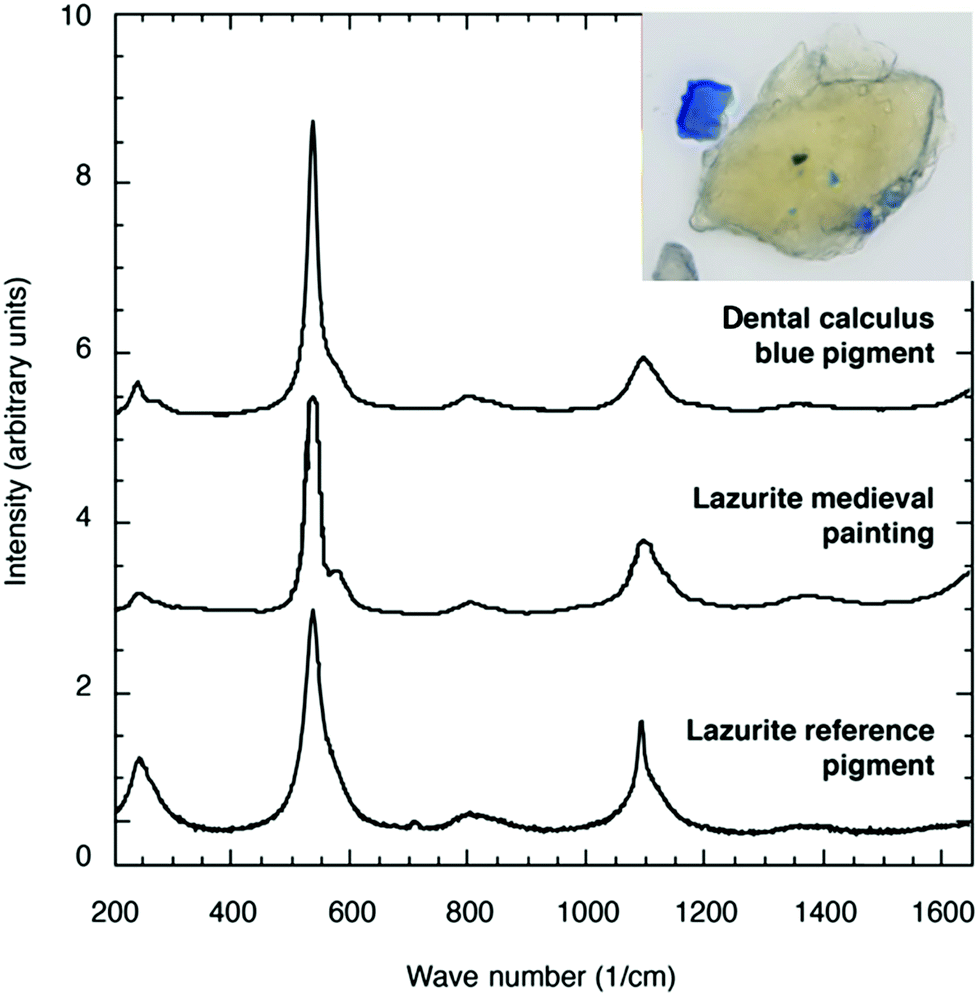 | ||
| Fig. 3 Comparison of micro-Raman spectra of the blue mineral found in the dental calculus of a medieval woman with reference lazurites. Inset shows the embedded blue mineral and a sample of it freed from the dental calculus (adapted with permission from Fig. 2 and 4 in ref. 35). | ||
The disulfide radical anion [S2]˙− is also well-characterized by IR/Raman, UV-visible and EPR spectra. However, despite numerous claims for the spectroscopic signature of the tetrasulfide radical anion [S4]˙− by various techniques, there is still some uncertainty about the identification of this important species (see Discussion in Sections 3.1–3.6).
3.1 Raman spectra
The S–S bonds of polysulfides are strong Raman scatterers due to the high polarizability of the bonding and lone pair electrons at the two-coordinate sulfur atoms. Therefore, laser Raman spectroscopy is a very useful technique for identifying individual polysulfides and polysulfide radical anions. However, assignments of lines in the spectra of polysulfide mixtures may be difficult owing to the coincidence of some lines for different molecular sizes and due to incidental degeneracies within one ion. In addition, the Raman effect is a low intensity phenomenon since only a tiny fraction of the incoming radiation is scattered inelastically. Consequently, a sufficient concentration is needed in solutions since several anion conformations may be present in equilibrium resulting in broad Raman signals from superposition of closely neighbouring lines. Fortunately, for highly coloured species such as some radical anions the enormous enhancement of Raman intensity caused by resonance effects helps to identify such species even at low concentrations. This requires that the wavelength of the laser radiation is close to the wavelength of an absorption band of the species to be detected.However, it should also be noted that S–S bonds in polysulfur compounds are light-sensitive since homolytic dissociation can be triggered by visible light of suitable wavelengths. Therefore, red or infrared laser radiation is recommended for Raman investigations. Sample cooling helps to reduce the thermal or photochemical decomposition and at the same time decreases the line widths resulting in larger and narrower peaks and hence better spectral resolution.
Vibrational wavenumbers of polysulfide anions can be calculated theoretically either from empirical force constants and geometrical parameters adopted from similar molecules and ions (preferred)36 and, with some restrictions, by quantum-chemical calculations (see also Section 4). As an example, the well-resolved Raman spectrum of solid cesium hexasulfide (Cs2S6) is shown in Fig. 4.21 The sample was prepared by reacting S8 with cesium carbonate at 120 °C in ethanol and was characterized by X-ray diffraction on single crystals. The helical anion is of C2 symmetry with SS bond lengths in the range 202.5–206.4 pm. The five stretching modes result in five well-separated lines between 410 and 510 cm−1, while the bending modes occur below 300 cm−1; the torsion modes are to be expected in the region below 100 cm−1, which has not been recorded in this case. The spectrum is in excellent agreement with the earlier Raman data of Ziemann and Bues of 1979;37a the observed stretching modes also agree very well with the wavenumbers predicted in 1977 for the free helical hexasulfide anion of C2 symmetry calculated from force constants and geometrical parameters adopted from S8 and other sulfur compounds such as [NH4]2[S5].36
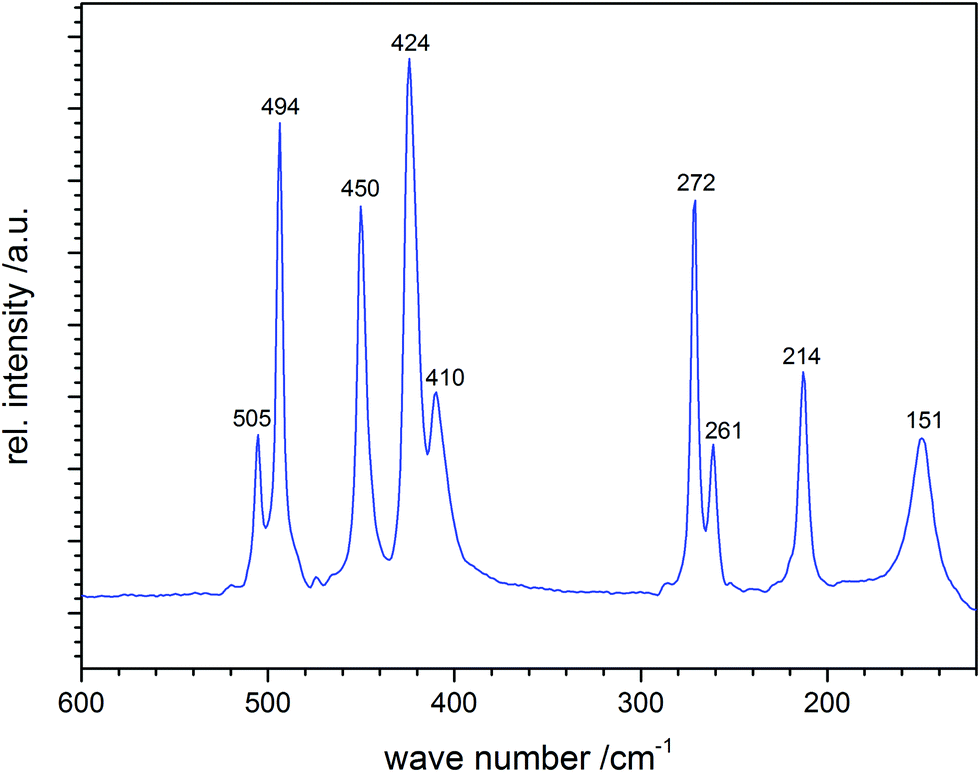 | ||
| Fig. 4 Raman spectrum of solid Cs2S6 (figure courtesy of A. Pfitzner). | ||
Interestingly, a high-temperature form of Cs2S3, which was predicted in 1979 on the basis of the additional Raman lines (splitting of stretching modes) observed when this trisulfide was heated to 125 °C followed by rapid cooling to 25 °C,37b was prepared from Cs2S2 and sulfur and structurally characterized forty years later (Section 2).19
All reliable Raman spectra of polysulfide dianions recorded with pure samples using red or infrared laser excitation and exclusion of oxygen to avoid autoxidation support the following statements:6,19
1. The stretching modes of unbranched dianions [Sn]2− occur only in the region 390–510 cm−1 and there are no fundamental modes in the region 300–390 cm−1.
2. The stretching modes are only slightly influenced by the cations or by solvents.
3. All dianions [Sn]2− with n = 3–9 in a large number of alkali-metal, ammonium, phosphonium and telluronium salts form unbranched chains of differing conformations as demonstrated by X-ray diffraction analyses of single crystals (a branched tetrasulfide anion has been identified in a zeolite structure; Section 2, Fig. 2)
4. There is no limitation to the chain-length n of the dianions [Sn]2−; in solutions and in melts even-membered as well as odd-membered chains exist in a dynamic equilibrium which is established rapidly (Section 3.4).
5. In organic solvents, but not in water, the alkali-metal polysulfides are also in equilibrium with dissolved S8 molecules and with radical anions (S8 is insoluble in water and can be present as a solid only).
6. Some alkali-metal polysulfides dissociate on melting with formation of radical anions which reversibly recombine on cooling and annealing of the melt to regenerate the original phase, provided the temperature has not been so high that elemental sulfur evaporates from the melt (the dissociation in solvents will be discussed below).
7. Autoxidation of polysulfide dianions produces thiosulfate and sulfate ions38 giving rise to novel Raman signals both above 500 cm−1 and below 400 cm−1.39
In salts with alkali-metal cations from Na+ to Cs+, as well as with organic ammonium and phosphonium cations, the polysulfide anions form chains of differing conformations which are located in a cavity formed by surrounding cations6 (Fig. 5). However, with lithium cations completely different structures are observed since quasi-molecular ion pairs are formed for n > 2. Besides Li2S2 no other binary lithium polysulfide has so far been characterized by X-ray structural analysis since these compounds crystallize from polar solvents with at least one solvent molecule attached to the cations in addition to the polysulfido ligand. The coordination number of Li is always 4. For example, crystals of [Li(TMEDA)]2[S6] consist of a bicyclic Li2S6 unit with one tetramethylethylenediamine (TMEDA) molecule chelated to each metal atom (see Fig. 6).41 Due to the small size of the naked Li cation and its high electric field strength, the Li–S interactions can be considered as partly covalent and partly ionic. Consequently, the Raman spectrum contains not only typical SS stretching modes in the region 390–500 cm−1, but also Li–S stretching modes (near and below 390 cm−1) which most probably are coupled to the SS stretching fundamentals of the neighbouring bonds. In addition, Li–N stretching modes are to be expected in the same region. With chelating triamines attached to Li ions the polysulfide unit forms a chain which is end-capped on both sides by the LiN3 unit.42
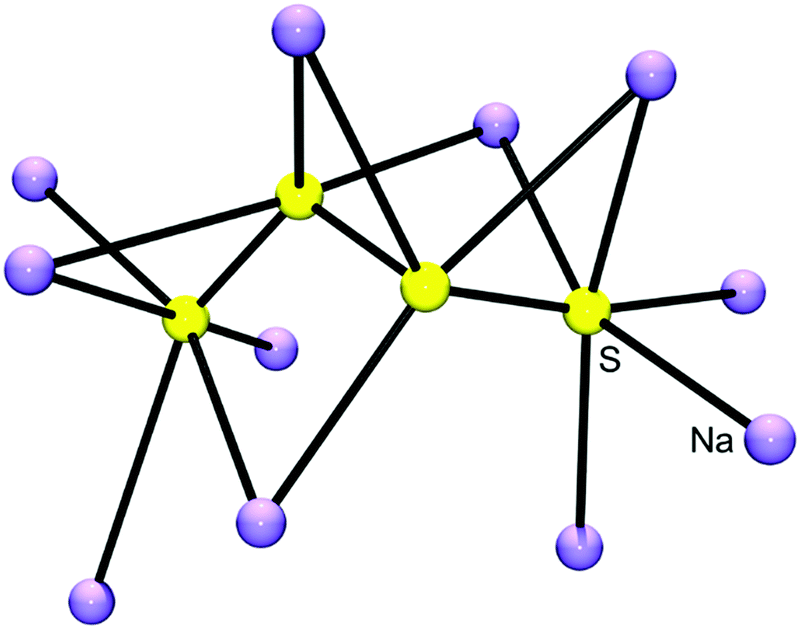 | ||
| Fig. 5 Cationic environment of the tetrasulfide anion (yellow) in the crystal structure of Na2S4 calculated from the atomic coordinates of Tegman;40 shown are all cations (violet) surrounding the [S4]2− anion with Na–Sterm distances of between 283 and 289 pm. The terminal sulfur atoms of the anion are approximately octahedrally coordinated by five Na and one S atom (figure courtesy of H. Hartl). | ||
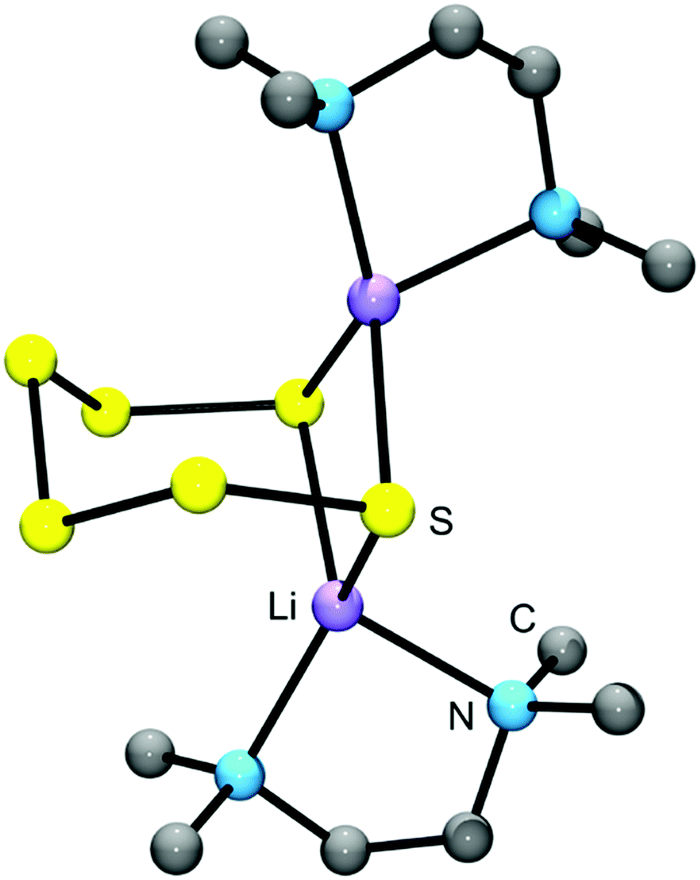 | ||
| Fig. 6 Solid-state structure of [Li(TMEDA)]2[S6] determined by X-ray crystallography (bond lengths: Li–S 249 pm, S–S: 204–208 pm; adapted from Tatsumi et al. 1990, ref. 41). | ||
Lithium polysulfides are expected to remain partly undissociated in organic solutions forming ion pairs such as [Li(solvent)Sn]−. In this way otherwise unstable, longer polysulfides are stabilized, since all polysulfide dianions are strong S-donor ligands and numerous metal complexes are known.43 The calculated structure of the ion pair [Li(THF)2Sn]− is shown in Fig. 7. Only ionic polysulfides with large complex cations like [K(crypt-2,2,2)]+, [R4N]+ and [R4P]+ (R = alkyl or phenyl group) do not engage in ion pair formation, since the positive charge is distributed over a large number of atoms in those cations. This important issue will be further discussed in Section 5.
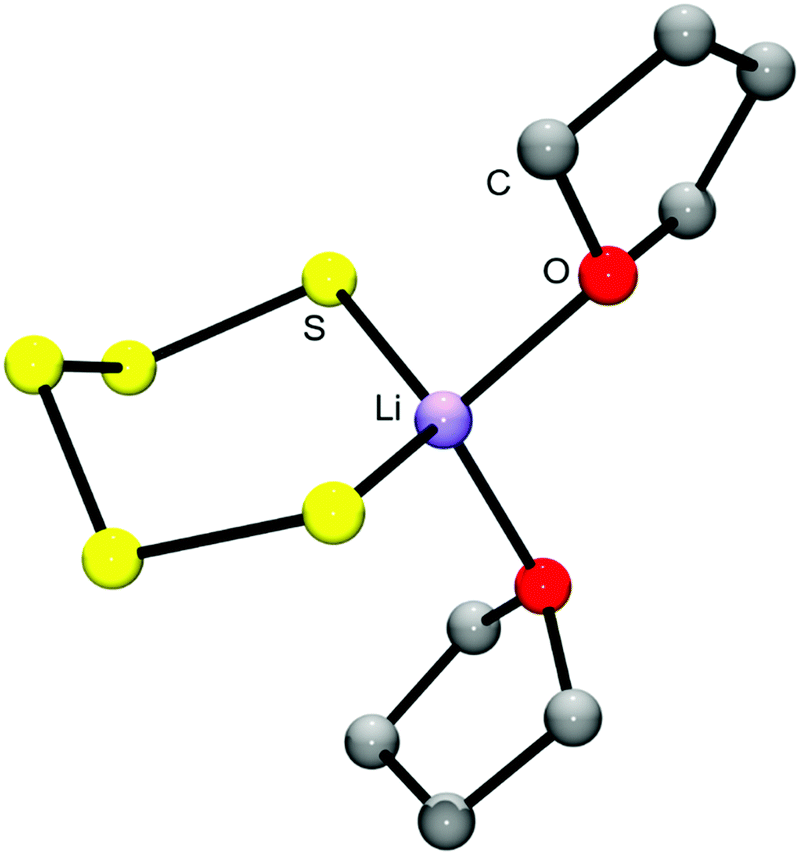 | ||
| Fig. 7 Molecular structure of the ion pair [Li(THF)2S5]− in the gas phase, calculated at the B3LYP/6-31G(d) level.44 The O2LiS5 fragment is approximately of Cs symmetry with bond lengths dSS = 210.6 and 212.1 pm, dLiS = 245.5 and 247.4 pm, dLiO = 204.0 and 205.2 pm. The fundamental vibrations in the region 310–600 cm−1 are all stretching modes of the O2LiS5 unit which are predicted as follows (in cm−1): 471 (SS), 463 (SS), 416 (SS), 401 (LiS), 386 (LiO), 363 (LiO/LiS/SS) and 361 (LiO/LiS) (adapted from ref. 44). | ||
The structures, vibrational spectra and thermodynamic properties of polysulfide anions and radical anions can also be predicted by quantum-chemical calculations both for the free ions (gas phase) as well as for the species in a polarizable medium of different dielectric constant simulating the influence of a solvent of a particular polarity (PCM calculations).44 However, free polysulfide dianions with up to 8 sulfur atoms are unstable in the gas phase or in vacuum since they either suffer spontaneous electron autodetachment or Coulomb explosion producing one or two radical monoanions, respectively, due to the strong Coulomb repulsion between the two excess electrons.45 Therefore, the electrospray mass spectra of sodium trisulfide, tetrasulfide and a mixture of potassium polysulfides, dissolved in ultra-pure water, recorded by Nagy et al. in 2019 in the negative ion mode have been found to be quite similar and showed only peaks due to radical anions with not more than three sulfur atoms (Section 3.3).46 However, longer-chain polysulfide ions with up to nine sulfur atoms were detected by negative ion mass spectrometry when the dianions were protonated to [HSn]− during electrospray using methanolic solutions with ammonium acetate buffer as reported by Gun et al. in 2004.47 In that case the overall charge of the gaseous anions is reduced by 50% and Coulomb explosion does not take place.
Penta- and hexasulfide anions are also unstable with respect to homolytic dissociation in solvents of low polarity such as THF (ε ≈ 8). However, all dianions [Sn]2− do become thermodynamically stable with respect to dissociation at one of the central bonds in solvents of higher polarity such as water (ε = 78).44 Therefore, solutions of Li2S6 in organic solvents of low polarity like tetrahydrofuran (THF), dimethoxyethane (DME) or tetraethyleneglycol dimethylether (TMGME) with dielectric constants of between 7.2 and 7.8 are of pale green to blue colour at low concentrations, while in dimethylacetamide (DMA, ε = 38) the intense blue colour of [S3]˙− is observed.48,49 The green colour may be explained by a mixture of [S2]˙− (yellow) and [S3]˙− (blue). The higher polarity and larger donor number of DMA (DN = 28) result in stronger cation–solvent interactions reducing the formation of ion pairs such as [LiS6]− in which the polysulfide dianion is stabilized against homolytic dissociation. Aqueous solutions of lithium polysulfides are yellow to orange-red depending on the chain length and the concentration, indicating a negligible (undetectable) radical concentration.
In general, the dissociation equilibrium of a polysulfide dianion to the corresponding monoanions depends on (a) the thermodynamic properties of the ion, (b) the temperature, (c) the concentration, (d) the dielectric constant of the solvent, (e) the ion strength of the solution, and (f) on the nature of the counterions.
From the most sophisticated calculations44 and reliable Raman as well as EPR spectra the following facts can be derived:
1. The small radical ions [Sn]˙− with n = 3–5 are chain-like while larger ions [Sn]˙− with n = 6–9 (and probably beyond) are cyclic but with one or two very long bonds.44,45
2. The Raman spectra of the polysulfide radical ions [Sn]˙− with n = 3–5 are characterized by fundamental modes in the region 330–600 cm−1.21,44,50
3. In solution the radical ions are in equilibrium with the corresponding dianions which are by far the dominant species at room temperature, but only in highly polar solvents and not in ether-based solvents as demonstrated by the concentration and temperature-dependence of the UV-vis absorption spectra;49 for Gibbs energies ΔG° of the homolytic dissociation reactions, see Section 4.
4. Electron paramagnetic resonance (EPR) spectra of lithium polysulfides dissolved in DMF exhibit a signal assigned to [S3]˙− and, at high sulfur content, a signal attributed to [S4]˙− is observed.51 A radical with an absorption maximum at 617 nm ([S3]˙−) in the UV-visible has also been detected by EPR spectroscopy in ether-based solvents.49
5. Low concentrations of the radical anions under equilibrium conditions can best be detected by resonance Raman spectroscopy exploiting their absorption bands near 400 nm ([S2]˙−) and, depending on the matrix, at 595–620 nm ([S3]˙−), respectively.8,52 Even the overtones 2ν1, 3ν1, 4ν1 and 5ν1 of [S3]˙− have been observed by resonance Raman spectroscopy.53,54
6. Stable concentrations of some of the radical anions can be obtained by matrix-isolation under non-equilibrium conditions, e.g. in ionic crystal structures such as NaI, KI, sodalite and other rigid matrices in which diffusion is not possible at room temperature.55
7. In molten alkali-metal polysulfides,50 as well as in high-temperature aqueous fluids containing potassium thiosulfate56 or other sulfur species under high pressure,53 trisulfide radical anions have been observed by Raman spectroscopy.
Calculated vibrational data for the four smallest polysulfide radical anions are summarized in Table 1 both for gaseous ions and for two solvents characterized by dielectric constants ε of 8 (e.g. tetrahydrofuran, THF) or 78 (e.g. propylene carbonate or water) using the polarizable continuum model (PCM).44
| Ion | Gas phase | PCM8-corrected | PCM78-corrected |
|---|---|---|---|
| [S2]˙− (D∞h) | 570 | 578 | 578 |
| [S3]˙− (C2v) | 559(b)/521(a)//222(a) | 544(b)/527(a)//222(a) | 538(b)/528(a)//222(a) |
| [S4]˙− (C2v) | 595(a)/570(b)/327(a)//281(b)/93(a)/87(a) | 589(a)/542(b)/344(a)//280(b)/109(a)/103(a) | 588(a)/533(b)/345(a)//279(b)/113(a)/105(a) |
| [S5]˙− (Cs) | 556(a′)/535(a′′)/424(a′)/332(a′′)//226(a′)/177(a′′)/155(a′)/60(a′′)/44(a′) | 548(a′)/495(a′′)/441(a′)/352(a′′)//244(a′)/176(a′)/175(a′′)/83(a′′)/45(a′) | 545(a′)/479(a′′)/444(a′)/356(a′′)//249(a′)/179(a′)/174(a′′)/87(a′′)/52(a′) |
Anharmonic vibrational frequencies of gaseous [S3]˙− have been calculated by two different methods at the RCCSD(T) level with the cc-pVQZ basis set as follows (values in cm−1): ν3 = 595/599(b2), ν1 = 543/543(a1), 2ν2 = 450/453(a1), ν2 = 225/226(a1).57 The calculated data are in excellent agreement with the following experimental wavenumbers (cm−1):
[S2]˙−: 590 (in sodalite58), 589 (in NaI) and 594 (in KI).6
[S3]˙−: 580–590 (ν3), 545–550 (ν1), 220–265 (ν2) in sodalite52,58 and in high-temperature high-pressure aqueous fluids;53 534 (ν1) in Li–S cells54,59 (Section 5.1.3).
While the radicals entrapped in natural minerals most probably result from reduction of sulfate ions by some carbon-containing material, the sulfur-doped alkali halides are prepared by strong heating of elemental sulfur with the crystalline halides. In this reaction an electron is transferred from a halide ion to a small sulfur molecule such as S2 or S3, which are components of sulfur vapour and of hot sulfur melts and which possess large electron affinities. The sulfur radical anion then takes the site of the former halide anion in the crystal structure.
The radical ion [S4]˙− has not yet been observed convincingly by Raman spectroscopy, but EPR spectra of [S4]˙− have been reported (Section 3.5).51 An absorption band at 520 nm previously assigned to [S4]˙− is in fact due to the red molecule S4 which is of C2v symmetry60 and characterized by a Raman line at 678 cm−1 which does not fit any vibrational wavenumber calculated for [S4]˙− (see Table 1). [S5]˙− and larger polysulfide radical ions with up to 25 atoms have been observed by mass spectrometry,61 but only [S6]˙− has been isolated in a crystal structure, namely with the rather innocent [Ph4P]+ cation9 and no vibrational spectra of this cyclic radical anion have been published.
In 2013 Tübke et al. reported vibrational wavenumbers for polysulfide dianions and monoanions with up to 8 sulfur atoms both for the gas phase and for THF solution, calculated at the B3PW91/6-311G(2df,p) level.62 Not all of these frequencies agree with well-established experimental data or with the statements given above. For example, the stretching modes of [S3]˙− have been observed at 584 (νas) and 550 cm−1 (νs),52 while Tübke et al. calculated these modes at 522 (νas) and 535 cm−1 (νs) which is the incorrect order. Furthermore, intense Raman signals were predicted by these authors for the dianions [Sn]2− (n = 6–8) in the region 300–400 cm−1, which contradicts experimental spectra of pure polysulfides recorded at room temperature as shown above. Since the authors did not publish the calculated geometries of the di- and monoanions, the reason for the deviation from experiments and other theoretical calculations cannot be identified. In Section 4 we will show that certain DFT calculations on thermodynamically unstable (gaseous) polysulfide anions may result in partly unreliable geometries and frequencies.
Since there is a mathematical relationship between SS stretching frequencies and the corresponding S–S bond lengths in polysulfur compounds,63 the geometrical parameters of the polysulfide dianions in crystalline salts6 can be used to demonstrate that SS stretching modes below 390 cm−1 are unlikely in chain-like anions [Sn]2− (n = 3–8).
3.2 UV-visible spectra
The colours of polysulfide dianions [Sn]2− range from pale-yellow for the disulfide ion via yellow-orange for the intermediate chain-length species to orange-red for the longer chains. In principle, therefore, UV-visible spectra should be a useful diagnostic tool for the identification of individual dianions. However, in electron-pair donating solvents, e.g. DMSO, DMF, THF and in aqueous solutions these species exist as an equilibrium mixture of various polysulfide dianions formed via disproportionation. The interpretation of the UV-visible spectra of such solutions is further complicated by the generation of highly coloured radical anions upon dilution via partial dissociation of the dianions in non-aqueous solvents. The most prevalent of these is the bright blue trisulfide radical anion [S3]˙− with a solvent-dependent absorption band in the range 595–620 nm, while the yellow [S2]˙− has λmax ≈ 400 nm.8 The reported assignment of an absorption band for the red [S4]˙− radical anion is controversial (vide infra). Consequently, UV-visible spectra of polysulfide solutions should be interpreted cautiously in conjunction with data obtained by other techniques, especially Raman spectra (Section 3.1) and derivatization experiments followed by chromatographic separation (Section 3.4).This caveat is clearly illustrated by several independent studies of the UV-visible spectra of lithium polysulfides prepared according to eqn (1) during the period 2012–2014.64–67 The consistent finding of these investigations is that, within the spectral range 300–600 nm, the absorption maxima for long-chain polysulfide dianions occur at higher wavelengths, whereas short-chain polysulfide dianions exhibit absorption bands closer to the UV part of the spectrum. Operando UV-visible spectroscopy has been used by Lu et al. in 2018 as part of an investigation of the redox chemistry that occurs in alkali metal–sulfur batteries with a view to understanding the influence of the alkali-metal cation.22 These authors had previously reported on the basis of a rotating-ring disk electrode study that the initial electrochemical reduction of cyclo-S8 produces the dianionic [S8]2− chain.68 The reduction was carried out in DMSO containing a M[ClO4] electrolyte (M = Li+, Na+, K+ or Cs+) and monitored by recording the UV-visible spectra. The following assignments of absorption bands for polysulfide dianions and radical anions were invoked based on previous data from the literature:15a [S8]2− (492 nm), [S6]2− (475, 350 nm), [S4]2− (420 nm), [S3]2− (340, 270 nm) and [S3]˙− (617 nm). The odd-membered species [S7]2− and [S5]2− were ignored in this study despite the fact that [S5]2− is a thermodynamically favored polysulfide dianion (Section 4). Interestingly, in contrast to the behaviour of polysulfide dianions in electron-pair donor solvents, Yellowlees and co-workers found that the formation of the blue trisulfide radical anion [S3]˙− only occurs to a small extent in the ionic liquid [C4mim][DCA] (C4mim = 1-butyl-3-methylimidazolium; DCA = dicyanamide).69 By contrast, Seddon and co-workers had shown earlier that elemental sulfur produces highly coloured solutions involving an equilibrium between [S6]2− and [S3]˙− ions in the ionic liquid tributylethylphosphonium diethylphosphate [Bu3EtP][(EtO)2PO2].8,70
The formation of the unbranched [SSNO]− anion from the reaction of NO with sulfide is a significant process in biological signaling and may involve the intermediate formation of polysulfide dianions (Section 5.3).71 A recent study provides a compelling illustration of the capability of UV-visible spectroscopy for monitoring reactions of polysulfide radical anions. In an elegant experiment Pluth and co-workers followed the reaction of the blue trisulfide radical ion [S3]˙− (617 nm) with NO by UV-visible spectroscopy.31 The reagent [S3]˙− was produced by homolytic dissociation of the polysulfide dianion in [Bu4N]2[S6], which was generated from the reaction of cyclo-S8 and [Bu4N][HS] in THF as described in Section 2. The formation of the red [SSNO]− anion (446 nm) in this reaction is depicted convincingly in Fig. 8.
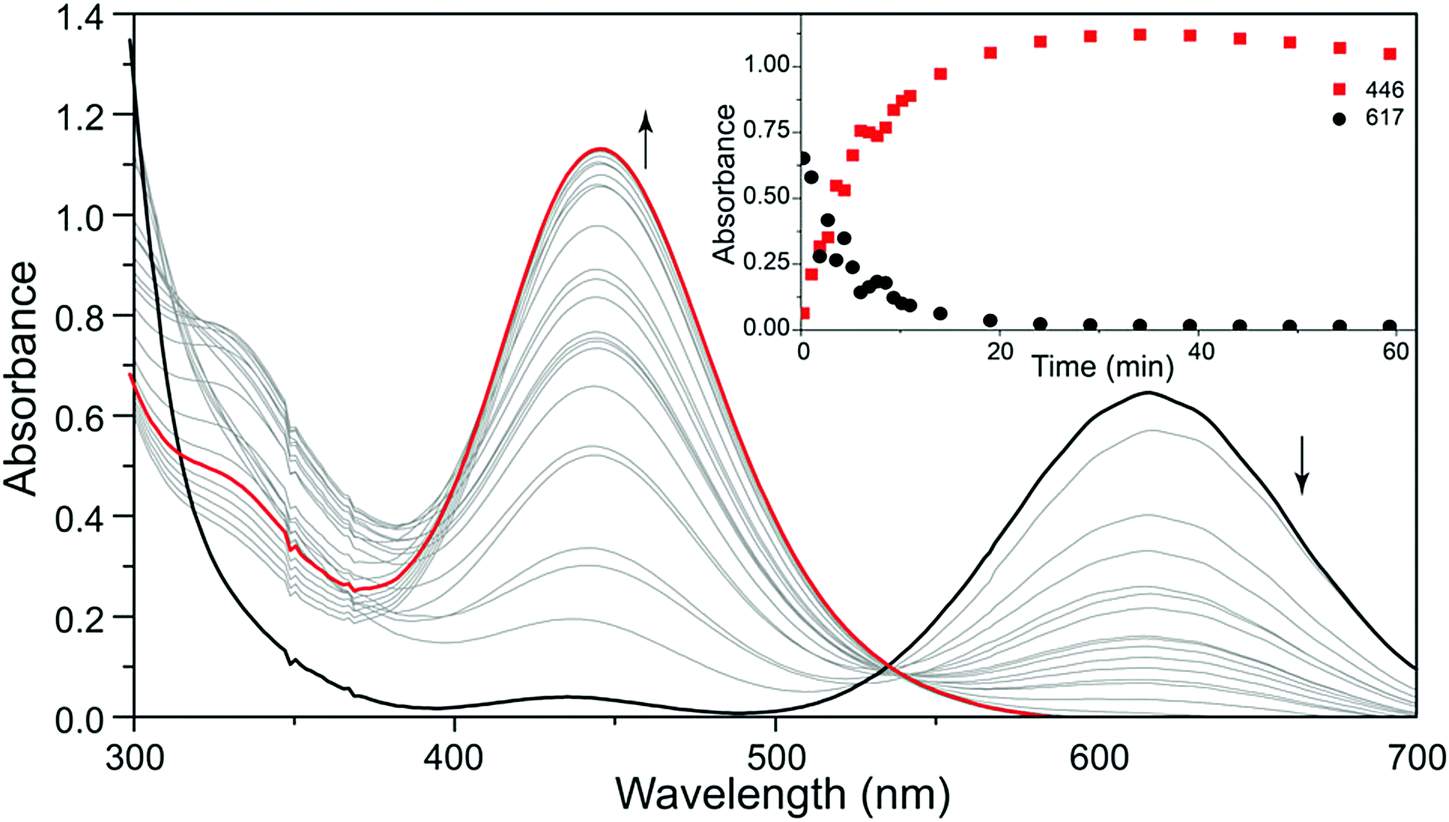 | ||
| Fig. 8 UV-visible spectra of the reaction of [S3]˙− with NO to give [SSNO]− (red curves). Reproduced from ref. 31 with permission. Copyright 2016, American Chemical Society. | ||
UV-visible spectroscopy has also been used by Akaike, Nagy et al. to estimate the variation in the concentrations of polysulfide radical anions [Sn]˙− (n = 2–4) generated from the reaction of cyclo-S8 and [Bu4N][HS] as a function of reaction stoichiometry and temperature. The reaction was carried out in various stoichiometries (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, 1:3 and 1:8) in dilute d8-THF/CD3CN solutions for the temperature range 20–60 °C.46 The authors concluded that (a) at room temperature [S4]˙− (490 nm) is the predominant species, (b) as the temperature is increased more [S3]˙− (610 nm) is formed, and (c) [S2]˙− (426 nm) is the primary species for the 1:8 stoichiometry, but the equilibrium is shifted to [S3]˙− as the temperature is increased for this stoichiometry. However, these conclusions should be viewed with circumspection owing to a potential overlap of the band attributed to [S4]˙− (490 nm) with that assigned to the formal dimer [S8]2− (492 nm) (vide supra). Furthermore, time-dependent DFT calculations by Fabian et al. in 2006 using different hybrid functionals predicted a very weak electronic transition in the visible region with an absorption maximum in the range 562–604 nm for [S4]˙−, whereas the same theoretical study predicted values of 608–617 nm for [S3]˙− in good agreement with the experimental data (vide supra).60
:1, 1:2, 1:3 and 1:8) in dilute d8-THF/CD3CN solutions for the temperature range 20–60 °C.46 The authors concluded that (a) at room temperature [S4]˙− (490 nm) is the predominant species, (b) as the temperature is increased more [S3]˙− (610 nm) is formed, and (c) [S2]˙− (426 nm) is the primary species for the 1:8 stoichiometry, but the equilibrium is shifted to [S3]˙− as the temperature is increased for this stoichiometry. However, these conclusions should be viewed with circumspection owing to a potential overlap of the band attributed to [S4]˙− (490 nm) with that assigned to the formal dimer [S8]2− (492 nm) (vide supra). Furthermore, time-dependent DFT calculations by Fabian et al. in 2006 using different hybrid functionals predicted a very weak electronic transition in the visible region with an absorption maximum in the range 562–604 nm for [S4]˙−, whereas the same theoretical study predicted values of 608–617 nm for [S3]˙− in good agreement with the experimental data (vide supra).60
3.3 Mass spectrometry
Electrospray mass spectrometry (ESI-MS) is a powerful, soft ionization tool that can potentially distinguish between polychalcogenides of different chain length in solutions. In 2004, both ion trap and electrospray time-of-flight mass spectrometry were applied to the analysis of aqueous solutions of sodium tetrasulfide Na2S4 in the pH range of 6–10 and in the presence of methanol and ammonium acetate for protonation of the gaseous dianions. Remarkably, the most abundant species detected in the negative ion mode was the hexasulfide [HS6]−, although significant amounts of other polysulfides [HSn]− (n = 4, 5, 7, 8, 9) were also evident. At higher capillary temperatures (>140 °C) disproportionation and redox processes took place in the ESI chamber,47 as predicted on the basis of chemical reasoning.72 Interestingly, the laser desorption ionization mass spectra of cyclo-S8 in the negative ion mode gives rise to peaks for the monoanions [Sn]˙− (n = 1–8, 10). The heavier clusters (n = 4–8, 10) appear at relatively low laser power, while [S3]˙− dominates the spectra at intermediate energies.11In 2015 high-resolution mass spectrometry was employed by Cortese-Krott et al. to confirm the presence of polysulfide radical anions [Sn]˙− (n = 2–4) in dilute (100 μM) aqueous solutions of Na2Sn (n = 3, 4).71 The most abundant species observed was the trisulfide radical anion [S3]˙− (m/z ≈ 96). In addition, peaks at m/z ≈ 64 and ≈128 indicated the possible presence of [S2]˙− and [S4]˙−. These tentative assignments were seemingly confirmed by high-resolution mass spectra obtained from triple quadrupole experiments, which gave the following m/z values (calculated masses are given in parentheses): [S2]˙− 63.9469 (63.9448), [S3]˙− 95.9170 (95.9168), [S4]˙− 127.9057 (127.8890). The assignment of these m/z values to polysulfide radical monoanions rather than dianions with twice the number of sulfur atoms was verified by inspection of the isotopic distribution patterns.71 However, the assignment of the peak at m/z = 127.9057 to [S4]˙− is probably in error since the almost isobaric ion [S3O2]˙− fits the observed mass much better (calcd 127.9060); the latter ion may result from the reaction of [S3]˙− with O2, which evidently has not been excluded carefully enough. The peaks observed at m/z 80 and 119 may be assigned to [SO3]− and [NaS3]−, respectively. The reaction of [S3]˙− with O2 will be further discussed in Section 5.3.
3.4 Chromatography and NMR spectra of polysulfide dianions after derivatization
The exact speciation of polysulfide dianions in solutions cannot be determined by vibrational spectroscopy due to the many overlapping signals and due to possible homolytic dissociation (Section 3.1). Since the various anions exist in a rapidly established dynamic equilibrium, direct chromatographic separation by either HPLC or ion chromatography is also not possible. Therefore, indirect analytical methods have been developed using rapid derivatization of the dianions to produce less reactive end-capped polysulfanes R–Sn–R, which then can be identified by liquid chromatography, mass spectrometry or 1H NMR spectroscopy.Protonation of aqueous polysulfide anions by cold concentrated hydrochloric acid yields an oily mixture of water-insoluble polysulfanes H2Sn (n = 1–25) as shown by 1H NMR spectroscopy.12 However, it is not always clear during derivatization reactions of this type whether exchange of sulfur atoms between anions or partly protonated ions takes place before full protonation and kinetic stabilization is achieved, e.g.:
| [HSx]− + [Sy]2− → [HSm]− + [Sn]2− (x + y = m + n) | (4) |
| 2H2Sn → H2Sn−x + H2Sn+x | (5) |
| H2Sn → H2Sn−8 + S8 (n > 8) | (6) |
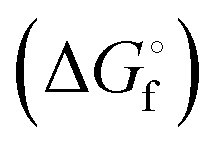 of the ions [HSn]− and of the sulfanes H2Sn (n = 2–8) see ref. 73 and 74.
of the ions [HSn]− and of the sulfanes H2Sn (n = 2–8) see ref. 73 and 74.
End-groups that are less reactive than hydrogen atoms have been developed taking advantage of the chemical and thermal stability of the carbon–sulfur bond. Numerous organic polysulfanes R–Sn–R with up to 13 sulfur atoms have been prepared as pure compounds and have been characterized by X-ray diffraction on single crystals; even much longer sulfur chains have been detected by HPLC techniques.13,75 Therefore, alkylation of polysulfide anions in solution seems to be a suitable derivatization procedure. A first attempt was made by Rethmeier et al. in 1997 using monobromobimane (BmBr) as a reagent to convert inorganic sulfur anions into derivatives which were suitable for fluorescence detection after HPLC analysis.76 Bimane (Bm) is a bicyclic diketone with a central NN bond and four methyl groups one of which has been brominated in BmBr; its main property is the intense fluorescence (excitation at 380 nm and emission at 480 nm). Unfortunately, the reaction time of the brominated derivative BmBr with nucleophiles such as sulfide, sulfite, thiosulfate and polysulfides (30 min at 25 °C) is quite high and specific polysulfides could not be identified by this method. Nevertheless, this method has been applied several times in microbiology for the quantitative trace analysis of certain sulfur compounds in bacterial cultures (Section 5.3), e.g.:
| 2Bm–Br + [HS]− → Bm–S–Bm + 2Br− + H+ | (7a) |
| 2C6H5CH2Cl + [Sn]2− → (C6H5CH2)2Sn + 2Cl− | (7b) |
Argyropoulos et al. in 2006 used dimethyl sulfate as an alkylating agent for an aqueous solution of nominal composition Na2S2.58 at 25 °C. However, the reaction time of 2 h together with the absence of a buffer to keep the pH constant (despite sulfuric acid production by hydrolysis of Me2SO4) may be responsible for the observation that mainly Me2S2, Me2S3 and Me2S4 were detected by 1H NMR spectroscopy, while the penta- and hexasulfane were present in trace concentrations only and longer-chain species Me2S>6 were completely absent in the derivatized polysulfide solutions.78
The formal disproportionation of polysulfide anions in solution is usually expressed by equations such as:
| [Sy]2− + [Sz]2− = 2[Sx]2− (with y + z = 2x) | (8) |
| [Sx]2− + [HS]− + [OH]− = [Sy]2− + [Sz]2− + H2O (x + 1 = y + z) | (9a) |
| S8 + [HS]− + [OH]− = [S9]2− + H2O | (9b) |
| [Sn]2− + 2CF3SO3CH3 → Me2Sn + 2[CF3SO3]− | (10) |
Reversed-phase HPLC was then used to separate and determine the dimethyl polysulfanes (Fig. 9).
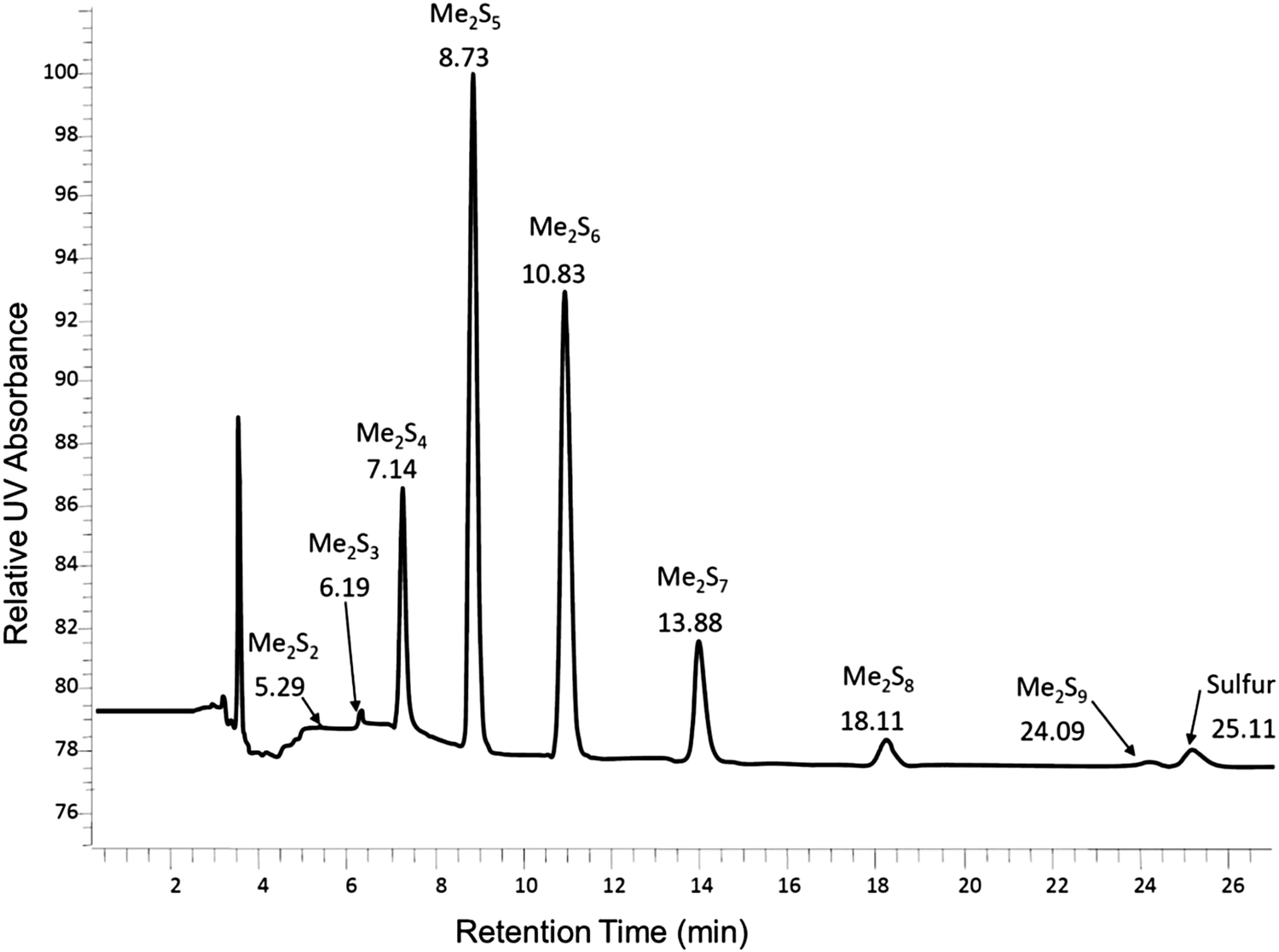 | ||
| Fig. 9 Chromatogram of the dimethyl polysulfane mixture obtained by derivatization of aqueous K2S5 by methyl triflate in a phosphate buffer at 25 °C (figure courtesy of A. Kamyshny, Jr).73 | ||
This sophisticated technique together with isotopic dilution experiments allowed the following facts to be established:73
1. When an aqueous solution of Na2S4 with natural abundance of isotopes and a solution of Na234S4 were mixed the isotopic exchange between the various anions was ca. 70% complete within 8 s.
2. When solid K2S5 was dissolved in water using a phosphate buffer the anions detected after derivatization with CF3SO3CH3 were (in the order of decreasing concentrations): [S5]2− ≫ [S6]2− > [S4]2− ≫ [S7]2− > [S3]2− > [S8]2− > [S2]2−.
3. In aqueous polysulfide solutions saturated with elemental sulfur (S8) the ratio of the activities of any two polysulfide species is constant at equilibrium and invariant to pH change, i.e. the distribution of the polysulfide dianions remains constant regardless of the overall concentration.
4. In unsaturated (dilute) polysulfide solutions decreasing pH values favour larger chain-lengths until sulfur precipitation occurs at pH < 7. S8 is thought to be ejected from either [S9]2− or [HS9]− by protonation and elimination of [HS]− or H2S (or from longer polysulfide chains).
The polysulfide derivatization by methyl triflate has later been modified to analyze polysulfides at low concentrations as in environmental samples.79,81 In addition, it was shown in 2006 that oxidation of aqueous hydrogen sulfide by H2O2 produces initially all polysulfide dianions from [S3]2− to [S8]2− before sulfate ions are formed eventually.81
When aqueous polysulfide solutions are heated, slow disproportionation of the “zero-valent” sulfur atoms (S0) within the chains and formation of thiosulfate are observed; the same holds for the disproportionation of elemental sulfur suspended in alkaline solutions:12
| 1/2S8 (or 4S0) + 4[OH]− → 2[HS]− + [S2O3]2− + H2O | (11) |
| [HS]−(aq.) + (n − 1)/8S8(s.) → [Sn]2−(aq.) + H+(aq.) | (12) |
In Table 2 the thermodynamic results for room temperature are summarized [using 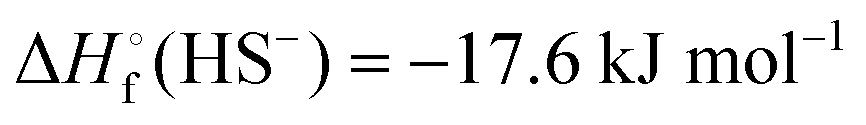 ]. The equilibrium constants (pK values) for the reactions in eqn (12) are: 11.5, 10.4, 9.7, 9.5, 9.7, 10.2 and 10.8 for n = 2 to 8.73
]. The equilibrium constants (pK values) for the reactions in eqn (12) are: 11.5, 10.4, 9.7, 9.5, 9.7, 10.2 and 10.8 for n = 2 to 8.73
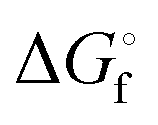 and enthalpies
and enthalpies 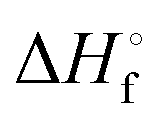 of formation (in kJ mol−1) as well as entropies S° (in J mol−1 K−1) of aqueous polysulfide dianions [Sn]2− with n = 2–8 derived from the analysis of the reaction products of solid elemental sulfur (S8) with aqueous hydrogen sulfide according to eqn (12) (adapted from ref. 74)
of formation (in kJ mol−1) as well as entropies S° (in J mol−1 K−1) of aqueous polysulfide dianions [Sn]2− with n = 2–8 derived from the analysis of the reaction products of solid elemental sulfur (S8) with aqueous hydrogen sulfide according to eqn (12) (adapted from ref. 74)
| Parametera | n = 2 | n = 3 | n = 4 | n = 5 | n = 6 | n = 7 | n = 8 |
|---|---|---|---|---|---|---|---|
a Slightly different 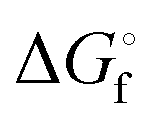 values have been published by Kamyshny et al. in 2004.73
b Extrapolated value. values have been published by Kamyshny et al. in 2004.73
b Extrapolated value.
|
|||||||
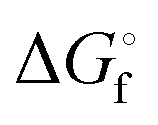
|
77.4 ± 1.3 | 71.1 ± 0.7 | 67.1 ± 0.1 | 66.0 ± 0.1 | 67.4 ± 0.1 | 70.7 ± 0.3 | 74.9 ± 0.5 |
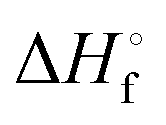
|
13.0b | 6.6 ± 0.1 | 9.0 ± 0.1 | 9.6 ± 0.1 | 13.3 ± 0.1 | 16.5 ± 0.1 | 23.8 ± 0.2 |
| S° | −22b | 9 ± 4 | 63 ± 1 | 100 ± 2 | 139 ± 1 | 171 ± 4 | 213 ± 8 |
From the data in Table 2 it follows that aqueous [S5]2− is thermodynamically favoured over other chain lengths and that the enthalpy change by consecutive addition of one sulfur atom at a time to a polysulfide chain gradually increases with the length of the sulfur chain. Thus, the disulfide to trisulfide transition is slightly exothermic, but since the reaction enthalpy is gradually increasing the conversion of pentasulfide to hexasulfide is already endothermic; the same holds for the higher polysulfides. While at low values of n the main driving force for further addition of a sulfur atom to the polysulfide chain is the exothermicity of the reaction, at high values of n the entropy gain becomes a more important factor in overcoming the endothermic nature of the reaction. This entropy increase results from the increasing number of rotational isomers as the chain-length grows due to the almost free rotation about the SS bonds and the many possible conformations of the larger anions; the torsional angle at each SS axis can be either positive or negative. The average length of the polysulfide chains in an equilibrium mixture increases with temperature, since species with n > 5 increase in concentration at the expense of the shorter chains.74
Zheng et al. used the fast derivatization of polysulfides by methyl triflate in 2016 to identify the anions obtained by electrochemical reduction of S8 in a Li–S battery.82 It was convincingly shown that in situ derivatization within the electrochemical cell gives results different from those of ex situ derivatizations as used by previous researchers. The details will be discussed in Section 5.1.3.
3.5 EPR spectra of polysulfide radical anions
Species with an unpaired electron give rise to a strong signal in the electron paramagnetic resonance (EPR) spectrum. Consequently, EPR spectroscopy is a valuable diagnostic technique for the detection of polysulfide radical anions [Sn]˙− (n = 2, 3 or 4) either in solution or in the solid state. As discussed in earlier reviews,6,8 the radical anions [Sn]˙− (n = 2, 3) have been identified by EPR spectroscopy in ultramarine pigments. The trisulfide radical anion [S3]˙− predominates in ultramarine blue, while [S2]˙− is also present in ultramarine green and it is the primary component in ultramarine yellow. There is controversy over the identity of the chromophore in ultramarine red as either the neutral S4 allotrope or the radical anion [S4]˙− (vide infra).Ultramarines are aluminosilicate sodalite minerals Na8[Al6Si6O24]Cl2 with a zeolitic architecture. The cavities in this framework incorporate Na+ cations and Cl− anions some of which may be replaced by [Sn]˙− radical anions. Early work attributed distinct EPR signals to two different trisulfide radical anions [S3]˙−. The most common one was an isotropic signal with g = 2.029, while other reports associated [S3]˙− with a rhombic g tensor with values of 2.005, 2.036 and 2.046. In order to explain this discrepancy, Goldfarb et al. carried out high-field EPR and ENDOR (electron nuclear double resonance) measurements on three blue pigments that were obtained by calcination of sodalite samples prepared from aluminum sulfate and [Me4N][OH] at high temperatures.83a On the basis of the spectroscopic results together with DFT calculations, two different [S3]˙− radicals, both of which have an open C2v structure, were identified. The ENDOR spectra showed that the number and location of 23Na+ cations around these species are different. A third [S3]˙− radical with an isotropic g value was also detected in some samples.83a Subsequently, Goslar et al. described combined EPR and electron spin echo studies of the structure and dynamics of [S3]˙− in ultramarines and determined g values of 2.016, 2.0355 and 2.0505 in the temperature range 4.2–50 K.83b Above 300 K re-positioning of [S3]˙− between 12 possible orientations in the sodalite cage with a very low energy barrier results in a single line with an average g value of 2.028.83b In 2011 Raulin and co-workers provided the first unambiguous EPR identification of [S2]˙− by using continuous wave and field-sweep echo detected EPR.83c The highly anisotropic g values were determined to be 2.69(6), 2.03(4) and 1.86(4).83c In 2018 Rejmak performed all-electron DFT calculations of [S3]˙− and [S2]˙− in ultramarine pigments with periodic structure optimization and showed that different orientations of [S3]˙− within a sodalite cage have little effect on the calculated g tensor components, which were in excellent agreement with experimental values.83d This author also found that embedded [S2]˙− ions are more sensitive to the local environment than [S3]˙−. Unfortunately, reliable values of the g tensor for [S2]˙− could not be obtained.83d
EPR spectra of solutions of lithium polysulfides Li2Sn in DMF show a strong signal for [S3]˙− radical anions when n < 6. For higher sulfur contents (n > 6) an additional EPR resonance attributed to [S4]˙− is observed.51 The concentration of this radical anion is very low and the equilibrium constant for the dissociation [S8]2− ↔ 2[S4]˙− was estimated to be about 10−9 M. The [S4]˙− radical anion has been considered for the identity of the chromophore in ultramarine red, but this assignment has been controversial (Section 3.2). In 2016 spectroscopic investigations of an ultramarine that was prepared by encapsulation of the red chromophore inside a microporous zeolitic imidazolate framework with Na2S4 as the sulfur source were reported.84 The lack of an EPR signal for the red solid product indicated the absence of any radical anions. However, the red solid exhibited a broad visible absorption band at ca. 525 nm (cf. ≈ 530 nm for ultramarine red), which was attributed to a neutral cis-S4 chain. This assignment is supported by time-dependent DFT calculations at different levels of theory, which yielded values in the range 542–564 nm for this isomer compared to 562–604 nm for the radical anion [S4]˙− (Section 3.2).71
During the past 4–5 years EPR spectroscopy has been employed to address the role of polysulfide radical anions in the redox chemistry that occurs in lithium–sulfur batteries as well as the use of the transient species [S3]˙− as a reagent in organic syntheses. Details of these applications are discussed in Sections 5.1 and 5.2, respectively.
3.6 Lithium and sodium NMR spectra of inorganic polysulfides
The limited natural abundance and quadrupole moment of 33S (I = 3/2, 0.76%) combined with the low symmetry of the sulfur centers render 33S NMR spectra uninformative for the characterization of polysulfide anions. On the other hand, both 7Li (I = 3/2, 92.6%) and 23Na (I = 3/2, 100%) have the potential to provide chemical shift data for the identification of individual alkali-metal polysulfides as long as the cations and anions are in contact. In this context several in situ7Li NMR studies have been carried out on the formation of lithium polysulfides during the discharge of lithium–sulfur batteries.85–88 In 2013 Nazar and co-workers reported the 7Li NMR spectra of solid lithium polysulfides prepared by the reduction of appropriate amounts of elemental sulfur with Li[Et3BH] in THF.85 However, with the exception of Li2S and Li2S6, which showed isotropic NMR shifts of 2.3 and 1.0 ppm, respectively, the other polysulfides with nominal compositions Li2Sn (n = 2, 4, 8) were found to be phase mixtures. In their 2017 study of the lithium–sulfur redox process Wang and co-workers were able to track the formation and evolution of lithium polysulfides by 7Li NMR spectroscopy and showed that it followed a four-step sequence (Section 5.1.3); however, 7Li chemical shifts for individual polysulfides could not be obtained.88By contrast, in 2016 Mali and co-workers employed solid-state 23Na NMR spectra, supported by X-ray diffraction patterns and ab initio computational predictions, to determine the 23Na chemical shifts of the different sodium crystallographic sites in the series Na2Sn (n = 1–5) and, hence, provide a quantitative estimate of the composition of samples of sodium polysulfides.18 The unit cells of known crystalline sodium polysulfides are shown in Fig. 10a and their 23Na magic angle spinning (MAS) NMR spectra are depicted in Fig. 10b, which reveals that the different crystalline sites in Na2Sn (n = 2, 4, 5) can be resolved. The capability of 23Na MAS NMR spectroscopy is demonstrated impressively by the spectrum of the Na2S3 sample, which clearly shows that this material is a mixture of almost equal amounts of β-Na2S2 and Na2S4.18
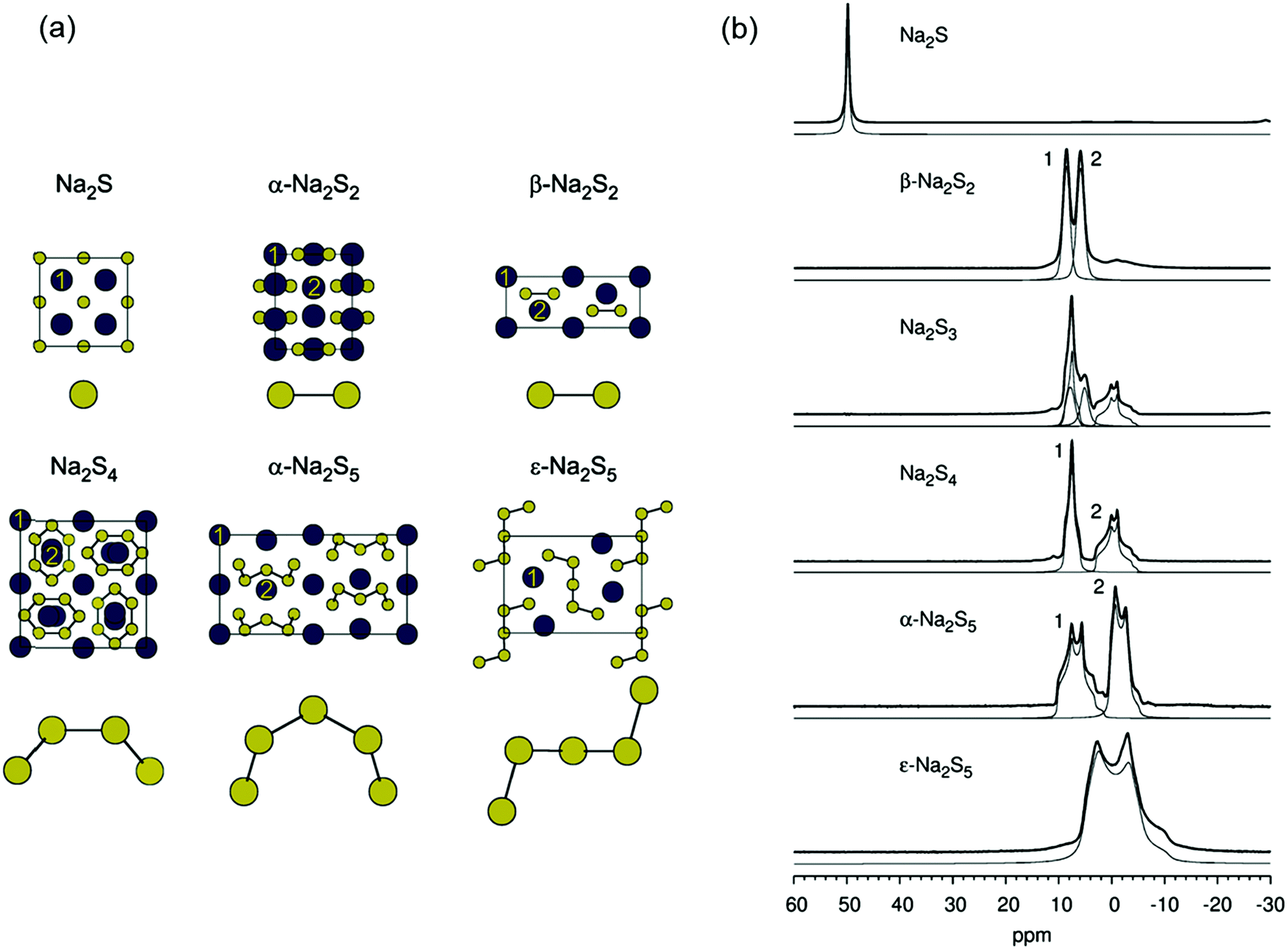 | ||
| Fig. 10 (a) Unit cells and (b) 23Na MAS NMR spectra of crystalline sodium polysulfides (blue circles = Na, yellow circles = S; crystallographically inequivalent Na sites are labeled 1 and 2. Reproduced from ref. 18 with permission from Wiley. Copyright 2016. | ||
3.7 X-ray absorption (XAS) and photoelectron spectra (XPS)
X-ray absorption spectroscopy (XAS) involves the determination of the energy of core electrons that are ejected when a sample absorbs X-rays. This technique can be applied to powders, liquids, solutions, polymeric materials or surfaces. XANES spectra are obtained by using synchrotron radiation with an energy of ca. 2500 eV. An X-ray absorption near edge structure (XANES) spectrum arises from excitation of core electrons (1s in the case of a K edge) to high-level vacant orbitals. For sulfur compounds the energy of the absorption depends on the oxidation state, which may vary from 2− in metal sulfides to 6+ in sulfate. In the specific case of polysulfides [Sn]2− different absorption lines are to be expected for the central and terminal sulfur atoms, since the negative charge resides predominantly on the latter (Section 4).89 Polysulfide radical anions [Sn]˙− (n = 2–4) may be expected to exhibit characteristic XANES spectra depending on the value of n. XANES is also well-suited to the in situ identification of sulfur species in biological samples that may contain long sulfur chains in the form of polythionates [O3S–Sn–SO3]2− as well as cyclo-S8 (Section 5.3).90Pioneering work by Fleet and co-workers on the sulfur K-edge XANES spectra of ultramarine pigments revealed a pre-peak at ca. 2468.0 eV, which was attributed to the presence of the unpaired electron in [S3]˙−. The intensity of this pre-peak is directly proportional to the depth of the blue colour of the mineral.91 Subsequent studies of sulfur K-edge XANES spectra of lithium polysulfides attributed the pre-edge to significant concentrations of [S3]˙−.92 However, this assignment has been questioned on the basis of DFT calculations, which revealed that the XAS of polysulfide dianions [Sn]2− (n = 2–8) are characterized by pre-edge and main-edge features in the same energy range.93,94 More recent studies dispute the assignment of the pre-edge feature in sulfur K-edge XAS of lithium polysulfides to [S3]˙−.95–97
In 2015, in order to provide a more solid foundation for the interpretation of XAS of lithium polysulfides, Pascal et al. conducted DFT-based simulations of the XAS of neutral sulfur allotropes and the corresponding radical anions and dianions ranging from two to eight sulfur atoms.98 They found that the source of the pre-edge feature near 2470 eV in the sulfur K-edge XAS is primarily due to core excitations of the terminal atoms of polysulfides to σ* orbitals. They also predicted a spectral feature that is unique to the radicals in the range 0.5–1 eV below the pre-edge that arises from 1s → σ* transitions of the terminal atoms. The energy of this feature for [S3]˙− was calculated to be 2468.5 eV, but its intensity is weak and it was suggested that it may only be observed for radical mol fractions >20%.98 This prediction was confirmed in the same year by Nazar et al. in an experimental study of dissolved lithium polysulfides in which [S3]˙− was identified by a pre-edge feature in the XAS near 2468.5 eV.99 In a contemporaneous study of intermediates produced in a lithium–sulfur battery, Gorlin and co-workers used XANES to determine the influence of solvent on the conversion of polysulfides to Li2S and found that this process proceeds more rapidly in a solvent with a low dielectric constant.100
Further to the earlier work of Fleet et al. (vide supra),91 sulfur K-edge XANES spectra have been used to determine the presence of polysulfide radical anions in a variety of solid-state materials, including the mineral lazurite from different geographical locations, slag-based materials such as mortars or concretes, and artists’ pigments. In 2016, Schmidt Patterson, Walton and co-workers analyzed samples of lazurite from Afghanistan, Russia, Chile, the USA, Iran, Tajikistan and Myanmar by XANES spectroscopy with a view to using this technique to determine the source of this mineral.101a In addition to a peak at 2482.5 eV attributed to sulfate, all averaged XANES spectra exhibited a peak at 2469.1 eV attributed to [S3]˙−, which was more prevalent than that of [S2]˙−. They also observed an envelope of peaks between 2470 and 2475 eV with a variable spectral profile. For example, samples from Chile showed two distinct peaks near 2471.7 and 2473.5 eV, whereas those from Afghanistan displayed an absorption maximum at 2472.5 eV implying a unique geological environment for the Chilean samples.
In an intriguing and somewhat related application of XANES spectroscopy carried out in 2018, samples taken during the extraction of the blue component, lazurite, from lapis lazuli employing the method used to generate a blue pigment for artists were analyzed.101b All spectra displayed a strong absorption peak at 2483 eV for sulfate, an envelope of peaks between 2470 and 2475 eV, and a pre-peak at 2469.1 eV attributable to [S3]˙−. This study elucidated the changes in composition of sulfur species that occur during the extraction process. Interestingly, an increase in the [S3]˙− content of the pigments was observed upon prolonged exposure to X-rays.101b
Slag-based materials such as mortars and concretes are known to exhibit a blue-green colour upon hydration. Analysis of slags activated with sodium silicate or Portland cement by XANES spectroscopy revealed that, in addition to major peaks for sulfate at 2482.5 eV and thiosulfate at 2481.0 and 2471.7 eV, a pre-peak at 2469.6 eV attributed to [S3]˙− was associated with the blue colour.102 The green colour of these slags is likely due to the additional presence of yellow [S2]˙−, cf. ultramarine green,8 but this species was not detected in the XANES study.102 The genesis of [S3]˙− can be attributed to the formation of polysulfides from sulfur(0) and sulfide (S2−), which were also detected in the XANES spectra. The simultaneous creation of aluminosilicates with a zeolitic structure provides a trap for the blue [S3]˙− radical anions.
X-ray photoelectron spectroscopy (XPS) also provides information about the energies of core electrons. Typically, Al Kα (1487 eV) excitation is used as the X-ray source and 2p binding energies in the region 160–172 eV have been determined for sulfur compounds. In 2015 Fantauzzi et al. utilized XPS for the identification of sulfides and polysulfides on surfaces.89 Commercial samples of anhydrous Li2S and Na2S, Na2S4 (90–95%; <5% H2O) and potassium polysulfide K2Sn were analyzed by this technique and it was possible to assign the S(2p) binding energies for sulfide S2− and the central and terminal S atoms in polysulfide chains. Sulfur in the terminal position is shifted by ca. 1.6 eV to lower binding energies compared to sulfur in a central position, confirming that the negative charge is mainly located at the end of polysulfide chains (Section 4).
In 2018 Cato et al. applied XPS to the determination of the ratio of polysulfide radical anions [S3]˙−/[S2]˙− in ultramarines ranging in colour from deep blue to green.103 The samples under investigation were swatches of synthetic ultramarine paints taken from the 1939 catalogue of a German manufacturer. Importantly, the same samples were also analyzed by Raman spectroscopy, which is the method of choice for determining [S3]˙−/[S2]˙− ratios (Section 3.1). The sulfur 2p spectra revealed peaks in the regions 161.6–161.9, 164.3–165.2 and 168.8–169.2 eV assigned to sulfide (S2−), sulfite (S4+) and sulfate (S6+), respectively. In addition, peaks at 162.9 and 163.8 eV were attributed to yellow [S2]˙− and blue [S3]˙− chromophores, respectively.103 The XPS measurements and the Raman spectra showed the same trend for the [S3]˙−/[S2]˙− ratio with markedly lower values for green compared to blue samples.
In the context of the assignments above for sulfur species in ultramarines, the interpretation of the sulfur 2p XPS spectrum of synthetic ultramarine red, prepared as described in Section 3.5,84 should be regarded as tentative except for the ubiquitous peak for sulfate, which is observed at 168.7 eV. In addition, a peak at 162.7 eV is assigned to polysulfide anions [Sn]2−, although the expected difference in energies for terminal and central S atoms (vide supra) is not mentioned. A third peak at 164.0 eV is attributed to a sulfur(0) species, possibly S4.84
In 2013 the high-resolution photoelectron spectrum using slow electron velocity-map imaging was reported for cryogenically cooled [S3]˙−.104 The electron affinity was found to be 2.6330(9) eV and the wavenumber of the bending mode ν2 was 260(2) cm−1, a somewhat lower value than determined for gaseous S3 (for more details on the vibrational spectrum see Section 3.1).
4 Computational studies of polysulfide dianions and radical anions
Reliable thermodynamic data on polysulfide di- and monoanions have only become available in recent years through high-level quantum-mechanical calculations taking the special requirements for second-row elements into account. As shown by Wong in 2003, higher levels of theory than those needed to obtain the underlying geometries are required to obtain satisfactory relative energies of isomeric structures as well as reliable activation energies. MP2 is the most economical and popular method to incorporate electron correlation. To predict thermochemical data with an accuracy of ±8 kJ mol−1, quantum-chemical calculations using high-level correlation methods such as QCISD(T) and CCSD(T) in conjunction with very large basis sets are required.61For sulfur compounds without highly electronegative substituents the Hartree–Fock (HF) method reproduces the experimental geometries reasonably well. Density Functional Theory (DFT) is nowadays the most frequently used method because of its relatively low cost. However, as shown below, HF methods yield more realistic geometries and vibrational frequencies of polysulfide dianions than DFT methods such as B3LYP, B3P86 and B3PW91. Nevertheless, the relatively new DFT methods G3X and G3X(MP2) in conjunction with B3LYP/6-31+G(2df,p) optimized geometries do yield reliable energies and enthalpies. The G3X(MP2) method represents a modification of the G3(MP2) method with the addition of a g polarization function to the G3Large basis set for the second-row atoms at the HF level.61 All three features are important for the proper description of sulfur-containing species including radicals and only data obtained at this or comparably high levels of theory will be discussed in this section.
The electron affinities of all neutral sulfur molecules Sn are positive, i.e. addition of an electron with formation of a radical anion is exothermic and exergonic both in the gas phase and in polarizable media with dielectric constants of between 8 (THF) and 78 (water). However, the addition of a second electron with formation of dianions is endothermic and endergonic in the gas phase, but exothermic and exergonic in the mentioned polarizable media.44 Therefore, the theoretical treatment of isolated anions [Sn]2− with n = 5–8 is problematic since their gas phase structures do not correspond to global minima on the potential energy hypersurface. Especially density functional theory does not always reproduce the experimental geometrical and vibrational data correctly as shown by the data for [S6]2− in Tables 3 and 4.
| S1–S2 | S2–S3 | S3–S4 | S4–S5 | S5–S6 | Ref. | |
|---|---|---|---|---|---|---|
| Cs2S6 (s.) | 203.7 | 204.3 | 206.3 | 206.4 | 202.5 | 21 |
| [S6]2− (g.) | 209.1 | 208.5 | 208.7 | 208.5 | 209.1 | 45 |
| [S6]2− PCM | 207.4 | 207.7 | 209.2 | 207.7 | 207.4 | 105 |
| [S6]2− (g.) | 207.4 | 209.4 | 215.8 | 209.7 | 207.4 | 44 |
| [S6]2− (ε = 8) | 207.5 | 208.3 | 213.2 | 208.3 | 207.5 | 44 |
| [S6]2− (ε = 78) | 207.6 | 208.2 | 213.0 | 208.2 | 207.6 | 44 |
In Table 3 the S–S bond lengths of Cs2S6 determined in 2015 by an X-ray crystallographic study are compared to the theoretical results obtained by different methods. Cederbaum et al. used a SCF Hartree–Fock technique with double-ζ basis set augmented with d-type polarization and s-type diffuse functions in 1998 (row 3).45 Evidently the calculated structural data agree reasonably well with the experimental results. The same holds for the bond lengths obtained by Tossell in 2012 with the cc-pvTZ CCSD method and PCM correction (row 4; without any specification of the dielectric constant).105 In contrast, the density functional calculations used later by other authors gave a too long central bond S3–S4 of the hexasulfide dianion (row 5) which, however, became shorter if the polarizability of the solvent was increased (rows 6 and 7).
The long central bond calculated for the hexasulfide anion by DFT methods also results in too low a wavenumber for the corresponding SS stretching mode, as shown in Table 4. This mode of a symmetry has been predicted by a normal coordinate analysis at 425 cm−1 using force constants from S8 and [NH4]2[S5] (row 4), but was predicted by DFT methods in the region 310–360 cm−1 (rows 5–7) while it has been observed by several authors near 409 cm−1 in solid Cs2S6 (rows 2 and 3).
Similar predictions using DFT methods have been made for the penta-, hepta- and octasulfide dianions for which one SS stretching mode each was calculated in the region 360–380 cm−1 (with ε = 8) which is silent in the Raman spectra of corresponding salts.44 Evidently, the cations in crystalline polysulfides withdraw enough electron density from the anions that the sulfur chain is stabilized and SS bond lengths shrink to normal single-bond values compared to the theoretical gas phase values as shown in Table 3.
As mentioned before, gaseous polysulfide dianions are subject to electron autodetachment and homolytic dissociation (Coulomb explosion). Symmetrical dissociations are more favourable thermodynamically than asymmetrical ones, but the activation energies for these processes are not known with certainty, especially for condensed phases. Vijayakumar et al. estimated the activation energies for the symmetrical cleavage of the hypothetical gaseous tetra- and hexasulfide dianions producing 2[S2]˙− and 2[S3]˙−, respectively.106 Using the functional PBE-D3 with all-electron TZ2P basis sets the cleavage of the central bonds was found to be slightly exothermic with activation energies of 1.25 eV (tetrasulfide) and 0.25 eV (hexasulfide). However, the overall reaction energies of ca. 0.20 eV do not agree with the results obtained at higher levels of theory which predict these reactions to be much more exothermic in the gas phase (see below, Table 5).
| Reaction | Symmetry of products |
|
|
|
|
|---|---|---|---|---|---|
| 1 | [S8]2− → 2[S4]˙− | C 2v | −109.1/−158.2 | +72.5/+22.2 (−23.5) | +96.7/+46.6 |
| 2 | [S8]2− → [S3]˙− + [S5]˙− | C 2v + Cs | −93.0/−139.9 | +79.7/+31.4 (−12.4) | +102.8/+54.8 |
| 3 | [S7]2− → [S3]˙− + [S4]˙− | C 2v + C2v | −143.8/−190.3 | +54.8/+6.4 (−37.3) | +97.1/+33.3 |
| 4 | [S6]2− → 2[S3]˙− | C 2v | −187.0/−230.7 | +34.2/−11.5 (−52.4) | +64.9/+19.0 |
| 5 | [S5]2− → [S2]˙− + [S3]˙− | D ∞h + C2v | −175.5/−215.0 | +63.1/−7.2 (−16.1) | +96.9/+ 55.6 |
| 6 | [S4]2− → 2[S2]˙− | D ∞h | −179.2/−215.0 | +86.5/+49.9 (−14.9) | +124.0/+87.2 |
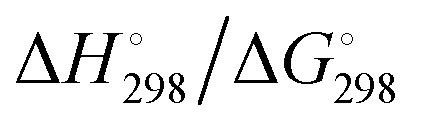
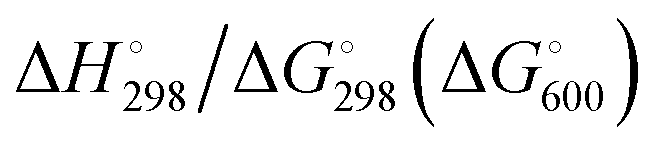
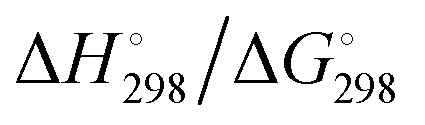
At the high-level G3X(MP2) theory with geometry optimization at the B3LYP/6-31+G(2df,p) level the dissociation enthalpies and Gibbs energies for the symmetrical and asymmetrical dissociation of penta- through octasulfide show that these reactions are all strongly exothermic and exergonic in the gas phase, but with two exceptions are endothermic and endergonic in a polarizable continuum (Table 5).44 In other words, the polarizable phase stabilizes these anions. The two exceptions are the slightly negative Gibbs free energies ΔG° for the homolytic dissociation of penta- and hexasulfide dianions in a continuum of ε = 8.0 (rows 4 and 5). This result is consistent with the observation that the trisulfide radical anion has been observed in many experiments and even in natural samples, unlike the di- and especially the tetrasulfide radical ions which have been observed more rarely. The value of 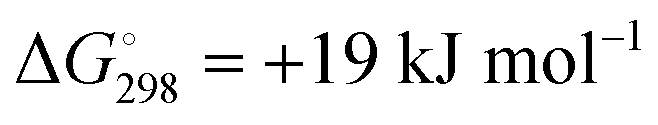 for ε = 78 for the dissociation of hexasulfide ions in water is in excellent agreement with the results reported by Tossell, who obtained
for ε = 78 for the dissociation of hexasulfide ions in water is in excellent agreement with the results reported by Tossell, who obtained 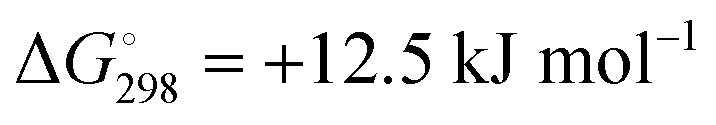 in 2012 by Hartree–Fock methods.105 The finding that the dissociation reactions are endergonic with ε = 78 indicates that dianions are more stabilized than the monoanions by this type of continuum.
in 2012 by Hartree–Fock methods.105 The finding that the dissociation reactions are endergonic with ε = 78 indicates that dianions are more stabilized than the monoanions by this type of continuum.
At 600 K all dissociation reactions listed in Table 5 are predicted to be exergonic in a polarizable continuum of ε = 8 (column 5) as in high-temperature Na–S batteries (see below). Solvents such as n-butyl acetate, ethylene glycol dimethyl ether (glyme), THF, trifluoroacetic acid, o-dichlorobenzene and even pyridine show dielectric constants at 25 °C in the range of 5 to 12, and the same holds for water at the critical point of 374 °C.
In the following parts of this section structural details and thermodynamic data of the radical monoanions and the related dianions, both for the free (gaseous) species and with the PCM corrections for the two polarizable phases ordered by increasing number of sulfur atoms, are discussed.
The diatomic species [S2]˙− and [S2]2− are of D∞h symmetry with the following calculated bond lengths: [S2]˙− 202.8 pm (gas), 202.7 pm (PCM8), 202.7 pm (PCM78); [S2]2− 218.1 pm (PCM8), 218.1 pm (PCM78).44 The experimental S–S bond length of the anion in α-Na2S2 is 215 pm which is in good agreement with the PCM-corrected value of 218 pm. The disulfide dianion cannot be calculated in the gas phase due to the expected electron autodetachment.
The triatomic ions [S3]˙− and [S3]2− are of C2v symmetry. The trisulfide radical anion has been most thoroughly studied by several theoretical methods to predict its structure, vibrational spectrum and UV-vis absorption wavelengths. The most elaborate calculation of this radical by Koch et al. using highly correlated MRCI and CCSD(T) wavefunctions together with large atomic natural orbital (ANO) basis sets confirmed the C2v structure as the global minimum on the potential energy surface and predicted the D3h isomer as higher in energy by ca. 170 kJ mol−1.107 The MRCI+Q/ANO6532 calculation yielded S–S bond lengths of 200.2 pm and an SSS bond angle of 115.1°. The computed electron affinity of S3 of 2.10 eV is in perfect agreement with photoelectron spectroscopic results (2.09 eV).107 Other methods gave similar geometrical values, for example the following bond lengths d (pm) and angles α obtained at the B3LYP/6-31+G(2df,p) level: [S3]˙− 201.8/115.8° (gas), 201.7/114.4° (PCM8), 201.7/114.1° (PCM78); [S3]2− 213.3/114.4° (gas), 211.9/111.2° (PCM78), 211.8/110.5° (PCM78). The NPA atomic charges of the terminal atoms of [S3]˙− and [S3]2− are −0.53 and −0.86 in the gas phase and −0.55 and −0.90, respectively, with both PCM8 or PCM78 correction.44 In solid K2S3 the experimental geometrical parameters of the anion are dSS = 208.3 pm and α = 105.4°,44 and in the room temperature form of Cs2S3: dSS = 212.6 pm and α = 106°.19
The unpaired electron of [S3]˙− resides in an antibonding π* orbital of b1 symmetry (SOMO) to which all atoms contribute almost equally.105 The dissociation energy as well as the equilibrium geometries and harmonic wavenumbers for six electronically excited states of [S3]˙− have been reported by Linguerri et al. in 2008 and the dissociation of [S3]˙− into S2 and S˙− was predicted to be strongly endothermic.57
Reliable experimental information on the tetrasulfide radical anion is scarce, but several theoretical calculations on its structure, fundamental vibrational modes, UV-vis spectrum and thermodynamics have been published.44,60,108,109 Contradictory results have been obtained since DFT calculations on sulfur compounds without additional energy refinements sometimes yield relative energies in conflict with results obtained at a higher level of theory. As shown above, molecular structures of dianions do not always agree with well-established experimental data such as X-ray structures. At the highest theoretical level applied so far [S4]˙− forms a cis-planar chain of C2v symmetry both in the gas phase and in a polarizable continuum of ε = 78. However, the trans-isomer of C2h symmetry with a centre of symmetry is only slightly less stable, by 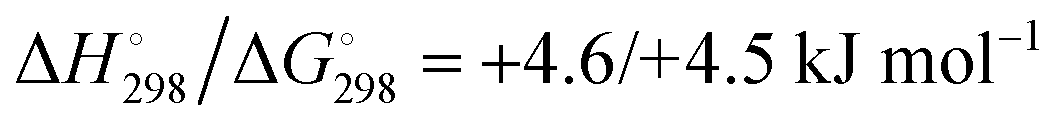 in the gas phase and by +15.3/+14.0 kJ mol−1 in the simulated aqueous phase (ε = 78).44 Geometrical parameters and atomic charges are given in Fig. 11.
in the gas phase and by +15.3/+14.0 kJ mol−1 in the simulated aqueous phase (ε = 78).44 Geometrical parameters and atomic charges are given in Fig. 11.
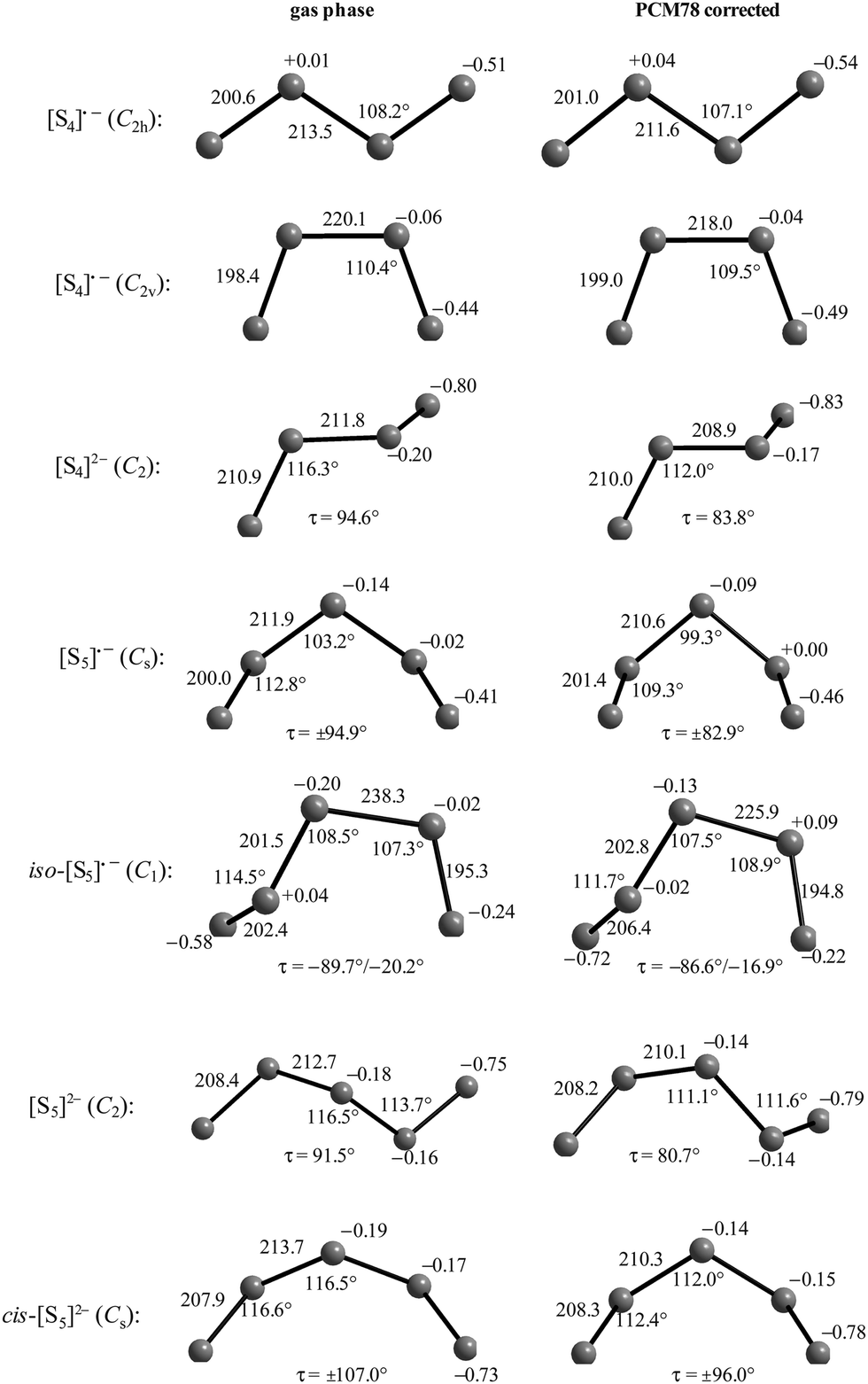 | ||
| Fig. 11 Calculated structures and NPA atomic charges of the more stable isomers of the anions [Sn]˙− and [Sn]2− (n = 4 and 5) in the gas phase (left) and in a polarizable continuum of ε = 78 (right). Bond lengths are in pm. Reproduced from ref. 44 with permission from Wiley. Copyright 2013. | ||
The most stable tetrasulfide dianion is of C2 symmetry as in the crystal structures of the corresponding alkali-metal salts.44 The PCM78-corrected bond lengths (d) and bond angles (α) calculated for [S4]2− (Fig. 11) are in better agreement with the experimental data for solid α-Na2S4 [d = 206.1 (twice) and 207.4 pm, α = 109.8°]40 than the calculated gas-phase geometry. An isomeric pyramidal tetrasulfide anion of C3v symmetry (not shown) in analogy to the isovalent sulfite ion [SO3]2− is by only 22.3 kJ mol−1 (gas phase), 10.3 kJ mol−1 (PCM8) or 8.9 kJ mol−1 (PCM78) less stable than the C2 symmetric chain structure. At 600 K the relative Gibbs energy of this C3v isomer has been predicted as 13.0 kJ mol−1. This isomer has in fact been observed in 2018, trapped in a cage of a zeolite structure and surrounded by sodium cations which evidently stabilize this novel species (see Fig. 2).32a
Calculated bond lengths, bond angles and torsion angles (τ) of the more stable pentaatomic polysulfide anions are given in Fig. 11. The addition of an electron to the cyclic molecules S5 to S8 is strongly exothermic in the gas phase and even more so in a polarizable continuum (Table 6), rows 1–4. The resulting radical anions are of either C2 or Cs symmetry (Fig. 11).
 and Gibbs energies ΔG° at 298 K (kJ mol−1) for the reduction of cyclic sulfur species both in the gas phase and with PCM corrections for ε = 8.0 and 78.0, calculated by the modified G3X(MP2) theory; product symmetries in the gas phase are also given. Reproduced from ref. 44 with permission from Wiley. Copyright 2013
and Gibbs energies ΔG° at 298 K (kJ mol−1) for the reduction of cyclic sulfur species both in the gas phase and with PCM corrections for ε = 8.0 and 78.0, calculated by the modified G3X(MP2) theory; product symmetries in the gas phase are also given. Reproduced from ref. 44 with permission from Wiley. Copyright 2013
| Entry | Reaction | Symmetry of products |
|
|
|
|---|---|---|---|---|---|
| 1 | S8 + e− → [S8]˙− | C 2 | −151.1/−162.4 | −294.5/−305.2 | −315.3/−326.1 |
| 2 | S7 + e− → [S7]˙− | C s | −162.1/−171.8 | −312.5/−321.4 | −334.1/−342.7 |
| 3 | S6 + e− → [S6]˙− | C 2 | −151.1/−160.4 | −308.7/−317.6 | −330.7/−339.6 |
| 4 | S5 + e− → [S5]˙− | C s | −177.9/−189.0 | −350.6/−360.0 | −375.1/−383.9 |
| 5 | [S8]˙− + e− → [S8]2− | C 2 | +16.2/+ 8.0 | −379.5/−385.1 | −432.5/−38.1 |
| 6 | [S7]˙− + e− → [S7]2− | C 2 | +39.8/+34.0 | −377.2/−381.1 | −432.9/−437.0 |
| 7 | [S6]˙− + e− → [S6]2− | C 2 | +68.2/+ 62.5 | −376.1/−380.2 | −435.9/−439.8 |
| 8 | S8 + 2e− → [S8]2− | C 2 | −134.9/−154.4 | −673.9/−690.3 | −747.8/−764.2 |
| 9 | S7 + 2e− → [S7]2− | C 2 | −122.2/−137.8 | −669.7/−702.5 | −767.0/−779.7 |
| 10 | S6 + 2e− → [S6]2− | C 2 | −82.9/−97.9 | −684.8/−697.8 | −766.5/−779.4 |
The further reduction of the cyclic radical anions to the helical polysulfide dianions is endothermic in the gas phase, but strongly exothermic and exergonic in polarizable media (Table 6, rows 5–7). The first step of a hypothetical two-electron reduction of the neutral homocycles (rows 1–4) is more exothermic than the second step is endothermic (rows 5–7); therefore, the overall enthalpy change for the formation of the dianions from neutral molecules Sn is surprisingly exothermic for n > 5 (rows 8–10). However, this formal stability of the gaseous polysulfides is questionable since they are all unstable in the gas phase with respect to homolytic dissociation at one of the central bonds producing two radical monoanions (Table 5). This homolytic reaction is only endothermic and endergonic at 298 K in polarizable media; at 600 K all six dissociation reactions shown in Table 5 are exergonic in a medium of ε = 8.0 (see the data in parentheses).
Neither the neutral cyclic S5 molecule nor the radical anion [S5]˙− have definitely been observed in solution or in the solid state. According to high-level quantum-chemical calculations their most stable isomers are both of Cs symmetry. However, S5 is cyclic while the pentasulfide radical anion [S5]˙− differs from S5 by the large internuclear distance of 473.5 pm (gas phase) or 392.0 pm (PCM78) between the terminal atoms of the sulfur chain (Fig. 11). Other chain-like [S5]˙− isomers of C1 and C2 symmetry are less stable. However, the structure of the novel species iso-[S5]˙− shown in Fig. 11 is by only 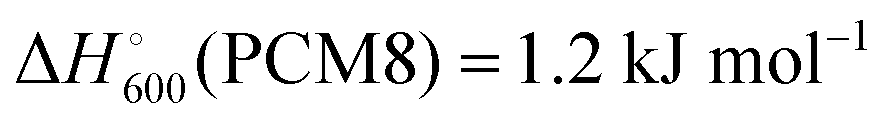 above the Cs symmetric global minimum structure, despite its unusual asymmetrical geometry (Table 7, entry 3). Because of its long S–S bond of >225 pm this isomer may be considered as an adduct between the radical [S3]˙− and triplet state S2(3Σg) in analogy to the adduct between [S3]˙− and O2(3Σg), which will be discussed below (Section 5.3). Polysulfide species like iso-[S5]˙− may be present in high temperature polysulfide melts and fluids.44
above the Cs symmetric global minimum structure, despite its unusual asymmetrical geometry (Table 7, entry 3). Because of its long S–S bond of >225 pm this isomer may be considered as an adduct between the radical [S3]˙− and triplet state S2(3Σg) in analogy to the adduct between [S3]˙− and O2(3Σg), which will be discussed below (Section 5.3). Polysulfide species like iso-[S5]˙− may be present in high temperature polysulfide melts and fluids.44
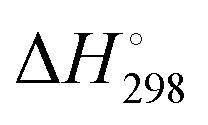 and Gibbs energies ΔG° at 298 K and – in parentheses – at 600 K (all in kJ mol−1) for the isomerization of some polysulfide anions both in the gas phase and with PCM corrections for ε = 8.0 and 78.0, calculated by the modified G3X(MP2) theory. Reproduced from ref. 44 with permission from Wiley. Copyright 2013
and Gibbs energies ΔG° at 298 K and – in parentheses – at 600 K (all in kJ mol−1) for the isomerization of some polysulfide anions both in the gas phase and with PCM corrections for ε = 8.0 and 78.0, calculated by the modified G3X(MP2) theory. Reproduced from ref. 44 with permission from Wiley. Copyright 2013
| Entry | Reaction |
|
|
|
|---|---|---|---|---|
| 1 | [S4]˙− (C2v)→ iso-[S4]˙− (C2h) | +4.6/+4.2 | — | +15.3/+14.0 |
| 2 | [S4]2− (C2) → [S4]2− (C3v) | +19.5/+22.3 | +7.8/+10.3 (+13.0) | +6.5/+8.9 |
| 3 | [S5]˙− (Cs)→ iso-[S5]˙− (C1) | +4.3/+0.7 | +3.5/+2.2 (+1.2) | +2.4/+1.0 |
| 4 | [S5]2− (C2) → cis-[S5]2− (Cs) | +12.1/+11.4 | +8.0/+7.6 (+7.1) | +6.1/+5.8 |
| 5 | [S5]2− (C2) → iso-[S5]2− (C1) | +31.3/+31.9 | +19.3/+20.5 (+21.9) | +18.2/+19.6 |
| 6 | [S5]2− (C2) → [S5]2− (Td) | +101.0/+109.1 | +64.5/+71.4 (+78.8) | +59.4/+66.2 |
| 7 | [S6]2− (C2) → iso1-[S6]2− (C2v) | +39.9/+35.2 | +7.7/+ 8.5 (+9.8) | +2.8/+4.2 |
| 8 | [S6]2− (C2) → iso2-[S6]2− (C1) | +27.6/+27.0 | +18.7/+19.5(+20.4) | +17.5/+18.2 |
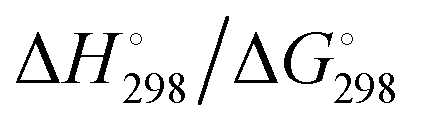
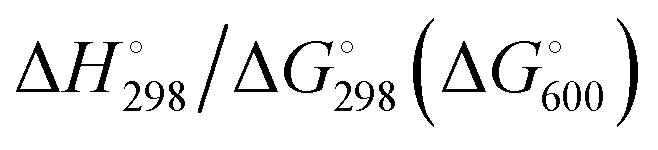
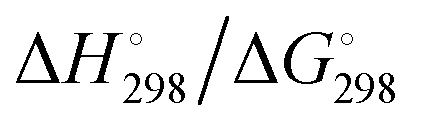
The pentasulfide dianion is of Cs symmetry in the crystal structure of α-Na2S5 but of C2 symmetry in crystalline ε-Na2S5, K2S5, Rb2S5 and Cs2S5.18,44 Both in the gas phase and with PCM corrections the helical C2 symmetric isomer represents the global minimum structure with torsion angles of 91.5° (gas) and 80.7° (PCM78) (Fig. 11). Since the rotational barriers of sulfur chains and the energy differences between rotational isomers are small, the Cs symmetric isomer with a relative Gibbs energy of +5.8 kJ mol−1 at 25 °C (PCM78-corrected) can be expected to exist in addition to the C2 isomer, both in pentasulfide melts and other polysulfide solutions (Table 7). The geometry of cis-[S5]2− calculated with the PCM78 correction (Fig. 11) is in much better agreement with the data of α-Na2S5 (d12 = 206.1 and d23 = 206.6 pm, α = 107.3° and 108.1°, τ = ±88.6°) than the calculated gas-phase geometry demonstrating the usefulness of the polarizable continuum model.44
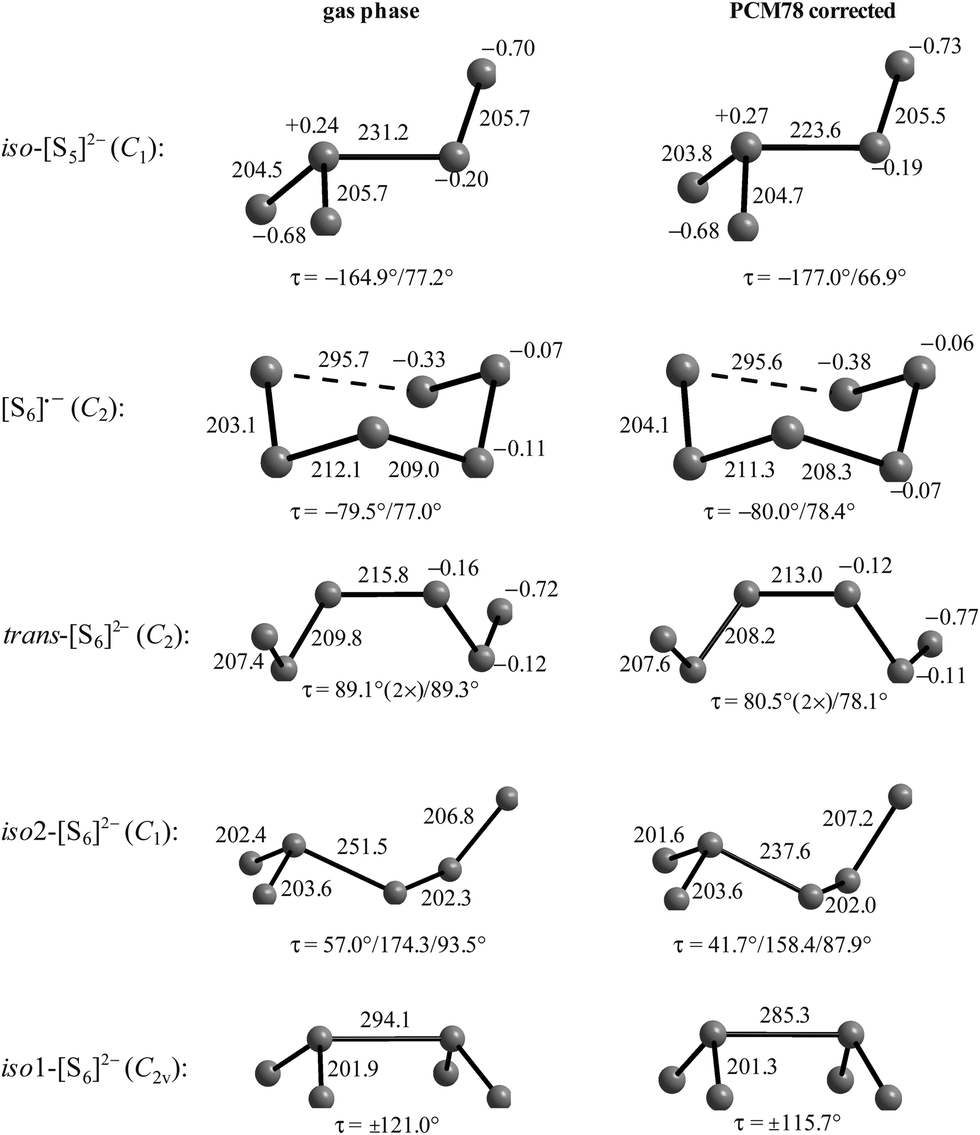 | ||
| Fig. 12 Calculated structures and NPA atomic charges of isomeric anions [Sn]˙− and [Sn]2− (n = 5 and 6) in the gas phase (left) and with PCM correction for ε = 78 (right); bond lengths in pm. Reproduced from ref. 44 with permission from Wiley. Copyright 2013. | ||
The hexasulfide radical anion [S6]˙− is the only polysulfide monoanion, which has been prepared in pure form and structurally characterized as the salt [Ph4P][S6].9 The cyclic anion has two long bonds of 263 pm connecting two S3 units and the average S–S bond length within the S3 fragments is 206 pm, cf. 205 pm in S8. Density functional calculations yielded a global minimum structure of C2 symmetry in which the two S3 fragments are connected by one normal (212.5 pm) and just one very long (294.3 pm) bond. However, ab initio MP2/6-31+G* computations favoured a chair conformation of C2h symmetry. At the B3LYP/6-31+G(2df,p) level the radical is of C2 symmetry with a conformation similar to that of the homocyclic S6 molecule (D3d), but again with one very long (“broken”) bond of 295.7 pm in the gas phase or 295.6 pm with the PCM78 correction, respectively.44 All other bonds have expected lengths of between 203 and 212 pm (Fig. 12). The difference between the experimental and calculated structures may be explained by a disorder of the anions in the crystal structure. In addition, it has been shown that the observed structure is a transition state in the gas phase (at the DFT level) which is only slightly less stable than the global minimum structure of C2 symmetry.9 Also, the anion-cation interaction may play a role for the relative stabilities of these conformers.
The experimental structure and vibrational spectrum of the helical dianion [S6]2− have been discussed in Section 3.1; its calculated structure is shown in Fig. 12. The branching of [S6]2− has also been studied computationally, since an isomeric hexasulfide anion [(S2)S–S(S2)]2− (iso1-[S6]2−) in analogy to the isovalent dithionite anion [S2O4]2− has been claimed as a constituent of molten Cs2S6 on the basis of Raman spectra taken after quenching the melt from 300 °C to 25 °C.37 At the G3X(MP2) level of theory this branched isomer is of C2v symmetry (Fig. 12) and was found to be less stable than the helical chain, by 35.2 (gas phase) and 8.5 kJ mol−1 (PCM8); at 600 K the relative Gibbs energy is 9.8 kJ mol−1 (Table 7, entry 7).44 Consequently, small concentrations of branched polysulfide anions can be expected in sulfur-rich polysulfide melts at temperatures near 300 °C (as in Na–S batteries). However, such melts contain the trisulfide radical anion which, on cooling, could recombine to the C2v symmetric hexasulfide dianion, as discussed below; the latter may return to the helical ground-state conformation on annealing.
The hepta- and octasulfide radical anions [S7]˙− and [S8]˙− are probably the most important species in the primary reduction processes occurring at the sulfur electrodes in sulfur-based batteries. So far, both radicals have been observed only by mass spectrometry. Surprisingly, their calculated structures strongly resemble those of the related homocycles. The neutral chair-like homocycle S7 is of Cs symmetry in the crystal structures of γ- and δ-S7,2a and G3X(MP2) calculations predicted the same symmetry for [S7]˙− both in the gas phase and with PCM corrections.44 The main geometrical difference between S7 and [S7]˙− is the large internuclear distance of 308 pm between the terminal atoms of the sulfur chain of [S7]˙− which could be considered as a “broken bond” since the van der Waals distance between sulfur atoms is 350 pm. In cyclo-S7 the longest bond is of length 217.5 pm; all other bonds and the torsion angles of [S7]˙− are very similar to those of S7. On further reduction of [S7]˙− the helical chain-like dianion [S7]2− is formed in which all bonds are of normal length: 206.4–212.5 pm in the gas phase and 207.2–210.6 pm with PCM78 correction (Fig. 13). The latter data are consistent with the range of bond lengths determined by X-ray crystallography of six ionic heptasulfides with large cations (196.3–209.8 pm).6
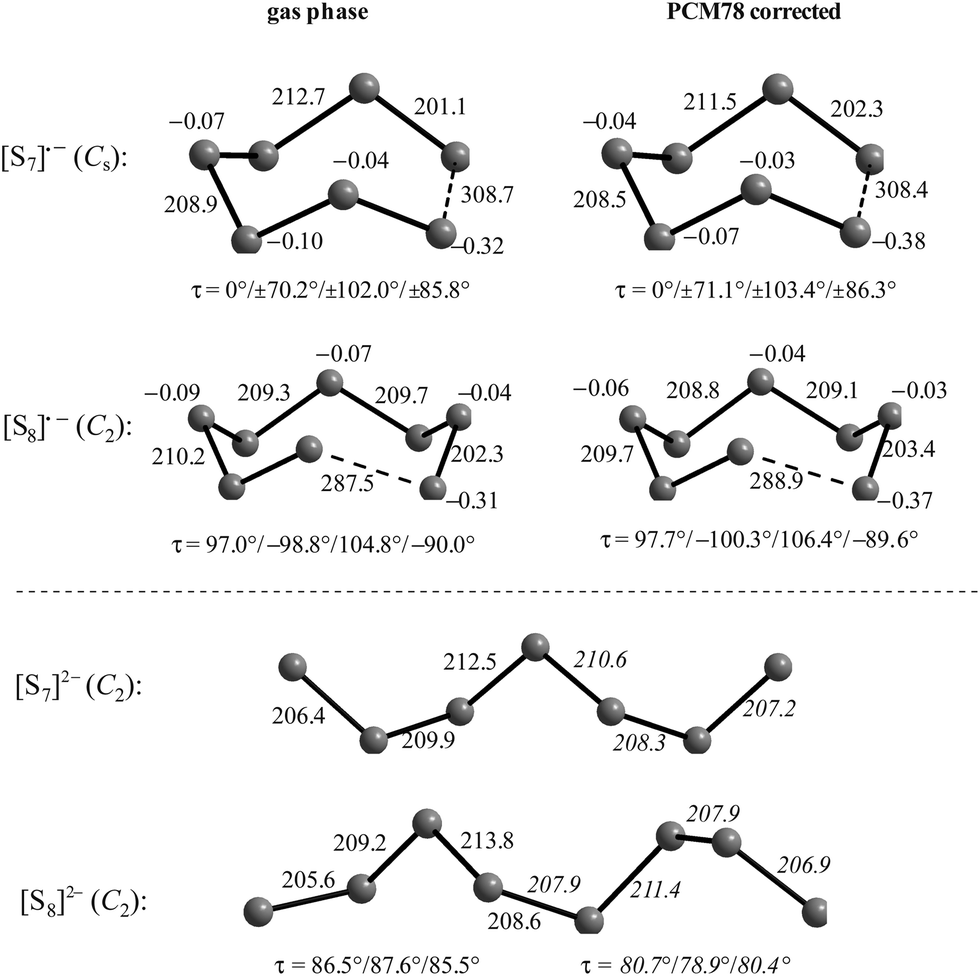 | ||
| Fig. 13 Calculated structures and NPA atomic charges of the anions [Sn]˙− (n = 7 and 8) in the gas phase (left) as well as with PCM78 correction (right). Bond lengths in pm. For the two helical dianions [Sn]2− (n = 7 and 8) shown at the bottom the data in italics are with PCM78 correction, otherwise for the gas phase. Reproduced from ref. 44 with permission from Wiley. Copyright 2013. | ||
In orthorhombic α-S8 the average experimental bond length is 204.6 pm (ideal symmetry D4d). One-electron reduction of S8 produces a radical of almost identical conformation, which is of C2 symmetry mainly due to one very long bond of 287.5 pm (gas phase), 288.2 pm (PCM8) or 288.9 pm (PCM78). The remaining bonds of [S8]˙− vary in length between 202.3 and 210.2 pm (Fig. 13) with the typical bond-length alternation frequently observed for cumulated sulfur–sulfur bonds. In contrast, the octasulfide dianion [S8]2− is most stable as a helical chain of C2 symmetry which has two relatively long symmetry-related bonds of 213.8 pm (gas phase), 211.5 pm (PCM8) and 211.4 pm (PCM78) at the B3LYP/6-31+G(2df,p) level (Fig. 13).44 This feature may be an indication of the ease of the exothermic thermal dissociation into the two radical monoanions [S3]˙− and [S5]˙− (Table 5). For the dianion of the salt [Et3NH]2[S8] the range of bond lengths has been determined crystallographically as 204.1–207.3 pm, which may however be influenced by hydrogen bonds to the cations; the motifs of the chiral anions in this salt are ++−++ and −−+−− (enantiomers of symmetry C2) (see Section 2 for an explanation of the nomenclature for these motifs).26b The thermodynamic properties of [S7]˙−, [S8]˙−, [S7]2− and [S8]2− will be discussed further in Section 5.1.
5 Significance and applications of polysulfides and polysulfide radical anions
5.1 Sulfur-based batteries
The reaction of alkali metals with elemental sulfur to produce polysulfides is highly exothermic, especially with a high metal-to-sulfur ratio resulting in mono- and disulfides which are characterized by large lattice energies. These reactions can be reversed by electrolysis, provided the polysulfides are either soluble or can be melted without decomposition. Enormous efforts have been undertaken in recent years to utilize such reactions for energy storage systems since lithium and sodium are environmentally friendly metals while elemental sulfur is an inexpensive by-product from refineries. Since the high-temperature solvent-free sodium–sulfur battery is the simplest device from a chemical point of view it will be discussed first followed by a critical evaluation of the rather complex polysulfide redox chemistry in room-temperature sodium–sulfur and lithium–sulfur batteries, which normally require a solvent and an additional electrolyte. The first descriptions of lithium– and sodium–sulfur secondary batteries have been published in the 1960s, and thousands of papers on these subjects have appeared since. The technical aspects of sulfur-based batteries have been outlined in detail in recent reviews.110–112 The use of magnesium113 as well as zinc114 as anode materials in sulfur-based batteries has also been reported, but these devices are less well-studied and will not be discussed here. Furthermore, polysulfides play an important role in certain redox-flow batteries.115 | ||
| Fig. 14 Schematic structure of a cell of high-temperature sodium–sulfur batteries. | ||
During discharge sodium cations are generated at the sodium electrode and migrate through the ceramic separator into the sulfur melt to compensate the charge of the polysulfide anions generated by the electrochemical reduction of sulfur. Therefore, the volume of liquid sodium decreases and the volume of the “sulfur/polysulfide electrode” increases during discharge. The formation of solid polysulfides such as high-melting Na2S2 must be avoided.
The energy density of the Na–S system is much higher than that of the lead–acid battery and large-scale batteries with more than 1000 single cells and weighing about 100 tons have been established in Japan, USA, China, Korea and elsewhere. The Na–S battery was the first commercialized battery system capable of megawatt-level electric power storage from solar devices and wind-powered electric generators. It has several superior features, including large capacity, high energy density, and long service life, thus enabling a high output of electric power for long periods of time (cell voltage 1.8–1.9 V). Na–S batteries may be charged at night when power demand is low and provide power at daytime to reduce peak power. Moreover, the Na–S battery system can be used as an emergency power supply during power outages and momentary drops in voltage. Disadvantages are the elevated operating temperature and the high manufacturing costs of the ceramic separator as well as corrosion and leakage problems.116 Therefore, the battery is restricted to stationary applications. The following discussion is mainly concerned with the polysulfide redox chemistry rather than the technology of this battery.
The underlying basic chemical reaction is the reversible formation of polysulfides from elemental sulfur by reduction with sodium metal, often simplified to the following equation:
| 2Na + 1/2S8 → Na2S4 | (13) |
At the sulfur electrode of Na–S batteries the various sulfur molecules present in liquid sulfur117 are expected to be reduced in a similar fashion as has been observed in solvents by cyclic voltammetry. Sulfur radical anions are likely to be the first products [eqn (14)].
| S8 + e− → [S8]˙− | (14) |
To the best of our knowledge, the adiabatic electron affinity of S8 is unknown but recent DFT calculations predicted a value of 1.57 eV at the G3X(MP2) level44 and 2.02 eV at the B3PW91/aug-cc-pVDZ level of theory109 for the gaseous molecule. For gaseous S7 slightly higher values were predicted. However, anions are stabilized in molten sodium polysulfides and in polar solvents. This effect has been modeled by PCM calculations using a dielectric constant ε of 8. Under these circumstances the electron affinity of S8 was predicted as 3.05 eV; with larger values of ε only slightly higher affinities were calculated.44
The octasulfide radical anion [S8]˙− formed in eqn (14) is thermodynamically unstable at the operating temperatures of NaS batteries since its disproportionation to S8 and [S8]2− has been predicted to be exergonic by  at 600 K. Even the dissociation of [S8]˙− into [S3]˙− and cyclo-S5 is exergonic by −33.4 kJ mol−1 at this temperature and the same holds for a dissociation into [S2]˙− and cyclo-S6
at 600 K. Even the dissociation of [S8]˙− into [S3]˙− and cyclo-S5 is exergonic by −33.4 kJ mol−1 at this temperature and the same holds for a dissociation into [S2]˙− and cyclo-S6 . The octasulfide dianion [S8]2− formed from [S8]˙− by either disproportionation or by addition of another electron is also thermodynamically unstable at 600 K since its dissociation into 2[S4]˙− or [S3]˙− plus [S5]˙− is exergonic, by −23.5 kJ mol−1 and −12.4 kJ mol−1, respectively (Table 5).44 Therefore, the polysulfide melt of Na–S batteries will contain a complex mixture of radical monoanions as well as dianions; sulfur radicals had in fact been detected in sodium polysulfide melts by Raman, EPR and UV-Vis spectroscopy already in the 1970s.6
. The octasulfide dianion [S8]2− formed from [S8]˙− by either disproportionation or by addition of another electron is also thermodynamically unstable at 600 K since its dissociation into 2[S4]˙− or [S3]˙− plus [S5]˙− is exergonic, by −23.5 kJ mol−1 and −12.4 kJ mol−1, respectively (Table 5).44 Therefore, the polysulfide melt of Na–S batteries will contain a complex mixture of radical monoanions as well as dianions; sulfur radicals had in fact been detected in sodium polysulfide melts by Raman, EPR and UV-Vis spectroscopy already in the 1970s.6
Owing to the high concentrations of cations and anions in polysulfide melts there will also be contact ion pairs like [NaSm]˙, [NaSn]− and, possibly, also neutral ion-triples [Na2Sn]. Such species have been studied computationally at the G3X(MP2) level and cyclic as well as cluster-like structures with chelating polysulfide ligands have been identified as global minimum geometries. The calculated structures of [NaSm]˙ (m = 2, 3) and [NaSn]− (n = 3–5) both for the gas phase and for a polarizable continuum with ε = 8 are shown in Fig. 15. None of the species [NaSn]˙ and [NaSn]− adopts a conformation similar to that of the sulfur homocycle of the same number of atoms and all are of relatively low symmetry: C2v and Cs for the smaller and C2 or Cs for the larger ion pairs. The formation of the species [NaS2]˙ and [NaSn]− (n = 3–6) from Na+ and the corresponding anions is expected to be strongly exergonic, even at 600 K and ε = 8.44
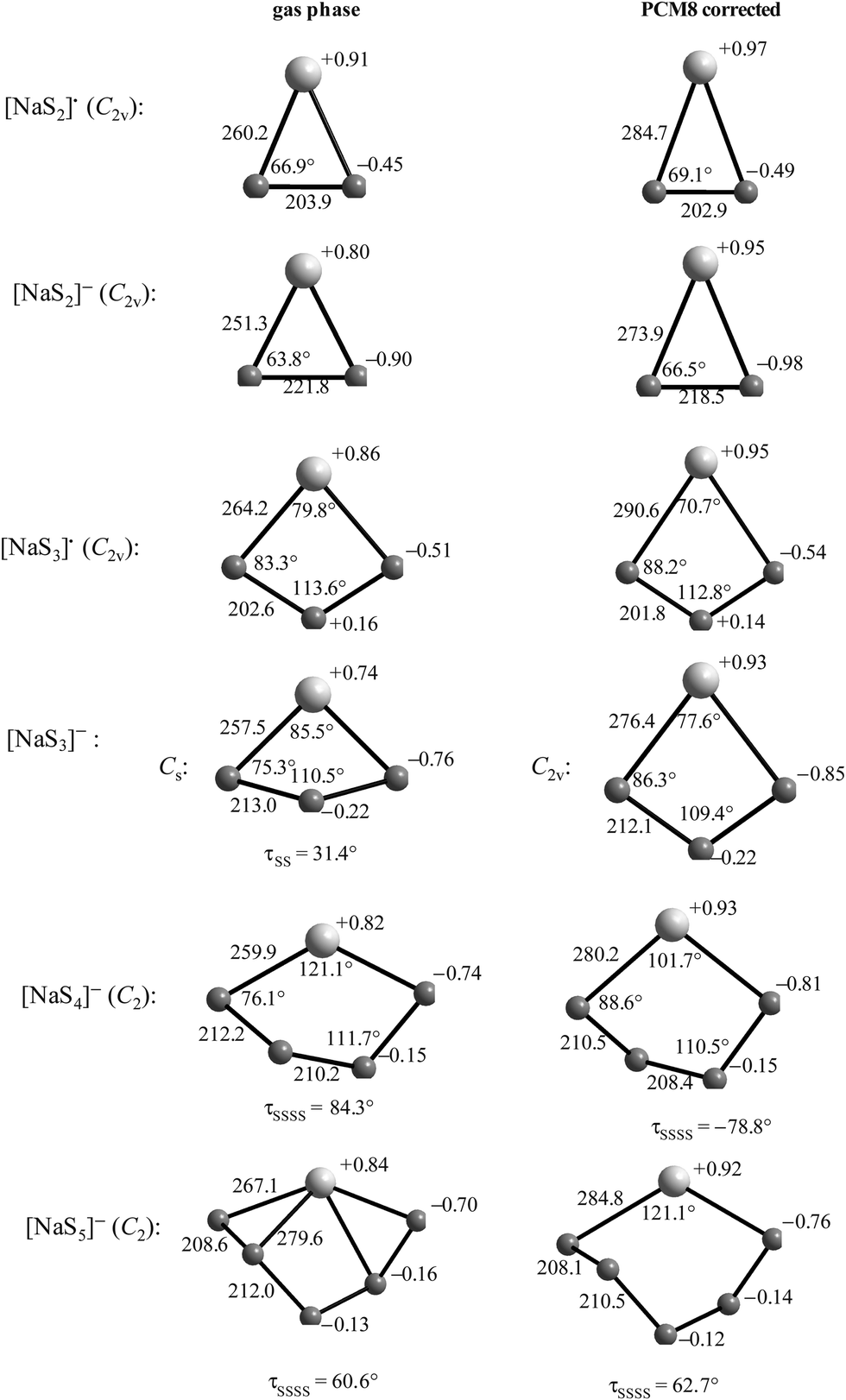 | ||
| Fig. 15 Structures and symmetries of the ion pairs [NaSn]˙ and [NaSn]− (n = 2–5) in the gas phase (left) and with PCM8 correction using ε = 8.0 (right), calculated at the B3LYP/6-31+G(2df,p) level of theory; bond lengths are given in pm and atomic charges in electrostatic units. Reproduced from ref. 44 with permission from Wiley. Copyright 2013. | ||
The formation of ion pairs [NaSn]− by one-electron reduction of the radicals [NaSn]˙ is also strongly exergonic both in the gas phase and in polarizable phases. Since the absolute values of the corresponding electron affinities increase with the molecular size, electron-transfer reactions of the type
| [NaSx]− + [NaSy]˙ → [NaSx]˙ + [NaSy]− (x < y) | (15) |
| [NaSx]− + 1/8S8 → [NaSx+1]− | (16) |
The Coulombic interaction between sodium cations and polysulfide dianions is very strong and the corresponding reaction energies in the gas phase are almost twice as large as for the monoanions. The Gibbs reaction energies in the polarizable phase with ε = 8 are much smaller but still negative, both at 298 and 600 K. It is interesting to note that the homolytic dissociation of [NaS6]− into [NaS3]˙ and [S3]˙− is predicted to be endergonic by +137.8 kJ mol−1 in the gas phase and by +29.8 kJ mol−1 with PCM8 correction at 300 K. This result is in agreement with the detection of the large gaseous ion pairs [NaSn]− (n = 7–9) by negative ion EI mass spectrometry of aqueous sodium polysulfide solutions although these measurements took place under non-equilibrium conditions.47 However, at 600 K the dissociation of [NaS6]− into [NaS3]˙ and [S3]˙− is calculated to be exergonic by −14.4 kJ mol−1 and charged ion pairs of the size similar to [NaS6]− are therefore predicted to be unimportant in HT Na–S batteries. In other words, polysulfide dianions larger than [S4]2− are not stabilized by ion-pair formation with Na+ at high temperatures.44
In the neutral ion triples [Na2Sn] the coordination numbers of all atoms are either 2 or 2 + 1. The bonding between the cations and the polysulfide ligands is mainly ionic as can be concluded from the NPA atomic charges on the sodium atoms, which decrease with increasing sulfur content from +0.92 in Na2S2 (+0.97 with PCM8 or PCM78 correction) to +0.86 in [Na2S8]. The coordination numbers of the cations increase with molecular size from 2 in [Na2Sn] (n = 2–4) to 2 + 1 in [Na2S5] and the larger species.44
The thermodynamically stable crystalline forms of sodium polysulfides with small unit cells have been calculated in 2016 using the crystal-structure prediction algorithm USPEX (Section 3.6). The formation energies per atom at zero pressure and temperature were found to increase linearly from penta- to monosulfide, while Na2S4 and ε-Na2S5 were predicted to be the most stable phases at 100 kbar. α-Na2S5 and ε-Na2S5 were identified as metastable at ambient temperature, transforming to Na2S4 and sulfur on heating.18 Solid Na2S3 is another metastable sodium polysulfide, and Na2S2 as well as Na2S4 also exist as several metastable phases.118
Nevertheless, there have been some demonstrations of RT sodium–sulfur batteries in the literature, most of which focus on improving the cycling stability on the sulfur cathode side.111,119 Examples include the use of polyacrylonitrile–sulfur composites, small sulfur molecules in microporous carbon, Nafion separators, carbon interlayers, polymer electrolytes, etc. The application of sodium polysulfide catholytes and sodium sulfide cathodes on high surface area carbon supports have also been demonstrated with some success. However, the overall cycling performance of RT sodium–sulfur batteries, in terms of their specific capacity, Coulombic efficiency and cycle life, still lag behind that of lithium–sulfur batteries. The key to achieve progress is to solve the crucial problems on the sodium metal anode side, which is the major bottleneck in the performance of RT sodium–sulfur batteries. Like in the case of lithium metal, sodium metal anodes are plagued with challenges of poor reversibility, dendrite growth and non-uniform solid electrolyte interfaces on the surface during repeated plating and stripping.
While the polysulfide chemistry of RT Na–S batteries is quite analogous to the high-temperature system discussed above, the thermodynamics at 298 K is slightly different from that at 600 K as will be outlined below. In addition, there is now a competition between polysulfide ligands and the solvent molecules for access to the sodium cations since organic carbonates are well-known O-donor ligands as are the anions of Na[ClO4] and Na[CF3SO3].
There has been a considerable debate in the literature whether the first step in the electroreduction of dissolved S8 molecules is a one- or a two-electron transfer producing either octasulfide radical anions [S8]˙− or octasulfide dianions [S8]2− as primary products. In a series of remarkable studies Levillain et al. have shown by cyclic voltammetry and spectro-electrochemical experiments in the 1990s that S8 dissolved in dimethylformamide (DMF, ε = 36.7 at 25 °C) yields two reduction waves, at −1.05 V and −1.80 V with a pre-wave at −1.30 V at 298 K versus Fc+/Fc. The first wave was assigned to the one-electron transfer producing [S8]˙− which subsequently disproportionates spontaneously to S8 and [S8]2−:44,51
| S8 + e− → [S8]˙− | (17) |
| 2[S8]˙− → S8 + [S8]2− | (18) |
| [S8]2− → 2[S4]˙− | (19) |
| [S8]2− + [S4]˙− → [S6]2− + [S6]˙− | (20) |
| [S6]2− → 2[S3]˙− | (21) |
Reaction (21) has been predicted to be slightly exergonic in solvents of ε = 8 at 298 K.44 The second wave at −1.80 V and the pre-wave at −1.30 V in the voltammograms have been explained by the reduction of polysulfide radical anions to the corresponding dianions, especially [S3]˙− (−1.80 V) and [S4]˙− (−1.30 V).51 However, according to G3X(MP2) calculations the homolytic dissociation of polysulfide dianions [Sn]2− with n = 7 and 8 is slightly endergonic in a polarizable continuum at 298 K, although this may change under the influence of the cations with formation of ion pairs [NaSn]−. The important message here is that dianions are not reducible electrochemically in solutions at 298 K, whereas monoanions are being reduced.44,51 Other possible reaction channels so far not discussed in the literature for production of radical anions are the following dissociative reductions of [S8]˙− at the cathode producing simultaneously two radical monoanions rather than [S8]2−:
| [S8]˙− + e− → [S3]˙− + [S5]˙− | (22) |
| [S8]˙− + e− → 2[S4]˙− | (23) |
After polysulfide dianions have been formed they are assumed to undergo several disproportionation as well as sulfurization reactions to produce other anionic polysulfides. However, the often-cited disproportionation reactions according to eqn (24)
| 2[Sx]2− → [Sy]2− + [Sz]2− (2x = y + z) | (24) |
| [Sx]2− + [Sy]˙− → [Sm]2− + [Sn]˙− (x < y; m > n; x + y = m + n) | (25) |
| [Sx]2− + [Sy]˙− → [Sx]˙− + [Sy]2− (x < y) | (26) |
Aqueous sulfide, hydrogensulfide and polysulfide ions are strong nucleophiles, which attack elemental sulfur at room temperature to yield mixtures of sulfur-rich polysulfides. In principle, sulfurization reactions as in eqn (27) may also occur with radical monoanions [eqn (28)].
| [Sn]2− + 1/8S8 → [Sn+1]2− | (27) |
| [Sn]˙− + 1/8S8 → [Sn+1]˙− | (28) |
On the other hand, the sulfurization reactions of the dianions according to eqn (27) are predicted to be exothermic and exergonic for all n < 7, both in the gas phase and in a polarizable medium of ε = 8.0 at 298 K. The corresponding Gibbs reaction energies become smaller as the length of the polysulfide chain [Sn]2− grows. For n = 6 and 7 the production of the hepta- and octasulfide dianions 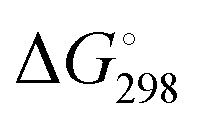 is already slightly positive if a dielectric constant of 78.0 is applied.44 This result is in perfect agreement with the speciation of anions in aqueous polysulfide solutions saturated with elemental sulfur in which the tri, tetra-, penta- and hexasulfide dianions dominate. Experimental thermodynamic data for the reactions discussed here are not available.
is already slightly positive if a dielectric constant of 78.0 is applied.44 This result is in perfect agreement with the speciation of anions in aqueous polysulfide solutions saturated with elemental sulfur in which the tri, tetra-, penta- and hexasulfide dianions dominate. Experimental thermodynamic data for the reactions discussed here are not available.
The thermodynamic considerations discussed in this section apply to solvated polysulfide di- and monoanions. However, ion pairs such as [NaSn]− and [NaSn]˙ may also be components of the electrolytes in RT Na–S batteries. For the smaller ion pairs (n < 5) the calculated structures are shown in Fig. 15 and those for the larger ones are given in Fig. 16. All species are cyclic for ε = 8 and 300 K with a coordination number of 2 or, in some cases, 2 + 2 for the sodium ions.
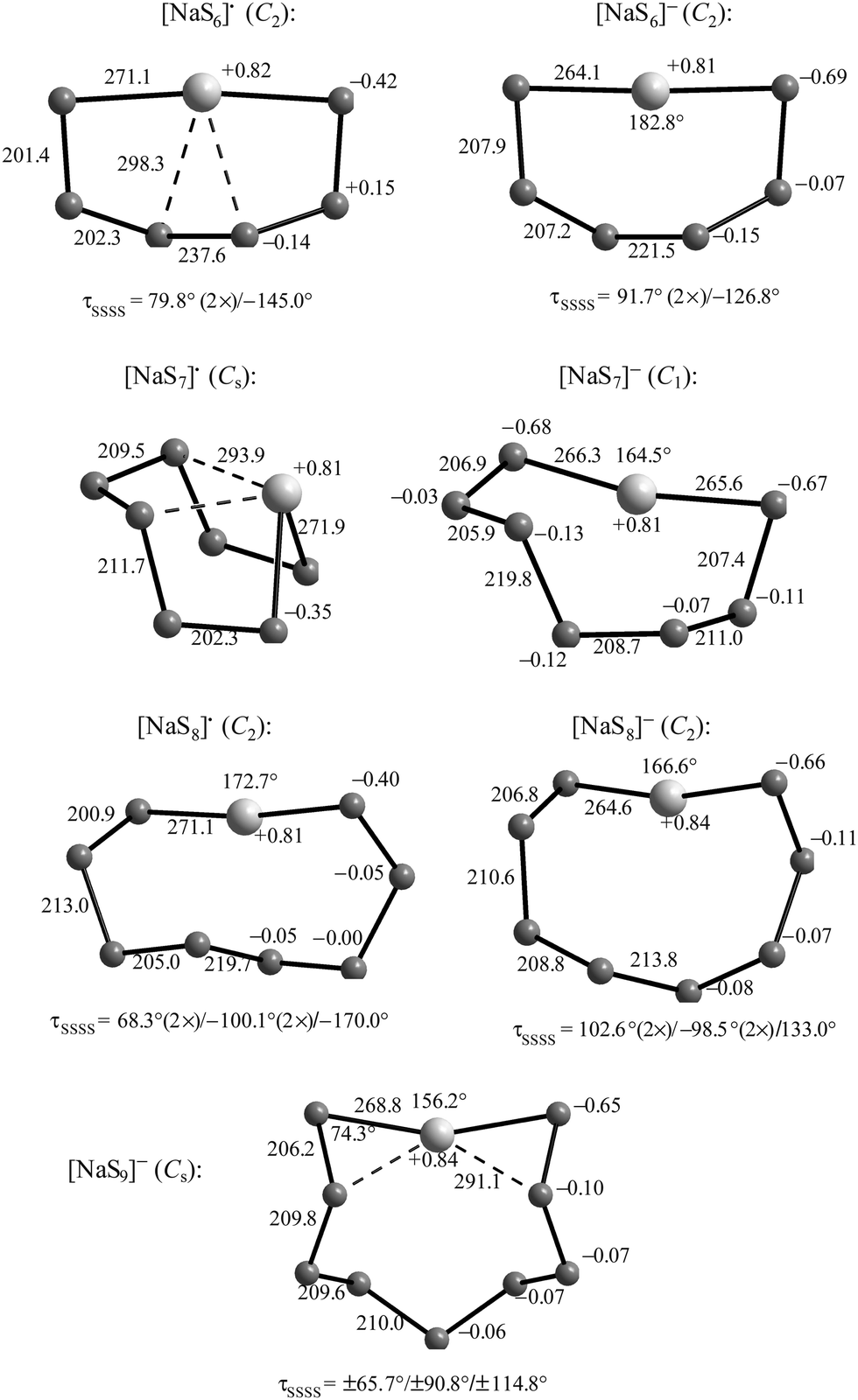 | ||
| Fig. 16 Structures and symmetries of the ion pairs [NaSn]˙ (left) and [NaSn]− (right) for n = 6–9 in the gas phase, calculated at the B3LYP/6-31+G(2df,p) level of theory; bond lengths are given in pm and atomic charges in electrostatic units. Reproduced from ref. 44 with permission from Wiley. Copyright 2013. | ||
The formation of ions [NaSn]− from Na+ and the corresponding polysulfide dianions is exothermic and exergonic for all values of n = 2–8 and at ε = 8 (Table 8).
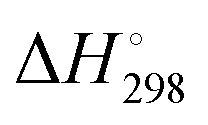 and Gibbs energies ΔG° at 298 K (in kJ mol−1) for the formation of the most stable sodium polysulfide ion pairs [NaSn]˙ and [NaSn]− as well as ion triples [Na2Sn] both in the gas phase and with PCM corrections for ε = 8.0, calculated at the modified G3X(MP2) level of theory (for more data, see ref. 44)
and Gibbs energies ΔG° at 298 K (in kJ mol−1) for the formation of the most stable sodium polysulfide ion pairs [NaSn]˙ and [NaSn]− as well as ion triples [Na2Sn] both in the gas phase and with PCM corrections for ε = 8.0, calculated at the modified G3X(MP2) level of theory (for more data, see ref. 44)
| Entry | Reaction |
|
|
|---|---|---|---|
| 1 | Na+ + [S2]˙− → [NaS2]˙ | −529.4/−505.3 | −50.9/−30.3 |
| 2 | Na+ + [S3]˙− → [NaS3]˙ | −514.9/−485.4 | −40.9/−15.3 |
| 3 | Na+ + [S4]˙− → [NaS4]˙ | −503.8/−476.5 | — |
| 4 | Na+ + [S6]˙− → [NaS6]˙ | −483.8/−457.1 | — |
| 5 | Na+ + [S7]˙− → [NaS7]˙ | −494.6/−460.5 | — |
| 6 | Na+ + [S8]˙− → [NaS8]˙ | −429.9/−404.1 | — |
| 9 | [NaS2]− + [NaS3]˙ → [NaS2]˙ + [NaS3]− | −61.3/−62.1 | −18.6/−19.2 |
| 10 | Na+ + [S2]2− → [NaS2]− | — | −110.0/−87.6 |
| 11 | Na+ + [S3]2− → [NaS3]− | −958.8/−928.4 | −104.7/−77.3 |
| 12 | Na+ + [S4]2− → [NaS4]− | −938.9/−905.5 | −103.8/−74.6 |
| 13 | Na+ + [S5]2− → [NaS5]− | −910.4/−873.0 | −98.7/−68.1 |
| 14 | Na+ + [S6]2− → [NaS6]− | −888.5/−853.9 | −86.7/−56.7 |
| 15 | Na+ + [NaS2]− → [Na2S2] | −600.1/−570.7 | −74.6/−48.6 |
| 16 | Na+ + [NaS3]− → [Na2S3] | −572.9/−539.4 | −65.4/−37.7 |
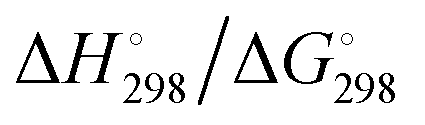
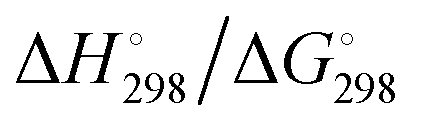
The question then arises whether ion-pair formation stabilizes the polysulfide dianions with respect to homolytic dissociation at one of the central S–S bonds. However, it turns out that the opposite is true. For example, the reaction in eqn (29)
| [NaS6]− → [NaS3]˙ + [S3]˙− | (29) |
One of the problems of metal–sulfur batteries working at room temperature is the decomposition of the solvent by either the bulk metal or by nucleophilic attack of the polysulfide dianions and radical anions. In addition, the self-discharge of the battery by diffusion of the polysulfide anions and radical anions to the metal anode followed by chemical reduction to shorter anions results in loss of capacity. This shuttle mechanism, shuttle effect or shuttle phenomenon will be further discussed in Section 5.1.3. The shuttle effect can best be suppressed using an ion-selective separator between the two half-cells which rejects any anions. However, such membranes are expensive. Other means are modified sulfur electrodes in which the sulfur is confined in a nanoporous carbon material either physically or chemically by additional, positively charged components to bind the sulfur anions by Coulombic forces. Numerous attempts of this type to solve the shuttle problem have been described in the literature.111,112
Another possible solution to the shuttle problem may be the use of an organic polysulfane R2Sn (n > 2) instead of elemental sulfur in which the strong C–S bonds would keep the terminal sulfur atoms in place with the hope that the remaining S atoms would bind on these terminal atoms on recharging the battery.121 However, the capacity of such batteries is unavoidably smaller than with just elemental sulfur since the organic matrix also needs some space; in all cases porous carbon materials are needed as a conducting additive.
A totally different approach to overcome the shuttle problem is the use of an all-solid-state sodium–sulfur battery at room temperature in which a glass ceramic based on sodium tetrathiophosphate Na3[PS4] serves as electrolyte. The sulfur is mixed with acetylene black as conductive additive and can be reduced to Na2S resulting in a very high capacity, but the voltage drops from initially 2.1 V to 1.5 V since the slow migration of the sodium cations through the solid electrolyte produces a higher than predicted overpotential.122 Therefore, a gel-type electrolyte consisting of a polymeric isocyanurate as a sodium cation conducting material has been proposed to suppress the shuttle effect resulting in a high reversible capacity and extended cycling stability.121
However, ambient-temperature Na–S batteries still face many intrinsic obstacles. These include the poor electrical conductivity of sulfur and its large volumetric variation during cycling, the high costs for the preparation of the sulfur electrode as a composite material and of the membrane, the poor kinetics of conversion from short-chain Na polysulfides or Na2S to long-chain polysulfides, the passivation of the anode by solid Na2S2, and the uncontrollable growth of Na dendrites during charging. More research is needed to overcome these problems before commercialization of RT Na–S batteries will be possible.123 On the other hand, sodium is much more abundant and, therefore, cheaper than lithium the reserves of which may not be sufficient to satisfy the demand of the rapidly growing world population.
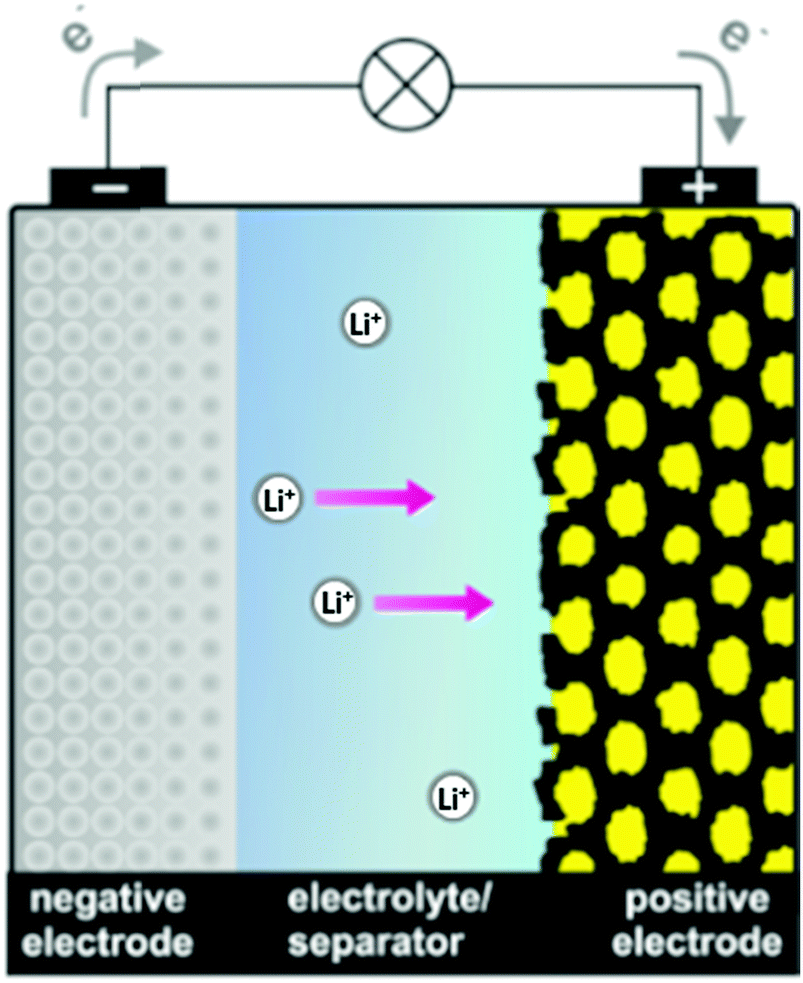 | ||
| Fig. 17 Schematic representation of a cell of a Li–S battery (adapted from Fig. 2 of ref. 110). | ||
In principle the polysulfide chemistry of Li–S batteries should be analogous to that of RT Na–S batteries discussed in Section 5.1.2, although the solubilities of lithium sulfide and polysulfides are different from those of their sodium counterparts. But the strong electric field of the naked lithium cation is responsible for the fact that lithium polysulfides dissolve not only as solvated ions but also as ion pairs of the type [LiSn]−, while neutral ion triples [Li2Sn] are less likely in solvents of medium or high polarity (ε > 10). However, the presence of ion pairs will also depend on the overall salt concentration. In general, the attractive force f between two opposite ionic charges qi at a distance d is given by Coulomb's law as f = q1 × q2/4πε0 × ε × d2 with ε0 being the dielectric constant of vacuum and ε the relative dielectric constant of the medium (solution). From this equation it follows that ion-pair formation is favored by the following factors:
(a) high ionic charges (dianions are more likely to form ion pairs than radical anions of same size),
(b) small ionic radii (Li+ is more likely to form ion pairs than Na+),
(c) a small dielectric constant of the medium (THF with ε ≈ 8 favors ion pair formation more than water with ε = 78),
(d) low temperatures, since ion-pair formation is exothermic, and
(e) high ion concentrations according to the law of mass action.
Remarkably, even LiCl is not completely dissociated in aqueous solution at 25 °C as indicated by the activity coefficient γ of less than 1.0 in the wide concentration range of 0.001 < c < 2.0 mol L−1 with a minimum value of γ = 0.73 at c = 0.5 mol L−1.
Salts dissolve in solvents only if the combined solvation enthalpies of cations and anions are absolutely larger than the lattice enthalpy of the crystalline material. Li2S and Li2S2 have high lattice enthalpies and therefore are insoluble in most solvents used in Li–S batteries. Crystal structures of lithium salts with more than two sulfur atoms are known only for solvated species and not for the binary compounds; consequently, the lattice energies of the binary species with more than two sulfur atoms are unknown (Section 2).
The reaction mechanisms for the redox processes of elemental sulfur and of polysulfides discussed even in the most recent literature are not always in agreement with the existing spectroscopic, voltammetric and thermodynamic data obtained in contemporary fundamental research. In particular, some authors still assume even-numbered polysulfide anions as the only important sulfur species in batteries while inorganic chemists have known for a long time that there is no fundamental difference between odd- and even-numbered dianions [Sn]2−.6 In Section 4 it has been shown that the aqueous pentasulfide dianion is thermodynamically favoured over ions of similar chain lengths since its Gibbs energy of formation is lower than those of other polysulfide dianions with between 2 and 8 atoms (Table 2).
The ideal overall reaction during discharge of Li–S batteries is:
| 16Li + S8 → 8Li2S | (30) |
On this basis the specific capacity of the Li–S battery would be as high as 1672 mA h g−1 and the theoretical energy density ca. 2600 W h kg−1, much higher than for lithium-ion batteries (both by volume and by weight). However, since Li2S and even Li2S2 are insoluble in suitable polar solvents, these values cannot be attained in experiments.
The technical aspects of Li–S batteries15d,110,123–131 and the available analytical tools15b,e,f have been reviewed previously and will not be repeated here. Instead, we present our view of the most probable pathways from elemental sulfur or sulfur-rich composites with S atoms in the oxidation state zero (S0) to products with sulfur atoms in the oxidation state 2− and back. To a certain extent this chemistry depends on the polarity of the solvent used since the solubility of the lithium salts is strongly influenced by the dielectric constant ε. Most frequently, ether-based solvents or solvent mixtures such as dimethoxyethane/dioxolane (DME/DOL; ε near 7) have been used. In addition, propylene, ethylene and diethylcarbonates, as well as high-ε solvents such as dimethylsulfoxide, ethylmethylsulfone, 1-methylimidazole, dimethylacetamide, dimethylformamide and tetramethylurea/dioxolane mixtures, have been investigated. For systematic studies of the influence of different solvents, see ref. 132a. Various lithium salts such as Li[N(SO2CF3)2], Li[PF6], Li[ClO4] and Li[CF3SO3] have been applied to increase the conductivity of the electrolyte; thus the Li+ cation concentration in such batteries is quite high, which favours ion-pair formation. It should be noted that the oxo-anions of these salts as well as of Li[NO3] also serve as potential ligands (O-donors) to Li+ ions.
The sulfur cathode is usually a composite of a carbon-rich (electrically conducting) material and elemental sulfur in the form of either S8, polymeric sulfur or an organic sulfur-rich covalent polysulfane R2Sn. Many publications deal with the suppression of the above-mentioned “shuttle effect” of polysulfide anions by designing special cathodes using carbon nanotubes, graphene, reduced graphene oxide, mesoporous carbon or hollow carbon spheres to confine or encapsulate the active material but sometimes even the carbon is replaced, e.g. by molybdenum carbide nanorods133 or other transition-metal substrates.134 Claims of encapsulation of small and highly endothermic sulfur molecules such as S2, S3 and S4 have been made, but have never been substantiated by direct detection and therefore are doubtful, especially since the carbon material of Li–S batteries is more like a sponge rather than a rock-solid matrix which would be needed to trap such high-temperature molecules at room temperature. Since the carbon–sulfur electrode changes its volume periodically due to the influx of cations during discharge and their exodus during recharge, the carbon material needs to be mechanically flexible in order to allow for diffusion. A nitrogen-doped carbon sponge has also been investigated as a cathode material with the aim that lithium cations bind to the pyridinic or pyrrolic nitrogen atoms and the polysulfide ions would then interact with the immobilized Li+ ions to suppress the shuttle effect.135
The anode consists of metallic lithium covered by a thin protecting layer, which helps to prevent dendrite growth and self-discharge due to polysulfide reduction (so-called solid electrolyte interface, SEI). Li[NO3] in rather high concentrations is sometimes used to improve the morphology of this layer which needs to be an electrical insulator but a Li+ conductor; many other compounds have been tried for this purpose.136 It should be noted, however, that nitrate ions are reduced both by lithium metal and by polysulfide dianions, which are strong reductants.137 Lithium bis(oxalate)borate has also been applied to give the lithium anode a smooth and dense surface morphology needed for a high reversible capacity.138 However, a dendrite-free Li0 anode has seldom been obtained under practical conditions, such as with large current densities (>1 mA cm−2) and high area capacity (>2 mA h cm−2). A working dendrite-free lithium anode could perhaps be realized through the use of novel additives.
To explain the shuttle effect, it has been demonstrated by HPLC analysis in 2016 that a solution of lithium polysulfides in dimethoxyethane (DME) being in contact with lithium metal changed its composition during several hours so that the concentrations of hexa- and heptasulfide, as well as of S8 decreased, while the concentration of tetrasulfide ions increased. The concentration of trisulfide remained low, probably due to the low solubility of Li2S3, and octasulfide was practically absent in the solution investigated. This chemical reduction of the higher polysulfides by Li metal could be slowed down by addition of lithium nitrate, but could not be completely prevented since the nitrate ions were irreversibly consumed over time.139 A separator or an ion-selective membrane may be applied between the two half-cells of a Li–S battery in order to hinder the diffusion of polysulfides to the lithium electrode.140
The electrochemical reduction of S8 and other sulfur-rich substrates in Li–S batteries proceeds via radical anions from which the polysulfide dianions may be generated by disproportionation, by recombination or by direct reduction at the cathode as shown in Scheme 3.
 | ||
| Scheme 3 Most likely reactions occurring during electrochemical reduction of S8 molecules in Li–S batteries on discharging. Most reactions are reversible, and the reversed processes may occur on recharging the battery. Reproduced from ref. 44 with permission from Wiley. Copyright 2013. | ||
As discussed in Section 5.1.2, the polysulfide radical ion [S8]˙− is likely to be the primary product formed during discharge of a Li–S battery loaded with S8. This highly reactive ion is predicted to disproportionate exothermically to S8 and [S8]2−, while its further electrochemical reduction most probably results in dissociation to either 2[S4]˙− or [S3]˙− + [S5]˙−. These smaller radical ions can be reduced directly to the corresponding dianions or they recombine with each other to form the whole range of polysulfide ions [Sn]2− (n = 3–8), which have been detected by HPLC analysis after in situ derivatization by methyl triflate CF3SO3CH3 (Section 3.4); dimethyl polysulfanes Me2Sn of medium chain-lengths had the highest concentrations, while only marginal amounts of Me2S2 and Me2S8 were detected (Fig. 9).82 A mixture of polysulfide dianions and radical anions with between three and seven sulfur atoms is expected to show at least 20 Raman lines in the region 390–510 cm−1 due to the SS stretching vibrations (Section 3.1). In addition, some of these ions are expected to form ion pairs with the lithium cations enhancing the complexity of the vibrational spectra even more. Most of the reactions shown in Scheme 3 are exothermic in polar solvents, but their rate constants and concentration-dependence will be different. Therefore, the product distribution is expected to depend on the current density as well as on the polarity and viscosity of the solvent mixture. On the surface of the metal anode additional reactions occur including the formation of mono- and disulfide ions by chemical rather than electrochemical reduction.
For the description of the reaction mechanisms during discharge and re-charge of Li–S batteries by chemical equations the use of mass balances with fractional molecules or electrons should be avoided. Also, imaginary ions such as [S8]4− are unacceptable from a chemical point of view. For example, the frequently cited equation
| [S8]2− → [S6]2− + 1/4S8 | (31) |
| [S8]2− → 2[S4]˙− | (32) |
| [S8]2− + [S4]˙− → [S6]2− + [S6]˙− | (33) |
| [S4]˙− + [S6]˙− → [S10]2− | (34) |
| [S10]2− → [S2]2− + cyclo-S8 | (35) |
| [S4]2− + [S8]2− → 2[S6]2− | (36) |
Several authors have tried to elucidate the sulfur redox reactions in Li–S batteries by in situ Raman spectroscopy54,59,141 but, besides the radical anion [S3]˙−, a convincing identification of specific polysulfide dianions has not been obtained since the composition of the solutions is too complex and the spectra of the dianions do not differ sufficiently from each other. Surprisingly, the most intense fundamental modes of free polysulfide dianions in the region 400–500 cm−1 have seldom been detected and the spectra of ion pairs have usually not been considered at all. Furthermore, anions with an odd number of sulfur atoms were often neglected in the modelling of the reaction sequence. However, the disappearance and reappearance of the strong Raman signal of S8 at 217 cm−1 during discharging and re-charging has been reported by Wu et al. in 2015.59 The assignment of a weak line at 518 cm−1 to [S4]˙− in the latter work is probably in error since it does not fit the predicted fundamental modes of this ion (Section 3.1). In addition, a band at 1066 cm−1 attributed by Wu et al. to the dithionite anion is most probably caused by the first overtone 2ν1 of the trisulfide radical, calculated at ca. 1060 cm−1 (Section 3.1, Table 1); this line appears simultaneously with the 534 cm−1 Raman line of [S3]˙−.59
In general, in situ Raman spectroscopy may not be the best method to analyze the redox reactions in Li–S cells, since the spectra also include lines originating from solvent molecules and from the anions of the Li salts added to increase the conductivity and to protect the lithium anode. In 2015, Hannauer et al. recorded Raman spectra in situ using a helium–neon laser emitting at 632.8 nm, which fits the absorption band of the trisulfide radical anion near 600 nm. Therefore, these authors observed a strong resonance enhancement of the symmetric stretching vibration ν1 of [S3]˙− and of the overtones 2ν1 and 3ν1, which together dominate the spectrum despite the probably low radical concentration in the tetraethyleneglycol dimethylether–dioxolane (1:1) solvent mixture. Two additional weak and rather broad signals at 369 and 429 cm−1 were assigned to a polysulfide dianion mixture, although the first line does not match the experimental Raman spectrum of any polysulfide dianion. These signals occur and disappear simultaneously with the signals of [S3]˙− on discharge and re-charge of the model battery.54 The signal at 369 cm−1 may therefore arise from the LiS stretching vibrations of [LiSn]− ion pairs and the line-width indicates a superposition of two or more closely neighbouring lines. Only the 429 cm−1 line indicates an SS stretching vibration of a polysulfide anion or a mixture thereof.
The disappearance of S8 and formation of [S3]˙− during discharge of an Li–S battery has also been monitored by XANES measurements in 2015,48 but EPR spectra had already revealed the formation of [S3]˙− in 2012.67 Dominko et al. reported in 2018 the disappearance of S8 during discharge and formation of long-chain polysulfides followed by short-chain polysulfides and finally formation of Li2S and Li2S2 by XAS (Section 3.7).142 However, it is not clear whether the observed signals stem from free polysulfide anions or – more likely – from the ion pairs discussed above. Similar results have been reported by Patel et al., but were discussed on the basis of even-membered polysulfide ions only.96
Prior to 2010 the active sulfur species in the redox reactions of Li–S batteries have mainly been assumed to be free polysulfide dianions together with S8 and the radical anion [S3]˙−. However, in more recent work small, undissociated molecule-like clusters [Li2Sn] have been assumed to be the active ingredients. Polysulfide dianions are in fact stronger ligands (S-donors) than most solvents used in Li–S batteries. But lithium polysulfide solutions conduct electricity and, therefore, must contain ions. Consequently, anionic ion pairs [LiSn]− are probably the dominant species, with the lithium cations additionally coordinated by solvent molecules (or O-donor oxoanions) to obtain a coordination number of 4. The equilibrium between Li+ and [Sn]2− and the ion pair [LiSn]− will depend on the nature of the solvent. It has in fact been reported in 2017 that the UV-vis spectra of lithium polysulfide solutions strongly depend on the nature of the solvent,15e and in 2018 it was found that the voltammetric results are also solvent-dependent.131 Molecular dynamics (MD) calculations on molecular clusters [Li2S6] and [Li2S8] in DME or DOL did not show any dissociation of these ion pairs, but the cell dimensions (15 × 10 × 10 Å) may have been too small to allow for the polarization of the solvent continuum by the expected cations and anions.143 This solvent polarization stabilizes polysulfide anions enormously.44
Several authors have tried to elucidate the sulfur redox reactions in Li–S batteries by in situ7Li NMR spectroscopy (Section 3.6).86–88,96 While the formation of lithium polysulfides and Li2S during discharge and their disappearance during charging can clearly be seen in the spectra from a broad signal extending from −260 to +100 ppm, specific polysulfides could not be identified. During discharge the total area under this peak gradually increases, reaching a maximum at about 2.0 V and then gradually decreases and reaches a minimum at nearly the fully discharged state. Therefore, this peak which could be deconvoluted into four subpeaks was assigned to a mixture of Li2Sn species. The most important result is, however, the likely in situ detection of neutral ion pairs [LiSn]˙ by the strong influence of the unpaired electron on the chemical shift of the lithium ion. A broad signal near 200 ppm has been tentatively assigned to this type of species.87
The structures of the ion pairs [LiSn]− (n = 4, 6) as well as of [LiSn]˙ (n = 2, 3), each solvated by two DME molecules, have been investigated by computational methods using DFT theory and ε = 7.0 (Fig. 18).87 However, whether these structures represent the global minimum geometries needs to be confirmed, since the polyether ligands may also function as bidentate ligands forming bis-chelate complexes of composition [(DME)LiSn]˙ and [(DME)LiSn]−. Chelate complexes are usually of higher thermodynamic stability due to the lower reaction entropy.
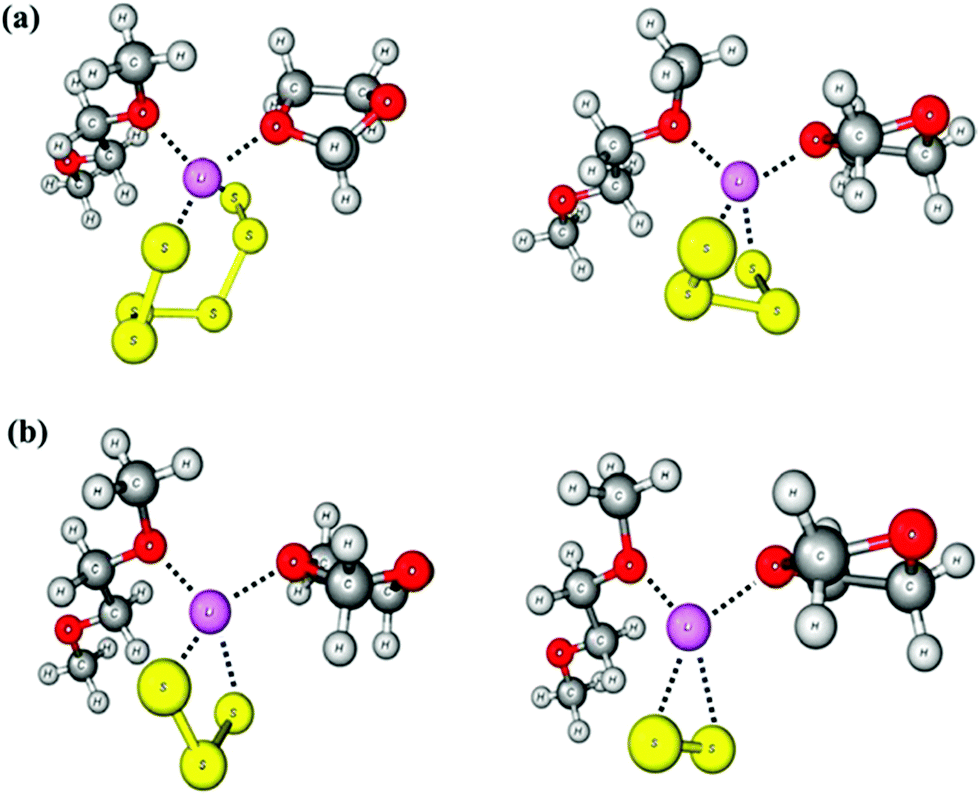 | ||
| Fig. 18 Molecular structures of the ion pairs (a) [(DME)2LiSn]− (n = 4 and 6) and (b) [(DME)2LiSn]˙ (n = 2, 3) solvated by dimethoxyethane molecules, calculated at the B3LYP/ccVQZ level using the COSMO correction for ε = 7 to simulate the solvent's influence. Reproduced from ref. 87 with permission. Copyright 2015, American Chemical Society. | ||
The Raman spectra of the various species [Sn]˙−, [Sn]2−, [LiSn]˙, [LiSn]− and cluster-like [Li2Sn], possibly present in the electrolytes of Li–S batteries, will be significantly different from each other, and Li–S stretching modes are to be expected in the region 200–350 cm−1.44 But with this complexity the spectroscopic determination of single species during discharge and recharge of Li–S batteries by any spectroscopic technique will be problematic. Unfortunately, anionic ion pairs of the type [LiSn]− have not been studied systematically by high-level quantum chemical calculations taking the solvation enthalpies into account in order to predict their thermodynamic properties. However, the analogous ion pairs of type [NaSn]− (n = 2–9) have been calculated and were shown to be cyclic with the polysulfide anions functioning as bidentate ligands (Fig. 15 and 16).
The influence of different alkali-metal cations M+ on the redox potentials of M–S batteries and their in situ UV-vis spectra has been studied by Zou et al. in 2018.121
Attempts have been made to elucidate the thermodynamics of neutral lithium polysulfide molecules [Li2Sn] in solution by high-level quantum-chemical calculations taking the solvation enthalpy into account.120 However, the structures of the [Li2Sn] clusters (n = 1–8) were calculated without coordinated solvent molecules, and some of the predicted clusters have puzzling geometries with Li–Li bonds. Experimental structures of solvate-free lithium polysulfides are unknown excepting Li2S2 for which a crystal structure determination is available.6 The reason is the very high electric field strength of the small naked Li+ ion resulting in strong coordinate bonds to polar solvent molecules, which function as chelating ligands. Therefore, lithium polysulfides always crystallize from solvents as solvates (Section 3.1, Fig. 6).
Finally, it should be mentioned that the decomposition of carbonate-type solvent molecules by nucleophilic attack of polysulfide dianions has been studied computationally.120 Ether-based solvents are more resistant to the polysulfide dianions and radical anions. However, a completely different approach to the solubility and shuttle problems has been reported by two groups who used either all lithium polysulfides as insoluble solids or all Li–S species completely dissolved. In 2017, Krossing, Nazar et al.144a replaced the conventional ether-based solvents by N,N-dimethyl trifluoromethylsulfonyl amine (DMT), which does not dissolve lithium polysulfides, and used the lithium salt of the particularly weakly coordinating anion [Al(OCHCF3)4]− at a concentration of 0.2 M as an ion carrier to suppress the dissolution of the lithium polysulfides formed during discharge at the carbon–sulfur cathode. In this way the shuttle phenomenon could be effectively suppressed, but the poor kinetics required an operating temperature of 50 °C. On the other hand, Yang et al.144b reported recently that all Li2Sn salts with n = 1–8 are soluble in a eutectic 1:1 mixture of ε-caprolactam and acetamide (m.p. −8 °C) up to a concentration of 0.7 M due to the high dielectric constant. This mixture has therefore been used as solvent for an inexpensive, almost non-flammable electrolyte together with the conducting salts Li[N(SO2CF3)2] and Li[NO3]. This type of battery not only avoids the massive volume expansion of the carbon electrode during discharge, but also uses the full energy capacity of the lithium–sulfur system since neither Li2S nor Li2S2 are precipitating. By addition of some DOL/DME to the solvent mixture the viscosity has been reduced to an acceptable level, and a high retention of the capacity during cycling is observed while growth of dendrites and shuttle effects are hardly noticeable. On the other hand, the detection of SH bonds by Raman and NMR spectroscopy indicated chemical reactions between the solvent mixture and the polysulfide dianions or radical anions. The authors stated that further studies are needed with high-mass loading of solid sulfur, the solubility of which is much lower than that of lithium sulfides.
Despite its extraordinary high specific energy, the Li–S battery in its present form (with ether-based solvent mixtures) can still not compete with the well-established lithium-ion battery for applications in the transport sector since the following inherent challenges still exist:
1. The formation of Li dendrites is a critical issue in most Li0-based rechargeable batteries. It leads to problems with safety, low Coulombic efficiencies, and short lifespans.
2. Solubility of active species and polysulfide (PS) shuttling: The redox shuttle phenomenon occurs as a result of the diffusion of soluble high-order PS such as Li2S6 formed on discharge through the separator and its reaction with the Li anode to form low-order and partially or completely insoluble lithium di- and monosulfide (Li2S2 and Li2S) in each cycle. This leads to the loss of active sulfur in the cathode and thereby lowers the degree of S utilization. The gradual accumulation of insulating sulfides can also seriously impede rapid access to the Li anode, thereby resulting in a poor rate capability. Furthermore, the PS could react negatively with the electrolyte components such as solvents, salt anions etc., depending on their functionality.
3. Insulating nature: S8 (10−30 S cm−1 at RT) and its various discharge products (Li2Sn, mainly Li2S (ca. 10−13 S cm−1 at RT) and Li2S2 exhibit poor electronic and ionic conductivities, which lead to an increase in the internal resistance of the Li–S cell, and thereby to high voltage polarization. This in turn results in a reduction in the Coulombic and energy efficiencies of the Li–S cell.136
4. The high costs of some of the lithium salts and of Nafion-type separators used in some cases are another limitation.
In order to overcome some of these problems bulk-type all-solid-state lithium–sulfur batteries without organic liquid electrolytes have been studied. Solid-state Li–S batteries are made up of negative/positive electrodes separated by a solid electrolyte resulting in a high energy density due to the compact structure. In addition, solid-state ionic electrolytes prevent polysulfide shuttling, are inert toward metallic lithium, can resist the growth of lithium dendrites, have excellent temperature stability and a wide stable electrochemical window that enables high voltage cathodes. But there are still many critical challenges to be overcome for large-scale application, including a large interfacial resistance between electrolyte and electrodes, low ionic conductivity of electrode materials, volume change of the cathode during cycling, and instability in contact with the anode.145
5.2 Organic syntheses
The use of inorganic polysulfides for the synthesis of organosulfur compounds (OSCs) either in the form of an ammonium salt or generated in situ in solutions of sulfur in liquid ammonia or amines has a long history, e.g. in the Willgerodt–Kindler reaction for the conversion of alkyl aryl ketones into thioamides.75 The formation of a blue colour when sulfur is treated with a base in water or acetone is also well-documented, although it was not until the early 1970s that this blue species was unequivocally identified as the trisulfur radical anion [S3]˙−.8,16 Solutions of elemental sulfur in combination with bases such as M2[CO3] (M = K, Cs), M[OtBu] (M = Na, K), K[OH] or Na[SH] in electron-pair donor solvents are now widely used in organic synthesis.75 Since this combination produces polysulfides and, hence, polysulfide radical anions as evidenced by a blue colour, it is surprising that more effort has not been made until very recently to target [S3]˙− as a reagent. This section will focus on advances in the last few years in the use of this radical anion in organic syntheses.In a 2013 study related to formation of organosulfur compounds (OSCs) under geological conditions, Said-Ahmed et al. reported that the reaction of elemental sulfur with 1-octene and water at 100–130 °C in the pH range 8.0–8.5 produces OSCs and 2-octanone.146a These authors also observed a pale blue colour (possibly [S3]˙−) for the reaction solution and proposed a radical mechanism for the transformation. However, in a re-investigation two years later, Ding and Adam challenged this proposal and suggested an alternative concerted mechanism to explain the experimental results.146b
This controversy implies that direct evidence for the involvement of polysulfide radical anions is essential to any mechanistic proposal involving the reactions of organic compounds with sulfur in the presence of a base. In that context, in 2014 Zhang and co-workers investigated the synthesis of thiophenes via the interaction of 1,3-diynes with sulfur and Na[OtBu] (Scheme 4a).147 In a separate experiment they showed that the blue solution formed upon treatment of sulfur with Na[OtBu] in DMF at room temperature displays the characteristic EPR signal for [S3]˙− (g = 2.0292) (Section 3.5) which, they suggested, plays an essential role as an intermediate in the formation of thiophenes. These authors also demonstrated that this cheap, environmentally attractive use of elemental sulfur provides a general route to both symmetrically and unsymmetrically 2,5-disubstituted thiophenes in 56–76% yields.147 Two years later Wang and co-workers described a related synthesis in which diarylannulated sulfides were constructed in high yields via sulfur–iodine exchange from the reaction of diaryliodonium salts with sulfur and Cs2[CO3] in DMSO at 120 °C (Scheme 4b).148 The formation of [S3]˙− was also demonstrated in that study by the EPR spectrum of a blue DMSO solution of sulfur and Cs2[CO3] when heated to 100 °C. The same reagent has been used to prepare thiazole derivatives by reaction with a mixture of ketoxime acetates and PhNCS.149
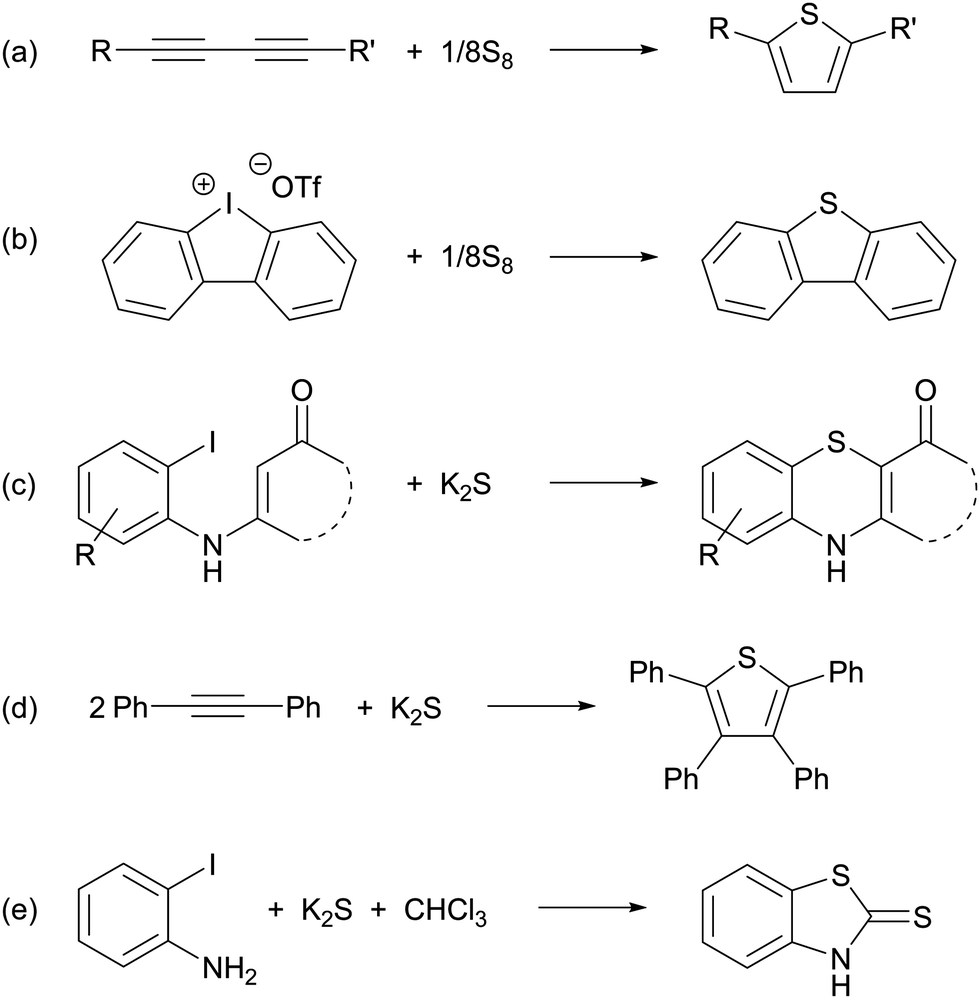 | ||
| Scheme 4 (a) Na[OtBu]/tBuOH, DMF, 25 °C; (b) Cs2[CO3], DMSO, 100 °C; (c) and (d) DMF, 130 °C; (e) Cu[OAc]2, 2,2′-dipyridyl, NaOH, NMP. | ||
Potassium sulfide K2S has also been used as a source of [S3]˙− for organic synthesis. Thus, Ji and co-workers obtained benzothiazine derivatives in high yields (47–96%) from reactions of K2S with enaminones in DMF at 130 °C (Scheme 4c).149 Evidence for the formation of [S3]˙− in this system was provided by the observation of a strong EPR signal, g = 2.02, for a DMF solution of K2S at room temperature. Furthermore, the presence of the trisulfide radical anion in such solutions was confirmed by the characteristic broad visible absorption band at ca. 615 nm (Section 3.2) and the symmetric S–S–S stretching vibration at 531 cm−1 in the Raman spectrum (Section 3.1). The authors proposed that “DMF activates the electron-donating agent K2S to give [S3]˙−”,150a but evidence for this suggestion is lacking and the generation of [S3]˙− must involve either the presence or formation of polysulfides. The same group has also employed [S3]˙−, generated in situ from K2S in DMF, for the synthesis of thiophenes via [1+2+2] cycloaddition (Scheme 4d).150b
In a further development of this synthetic strategy, Jiang and co-workers used a combination of K2S and chloroform as an alternative to carbon disulfide for the synthesis of a library of benzothiazine-thiones from N-substituted-2-iodobenzylamines in N-methylpyrrolidone (NMP) in the presence of a Cu(II) catalyst.64 Of particular interest is the application of this protocol to the synthesis of 2-mercaptobenzothiazole, which is manufactured on a multi-ton scale for use as an accelerator in rubber vulcanization as well as an antioxidant stabilizer and metal corrosion inhibitor for rubber and plastics (Scheme 4e). The authors established the presence of [S3]˙− in solutions of K2S in NMP via characteristic UV-visible and EPR spectroscopic signatures (vide supra) and concluded that the radical anion plays a key role in the final thiocarbonyl formation.64
In an investigation of the use of sulfur–amine solutions for the production of metal sulfide nanocrystals, Ozin and co-workers provided insights relevant to the application of these reagents in organic synthesis.151 These authors confirmed earlier observations that solutions of sulfur in octylamine form octylammonium polysulfides at low temperature and, on warming to 130 °C, generate H2S which reacts with the amine to produce thioamides [eqn (37)]. Although the possibility of sulfur radical anions in this transformation was recognized, the specific role of [S3]˙− was not pursued. However, it was shown that thioamides exhibit more rapid kinetics than sulfur–alkylamine solutions in the synthesis of metal sulfide nanocrystals.151
C7H15CH2NH2 + [Sn]2− → C7H15(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)NH2 + H2S + [Sn−2]2− S)NH2 + H2S + [Sn−2]2− | (37) |
5.3 Biological chemistry of polysulfides
Sulfur compounds of oxidation states ranging from 2− to 6+ are abundant on Earth and are essential for life. Therefore, sulfur is the fourth most abundant element in artificial fertilizers, after N, P and K. As for other elements a natural cycle also exists for sulfur, and microorganisms play a critical role in driving sulfur cycling by sulfide oxidation to sulfate in one habitat and by reduction of the latter back to sulfide in another environment.72 The corresponding oxidation, reduction and disproportionation reactions include intermediate species such as polysulfides [Sn]2−, elemental sulfur (S0), thiosulfate [S2O3]2− and polythionates [SnO6]2−.Polysulfide anions are the key reactants in a large number of important biological and microbiological oxygen-poor systems.46,152 They can be generated during mitochondrial oxidative catabolism,153–155 by metalloprotein-catalyzed oxidation of sulfide156–158 or by reaction of sulfide with NO.159 However, direct oxidation of sulfide by reactive oxygen species (ROS) is relatively slow in many cases.160
The mechanism of polysulfide formation by transition-metal catalyzed oxidation of monosulfide anions is well understood.161 Removal of an electron from [HS]− ions gives hydrogen sulfide radicals which are relatively strong acids and therefore are deprotonated in water at pH values near or above 7; the product radicals then dimerize to disulfide dianions:
| [HS]−– e− → [HS]˙ | (38) |
| [HS]˙ + H2O → S˙− + [H3O]+ | (39) |
| 2S˙− → [S2]2− | (40) |
| [S2]2−– e− → [S2]˙− | (41) |
| 2[S2]˙− → [S4]2− | (42) |
| [S4]2−– e− → [S4]˙− | (43) |
| 2 [S4]˙− → [S8]2− | (44) |
In some sulfur bacteria the growth of a chain of sulfur atoms may begin with the cleavage of the sulfur–sulfur bond of cystine (R2S2) as a component of an enzyme by the nucleophilic attack of sulfide ions and may proceed stepwise by a series of oxidation and chain-cleavage reactions, possibly catalyzed by a transition metal (see below), e.g.:
| R–S–S–R + [HS]− → [R–S2]− + [R–S]− + H+ | (45) |
| [R–S2]−– e− → [R–S2]˙ | (46) |
| [R–S2]˙ + S˙− → [R–S3]− | (47) |
| [R–Sn]− → cyclo-S8 + [R–Sn−8]− (n > 8) | (48) |
| R–S2˙ + [HS]− → [R–S]− + [S2]˙− + H+ | (49) |
| 2[S2]˙− → [S4]2− | (50) |
The electrons formally released in the above reactions reduce the transition metal (usually iron) from Fe3+ to Fe2+ which is then re-oxidized to Fe3+ by O2. The latter is reduced to [O2]˙− from which H2O2 and its anion [HO2]− are formed by disproportionation:157
| Fe3+–OH2 + [HS]− → Fe2+–SH + H2O | (51) |
| Fe2+–OH2 + O2 → Fe2+–O2 + H2O | (52) |
| Fe2+–O2 + [HS]− → Fe3+–SH + [O2]˙− | (53) |
| Fe3+–SH + O2 → Fe2+−O2 + [HS]˙ | (54) |
| 2[O2]˙− + H2O → HO2− + [OH]− + O2 | (55) |
Polysulfide ions in biological samples have been notoriously challenging to quantify due to their reactivity, sensitivity to sample handling and the analytical complexity associated with their measurement. Some authors tried to identify these ions during enzymatic sulfide oxidation by derivatization with bromobimane but, as described in Section 3.4, this reaction is too slow to yield reliable results. However, the appearance and disappearance of polysulfide ions has definitely been observed during bacterial oxidation of sulfide to elemental sulfur (forming “globules”), and for the further oxidation of the “globules” to sulfate after all sulfide has been depleted. The polysulfides are also essential to solubilize the water-insoluble elemental sulfur (globule degradation).162–164
Marnocha et al. reported in 2016 that a standard polysulfide solution prepared from sulfide and S8 yielded four peaks in the HPLC chromatogram after derivatization by monobromobimane, but the sulfur chain-lengths of the suspected Bm2Sx species were not determined.164 Recently, Nagy46 studied the polysulfide speciation in aqueous solution using the cell-permeable alkylating agents iodoacetamide (I–CH2–CO–NH2) and N-ethylmaleimide [Et–N(CO)2C2H2] for the derivatization procedure, but observed that the derivatization changed the speciation of the polysulfide mixture substantially. This may be due to the amine-type nitrogen atoms in these two reagents, since amines like ammonia are well-known catalysts for the interconversion of polysulfide and polysulfane compounds by nucleophilic attack.13 Nevertheless, polysulfide ions have definitely been detected in biological samples, but the exact quantitative distribution needs to be demonstrated (for references, see ref. 46). The formation of chain-like sulfur compounds during enzymatic sulfide oxidation has also been demonstrated by XANES measurements, which revealed differing chain-terminating groups depending on the organism.90
An unresolved question seems to be the reaction of polysulfide ions with molecular oxygen in microbiological samples, although the autoxidation of aqueous polysulfide yielding thiosulfate and sulfur has been studied extensively.38 However, the formation of sulfur-chains via condensation of a sulfenic acid derivative of cysteine with H2S or thiols has been discussed in 2017.167 The sulfenic acids may well be reactive intermediates in the oxidative metabolism of sulfur bacteria, but solid evidence seems to be missing.
| –S–OH + H–S– → –S–S– + H2O | (56) |
| [S3]˙− + O2˙˙ → [SSSOO]˙− | (57) |
The validity of eqn (57) can be inferred from the well-studied reaction of [PenSS]˙ radicals with molecular oxygen (Pen = penicillamine).168 This reaction was monitored by following the decay kinetics of [PenSS]˙ after pulse irradiation of aqueous solutions containing 1 × 10−3 M PenSSSPen, 1.0 M tert-butanol and different O2 concentrations at pH = 5.9. In the presence of molecular oxygen, the pure second-order decay kinetics of [PenSS]˙ (due to dimerization) observed in N2-saturated solutions, changed to pseudo-first-order kinetics showing a linear decrease of the [PenSS]˙ lifetimes with increasing oxygen concentrations:
| [PenSS]˙ + O2 → [PenSSOO]˙ | (58) |
5.4 Geochemical transformations
Inorganic polysulfides are salient reagents in numerous geochemical processes in sediments and oxygen-poor systems.152,169–171 For example, both the diagenetic formation of pyrite [iron(II) disulfide] and the production of organosulfur compounds have been attributed in part to reactions of polysulfides with metal ions and unsaturated organic substrates, respectively.172,173 Polysulfides are also the possible source of malodorous, volatile sulfur compounds such as organic polysulfanes R–Sn–R via reactions with chlorinated pollutants.174–176 Many metals have a high affinity for polysulfide ligands and the role of polysulfide radical anions in the transport of economically important metals such as Cu, Au and Pt at elevated temperatures and pressures has been the subject of recent studies (vide infra).Traditional studies of sulfur speciation in geological fluids have focused on dissolved sulfide and sulfate, since these anions predominate in sulfur-bearing minerals. In 2011 Pokrovski and Dubrovinsky reported the identification of the trisulfur radical anion [S3]˙− in aqueous solutions at elevated temperatures and pressures (>250 °C and 5–35 kbar) over a wide pH range by in situ Raman spectroscopy using a diamond anvil cell.8,177 As pointed out by these authors56 and by Manning,177 this ground-breaking discovery has important implications for the nature of geochemical processes such as metal transport and sulfur-isotope fractionation. The role of [S3]˙− in these processes has subsequently been investigated extensively by both experimental and computational approaches.
In a further illustration of the capability of in situ Raman spectroscopy for the detection of [S3]˙− in sulfur-rich hydrothermal fluids, it was shown that the formation of this radical anion commences in the temperature range 200–300 °C and grows up to (at least) 500 °C and 1 kbar.53 In addition, this process was found to be reversible, since the spectroscopic signature for [S3]˙− disappears on cooling and reappears on heating. The observation of [S3]˙− under these conditions suggests that this sulfur species may serve as a ligand for Cu–Au–Mo deposits. The long-held belief that H2S and Cl− ions are the ligands responsible for gold transport and precipitation in the Earth's crust has been challenged by the results of recent investigations by Pokrovski and co-workers using in situ X-ray absorption spectroscopy.178 By the use of this technique, in combination with first-principles molecular dynamics, these authors established that [S3]˙− makes a major contribution to gold solubility in aqueous solutions at elevated temperatures (>250 °C) and pressures (>100 bar) and [Au(SH)S3]− was identified as the most stable complex.178 Experimental results on metal complexes of polysulfide radical anions [Sn]˙− (n = 2, 4) are described in Section 5.5.
Thermochemical sulfate reduction (TSR) plays a critical role in the global sulfur cycle and may affect measurements of sulfur isotope fractionation. In contrast to sulfate reduction induced by bacteria, TSR generally occurs at high temperatures (T > 150 °C), while abiogenic reduction may occur at lower temperatures in some geological environments. Based on in situ Raman spectra, in 2014 Truche and co-workers demonstrated that [S3]˙− is the dominant intermediate sulfur species involved in abiogenic sulfate reduction by H2S in the temperature range 100–350 °C.179 This finding necessitates a re-evaluation of the sulfur isotope exchange mechanisms between sulfate and sulfide in order to include the possible role of [S3]˙−. Three years later Barré et al. disclosed the formation of [S3]˙− (and polysulfide dianions) above 100 °C in natural fluids from a typical buried sedimentary environment where TSR occurred; only sulfide and sulfate were observed at 25 °C.180 They estimated the [S3]˙− concentration to be 0.2–3.0% of total sulfur in the temperature range 200–300 °C.
Having established the existence and potential importance of [S3]˙− in aqueous sulfur-bearing fluids at high temperatures, in 2015 Pokrovsky and Dubessy turned their attention to the quantification of the stability and abundance of this ephemeral species.181In situ Raman spectra were obtained on model thiosulfate, sulfide and sulfate aqueous solutions for a wide range of sulfur concentrations (0.5–10.0 wt%), acidities (pH 3–8), temperatures (200–500 °C) and pressures (15–1500 bar). They found that [S3]˙− represents up to 10% of dissolved sulfur at 300–500 °C and more than 50% at 600–700 °C in sulfur-rich fluids. The disulfur radical anion [S2]˙− was also tentatively identified at 450–500 °C via observation of a small peak at 580 ± 5 cm−1 accompanied by weak overtone bands at 1160 ± 10 cm−1 and 1740 ± 15 cm−1.181
5.5 Metal complexes
The chemistry of both main-group and transition-metal complexes with polysulfido ligands [Sn]2− (n = 2–9) has a long history, which has been discussed extensively in the review literature.43 These homoatomic dianionic ligands usually undergo S,S′-chelation to the metal in mononuclear complexes or act as bridging ligands in dinuclear complexes. Information on metal complexes of polysulfide radical anions is relatively sparse and has mostly appeared in the last 15 years. The discussion in this section will be limited to complexes of [Sn]˙− (n = 2, 3, 4) ligands in view of their novelty and relevance both to computational studies (Section 4) and metal transport in geochemical systems (Section 5.4).Transition-metal complexes that may contain the end-on supersulfide radical anion [S2]˙− have been known for more than 45 years, e.g. [Cp(EtS)FeSSFe(SEt)Cp] and [(NH3)5RuSSRu(NH3)5]Cl4·2H2O.182 The nature of the bridging S2 ligand in these dinuclear complexes depends on the assignment of oxidation states to the two metal centers and may fall between the monoanion [S2]˙− and the dianion [S2]2−. For example, on the basis of structural and spectroscopic evidence, Elder and Trkula described the ruthenium complex as primarily a mixed-valent [Ru(II)/Ru(III)] species with a bridging [S2]˙− ligand accompanied by a minor contribution from the corresponding Ru(III)/Ru(III) combination bridged by a [S2]2− moiety.182b The SS stretching vibration in this complex is 519 cm−1,182bcf. ν(SS) = 594–623 cm−1 for the [S2]˙− radical anion in alkali-metal halide matrices,183 while the S–S distance is 202.4 (1) pm, cf. d(S–S) = 199.5–202.3 pm in complexes with a central M–S–S–M unit.43 In 2005 Tolman and co-workers characterized the binuclear complex [LCu(μ-S2)CuL][OTf]2 (L = tmeda; OTf = CF3SO3), which exhibited ν(SS) = 613 cm−1 and d(S–S) = 195.0(1) pm, leading to the conclusion that it is a Cu(II)/Cu(II) complex with two bridging [S2]˙− ligands.184
Three years later Driess and co-workers characterized an interesting nickel(II) complex of the [S2]˙− ligand, [LNi(η2-S2)]n with L = [CH{(CMe)(2,6-iPr2C6H3N)}2]−, which exists as a centrosymmetric dimer (n = 2) with a four-membered rectangular S4 ring in the solid state [d(S–S) = 194.4(2) and 277.7(2) pm].185 In solution, the 1H NMR and EPR spectra reveal the partial dissociation of this complex into the paramagnetic monomer (n = 1) (Scheme 5). DFT calculations of this monomer reveal that the HOMO is a singly occupied π* orbital (SOMO) completely located on the disulfur ligand, consistent with a complex of [S2]˙−. The dimer is formed from a π*(SOMO)–π*(SOMO) interaction to give a four-center two-electron bond.185 The reaction of [LNi(η2-S2)]n with Ph3P produces (LNi)(μ-η2:η2–S2), a side-on complex of the dianion [S2]2− with d(S–S) = 205.1(1) pm (Scheme 5).185
 | ||
| Scheme 5 Monomer–dimer equilibrium for [LNi(η2-S2)]n and reaction with PPh3. | ||
Metal complexes of the trisulfur radical anion are of interest in view of the possible role of [S3]˙− in the transport of coinage (and other) metals in geological systems (Section 5.4). Based on calculated free energies, Tossell found that [S3]˙− forms complexes with Cu+ that are similar in stability to Cu+ complexes with [HS]− and he predicted bidentate (S,S′) ligation of the trisulfide radical anion in the complexes [CuS3(OH2)] and [Cu(S3)2]−.105 Similarly, Mei and co-workers reported that the ligands [S3]˙− and [HS]− are of comparable strengths for Au+ as determined by ab initio molecular dynamics simulations.186 In experimental work Pokrovski et al. have provided evidence for the formation of the stable complex [Au(SH)S3]− at elevated temperatures and pressures from the interpretation of X-ray absorption spectra (Section 5.4).178 Although no molecular complexes of this important sulfur ligand have been isolated and characterized, Cummins and co-workers have described the reversible generation of [S3]˙− from a di-tin complex with bridging pentasulfido and monosulfido ligands [(μ-S5)Sn2(μ-S)(mBDCA-5t)]2− (Scheme 6).187 Solutions of this yellow polysulfido complex in DMF or DMSO are dark blue-green and exhibit the characteristic spectroscopic signatures of [S3]˙− (a strong absorption at λmax = 617 nm in the UV-visible spectrum and a broad signal centred at g = 2.0290 in the EPR spectrum). 187 The amount of [S3]˙− in DMF solution increases as (a) the concentration of the di-tin complex is diluted or (b) the temperature is increased from 20 to 85 °C. The formation of the neutral di-tin complex [(μ-S5)Sn2(μ-S)(mBDCA-5t)] as a result of this dissociation was proposed, but not confirmed.187
 | ||
| Scheme 6 A polysulfido di-tin complex as a reversible source of [S3]˙−. | ||
A metal complex of the tetrasulfide radical anion [S4]˙− was first proposed in the mid-1990s by Isobe and co-workers for the tetranuclear rhodium dication [{Rh2Cp*2(μ-CH2)2}2(μ-S4)]2+.188a The rectangular S4 unit in this complex has two short and two long S–S distances of 197.9(1) and 270.2(1) pm, respectively. The suggested radical monoanion formulation was based on a population analysis, which gave a charge of −0.7e on the S4 ligand.188b In 2004 the same group structurally characterized a related tetranuclear iridium dication [{Ir2Cp*2(μ-CH2)2}2(μ-S4)]2+ with S–S bond lengths of 204.9(3) and 289.5(3) pm.188c In the case of the iridium complex, however, the shorter S–S bonds lie parallel to the Ir–Ir bonds and the longer ones are perpendicular to the Ir2 backbone, whereas the reverse is true in the corresponding Rh4 complex (Fig. 19). Hofmann and co-workers have addressed this structural dichotomy for tetranuclear group 9 complexes of the rectangular [S4]2− ligand by using DFT and extended Hückel calculations.189 They found that either orientation is attainable for both metals with a barrier for interconversion of ca. 46 kcal mol−1 (192 kJ mol−1). The viability of both orientations is attributed to a Jahn–Teller distortion from a square, which is favoured by a −2 charge. In both arrangements the [S4]2− rectangle has two S–S σ bonds and two three-electron S–S bonds.189
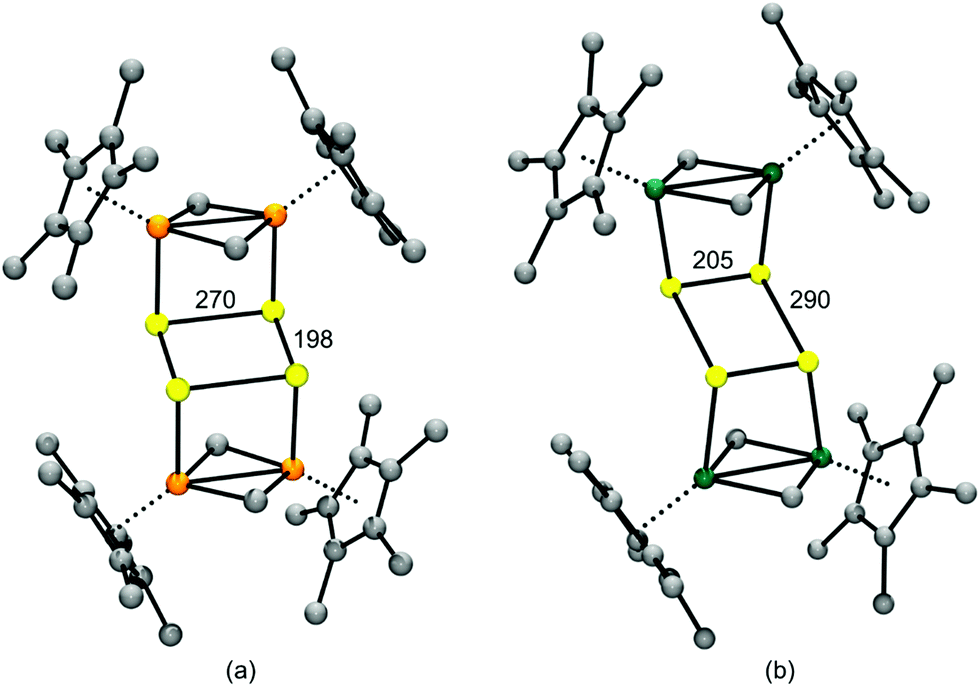 | ||
| Fig. 19 Chair-like structures of [{M2Cp*2(μ-CH2)2}2(μ-S4)]2+ with rectangular S4 units (a) M = Rh (b) M = Ir; bond lengths are given in pm (adapted from Fig. 1 in ref. 189). | ||
The first structurally characterized complex of [S4]˙− was reported in 2017 by Coles and co-workers as part of an investigation of the reactions of bismuth(II) radicals of the type Bi(NONR) (NONR = [O(SiMe2NR)2]2−).10 They found that treatment of ˙Bi(NONAr*) (Ar* = 2,6-[(C6H5)2CH]2-4-tBuC6H2), with cyclo-S8 gave yellow crystals of the dimer [Bi(NONAr*)]2(μ-S8) (Scheme 7). The [S8]2− ligand in this Bi(III)/Bi(III) complex is quite different from that in dinuclear coinage-metal complexes, e.g. [(S6)Cu(μ-S8)Cu(S6)]4−,190 or in the ion-separated salts [Ni(N-MeIm)6]S826a and [HNEt3][S8]26b (Fig. 1). In the centrosymmetric bismuth complex two Bi(NONAr*)(S4) monomer units with S–S bond lengths in the range 202.1(3)–211.3(3) pm are linked by weak S⋯S interactions [2.758(4)–321.3(4) pm]. Thus, the bismuth complex can be viewed as a Bi(III) complex of the elusive [S4]˙− radical anion which, according to DFT calculations, dimerizes via a π*(SOMO)–π*(SOMO) interaction similar to that described above for the Ni(II) complex of the [S2]˙− ligand [LNi(η2-S2)].185
 | ||
| Scheme 7 Formation of a Bi(III) complex of [S4]˙− from Bi(II) radical ˙Bi(NONAr*) and cyclo-S8. | ||
5.6 Atmospheric chemistry
In contrast to the salts of the hydrosulfide ion [HS]− with organic cations, e.g. [NBu4][HS], which are used to prepare soluble long-chain polysulfides (Section 2), ammonium hydrosulfide [NH4][HS] is unstable at ambient temperatures with respect to the formation of H2S and NH3. However, condensed [NH4][HS] is thought to be present in the cold atmospheres of planets, notably in Jovian clouds. The identity of the chromophore(s) responsible for Jupiter's Great Red Spot remains an enigma. Since Jupiter's atmosphere is subject to energetic particle bombardment, Loeffler and co-workers have investigated the effect of ≈0.9 MeV proton irradiation on [NH4][HS] at 10–160 K by using IR and UV-visible spectroscopy to identify the products.191 In the IR study both natural abundance and 15N-enriched (98%) [NH4][HS] was used, which enabled the identification of binary sulfur-nitrogen anions [NSn]− (n = 3, 4) based on the isotopic shift of SN stretching vibrations.191c,192 In addition, the IR spectra exhibited strong bands at 566 and 491 cm−1 attributed to [S3]˙−and the polysulfides [Sn]2−/[HSn]−, respectively.191c The observation of an absorption band at 610 cm−1 in the UV-visible spectrum, which is more pronounced with an increase in temperature or radiation dose, is consistent with the formation of [S3]˙− as is the change in the colour of the sample from yellow to red and then to green. These observations are reminiscent of the behaviour of solutions of sulfur in liquid ammonia, which are red at lower temperatures and green or blue at room temperature and have been shown to contain [NS4]− and [S3]˙− ions, both of which are blue species, and yellow-orange polysulfide dianions.1936 Summary and conclusions
The synthesis and X-ray structures of polysulfide dianions [Sn]2− with alkali or alkaline earth metal cations have been studied for at least 70 years. With the exception of the recently identified branched isomer of the tetrasulfide, pyramidal [SS3]2− (C3v), these dianions exist as chain-like species with up to 13 atoms and the negative charges concentrated on the terminal atoms. Odd- and even-membered dianions are equally important and the pentasulfide dianion is thermodynamically favoured in water. The entrapment of polysulfide radical anions [Sn]˙− in a zeolitic matrix, e.g. in ultramarines, and their characterization by spectroscopic methods also has a long history. However, the importance of these simple homoatomic anionic species across a broad range of chemical, physical and biological sciences has only come to the forefront in recent years. In this review we have discussed critically the significant advances that have been made during the past ca. 15 years in the synthesis, characterization by physical and computational techniques, reactions and applications of these pervasive chalcogenide anions.The contemporary emphasis in synthetic work is on the generation of pure, soluble salts of polysulfide dianions in order to determine their function in alkali-metal sulfur batteries and biological systems. The long-known reaction of hydrosulfide ions [HS]− with cyclo-S8 has been employed effectively for this purpose using large organic cations, e.g. tetraalkylammonium or pyrrolidinium in the case of [Me3SiS]− salts. The counter-cation has a notable influence on the stability of [HS]− salts. Thus, while [NBu4][HS] is stable under ambient conditions, the ammonium salt [NH4][HS] must be cooled to avoid decomposition. Spectroscopic evidence (IR/Raman, UV-visible) indicates that irradiation of cold [NH4][HS], which is present in planetary atmospheres, produces a mixture of polysulfides [Sn]2−, [HSn]−, [S3]˙− as well as binary sulfur–nitrogen anions [SnN]− (n = 3, 4), which contribute to the chromophore of Jupiter's Great Red Spot.
The identification of individual polysulfides [Sn]2− in solution is of utmost importance in the context of lithium–sulfur batteries and in biological and geological settings, but difficult owing to the existence of equilibria between different dianions as well as the partial dissociation or disproportionation to give polysulfide radical anions. In aqueous solutions, isotopic exchange between the various dianions is ca. 70% complete within 8 s. The polarity of the solvent has a strong influence on the extent of S–S homolytic dissociation of [Sn]2− chains, which are thermodynamically stable in an aqueous environment at 25 °C, but dissociate in solvents of low polarity, e.g. THF, and in the gas phase. The best approach to the identification of dissolved polysulfide dianions is rapid derivatization with methyl triflate (CF3SO3CH3) followed by characterization of the polysulfanes Me–Sn–Me so formed by HPLC and 1H NMR spectra. Solid and dissolved polysulfides also undergo autoxidation to give elemental sulfur and sulfur oxo-anions such as thiosulfate and sulfate; the novel adduct [S3O2]2−, formed by reaction of [S3]˙− with triplet state O2 and detected by mass spectrometry, is probably the primary reaction product in solution.
In the gas phase polysulfide dianions [Sn]2− (n < 7) are unstable thermodynamically owing to Coulomb explosion. Consequently, the use of density functional methods to calculate vibrational modes has to be applied with caution, whereas Hartree–Fock and valence force field calculations yield reliable results. In solution, the cations of polysulfide salts are coordinated to solvent molecules or to the polysulfide dianions, depending on the donor strength. This effect is particularly important with Li+ ions due to their extremely high field strength resulting in ion pairs such as [Li(solvent)Sn]− (n = 2–8) with cyclic or cluster-like structures, which are probably the active species in lithium–sulfur batteries. However, their thermodynamic properties under realistic conditions still need to be explored, e.g. by high-level quantum-chemical calculations. In contrast, the chemical reactions in solvent-free high-temperature Na–S batteries and their thermodynamics are well understood and dominated by polysulfide dianions and radical monoanions, as well as cyclic ion pairs [NaSn]−, the thermodynamic properties of which have been investigated by high-level quantum-chemical calculations. Computational studies show that the Gibbs energy of the pyramidal isomer [SS3]2− is only ca. 13.0 kJ mol−1 higher than for the [S4]2− chain suggesting that branched isomers could be present in polysulfides at high temperatures, e.g. in HT sodium–sulfur batteries.
Spectroscopic techniques, especially Raman, UV-visible and, in the case of the radical anions, EPR, are potentially useful for the identification of polysulfide species. The highly coloured radical anions [Sn]˙− are readily distinguished by their distinctive strong visible absorption bands at ca. 410 nm (n = 2) and 595–620 nm (n = 3). On the other hand, the absorption maxima for the series of dianions [Sn]2− (n = 2–8) lie close together and the interpretation of UV-visible spectra may be complicated by the additional presence of radical anions. Consequently, the use of a combination of spectroscopic techniques is advisable for a definitive identification of these homoatomic species. Raman spectra of solid and dissolved alkali metal polysulfides M2Sn are characterized by SS stretching modes in the region 390–510 cm−1, and bending and torsion modes occur below 300 cm−1. The polysulfide radical anions [Sn]˙− (n = 2,3) show characteristic Raman bands that are enhanced in the Resonance Raman spectra so that overtones can be observed. In a very recent example, Raman spectroscopy supplied the unambiguous identification of [S3]˙− in the blue mineral (lapis lazuli) recently found in a monastery in Germany in the teeth of a medieval woman, who was probably working on illuminated manuscripts. EPR spectra have been used to show that the bent trisulfide species [S3]˙− adopts different orientations within the sodalite cage of ultramarines as well as to provide a definitive characterization of the homologue [S2]˙−.
The occurrence of the tetrasulfide radical anion [S4]˙− has been proposed frequently on the basis of solitary spectroscopic evidence, e.g. EPR, UV-visible or Raman spectra, but the definitive characterization of this species is still elusive. In some cases, the possibility of an alternative species, e.g. neutral S4, the dianion [S8]2− or even the isobaric species [S3O2]2−, cannot be ruled out. A combined spectroscopic study of dilute solutions of a [S8]2− salt in electron-pair donor solvents may elucidate this conundrum. With the exception of the [PPh4]+ salt of cyclo-[S6]˙−, the larger polysulfide radical anions [Sn]˙− (n = 5, 7, 8) have not been characterized experimentally in condensed phases. The six-membered ring in [S6]˙− incorporates two long S–S bonds (263 pm) and computational studies for [Sn]˙− (n = 7, 8) predict “broken ring” structures with even longer S–S bonds. Electrochemical reduction of S8 produces [S8]˙− as the primary product, which, however, is unstable and disproportionates to S8 and [S8]2− and can also be further reduced with simultaneous dissociation to [S3]˙− + [S5]˙− or 2[S4]˙−. These exothermic reactions are proposed to occur in room-temperature alkali metal–sulfur batteries. The main problems with these batteries are the growth of dendrites at the metal anode, the destruction of the sulfur-based cathode by massive volume changes during charging and discharging, the reaction of the solvent(s) with the sulfur species and the self-discharge owing to the shuttle effect (diffusion) of the anions and their chemical reduction by the metal. During charging of batteries, as well as during oxidation of sulfide by sulfur bacteria, S8 is formed from long-chain polysulfide dianions by intramolecular nucleophilic attack with extrusion of the sulfur homocycle.
Ammonium polysulfides [NH4]2[Sn] or solutions of cyclo-S8 in ammonia or amines have been used as reagents in organic synthesis for >100 years, e.g. for the transformation of carbonyls into thiocarbonyls. Although such solutions are known to contain the trisulfide radical anion [S3]˙, the targeted use of this reagent for the preparation of organosulfur compounds is a recent development. In these applications [S3]˙− is generated from solutions of cyclo-S8 with a base, e.g. Na[OtBu], Cs2[CO3] or Na[SH] in electron-pair donor solvents or from K2S in DMF, and its presence is demonstrated by UV-visible and EPR spectroscopy. Reactions of [S3]˙− generated in this way give high yields of a variety of heterocyclic organosulfur compounds, including thiophenes and the industrially important 2-mercaptobenzothiazole. The pathway for the production of [S3]˙− in solutions of K2S in DMF has not been elucidated, but it must involve the formation of polysulfides [Sn]2−, presumably by autoxidation. Very recently, the combination of S8 and NaOH followed by oxidation with H2O2 has been used to generate sulfur quantum dots with unique optical properties.194 While this process undoubtedly involves the initial formation of polysulfide dianions [Sn]2−,194a the suggestion that these are limited to the di- and octa-sulfides (n = 2, 8)194a should be re-visited in the light of the information presented in this review.
In situ Raman spectroscopy has also been pivotal in investigations of the formation of [S3]˙− in hydrothermal fluids at elevated temperatures and pressures and the role that polysulfide radical ions [Sn]˙− (n = 2, 3) play in the transport of metals such as Cu and Au. Significantly, the gold(I) species [Au(SH)S3]− has been identified by X-ray absorption spectroscopy as the most stable complex under these conditions. Complexes of the disulfide radical anion [S2]˙− with transition metals such as Cu and Ni have been structurally characterised. In addition, the first complex of the tetrasulfide radical anion [S4]˙−, in the form of a dimeric Bi(III) species, was reported recently. To date, however, no metal complexes of the trisulfide radical anion [S3]˙− are known. In view of their geological significance, such complexes, e.g. with Cu or Au, represent a worthwhile challenge for inorganic chemists.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from NSERC (Canada) is gratefully acknowledged. The authors are thankful to Mr Chris Gendy and Dr Yana Steudel for the preparation of figures and schemes. The support of Profs. A. Pfitzner and H. Hartl for providing Fig. 4 and 5, respectively, and Dr A. Kamyshny Jr for Fig. 9 is also acknowledged.References
- For the correct spelling of sulfur, see the editorial entitled “So long Sulphur”:, Nat. Chem., 2009, 1, 333 Search PubMed.
- (a) R. Steudel and B. Eckert, Top. Curr. Chem., 2003, 230, 1–79 CrossRef CAS; (b) I. Krossing, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova and W.-W. du Mont, RSC Publishing, 2nd edn, 2013, vol. 1, ch. 7.1, pp. 349–381 Search PubMed.
- H. S. Low and R. A. Beaudet, J. Am. Chem. Soc., 1976, 98, 3849–3852 CrossRef CAS.
- I. Krossing and J. Passmore, Inorg. Chem., 2004, 43, 1000–1011 CrossRef CAS PubMed.
- J. Derendorf, C. Jenne and M. Keßler, Angew. Chem., Int. Ed., 2017, 56, 8281–8284 CrossRef CAS PubMed.
- R. Steudel, Top. Curr. Chem., 2003, 231, 127–152 CrossRef CAS , and references cited therein.
- W. S. Sheldrick, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova and W.-W. du Mont, RSC Publishing, 2nd edn, 2013, vol. 1, ch. 9.2, pp. 514–545. This review includes polyselenides and polytellurides and emphasizes synthetic and structural aspects Search PubMed.
- T. Chivers and P. J. W. Elder, Chem. Soc. Rev., 2013, 42, 5996–6005 RSC , and references cited therein.
- B. Neumüller, F. Schmock, R. Kirmse, A. Voigt, A. Diefenbach, F. M. Bickelhaupt and K. Dehnicke, Angew. Chem., Int. Ed., 2000, 39, 4580–4582 CrossRef.
- R. J. Schwamm, M. Lein, M. P. Coles and C. M. Fitchett, J. Am. Chem. Soc., 2017, 139, 16490–16493 CrossRef CAS PubMed.
- A. K. Hearley, B. F. G. Johnson, J. S. McIndoe and D. G. Tuck, Inorg. Chim. Acta, 2002, 334, 105–112 CrossRef CAS.
- R. Steudel, Top. Curr. Chem., 2003, 231, 99–125 CrossRef CAS.
- R. Steudel, Chem. Rev., 2002, 102, 3905–3945 CrossRef CAS PubMed.
- Sulfur: Its Significance for Chemistry, for the Geo-, Bio- and Cosmosphere and Technology, ed. A. Müller and B. Krebs, Elsevier, Amsterdam, 1984 Search PubMed.
- For recent reviews, see (a) K. H. Wujcik, D. R. Wang, A. A. Teran, E. Nasybulin, T. A. Pascal, D. Prendergast and N. P. Balsara, in Electrochemical Engineering: From Discovery to Product, ed. R. C. Alkire, P. N. Bartlett and M. Kopev, John Wiley and Sons, 2018, vol. 10, ch. 3, pp. 41–74 Search PubMed; (b) X. Chen, T. Hou, K. A. Persson and Q. Zhang, Mater. Today, 2019, 22, 142–158 CrossRef CAS; (c) E. Zhao, K. Nie, X. Yu, Y.-S. Hu, F. Wang, J. Xiao, H. Li and X. Huang, Adv. Funct. Mater., 2018, 28, 1707543 CrossRef; (d) D. Zheng, G. Wang, D. Liu, J. Si, T. Ding, D. Qu, X. Yang and D. Qu, Adv. Mater. Technol., 2018, 3, 1700233 CrossRef; (e) G. Zhang, Z.-W. Zhang, H.-J. Peng, J.-Q. Huang and Q. Zhang, Small Methods, 2017, 1, 1700134 CrossRef; (f) R. Xu, J. Lu and K. Amine, Adv. Energy Mater., 2015, 5, 1500408 CrossRef; (g) M. Wild, L. O’Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 RSC.
- (a) T. Chivers, Nature, 1974, 252, 32 CrossRef CAS; (b) T. Chivers, in Homoatomic, Rings, Chains and Macromolecules of Main-Group Elements, ed. A. L. Rheingold, Elsevier, Amsterdam, 1977, ch. 22, pp. 499–537 Search PubMed.
- T. Takata, D. Saeki, Y. Makita, N. Yamada and N. Kihara, Inorg. Chem., 2003, 42, 3712–3714 CrossRef CAS PubMed.
- G. Mali, M. U. M. Patel, M. Mazaj and R. Dominko, Chem. – Eur. J., 2016, 22, 3355–3360 CrossRef CAS PubMed.
- P. Stüble, A. Berroth, F. Wortelkamp and C. Röhr, Z. Naturforsch., B: J. Chem. Sci., 2019, 74, 33–47 Search PubMed.
- (a) F. Dornhaus, M. Bolte, M. Wagner and H.-W. Lerner, Z. Anorg. Allg. Chem., 2007, 633, 425–428 CrossRef CAS; (b) J. Getzschmann and S. Kaskel, Z. Anorg. Allg. Chem., 2014, 640, 905–906 CrossRef CAS.
- D. Friedrich, F. Pielnhofer, M. Schlosser, R. Weihrich and A. Pfitzner, Chem. – Eur. J., 2015, 21, 1811–1817 CrossRef CAS PubMed , and references cited therein.
- Q. Zou, Z. Liang, G.-Y. Du, E. Y. Li and Y.-C. Lu, J. Am. Chem. Soc., 2018, 140, 10740–10745 CrossRef CAS PubMed.
- R. D. Rauh, F. S. Shuker, J. M. Marston and S. B. Brummer, J. Inorg. Nucl. Chem., 1977, 39, 1761–1766 CrossRef CAS.
- N. A. Cañas, D. N. Fronczek, N. Wagner, A. Latz and K. A. Friedrich, J. Phys. Chem. C, 2014, 118, 12106–12114 CrossRef.
- N. Saqib, C. J. Silva, C. M. Maupin and J. M. Porter, Appl. Spectrosc., 2017, 71, 1593–1599 CrossRef CAS PubMed.
- (a) S. Dev, E. Ramli, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 1991, 30, 2514–2519 CrossRef CAS; (b) A. Schliephake, H. Fallius, H. Buchkremer-Hermanns and P. Böttcher, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 21–24 CAS.
- T. Chivers, F. Edelmann, J. F. Richardson and K. J. Schmidt, Can. J. Chem., 1986, 64, 1509–1513 CrossRef CAS.
- G. Bieker, J. Wellmann, M. Kolek, K. Jalkanen, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2017, 19, 11152–11162 RSC.
- M. D. Hartle, D. J. Meininger, L. N. Zakharov, Z. J. Tonzetich and M. D. Pluth, Dalton Trans., 2015, 44, 19782–19785 RSC.
- (a) L. H. Finger, F. Wohde, E. I. Grigoryev, A.-K. Hansmann, R. Berger, B. Roling and J. Sundermeyer, Chem. Commun., 2015, 51, 16169–16172 RSC; (b) L. H. Finger and J. Sundermeyer, Chem. – Eur. J., 2016, 22, 4218–4230 CrossRef CAS PubMed; (c) L. H. Finger, B. Scheibe and J. Sundermeyer, Inorg. Chem., 2015, 54, 9568–9575 CrossRef CAS PubMed.
- T. S. Bailey, H. A. Henthorn and M. D. Pluth, Inorg. Chem., 2016, 55, 12618–12625 CrossRef CAS PubMed.
- (a) H. S. Lim, N. H. Heo and K. Seff, J. Phys. Chem. C, 2018, 122, 28133–28141 CrossRef CAS; (b) the anion in the black solid formed upon melting Cs2S6 has been tentatively identified by vibrational spectra as [S2S4]2−, a branched isomer of the [S6]2− chain, cf. dithionite [S2O4]2−, see ref. 6.
- H. Ohtsu, W. Choi, N. Islam, Y. Matsushita and M. Kawano, J. Am. Chem. Soc., 2013, 135, 11449–11452 CrossRef CAS PubMed.
- T. Chivers and I. Drummond, Inorg. Chem., 1972, 11, 2525–2527 CrossRef CAS.
- A. Radini, M. Tromp, A. Beach, E. Tong, C. Speller, M. McCormick, J. Dudgeon, M. J. Collins, F. Rühli, R. Kröger and C. Warinner, Sci. Adv., 2019, 5, eaau7126 CrossRef CAS PubMed.
- R. Steudel and F. Schuster, Z. Naturforsch., A: Phys. Sci., 1977, 32, 1313–1319 Search PubMed.
- (a) H. Ziemann and W. Bues, Z. Anorg. Allg. Chem., 1979, 455, 69–87 CrossRef CAS; (b) W. Bues and H. Ziemann, Z. Anorg. Allg. Chem., 1979, 456, 54–60 CrossRef CAS.
- W. E. Kleinjan, A. de Keizer and A. J. H. Janssen, Water Res., 2005, 39, 4093–4100 CrossRef CAS PubMed.
- S. A. Khan, R. W. Hughes and P. A. Reynolds, Vib. Spectrosc., 2011, 56, 241–244 CrossRef CAS.
- R. Tegman, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1463–1469 CrossRef CAS.
- K. Tatsumi, Y. Inoue, A. Nakamura, R. E. Cramer, W. VanDoorne and J. W. Gilje, Angew. Chem., Int. Ed. Engl., 1990, 29, 422–424 CrossRef.
- K. Tatsumi, H. Kawaguchi, K. Inoue, K. Tani and R. E. Cramer, Inorg. Chem., 1993, 32, 4317–4323 CrossRef CAS.
- For a review, see N. Takeda, N. Tokitoh and R. Okazaki, Top. Curr. Chem., 2003, 231, 153–202 CrossRef CAS.
- R. Steudel and Y. Steudel, Chem. – Eur. J., 2013, 19, 3162–3176 CrossRef CAS PubMed and references cited therein.
- V. Berghof, T. Sommerfeld and L. S. Cederbaum, J. Phys. Chem. A, 1998, 102, 5100–5105 CrossRef CAS.
- V. Bogdándi, T. Ida, T. R. Sutton, C. Bianco, T. Ditrói, G. Koster, H. A. Henthorn, M. Minnion, J. P. Toscano, A. van der Vliet, M. D. Pluth, M. Feelisch, J. M. Fukuto, T. Akaike and P. Nagy, Br. J. Pharmacol., 2019, 176, 646–670 CrossRef PubMed.
- J. Gun, A. D. Modestov, A. Kamshny Jr., D. Ryzkov, V. Gitis, A. Goifman, O. Lev, V. Hultsch, T. Grischek and E. Worch, Microchim. Acta, 2004, 146, 229–237 CrossRef CAS.
- M. Cuisinire, C. Hart, M. Balasubramanian, A. Garsuch and L. F. Nazar, Adv. Energy Mater., 2015, 5, 140181 Search PubMed.
- K. H. Wujcik, D. R. Wang, A. Raghunathan, M. Drake, T. A. Pascal, D. Prendergast and N. P. Balsara, J. Phys. Chem. C, 2016, 120, 18403–18410 CrossRef CAS.
- D. P. Shoemaker, D. Y. Chung, J. F. Mitchell, T. H. Bray, L. Soderholm, P. J. Chupas and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 9456–9463 CrossRef CAS PubMed.
- E. Levillain, P. Leghié, N. Gobeltz and J. P. Lelieur, New J. Chem., 1997, 21, 335–341 CAS.
- B. Ledé, A. Demortier, N. Gobeltz-Hautecoeur, J.-P. Lelieur, E. Picqenard and C. Duhayon, J. Raman Spectrosc., 2007, 38, 1461–1468 CrossRef.
- N. Jacquemet, D. Guillaume, A. Zwick and G. S. Pokrovski, Am. Mineral., 2014, 99, 1109–1118 CrossRef.
- J. Hannauer, J. Scheers, J. Fullenwarth, B. Fraisse, L. Stievano and P. Johansson, ChemPhysChem, 2015, 16, 2755–2759 CrossRef CAS PubMed.
- Q. Gao, Y. Xiu, G.-D. Li and J.-S. Chen, J. Mater. Chem., 2010, 20, 3307–3312 RSC.
- G. S. Pokrovski and L. S. Dubrovinsky, Science, 2011, 331, 1052–1054 CrossRef CAS PubMed.
- R. Linguerri, N. Komiha, J. Fabian and P. Rosmus, Z. Phys. Chem., 2008, 222, 163–176 CrossRef CAS.
- N. Gobeltz-Hautecoeur, A. Demortier, B. Lede, J. P. Lelieur and C. Duhayon, Inorg. Chem., 2002, 41, 2848–2854 CrossRef CAS PubMed.
- H.-L. Wu, L. A. Huff and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2015, 7, 1709–1719 CrossRef CAS PubMed.
- J. Fabian, N. Komiha, R. Linguerri and P. Rosmus, J. Mol. Struct. THEOCHEM, 2006, 801, 63–69 CrossRef CAS , and references cited therein.
- M. W. Wong, Top. Curr. Chem., 2003, 231, 1–29 CrossRef CAS.
- M. Hagen, P. Schiffels, M. Hammer, S. Dörfler, J. Tübke, M. J. Hoffmann, H. Althues and S. Kaskel, J. Electrochem. Soc., 2013, 160, A1205–A1214 CrossRef CAS.
- B. Eckert and R. Steudel, Top. Curr. Chem., 2003, 231, 31–98 CrossRef CAS.
- W. Tan, C. Wang and X. Jiang, Org. Chem. Front., 2018, 5, 2390–2394 RSC , and references cited therein.
- Q. Zou and Y.-C. Lu, J. Phys. Chem. Lett., 2016, 7, 1518–1525 CrossRef CAS PubMed.
- (a) M. U. M. Patel and R. Dominko, ChemSusChem, 2014, 7, 2167–2175 CrossRef CAS PubMed; (b) M. U. M. Patel, R. Demir-Cakan, M. Morcrette, J.-M. Tarascon, M. Gaberscek and R. Dominko, ChemSusChem., 2013, 6, 1177–1181 CrossRef CAS PubMed.
- C. Barchasz, F. Molton, C. Duboc, J.-C. Lepretre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84, 3973–3980 CrossRef CAS PubMed.
- Y.-C. Lu, Q. He and H. A. Gasteiger, J. Phys. Chem. C, 2014, 118, 5733–5741 CrossRef CAS , and references cited therein.
- N. S. A. Manan, L. Aldous, Y. Alias, P. Murray, L. J. Yellowlees, M. C. Lagunas and C. Hardacre, J. Phys. Chem. B, 2011, 115, 13873–13879 CrossRef CAS PubMed , and references cited therein.
- E. Boros, M. J. Earle, M. A. Gilea, A. Metlen, A.-V. Mudring, F. Rieger, A. J. Robertson, K. R. Seddon, A. A. Tomaszowska, L. Trusov and J. S. Vyle, Chem. Commun., 2010, 46, 716–718 RSC.
- M. M. Cortese-Krott, G. G. C. Kuhnle, A. Dyson, B. O. Fernandez, M. Grman, J. F. DuMond, M. P. Barrow, G. McLeod, H. Nakagawa, K. Ondrias, P. Nagy, S. B. King, J. E. Saavedra, L. K. Keefer, M. Singer, M. Kelm, A. R. Butler and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4651–E4660 CrossRef CAS PubMed.
- R. Steudel, in Environmental Technologies to Treat Sulfur Pollution, ed. P. N. L. Lens and L. H. Pol, IWA, London, 2000, pp. 1–31 Search PubMed.
- A. Kamyshny Jr., A. Goifman, J. Gun, D. Rizkov and O. Lev, Environ. Sci. Technol., 2004, 38, 6633–6644 CrossRef.
- A. Kamyshny Jr., J. Gun, D. Rizkov, T. Voitsekovski and O. Lev, Environ. Sci. Technol., 2007, 41, 2395–2400 CrossRef.
- For a comprehensive recent review of the applications of elemental sulfur in synthetic organic chemistry, see T. B. Nguyen, Adv. Synth. Catal., 2017, 359, 1066–1130 CrossRef CAS.
- J. Rethmeier, A. Rabenstein, M. Langer and U. Fischer, J. Chromatogr. A, 1997, 760, 295–302 CrossRef CAS.
- A. Kawase, S. Shirai, Y. Yamoto, R. Arakawa and T. Takata, Phys. Chem. Chem. Phys., 2014, 16, 9344–9350 RSC.
- I. Filipponen, A. Guerra, A. Hai, L. A. Lucia and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2006, 45, 7388–7392 CrossRef.
- (a) A. Kamyshny Jr., A. Goifman, D. Rizkov and O. Lev, Aquat. Geochem., 2003, 9, 291–304 CrossRef; (b) K. Avetisyan, T. Buchshtav and A. Kamyshny Jr., Geochim. Cosmochim. Acta, 2019, 247, 96–105 CrossRef CAS.
- D. Rizkov, O. Lev, J. Gun, B. Anisimov and I. Kuselman, Accredit. Qual. Assur., 2004, 9, 399–403 CrossRef CAS.
- A. Kamyshny Jr., I. Ekeltchik, J. Gun and O. Lev, Anal. Chem., 2006, 78, 2631–2639 CrossRef PubMed.
- D. Zheng, X. Zhang, J. Wang, D. Qu and X. Yang, J. Power Sources, 2016, 301, 312–316 CrossRef CAS.
- (a) D. Arieli, D. E. W. Vaughan and D. Goldfarb, J. Am. Chem. Soc., 2004, 126, 5776–5788 CrossRef CAS PubMed , and references cited therein; (b) J. Goslar, S. Lijewski, K. Stanislaw, A. Jankowska and S. Kowalak, J. Chem. Phys., 2009, 130, 204504 CrossRef PubMed; (c) K. Raulin, N. Gobletz, H. Vezin, N. Touati, B. Ledé and A. Moissette, Phys. Chem. Chem. Phys., 2011, 13, 9253–9259 RSC; (d) P. Rejmak, J. Phys. Chem. C, 2018, 122, 29338–29349 CrossRef CAS.
- S. A. Basnayake, K. Tan, M. Leonard, Y. Chabal and K. J. Balkus, Microporous Mesoporous Mater., 2016, 219, 172–177 CrossRef CAS.
- M. Cuisinier, P.-E. Cabelguen, S. Evers, G. He, M. Kolbeck, A. Garsuch, T. Bolin, M. Balasubramanian and L. F. Nazar, J. Phys. Chem. Lett., 2013, 4, 3227–3232 CrossRef CAS.
- K. A. See, M. Leskes, J. M. Griffin, S. Britto, P. D. Matthews, A. Emly, A. van der Ven, D. S. Wright, A. J. Morris, C. P. Grey and R. Seshadri, J. Am. Chem. Soc., 2014, 136, 16368–16377 CrossRef CAS PubMed.
- J. Xiao, J. Z. Hu, H. Chen, M. Vijayakumar, J. Zheng, H. Pan, E. D. Walter, M. Hu, X. Deng and J. Feng, Nano Lett., 2015, 15, 3309–3316 CrossRef CAS PubMed.
- H. Wang, N. Sa, M. He, X. Liang, L. F. Nazar, M. Balusubramanian, K. G. Gallagher and B. Key, J. Phys. Chem. C, 2017, 121, 6011–6017 CrossRef CAS.
- M. Fantauzzi, B. Elsener, D. Atzei, A. Rigoldi and A. Rossi, RSC Adv., 2015, 5, 75953–75963 RSC.
- A. Prange, R. Chauvistré, H. Modrow, J. Hormes, H. G. Trüper and C. Dahl, Microbiology, 2002, 148, 267–276 CrossRef CAS PubMed.
- (a) M. E. Fleet, X. Liu, S. L. Harmer and H. W. Nesbitt, Can. Mineral., 2005, 43, 1589–1603 CrossRef CAS; (b) M. E. Fleet and X. Liu, Spectrochim. Acta, Part B, 2010, 65, 75–79 CrossRef.
- (a) M. A. Lowe, J. Gao and H. D. Abruña, RSC Adv., 2014, 4, 18347–18353 RSC; (b) J. Gao, M. A. Lowe, Y. Kiya and H. D. Abruña, J. Phys. Chem. C, 2011, 115, 25132–25137 CrossRef CAS.
- M. Vijayakumar, N. Govind, E. Walter, S. D. Burton, A. Shukla, A. Devaraj, J. Xiao, J. Liu, C. Wang and A. Karim, Phys. Chem. Chem. Phys., 2014, 16, 10923–10932 RSC.
- T. A. Pascal, K. H. Wujcik, J. J. Velasco-Velez, C.-H. Wu, A. A. Teran, M. Kapilashrami, J. Cabana, J. Guo, M. Salmeron and N. P. Balsara, J. Phys. Chem. Lett., 2014, 5, 1547–1551 CrossRef CAS PubMed.
- L. Nazar, M. Cuisinier, P.-E. Cabelguen, B. Adams, A. Garsuch and M. Balasubramanian, Energy Environ. Sci., 2014, 7, 2697–2705 RSC.
- M. U. M. Patel, I. Arčon, G. Aquilanti, L. Stievano, G. Mali and R. Dominko, ChemPhysChem, 2014, 15, 894–904 CrossRef CAS PubMed.
- K. H. Wujcik, J. J. Velasco-Velez, C.-H. Wu, T. A. Pascal, A. A. Teran, M. A. Marcus, J. Cabana, J. Guo, D. Prendergast and M. Salmeron, J. Electrochem. Soc., 2014, 161, A1100–A1106 CrossRef CAS.
- T. A. Pascal, C. D. Pemmaraju and D. Prendergast, Phys. Chem. Chem. Phys., 2015, 17, 7743–7753 RSC.
- M. Cuisinier, C. Hart, B. Adams, M. Balasubramanian, A. Garsuch and L. F. Nazar, Adv. Energy Mater., 2015, 5, 1401801 CrossRef.
- Y. Gorlin, A. Siebel, M. Piana, T. Huthwelker, H. Jha, G. Monsch, F. Kraus, H. A. Gasteiger and M. Tromp, J. Electrochem. Soc., 2015, 162, A1146–A1155 CrossRef CAS.
- (a) A. A. Gambardella, C. M. Schmidt Patterson, S. M. Webb and M. S. Walton, Microchem. J., 2016, 125, 299–307 CrossRef CAS; (b) M. Ganio, E. S. Pouyet, S. M. Webb, C. M. Schmidt Patterson and M. S. Walton, Pure Appl. Chem., 2018, 90, 463–475 CAS.
- M. Chaouche, X. X. Gao, M. Cyr, M. Cotte and L. Frouin, J. Am. Ceram. Soc., 2017, 100, 1707–1716 CrossRef CAS.
- E. Cato, A. Rossi, N. C. Scherrer and E. S. B. Ferreira, J. Cult. Herit., 2018, 29, 30–35 CrossRef.
- J. B. Kim, C. Hock, T. I. Tacovitch and D. M. Neumark, J. Phys. Chem. A, 2013, 117, 8126–8131 CrossRef CAS PubMed.
- J. A. Tossell, Geochim. Cosmochim. Acta, 2012, 95, 79–92 CrossRef CAS.
- M. Vijayakumar, N. Govind, E. Walter, S. D. Burton, A. Shukla, A. Devaraj, J. Xiao, J. Liu, C. Wang, A. Karim and S. Thevuthasan, Phys. Chem. Chem. Phys., 2014, 16, 10923–10932 RSC.
- W. Koch, J. Natterer and C. Heinemann, J. Chem. Phys., 1995, 102, 6159–6167 CrossRef CAS.
- H. Sormova, R. Linguerri, P. Rosmus, J. Fabian and N. Komiha, Collect. Czech. Chem. Commun., 2007, 72, 83–99 CrossRef CAS.
- Y. Jin, G. Maroulis, X. Kuang, L. Ding, C. Lu, J. Wang, J. Lv, C. Zhang and M. Ju, Phys. Chem. Chem. Phys., 2015, 17, 13590–13597 RSC.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, Beilstein J. Nanotechnol., 2015, 6, 1016–1055 CrossRef CAS PubMed.
- Y.-X. Wang, B. Zhang, W. Lai, Y. Xu, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef.
- D. Kumar, S. K. Rajouria, S. B. Kuhar and D. K. Kanchen, Solid State Ionics, 2017, 312, 8–16 CrossRef CAS.
- Z. Zhao-Karger and M. Fichtner, MRS Commun., 2017, 7, 770–784 CrossRef CAS.
- M. M. Gross and A. Manthiram, ACS Appl. Mater. Interfaces, 2018, 10, 10612–10617 CrossRef CAS PubMed.
- (a) P. Leung, X. Li, C. P. de León, L. Berlouis, C. T. J. Lowa and F. C. Walshab, J. Power Sources, 2009, 189, 1220–1230 CrossRef; (b) F. Pan and Q. Wang, Molecules, 2015, 20, 20499–20517 CrossRef CAS PubMed.
- Z. Wen, Y. Hu, X. Wu, J. Han and Z. Gu, Adv. Funct. Mater., 2013, 23, 1005–1018 CrossRef CAS.
- R. Steudel, Top. Curr. Chem., 2003, 230, 81–116 CrossRef CAS.
- (a) J. Sangster and A. D. Pelton, J. Phase Equilib., 1997, 18, 89–96 CrossRef CAS; (b) X. Yu and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 1943–1947 CrossRef CAS PubMed; (c) Y. Wang, Y. Hao, L.-C. Xu, Z. Yang, M.-Y. Di, R. Liu and X. Li, J. Phys. Chem. C, 2019, 123, 3988–3995 CrossRef CAS.
- (a) C. W. Park, J. H. Ahn, H. S. Ryu, K. W. Kim and H. J. Ahn, Electrochem. Solid-State Lett., 2006, 9, A123–A125 CrossRef CAS; (b) H. Ryu, T. Kim, K. Kim, J. H. Ahn, T. Nam, G. Wang and H. J. Ahn, J. Power Sources, 2011, 196, 5186–5190 CrossRef CAS.
- R. S. Assary, L. A. Curtiss and J. S. Moore, J. Phys. Chem. C, 2014, 118, 11545–11548 CrossRef CAS . The cyclic geometry of neutral S3 (D3h) obtained in this work is in conflict with the experimental acyclic structure (C2v), and the geometry of [S8]˙− is determined to be a chain while other authors found a ring with one long bond (Section 4).
- (a) D. Zhou, Y. Chen, B. Li, H. Fan, F. Cheng, D. Shanmukaraj, T. Rojo, M. Armand and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 10168–10172 CrossRef CAS PubMed; (b) P. Sang, Y. Si and Y. Fu, Chem. Commun., 2019, 55, 4857–4860 RSC; (c) H. Zhou, F. Yu, M. Wei, Y. Su, Y. Ma, D. Wang and Q. Shen, Chem. Commun., 2019, 55, 3729–3732 RSC.
- N. Tanibata, M. Deguchi, A. Hayasi and M. Tatsumisago, Chem. Mater., 2017, 29, 5232–5238 CrossRef CAS.
- S. Choudhury, S. Wei1, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Tu, J. H. Shin, P. Nath, A. Agrawal, L. F. Kourkoutis, T. A. Arias and L. A. Archer, Nat. Commun., 2017, 8, 898 CrossRef PubMed.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- A. Manthiram, S.-H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- W. Kang, N. Deng, J. Ju, Q. Li, D. Wu, X. Ma, L. Li, M. Naebe and B. Cheng, Nanoscale, 2016, 8, 16541–16588 RSC.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- H.-J. Peng, J.-Q. Huang and Q. Zhang, Chem. Soc. Rev., 2017, 46, 5237–5288 RSC.
- (a) Q. He, Y. Gorlin, M. U. M. Patel, H. A. Gasteiger and Y.-C. Lu, J. Electrochem. Soc., 2018, 165, A4027–A4033 CrossRef CAS; (b) A. Gupta, A. Bhargav and A. Manthiram, Adv. Energy Mater., 2019, 9, 1803096 CrossRef; (c) Y. Jung, S. Kim, B.-S. Kim, D.-H. Han, S.-M. Park and J. Kwak, J. Electrochem. Sci. Eng., 2008, 3, 566–577 CAS.
- Z. Wang, J. Liu, L. Sun, Y. Zhang, Q. Fu, H. Xie and H. Sun, Chem. – Eur. J., 2018, 24, 14154–14161 CrossRef CAS PubMed.
- C. Ye, Y. Jiao, H. Jin, A. D. Slattery, K. Davey, H. Wang and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 16703 CrossRef CAS PubMed.
- M. Zhen, J. Wang, X. Wang and C. Wang, Chem. – Eur. J., 2018, 24, 5860–5867 CrossRef CAS PubMed , and references cited therein.
- M. Armand, Angew. Chem., Int. Ed., 2018, 57, 15002–15027 CrossRef PubMed.
- S. S. Zhang, J. Power Sources, 2016, 322, 99–105 CrossRef CAS.
- D. Bresser, S. Passerini and B. Scrosati, Chem. Commun., 2013, 49, 10545–10562 RSC.
- D. Zheng, X.-Q. Yang and D. Qu, ChemSusChem, 2016, 9, 2348–2350 CrossRef CAS PubMed.
- W. Lu, Z. Yuan, Y. Zhao, H. Zhang, H. Zhang and X. Li, Chem. Soc. Rev., 2017, 46, 2199–2236 RSC.
- J.-J. Chen, R.-M. Yuan, J.-M. Feng, Q. Zhang, J.-X. Huang, G. Fu, M. S. Zheng, B. Ren and Q.-F. Dong, Chem. Mater., 2015, 27, 2048–2055 CrossRef CAS.
- R. Dominko, A. Vizintin, G. Aquilanti, L. Stievano, M. J. Helen, A. R. Munnangi, M. Fichtner and I. Arcon, J. Electrochem. Soc., 2018, 165, A5014–A5019 CrossRef CAS.
- E. P. Kamphaus and P. B. Balbuena, J. Phys. Chem. C, 2017, 121, 21105–21117 CrossRef CAS.
- (a) A. Shyamsunder, W. Beichel, P. Klose, Q. Pang, H. Scherer, A. Hoffmann, G. K. Murphy, I. Krossing and L. F. Nazar, Angew. Chem., Int. Ed., 2017, 56, 6192–6197 CrossRef CAS PubMed; (b) Q. Cheng, W. Xu, S. Qin, S. Das, T. Jin, A. Li, A. C. Li, B. Qie, P. Yao, H. Zhai, C. Shi, X. Yong and Y. Yang, Angew. Chem., Int. Ed., 2019, 58, 5557–5561 CrossRef CAS PubMed.
- R. Xu, S. Zhang, X. Wang, Y. Xia, X. Xia, J. Wu, C. Gu and J. Tu, Chem. – Eur. J., 2018, 24, 6007–6018 CrossRef CAS PubMed.
- (a) W. Said-Ahmad, A. Amrani and Z. Aizenshtat, Org. Geochem., 2013, 59, 82–86 CrossRef CAS; (b) K. Ding and P. Adam, Org. Geochem., 2015, 8, 133–136 CrossRef.
- G. Zhang, H. Yi, C. Bian, C. Liu and A. Lei, Org. Lett., 2014, 16, 6156–6159 CrossRef CAS PubMed.
- M. Wang, Q. Fan and X. Jiang, Org. Lett., 2016, 18, 5756–5759 CrossRef CAS PubMed.
- H. Huang, Z. Qu, X. Ji and G.-J. Deng, Org. Chem. Front., 2019, 1146–1150 RSC , and references cited therein.
- (a) Z.-Y. Gu, J.-J. Cao, S.-Y. Wang and S.-J. Ji, Chem. Sci., 2016, 7, 4067–4072 RSC; (b) J.-H. Li, Q. Huang, S.-Y. Wang and S.-J. Ji, Org. Lett., 2018, 20, 4704–4708 CrossRef CAS.
- J. W. Thomson, K. Nagashima, P. M. Macdonald and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5036–5041 CrossRef CAS PubMed.
- A. J. Findlay, FEMS Microbiol. Lett., 2016, 363, fnw103 CrossRef PubMed.
- F. Bouillaud and F. Blachier, Antioxid. Redox Signaling, 2011, 15, 379–391 CrossRef CAS PubMed.
- C. Szabo, C. Ransy, K. Modis, M. Andriamihaja, B. Murghes, C. Coletta, G. Olah, K. Vanagi and F. Bouillaud, Br. J. Pharmacol., 2014, 171, 2099–2122 CrossRef CAS PubMed.
- T. V. Mishanina, M. Libiad and R. Banerjee, Nat. Chem. Biol., 2015, 11, 457–464 CrossRef CAS PubMed.
- Z. Palinkas, P. G. Furtmuller, A. Nagy, C. Jakopitsch, K. F. Pirker, M. Magierowski, K. Jasnos, J. L. Wallace, C. Obinger and P. Nagy, Br. J. Pharmacol., 2015, 172, 1516–1532 CrossRef CAS PubMed.
- T. Bostelaar, V. Vitvitsky, J. Kumutima, B. E. Lewis, P. K. Yadav, T. C. Brunold, M. Filipovic, N. Lehnert, T. L. Stemmler and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 8476–8488 CrossRef CAS PubMed.
- K. R. Olson, Y. Gao, E. R. DeLeon, M. Arif, F. Arif, N. Arora and K. D. Straub, Redox Biol., 2017, 12, 325–339 CrossRef CAS PubMed.
- M. M. Cortese-Krott, G. G. C. Kuhnle, A. Dyson, B. O. Fernandez, M. Grman, J. F. DuMond, M. P. Barrow, G. McLeod, H. Nakagawa, K. Ondrias, P. Nagy, S. B. King, J. E. Saavedra, L. K. Keefer, M. Singer, M. Kelm, A. R. Butler and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4651–E4660 CrossRef CAS PubMed.
- S. Carballal, M. Trujillo, E. Cuevasanta, S. Bartesaghi, M. N. Möller, L. K. Folkes, M. A. Garcia-Bereguiain, C. Gutiérrez-Merino, P. Wardman, A. Denicola, R. Radi and B. Alvarez, Free Radical Biol. Med., 2011, 50, 196–205 CrossRef CAS.
- R. Steudel, Ind. Eng. Chem. Res., 1996, 35, 1417–1423 CrossRef CAS.
- A. Prange, H. Engelhardt, H.-G. Trüper and C. Dahl, Arch. Microbiol., 2004, 182, 165–174 CrossRef CAS PubMed.
- B. Franz, T. Gehrke, H. Lichtenberg, J. Hormes, C. Dahl and A. Prange, Microbiology, 2009, 155, 2766–2774 CrossRef CAS PubMed.
- C. L. Marnocha, A. T. Levy, D. H. Powell, T. E. Hanson and C. S. Chan, Microbiology, 2016, 162, 1125–1134 CrossRef CAS PubMed.
- J. S. Berg, A. Schwedt, A.-C. Kreutzmann, M. M. M. Kuypers and J. Milucka, Appl. Environ. Microbiol., 2014, 80, 629–636 CrossRef PubMed.
- C. Dahl, in Sulfur Metabolism in Phototrophic Bacteria, ed. P. C. Hallenbeck, Modern Topics in Phototrophic Prokaryotes, Springer, Berlin, 2017, pp. 27–66 Search PubMed.
- V. Vitvitsky, P. K. Yadav, S. An, J. Seravalli, U.-S. Cho and R. Banerjee, J. Biol. Chem., 2017, 292, 5584–5592 CrossRef CAS PubMed.
- S. A. Everett, C. Schöneich, J. H. Stewart and K.-D. Asmus, J. Phys. Chem., 1992, 96, 306–314 CrossRef CAS.
- A. Kamyshny Jr., C. G. Borkenstein and T. G. Ferdelman, Geostand. Geoanal. Res., 2009, 33, 415–435 CrossRef , and references cited therein.
- A. Kamyshny Jr., M. Zilberbrand, I. Ekeltchik, T. Voitsekovski, J. Gun and O. Lev, Aquat. Geochem., 2008, 14, 171–192 CrossRef.
- G. W. Luther, B. Glazer, S. F. Ma, R. Trouborst, B. R. Shultz, G. Drischel and C. Kraiya, Aquat. Geochem., 2003, 9, 87–110 CrossRef CAS.
- A. Amrani and Z. Aizenshtat, Org. Geochem., 2004, 35, 909–921 CrossRef CAS.
- A. Amrani, J. W. Turner, Q. Ma, Y. Tang and P. G. Hatcher, Geochim. Cosmochim. Acta, 2007, 71, 4141–4160 CrossRef CAS.
- B. Ginzburg, L. Dor, I. Chalifa, O. Hadas and O. Lev, Environ. Sci. Technol., 1999, 33, 571–579 CrossRef CAS.
- A. Heitz, R. I. Kagi and R. Alexander, Water Sci. Technol., 2000, 41, 271–278 CrossRef CAS.
- P. D. Franzmann, A. Heitz, L. R. Zappia, J. E. Wajon and K. Xanthis, Water Res., 2001, 35, 1730–1738 CrossRef CAS PubMed.
- C. E. Manning, Science, 2011, 331, 1018 CrossRef PubMed.
- G. S. Pokrovski, M. A. Kokh, D. Guillaume, A. Y. Borisova, P. Gisquet, J.-L. Hazemann, E. Lahera, W. Del Net, O. Proux, D. Testemale, V. Haigis, R. Jonchiere, A. P. Seitsonen, G. Ferlat, R. Vuilleumier, A. M. Saitta, M.-C. Boiron and J. Dubessy, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13484–13489 CrossRef CAS PubMed.
- L. Truche, E. F. Bazarkina, G. Barré, E. Thomassot, G. Berger, J. Dubessy and P. Robert, Earth Planet. Sci. Lett., 2014, 396, 190–200 CrossRef CAS.
- G. Barré, L. Truche, E. F. Bazarkina, R. Michels and J. Dubessy, Chem. Geol., 2017, 462, 1–14 CrossRef.
- G. Pokrovski and J. Dubessy, Earth Planet. Sci. Lett., 2015, 411, 298–309 CrossRef CAS.
- (a) A. Terzis and R. Rivest, Inorg. Chem., 1973, 12, 2132 CrossRef CAS; (b) R. C. Elder and M. Trkula, Inorg. Chem., 1977, 48, 4551–4554 Search PubMed.
- H. Fabian and F. Fischer, J. Raman Spectrosc., 1989, 20, 639–643 CrossRef CAS.
- J. T. York, E. C. Brown and W. B. Tolman, Angew. Chem., Int. Ed., 2005, 44, 7745–7777 CrossRef CAS PubMed.
- S. Yao, C. Milsmann, E. Bill, K. Wieghardt and M. Driess, J. Am. Chem. Soc., 2008, 130, 13536–13537 CrossRef CAS PubMed.
- Y. Mei, D. M. Sherman, W. Liu and J. Brugger, Chem. Geol., 2013, 347, 34–42 CrossRef CAS.
- J. M. Stauber, P. Müller, Y. Dai, G. Wu, D. G. Nocera and C. C. Cummins, Chem. Sci., 2016, 7, 6928–6933 RSC (Note 15 in this paper defines the abbreviation for the cryptand ligand[mBCDA-5t]6–).
- (a) K. Isobe, Y. Ozawa, A. Vazquez de Miguel, T.-W. Zhu, K.-M. Zhao, T. Nishioka, T. Ogura and T. Kitigawa, Angew. Chem., Int. Ed. Engl., 1994, 33, 1882–1883 CrossRef; (b) A. M. Mebel, K. Morokuma and K. Isobe, Inorg. Chem., 1995, 34, 1208–1211 CrossRef CAS; (c) T. Nishioka, H. Kitayama, B. K. Breedlove, K. Shiomi, I. Kinoshita and K. Isobe, Inorg. Chem., 2004, 43, 5688–5697 CrossRef CAS PubMed.
- A. Poduska, R. Hoffmann, A. Ienco and C. Mealli, Chem. – Asian J., 2009, 4, 302–313 CrossRef CAS PubMed , and references cited therein.
- A. Müller, F.-W. Baumann, H. Bögge, M. Römer, E. Krickemeyer and K. Schmitz, Angew. Chem., Int. Ed. Engl., 1984, 23, 632–633 CrossRef.
- (a) M. J. Loeffler and R. L. Hudson, Icarus, 2018, 302, 418–425 CrossRef CAS; (b) M. J. Loeffler, R. L. Hudson, N. J. Chanover and A. A. Simon, Icarus, 2016, 271, 265–268 CrossRef CAS; (c) M. J. Loeffler, R. L. Hudson, N. J. Chanover and A. A. Simon, Icarus, 2015, 258, 181–191 CrossRef CAS.
- T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2017, 46, 5182–5192 RSC , and references cited therein.
- T. Chivers and C. Lau, Inorg. Chem., 1982, 21, 453–455 CrossRef CAS.
- (a) H. Wang, Z. Wang, Y. Xiong, S. V. Kershaw, T. Li, Y. Wang, Y. Zhai and A. L. Rogach, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201902344; (b) L. Shen, H. Wang, S. Liu, Z. Bai, S. Zhang, X. Zhang and C. Zhang, J. Am. Chem. Soc., 2018, 140, 7878–7884 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |